

Abstract
Intervertebral disc degeneration is a ubiquitous condition closely linked to chronic low-back pain. The health of the avascular nucleus pulposus (NP) plays a crucial role in the development of this pathology. We tested the hypothesis that a network comprising HIF-1α, Carbonic anhydrase (CA) 9 and 12 isoforms, and sodium-coupled bicarbonate cotransporters (NBCs) buffer intracellular pH through coordinated bicarbonate recycling. Contrary to the current understanding of NP cell metabolism, analysis of metabolic-flux data from Seahorse XF analyzer showed that CO2 hydration contributes a significant source of extracellular proton production in NP cells, with a smaller input from glycolysis. Since enzymatic hydration of CO2 is catalyzed by plasma membrane-associated CAs we measured their expression and function in NP tissue. NP cells robustly expressed isoforms CA9/12 which were hypoxia-inducible. In addition to increased mRNA stability under hypoxia, we observed binding of HIF-1α to select hypoxia-responsive elements on CA9/12 promoters using genomic chromatin immunoprecipitation. Importantly, in vitro loss of function studies and analysis of discs from NP-specific HIF-1α null mice confirmed the dependency of CA9/12 expression on HIF-1α. As expected, inhibition of CA activity decreased extracellular acidification rate independent of changes in HIF activity or lactate/H+ efflux. Surprisingly, CA inhibition resulted in a concomitant decrease in intracellular pH which was mirrored by inhibition of sodium-bicarbonate importers. These results suggested that extracellular bicarbonate generated by CA9/12 is recycled to buffer cytosolic pH fluctuations. Importantly, long-term intracellular acidification from CA inhibition lead to compromised cell viability, suggesting that plasma-membrane proton extrusion pathways alone are not sufficient to maintain homeostatic pH in NP cells. Taken together, our studies show for the first time that bicarbonate buffering through the HIF-1α- Carbonic anhydrase axis is critical for NP cell survival in the hypoxic niche of the intervertebral disc.
Keywords: Cell/Tissue Signaling- Transcription Factors, Chondrocyte and Cartilage Biology, Intervertebral Disc, Nucleus Pulposus, Carbonic anhydrase, pH regulation, HIF-1
INTRODUCTION
Low back pain is the leading cause of years lived with disability in the United States and the second leading cause of disability adjusted life years (1, 2). LBP is caused by dysregulation and degeneration of the core structures of the spinal column: vertebrae and intervertebral disc. The disc comprises an outer fibrocartilaginous annulus fibrosus (AF), surrounding a gelatinous notochord-derived nucleus pulposus (NP), and cartilaginous endplates on the superior and inferior junctions with the vertebral bodies.
Due to a lack of tissue vasculature, cells of the NP reside in a unique hypoxic, acidic, and nutrient-limiting environment (3–6). Oxygen and nutrients diffuse from vasculature in the surrounding vertebrae, through the cartilaginous endplates and NP cell matrix, to reach the NP cells in the center of the disc. Metabolites cross the reverse path to be cleared from the NP compartment. Given that NP cells reside in a physiologically hypoxic niche and rely primarily on glycolysis for ATP generation, it is thought that these cells produce lactic acid which contributes to their acidic milieu (7–10). NP cells have a relatively low intracellular pH (measured at 6.7 in bovine cells) and studies suggest that the extracellular pH during degeneration can fall below 6.5 (11–13). Importantly, with degeneration, poor solute clearance through sclerotic endplates coupled with a drop in intracellular pH may create an environment that predisposes NP cells for considerable cellular stress. Pathologically acidic pH in NP cells has been shown to lead to matrix catabolism and decreased glucose uptake (14–16).
NP cells must therefore rely on robust pH-regulating systems to maintain the optimal pH for cellular metabolism, matrix production, and survival. It is widely accepted that pH regulation involves membrane-associated ion transporters and cytosolic as well as plasma membrane-associated carbonic anhydrases (CAs) to sense and balance levels of H+, HCO3−, and CO2. However, the intracellular pH (pHi) regulating capabilities of NP cells, including both plasma membrane H+-extrusion and HCO3− buffering pathways (including pathways of CO2 production), are largely unexplored. In regards to H+ extrusion, a study has detected the presence of Na+/H+ exchangers and H+-ATPases in NP cells that both enable H+ efflux as a means of regulating intracellular pH (11). On the other hand, the mechanisms contributing to HCO3− buffering in NP cells remain unknown.
The general model of pHi regulation via the HCO3− buffering capabilities of carbonic anhydrases is extensively studied in other cell types (17–22). CAs are a family of 16 proteins which catalyze the reversible hydration of CO to produce equal parts HCO3− (basic) and H+ (acidic) units at an exceedingly efficient rate of up to 106 s−1 at 37°C (23). However, the specific function of the various isoforms is not redundant; their functional relevance is determined by cellular localization (plasma membrane vs. cytosol vs. mitochondrial membrane) and the directionality of the enzymatic reaction (favoring production of HCO3−/H+ or CO2/H2O). In fact, contrary to the general understanding, rates of CO2 hydration by CAs are substantial enough to contribute up to 100% of total extracellular proton production, a metric often erroneously associated solely with glycolytic lactate/H+ production (24). Interestingly, our published RNA-Seq (GEO Accession #GSE86552) and microarray data (#GSE42611) along with other expression studies (GSE49560, GSE24612) show that levels of CA9, CA12 and CA3 expression are the highest in rat NP cells (25–28). These studies also show that NP cells express CA2 (cytosolic) and CA5b (mitochondrial) at lower levels while the remaining CA isoforms were undetectable.
Recently it has been found that the membrane-associated and extracellularly facing CA9 and CA12 isoforms are rapidly induced by hypoxia inducible factor-1α (HIF) under hypoxic conditions in select cell types (29–31). While these isoforms show broader expression in embryonic tissues, their expression is restricted to few adult tissues and elevated in hypoxic tumors (32, 33). In cancer cells, they are shown to be robust pHi regulators by generating HCO3− that is recycled into the cell through the coordinated actions of HCO3− transporters that includes SLC4 and SLC26 families of proteins (34, 35). The electrogenecity and directionality of HCO3− transport is varied for each isoform. However, important to this work, the Na+-coupled/HCO3− cotransporters (NBCs) are all capable of bicarbonate influx and are sensitive to inhibition by the stilbene derivative, SITS (36).
NP cells adapt to their unique hypoxic environment through the essential activities of the hypoxia-inducible factor (HIF) homologues (4) which is regulated in a unique fashion in the NP (5, 37–41). HIF-1α, in particular, regulates the expression of many genes critical to the survival of NP cells- including enzymes controlling glycolysis (10). Likewise, HIF-1 is implied to control CA12 expression in NP cells, however, in vivo evidence and mechanistic insights of this regulation are lacking (42, 43). We hypothesize that the HIF-1α- CA9/12 axis is critical for pHi regulation in NP cells. We clearly show that the inhibition of CA9 and CA12 in NP cells not only leads to decreased ECAR but also intracellular pH, implying a contribution to extracellular HCO3− generation and its cytosolic retrieval through NBCs. Importantly, long-term cytosolic acidification of NP cells due to catalytic inhibition of CA9 and CA12 results in increased incidence of cell death. Taken together, our studies show for the first time that bicarbonate buffering through the HIF-1α- Carbonic anhydrase axis is critical for NP cell survival in the hypoxic niche of the intervertebral disc.
MATERIALS AND METHODS
Isolation of NP cells, cell treatments and hypoxic culture
Rat NP cells were isolated and characterized as previously reported (4). Cells were maintained in Dulbecco’s Modification of Eagle’s Medium (DMEM) and 10% FBS supplemented with antibiotics. To investigate the effects of carbonic anhydrase inhibition, cells were treated with Acetazolamide (AZA; 100nM, 500 nM), Methazolamide (MZA; 500 nM), and U-104 (500 nM, 2 μM) for 18 hours after 24 pre-treatment in hypoxia. MZA and AZA are pan-CA inhibitors, whereas U-104 was designed to specifically inhibit CA9/12. It is selective for CA9 and CA12 (Ki = 45 nM and 4.5 nM, respectively) with very low inhibition for CA1 and CA2 (Ki = 5080 nM and 9640 nM, respectively) (44). Cells were cultured in a Hypoxia Work Station (Invivo2 300, Ruskinn, UK) with a mixture of 1% O2, 5% CO2 and 94% N2. To investigate the effect of NBC inhibition, we treated NP cells with SITS (100, 250, 500 μM) for 18 hours after 24 pre-treatment in hypoxia. To investigate the effect of HIF-1α stabilization on CA9/12 expression under normoxia, we inhibited PHD activity with Dimethyloxalylglycine (DMOG) (2mM) for 8 and 24 hours.
Seahorse XF Analyzer Respiratory Assay
The Seahorse XF24 instrument was used to measure extracellular acidification rate (ECAR) and O2 consumption rate (OCR), as reported by Csordás et al (45). Rat NP cells were seeded on 24-well XF Analyzer plate at 15,000 cells per well. Cells were incubated in hypoxia for 24 h prior to treatment with three carbonic anhydrase inhibitors for 18 h and new assay media was added (DMEM, 5mM glucose, 4 mM glutamine, pH 7.4 @ 37C). While the microplate was incubating for 1 h at 37°C, no CO2 and in normoxia, the antimycin-filled sensor cartridge was loaded into the Seahorse XF Analyzer for calibration. The assay protocol was a repeated sequence of 2-min mix, 2-min wait, and 2-min measurement. The measurement phase involves the lowering of the probe array to create a minimally oxygen-impermeable microchamber that allows amplification of O2 changes due to cellular processes. The OCR was calculated using the Akos algorithm, a standard algorithm which we determined was appropriate even for the low OCR readings that we recorded in NP cells; appropriateness was based on the approximate linearity of the pO2 vs time traces. ECAR was measured from readings of H+ concentration. After each measurement, the probe array rises, after which the solution in each well was mixed for 2 min (by gently moving the probe array up and down) to remove O2 and metabolite gradients, followed by a 2-min waiting period before the next measurement phase (i.e., lowering of the probe). Our experiments included 8 OCR and ECAR measurements to create a baseline, followed by the injection of Antimycin A (Sigma Aldrich). 3 OCR and ECAR measurements were then made. All measurements were normalized to total protein concentration using a standard BCA assay. Mitochondrial OCR was calculated by subtracting the final OCR value (pMoles O2/min) after Antimycin A treatment from the average of the 3 OCR values before Antimycin A treatment. In a separate assay, cells were treated with oligomycin, FCCP (200, 400, 800, 1000 nM), and Antimycin A to determine total electron transport chain capacity.
Real Time RT-PCR Analysis
Total RNA was extracted from NP cells using RNAeasy mini columns (Qiagen). Before elution from the column, RNA was treated with RNase-free DNase I (Qiagen). Purified, DNA-free RNA was converted to cDNA using EcoDry™ Premix (Clontech). Template cDNA and gene-specific primers were added to the SYBR Green master mixture (Applied Biosystems) and mRNA expression was quantified using the Step One Plus Real-time PCR System (Applied Biosystems). HPRT was used to normalize gene expression. Melting curves were analyzed to verify the specificity of the RT-PCR and the absence of primer dimer formation. Each sample was analyzed in duplicate and included a template- free control. All primers used were synthesized by Integrated DNA Technologies, Inc.
Actinomycin D Chase
To assess mRNA stability, cells were treated with Actinomycin D (5 μg/mL) for 0, 4, 8 and 24 hours. Total RNA was extracted from cells as described above. Relative mRNA expression was quantified and data was fit to a regression curve to calculate Car9/12 mRNA half-life (t1/2) under normoxic and hypoxic culture conditions.
Protein extraction, Immunoprecipitation, and Western Blotting
Cells were placed on ice immediately following treatment and washed with ice-cold PBS. Wash buffer and lysis buffer contained 1x protease inhibitor cocktail (Thermo Scientific), NaF (4 mM), Na3VO4 (20 mM), NaCl (150 mM), b-glycerophosphate (50 mM), and DTT (0.2 mM). Total cell proteins were resolved on 10% SDS-polyacrylamide gels and transferred to PVDF membranes (Fisher Scientific). Membranes were blocked with 5% nonfat dry milk in TBST (50 mM Tris pH 7.6, 150 mM NaCl, 0.1% Tween 20) and incubated overnight at 4°C in 5% nonfat dry milk in TBST with the anti-HIF-1α (1:500, R&D Systems); anti-CAIX (1:1000, Novus Biologicals), anti-CAXII (1:1000, Cell Signaling), anti-MCT4 (1:1000, Novus Biologicals), or anti-®-tubulin (1:5000, DSHB) antibodies. Specificity of all antibodies has been validated by the manufacturers using siRNA or negative control IgG. Immunolabeling was detected using ECL reagent (LAS4000, GE Life Sciences). Densitometric analysis was performed using ImageQuant TL (GE Life Sciences).
Immunohistological analysis
17-week old mouse tails were harvested and fixed in 4% PFA for 24 hours and decalcified in 12.5% EDTA at 4 °C for 6 weeks prior to paraffin embedding. Coronal sections, 7 μm in thickness, were deparaffinized, rehydrated through graded alcohols, and antigens were retrieved with citrate buffer pH 6 for 20 minutes. Slides were blocked in 5% normal donkey serum in PBST (1X PBS, 0.4% Triton-X) for 1 h at room temperature. Sections were then sequentially incubated with antibodies CAIX (1:500, Novus Biologicals) and CAXII (1:500, Cell Signaling) with an NP marker, Keratin 19 (1:3, DHSB Hybridoma Product TROMA-III), a in 5% normal donkey serum in PBST at 4°C overnight (46). After thoroughly washing the sections, the bound primary antibodies were incubated with Alexa Fluor-594 AffiniPure F(ab’)2 conjugated anti-rabbit or anti-rat secondary antibodies (Jackson ImmunoResearch) for 1 h at room temperature. Sections were visualized using a fluorescence microscope (AxioImager A2, Zeiss) fitted with a monochromatic camera (AxioCam MRm, Zeiss).
Generation of NP-specific HIF-1α knockout mice
NP-specific HIF-1α knockout mice were generated by the strategy previously reported by Merceron et al. 2014 (47). Briefly, Foxa2Cre knock-in males were bred with homozygous HIF-1α floxed (HIF-1αf/f) females to generate heterozygous floxed Foxa2Cre;HIF-1αf/+ animals. Heterozygous males (Foxa2Cre;HIF-1αf/+) were crossed with homozygous floxed HIF-1αf/f females to generate Foxa2Cre;HIF-1αf/f mutant mice, Foxa2Cre;HIF-1αf/+ and HIF-1αf/f control mice. Embryos were collected at E15.5, sectioned as previously reported, and analyzed for CA expression as described above. CAIX/XII staining intensity density was quantified using Fiji (ImageJ) Software. A region of interest was specifically drawn to include only the NP tissue compartment; staining intensity was determined by measuring integrated density per area.
Plasmids and Reagents
LV-shHIF-1α (#232222 designated clone 1, #54450 designated clone 2) and control pLKO.1 were purchased from Sigma-Aldrich. siHIF-1α was a gift from Dr. Connie Cepko (Addgene). The following plasmids were obtained from the Addgene repository: HRE-Luc (#26731) developed by Navdeep Chandel, HA-HIF1α (#18949) developed by Dr. William Kaelin, psPAX2 (catalogue no. 12260) and pMD2G (catalogue no. 12259) developed by Dr. Didier Trono.
Lentiviral Particle Production and Viral Transduction
HEK293T cells were seeded in 10-cm plates (1.3 × 106 cells/plate) in Opti-MEM (Life Technologies) with 2% FBS 2 days before transfection. The cells were transfected with 9 μg of control shRNA (pLKO.1) and shRNA against HIF-1α (shHIF-1α) plasmids along with 6 μg of psPAX2 and 3 μg of pMD2.G. After 16 h, the transfection medium was removed and replaced with DMEM with 10% heat-inactivated FBS and penicillin-streptomycin. Lentiviral particles were harvested at 48 h and 60 h post-transfection. NP cells were plated in DMEM with 10% heat-inactivated FBS 1 day before transduction. Cells in 10-cm plates were transduced with 5 ml of conditioned media containing viral particles along with 8 μg/ml Polybrene. After 24 h, media was removed and replaced with DMEM with 10% FBS and continued for 3 days. The cells were cultured in hypoxia or normoxia for additional 24 h and harvested for protein and mRNA extraction 5 days after transduction.
Bioinformatics Analysis
The nucleotide sequence and chromosomal location of the 2kb proximal promoter of rat Car9 and Car12 were found using the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables). Evolutionary conservation of the 2kb proximal promoter was determined using the ECR Browser (http://ecrbrowser.dcode.org/) with a threshold of 77%. Putative HRE consensus sequences (5′-[A/G]CGTG-3′) were determined using the JASPAR Core Database (http://jaspar.genereg.net/) with a relative score threshold of 0.85 (48). Multiz alignment of HRE motifs was performed using the Ensembl Lastz Database (http://www.ensembl.org/index.html).
Chromatin Immunoprecipitation
Rat NP cells were plated in 10-cm plates and either cultured in normoxia or hypoxia for 24 hours. ChIP assay was performed using ChIP-IT® high sensitivity kit (Active Motif, Carlsbad, CA) according to the manufacturer’s recommendations. Briefly, chromatin was sheared by sonication, and input DNA was generated by treating aliquots with RNase, proteinase K, and heat, followed by ethanol precipitation. DNA complexes were immunoprecipitated by incubation with anti-HIF-1α antibody (Cell Signaling) overnight at 4°C followed by binding to protein G-agarose beads for 3 h at 4°C. Cross-links were reversed by treatment with proteinase K and heat for 2.5 h, and DNA was purified using DNA purification elution buffer (Active Motif). Real time PCR analysis was performed using ChIP-IT® quantitative PCR analysis kit (Active Motif) using the following primer pairs for putative HRE sites (from JASPAR database (48)) as shown in supplementary table 1.
Negative control primers and standard curve primers used were provided with kit. Real time PCR was performed with Power SYBR® Green PCR Master Mix (Applied Biosystems). The Ct values were recorded, and the data were normalized based on primer efficiency, input DNA Ct values, amount of chromatin, and resuspension volume, based on manufacturer’s recommendations.
Cell Viability Assay
Rat NP cells were plated in a 96-well plate at 10,000 cells per well. Cells were incubated in hypoxia for 24 h prior to treatment with three carbonic anhydrase inhibitors for 18 h, 48 h, and 96 h. Viability was measured using Calcein-AM and Ethidium homodimer -1 dyes following standard protocol. Negative control (dead) cells were fixed with 70% Methanol for 30 mins at 37C.
Mitochondrial Oxidoreductase Activity Measurement
Rat NP cells were plated in a 96-well plate at 5,000 cells per well. Cells were incubated in hypoxia for 24 h prior to treatment with three carbonic anhydrase inhibitors for 18 h. Oxidoreductase activity was measured using MTT dye following standard protocol.
Extracellular Lactate Measurement
Rat NP cells were plated in duplicate in a 6-well plate at 200,000 cells per well. Cells were incubated in hypoxia for 24 h prior to treatment with three carbonic anhydrase inhibitors for 18 h. Lactate concentrations were measured in each sample using a clinical lactate measurement kit from Trinity Biotech, complete with reagents and standard curve set.
Intracellular pH Measurement
Rat NP cells were plated in a 96-well plate at 10,000 cells per well. Cells were incubated in hypoxia for 24 h prior to treatment with three carbonic anhydrase inhibitors for 18 h. Intracellular pH was measured following the pHrodo Red AM Intracellular pH Indicator (ThermoFisher) protocol. A standard curve was calculated by clamping the intracellular pH with extracellular pH buffers at 4.5, 5.5, 6.5, and 7.5 after treatment with a 10 μM valinomycin/nigericin ionophore cocktail.
Statistical analysis
All experiments were performed in triplicate at minimum, and data are presented as mean ± S.E. Differences between groups were analyzed by the Student’s t test and one-way ANOVA; p<0.05.
RESULTS
Hydration of CO2, rather than glycolytic production of lactate, contributes more to the extracellular proton production in NP cells
In order to elucidate the baseline metabolism of NP cells we measured extracellular acidification (ECAR) and oxygen consumption rates (OCR) of primary NP cells with a Seahorse XF Analyzer after 24 hours of hypoxic culture (Figs. 1A, B). It was apparent from the ECAR and OCR time-courses that basal metabolic rates in NP cells were relatively low; especially OCR which is 10-fold lower than those seen in other cell types (49, 50). Interestingly, inhibiting the electron transport chain with Antimycin A had no effect on ECAR or OCR, confirming that NP cells relied minimally on mitochondria for energy generation (Figs. 1A, B) (10).
Figure 1. Relative contribution of glycolysis and CO2-hydration on rates of total extracellular proton production; expression of plasma-membrane associated extracellularly-facing CA isoforms in the intervertebral disc.
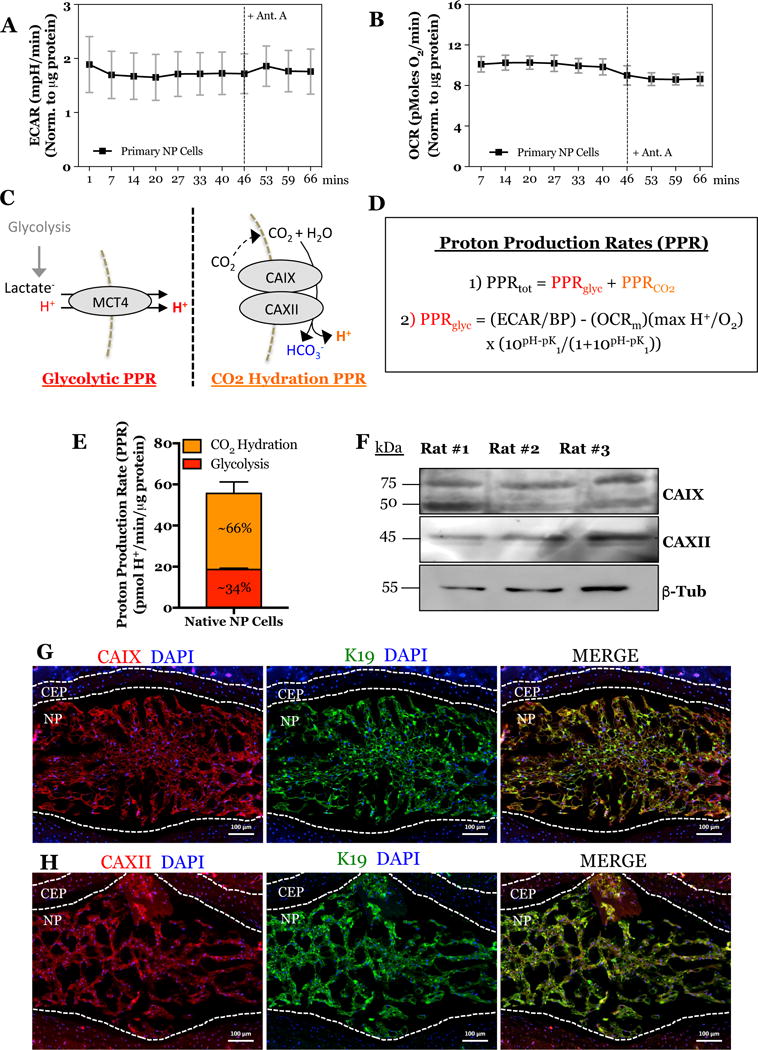
A) ECAR time-course showing proton production rate in primary NP cells before and after Antimycin A treatment. B) OCR time-course showing total oxygen consumption in primary NP cells before and after Antimycin A treatment. C) Schematic of two major cellular pathways contributing to extracellular proton production measured by the Seahorse XF Analyzer. D) Equations derived from Mookerjee et. al. to determine the relative contribution of glycolysis and CO2 hydration on total extracellular proton production rate. E) Graphical representation of glycolytic and CO2-derived proton production rates in the primary NP cells. F) Representative Western blot analysis of CAIX and CAXII protein expression from NP tissue of 3 adult rats. F) Representative histological image of a mid-coronal tail section of a 17 week-old mouse denoting the distinct IVD tissue morphology of the NP, AF, and CEP. G, H) Immunofluorescence detection of CAIX (G), CAXII (H), and K19 (an NP cell marker) in the NP and CEP of tail discs. All quantitative data is represented as mean ± SE, n=3 independent biological replicates with 4 technical replicates per biological replicate. Scale bar is 100 μm. 3 independent biological replicates were analyzed for immunofluoresence.
Recently, Mookerjee and colleagues have elegantly shown that there are two major sources of extracellular production that contribute to ECAR measurements (24). While ECAR measurements are generally used to solely report the glycolytic production of lactate/H+, CO2 produced from TCA cycle can be enzymatically hydrated into HCO3− and H+ units by membrane-associated and extracellularly-facing carbonic anhydrases (Fig. 1C). Although we have previously shown that that NP cells do not rely on mitochondria for ATP production, it is well known that the TCA cycle is necessary for generating the metabolic precursors for fatty acid, nucleotides, and amino acid synthesis (10). With this understanding, we sought to determine the proportion of total proton production rate (PPRtot) generated from glycolysis (PPRglyc) and CO2 hydration (PPRCO2) according to the equations derived and modeled by Mookerjee et. al. (24) (Fig. 1D):
| 1) |
| 2) |
We determined the glycolytic proton production rate in primary NP cells (using glucose and glutamine as substrates) to be 18.8-19.2 pmol H+/min/μg protein. Given that the total proton production rate of 56 ± 5 pmol H+/min/μg protein, the percentage of total proton production generated from glycolysis is 34 ± 3% and from CO2 hydration is 66 ± 3% (Fig. 1E). These results suggested that CO2 hydration by extracellularly-facing carbonic anhydrases should be critical in NP cell physiology.
Carbonic anhydrase isoforms 9 and 12 are expressed in intervertebral disc tissue in vivo
In order to confirm the presence of carbonic anhydrases 9 and 12 in intervertebral disc tissue, we isolated total protein from the NP tissue of adult rats. Western blot analysis confirmed the robust expression of both CAIX and CAXII protein in the native NP tissue (Fig. 1F). Furthermore, intervertebral disc sections were immunolabeled with antibodies against CAIX and CAXII, and cytoskeletal Keratin-19 (K19), a known NP phenotypic marker, to elucidate their tissue localization in vivo (46). CAIX (Fig. 1G) and CAXII (Fig. 1H) were predominantly located in the NP with lower level expression in the AF and endplates (Figs. S1). Furthermore, most cells in the NP compartment were visibly immuno-positive for CAIX and CAXII antibody staining, respectively, implying that both isoforms are co-expressed (Fig. S1). These results suggested that the expression of these two CA isoforms is enriched in the NP compartment of the adult intervertebral disc and may play an important role in tissue physiology.
Expression of carbonic anhydrase isoforms is hypoxia-inducible in NP cells
Given the robust expression of CA9 and CA12 in the NP tissue, we investigated the effect of oxygen tension on their mRNA and protein levels. We cultured primary rat NP cells under hypoxic conditions for 8-72 hours and measured mRNA and protein expression by qRT-PCR and Western blot. Car9 and Car12 mRNA showed significant hypoxic induction as early as 8 hours and persisted until 72 hours (Fig. 2A, B). Amongst both CA isoforms, the magnitude of induction was highest for Car12; levels were ~15-fold after 8 hours and remained elevated (~10-fold) at 72 hours in hypoxia (Fig. 1B). Additionally, an Actinomycin D chase assay showed that Car9 and Car12 mRNA had enhanced stability under hypoxia. The half-life (t1/2) of Car9 mRNA increased from 13.6 to 19.7 hours in hypoxia. Likewise, the half-life of Car12 mRNA increases from 11.5 to 16.8 hours (Fig. 2C, D). These results imply that Car9 and Car12 mRNA is stabilized by post-transcriptional regulation under hypoxia. Likewise, CAIX (Fig. 2E, F), and CAXII protein (Fig. 2E, G) levels were also elevated after 72 and 24 hours in hypoxia, respectively. Overall, it is evident that CA9 and CA12 are sensitive to changes in oxygen tension and their expression was up-regulated under hypoxic conditions. Moreover, we show here for the first time that CA9, in addition to CA12, is hypoxia sensitive in NP cells (43).
Figure 2. Effect of hypoxia on CA9 and CA12 expression, bioinformatic analysis of HRE motifs in Car9 and Car12 promoters, and ChIP analysis.
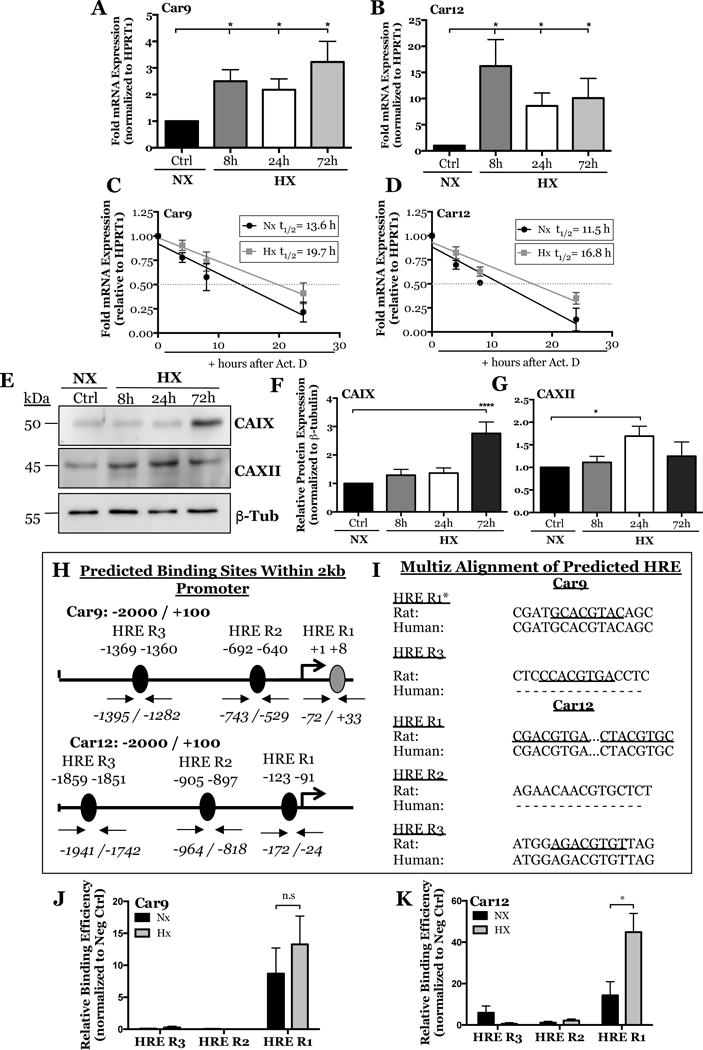
A, B) qRT-PCR analysis of Car9 (A), and Car12 (B) mRNA expression in rat NP cells cultured under hypoxia (1% O2) for up to 72 h. C, D) Actinomycin D chase assay showing regression curve of Car9 (B) and Car12 (C) mRNA at 0, 4, 8, and 24 hours after Actinomycin D treatment (5μg/mL). An increase in mRNA half-life (t1/2) is seen for both genes in hypoxia. E) Western blot analysis of CAIX and CAXII protein expression in rat NP cells cultured under hypoxia for up to 72 h. F, G) Densitometric analysis of CAIX (F) and CAXII (G) from Western blot experiment shown in (E). H) JASPAR predicted HRE regions (5′-[A/G]CGTG-3′) within -2000/+100 bp into the rat Car9 and Car12 promoters with a relative score threshold of 0.85. Predicted HRE regions; black, conserved binding regions validated in human promoter; gray. I) Multiz alignment from Ensembl lastz database. Core matrix binding locations are underlined, inability to match shown by (-), * sequences are from the negative strand. J, K) Relative binding efficiency of HIF-1α to putative HRE regions in the rat Car9 (J) and Car12 (K) promoters determined by ChIP. Statistical analysis performed on HRE regions with ≥ 5 HIF-1α binding events/1,000 cells and binding efficiency 5-fold greater than negative control. Data represented as mean ± SE, n ≥ 3 biological replicates, p<0.05.
HIF-1α binds to select HRE binding regions in the promoters of Car9 and Car12 in hypoxic NP cells
HIF-1α binds hypoxia-response elements (HREs) in the gene promoters of downstream targets for transactivation. The precise locations of HRE consensus sequence (5′-[A/G]CGTG-3′) within rat Car9 and Car12 promoters were predicted using a transcription factor binding databases JASPAR CORE database (48). Analysis identified several putative HRE binding sites on the promoters of Car9 and Car12 (Fig. 2H). Next, the sequence conservation of the putative HRE motifs was analyzed using Multiz alignment tool from Ensembl lastz database (http://www.ensembl.org/index.html) which showed 100% conservation at HRE R1 in Car9 and HRE R1 and R3 in Car12 promoters (Fig. 2I). We experimentally validated HIF-1α binding to these predicted HRE regions on Car9 and Car12 promoters in rat NP cells using genomic chromatin immunoprecipitation (ChIP). Of the three putative HRE binding regions on the Car9 promoter, the region with the highest conservation and closest to the transcriptional start site showed binding independent of oxemic tension (Fig. 2J). This result was not unexpected for NP cells, which constitutively express HIF-1α even under normoxic conditions (4, 5). Interestingly, of three putative HRE binding regions on the Car12 promoter, binding was seen only at HRE R1 which also showed enrichment in hypoxia (Fig. 2K).
CA9 and CA12 expression is regulated by HIF-1α in NP cells in vitro
We have previously shown the importance of HIF-1α for maintaining NP cell function and phenotype within the hypoxic niche of the intervertebral disc (10). Unique to NP, HIF-1α shows stabilized expression even under normoxic conditions (4, 5). Since expression of CA isoforms in NP cells is hypoxia sensitive and HRE regions in the Car9 and Car12 promoters bind HIF-1α, we sought to delineate their relationship using HIF-1α gain and loss of function approaches. Stabilizing HIF-1α protein with DMOG treatment for 8 hours showed a small increase in Car9 mRNA levels, induction was lost at 24 h (Fig. 3A). Whereas, Car12 mRNA levels were significantly increased (Fig. 3B). Importantly, we also transduced rat NP cells with lentiviruses expressing two HIF-1α shRNA sequences. This silencing approach resulted in a stable ~90% knock- down of HIF-1α (Figs. 3C, F, G). CA9 mRNA and protein expression was significantly reduced by HIF-1α silencing under both normoxia and hypoxia (Figs. 4D, F, H). Likewise, HIF-1α silencing resulted in a significant decrease in CA12 mRNA and protein expression in hypoxia (Figs. 4E, F, I). Taken together, our loss of function studies implied that expression of CA9 and CA12 is regulated by HIF-1α in NP cells.
Figure 3. HIF-1α gain and loss of function in NP cells effects CA9 and CA12 expression.
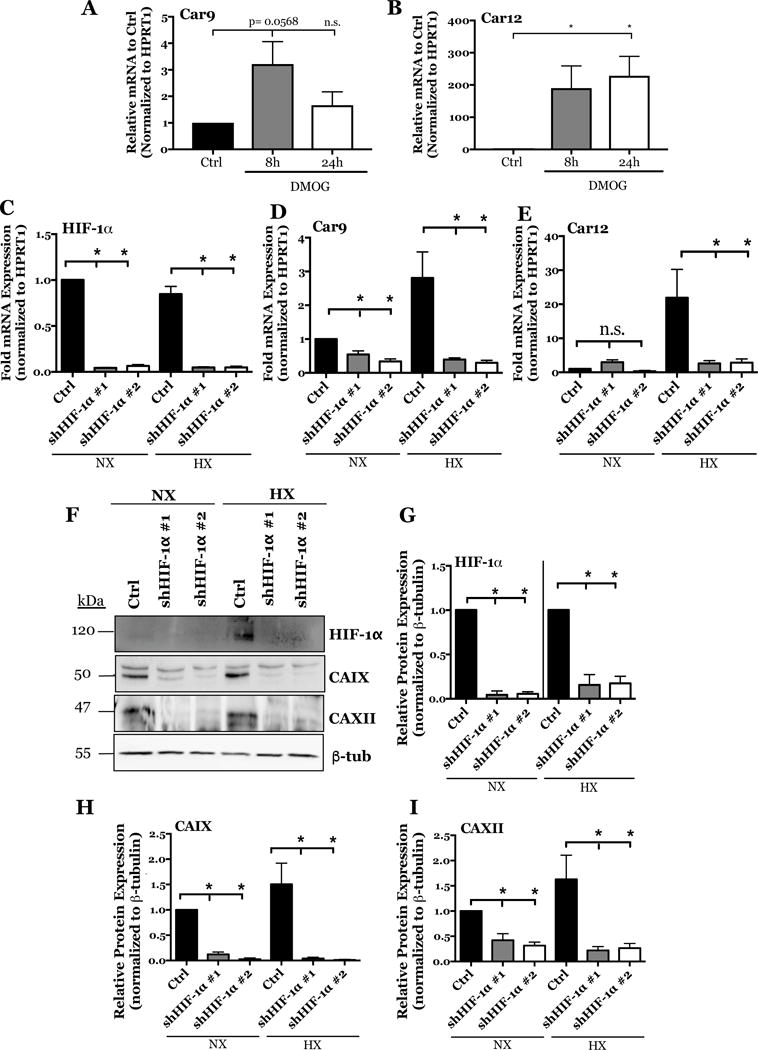
A, B) Effect of stabilizing HIF-1α protein with DMOG treatment for 8 and 24 hours on Car9 (A) and Car12 (B) expression. C-E) qRT-PCR analysis of HIF-1α (C), Car9 (D), and Car12 (E) mRNA expression in rat NP cells after silencing HIF-1α with two lentiviruses each expressing an independent HIF-1α targeting shRNA (shHIF-1α #1 and shHIF-1α #2). F) Representative Western blot analysis of HIF-1α, CAIX, and CAXII protein expression in rat NP cells after silencing HIF-1α with two independent shRNAs. G-I) Densitometric analysis of Western blot showed in (F) above. Data is represented as mean ± SE, n ≥ 4 independent biological replicates, p<0.05.
Figure 4. Mice with NP-specific deletion of HIF-1α show decreased expression of CAIX and CAXII in NP tissue.
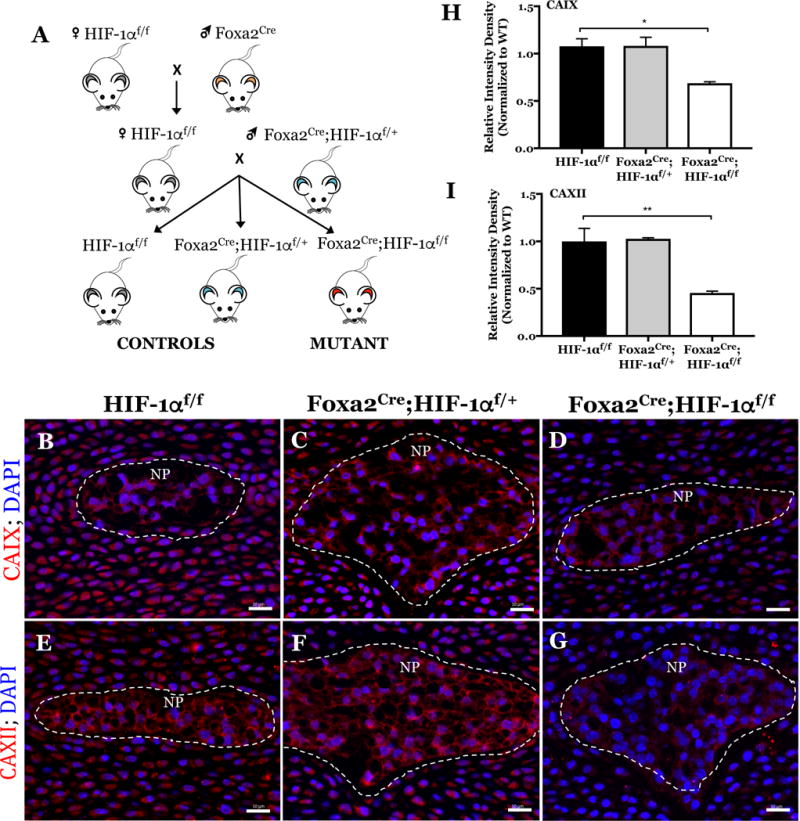
A) Schematic breeding diagram for generation of NP-specific HIF-1α knockout mice (Foxa2Cre;HIF-1αf/f) from heterozygous Foxa2Cre and control HIF-1αf/f mice. B-G) Representative immunofluorescence images of CAIX (B, C, D) and CAXII (E, F, G) expression in E15.5 control (B, E: HIF-1αf/f), heterozygous (C, F: Foxa2Cre; HIF-1αf/+), and HIF-1α mutant (D, G: Foxa2Cre;HIF-1αf/f) littermate mice imaged at 20x magnification. NP compartment denoted by white dashed line. H, I) Quantification of CAIX (H) and CAXII (I) staining intensity density (integrated density per NP tissue area) in control and HIF-1α cKO mice. 3 conditional null and littermate controls were analyzed. Scale bar; 20 μm.
CA9 and CA12 expression is reduced in the discs of NP-specific HIF-1α knockout mice
Our in vitro results clearly suggest that CA9 and CA12 expression is HIF-1α dependent in NP cells. To further support this observation, we investigated the effect of HIF-1α deletion on CA9 and CA12 expression in vivo using an NP-specific HIF-1α knockout mouse, where HIF-1α deletion was achieved by constitutive Cre expression driven by a notochord-specific Foxa2 promoter/enhancer (Fig. 4A) (47). We chose to analyze CA expression at E15.5 since, at this stage the NP is completely formed and, in null mice, NP cells undergo apoptosis at birth (47). We labeled disc sections of E15.5 HIF-1α mutant mice (Foxa2Cre;HIF-1αf/f) and control littermates with antibodies against CAIX and CAXII and analyzed expression with fluorescent microscopy (Fig. 4B–G). We observed that the expression of both CAIX (Fig. 4B–D) and CAXII (Fig. 4E–G) was reduced in the NP (indicated by the white, dotted line) of HIF-1α mutant mice (Fig. 4D, G) compared to heterozygous (Foxa2Cre;HIF-1αf/+) (Fig. 4C, F) and wild-type (HIF-1αf/f) (Fig. 4B, E) littermate controls. A significant decrease in CAIX (Fig. 4H) and CAXII (Fig. 4I) staining intensity was confirmed in the HIF-1α mutant mice by measuring integrated staining density per NP area. These results confirm the role of HIF-1α in maintaining levels of CAIX and CAXII in NP tissue.
Inhibition of CAs reduces extracellular acidification rate (ECAR) and total oxygen consumption rate (OCR) in NP cells without affecting mitochondrial OCR
The precise contribution of carbonic anhydrases in NP cell physiology is still unknown. To elucidate the role of CA9 and CA12 in NP cell metabolism and acid-base homeostasis we measured changes in ECAR and OCR with a Seahorse Analyzer following treatment of NP cells with specific CA inhibitors in hypoxia for 18 hours. While, methazolamide (MZA) and acetazolamide (AZA) are pan-carbonic anhydrase inhibitors, at the concentrations used in this study, they specifically inhibited CA9 and CA12 catalytic function in NP cells (51); whereas U-104 is a specific inhibitor of CA9 and CA12 (Ki = 45 nM and 4.5 nM, respectively) with very low inhibition for CA1 and CA2 (Ki = 5080 nM and 9640 nM, respectively) (52). When this inhibitor is used, the cytosolic CA isoforms expressed in the disc (CA2/3) will not be affected, given the high Ki for CA2 and known inability of CA3 to be effectively inhibited with small molecules (53). First, we confirmed that changes in cell viability did not confound metabolic profiling; inhibition of CA function for 18 hours did not significantly affect cell viability (Fig. 5A). However, cells treated with MZA, AZA, and U-104 experienced significant decreases in ECAR over time (Figs. 5B, C, D) and on average (Fig. 5E); treatment with antimycin A had no effect on ECAR, again suggesting minimal dependency on electron transport chain in NP cells (Fig. 5B–E). Overall, these results demonstrate that catalytic inhibition of CA9/12 decreased total extracellular proton production, confirming that extracellular acidification in NP cells is partially due to CA9/12-dependent hydration of CO2.
Figure 5. Inhibition of CA9 and CA12 in rat NP cells leads to reduced ECAR.
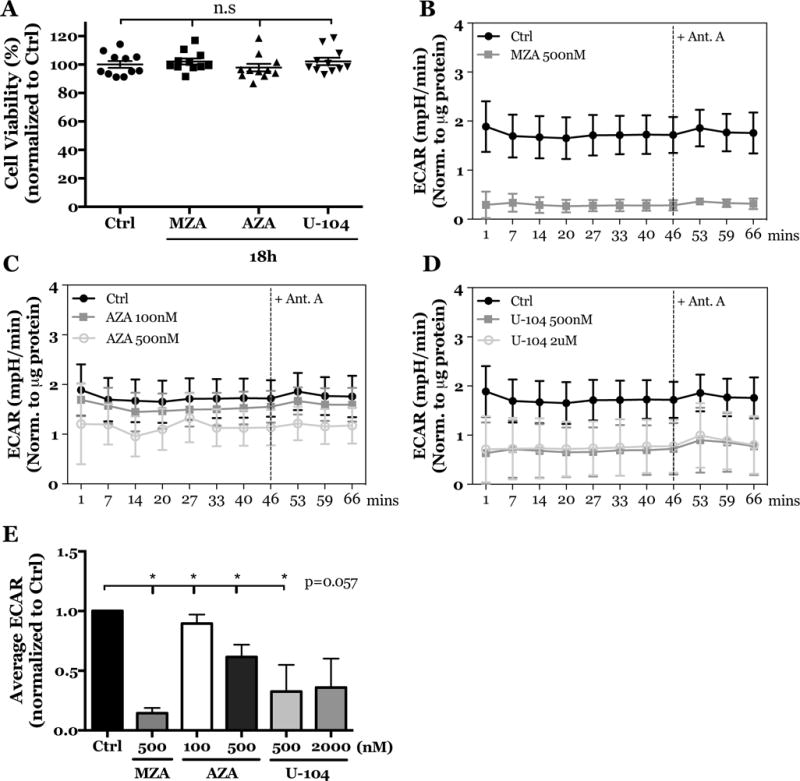
A) Treatment of rat NP cells with MZA (500 nM), AZA (500 nM), and U-104 (2 μM) for 18 h has no effect on cell viability. B-D) ECAR was reduced in rat NP cells treated with MZA (500 nM) (B), AZA (100 nM, 500 nM) (C) and U-104 (500 nM, 2 μM) (D) before and after treatment with antimycin A. E) Average ECAR was significantly decreased before addition of antimycin A. All quantitative data is represented as mean ± SE, n=3 independent biological replicates with 4 technical replicates per biological replicate, p<0.05.
Likewise, treatment with MZA, AZA, and U-104 significantly decreased total OCR over time (Figs. 6A–C) and on average (Fig. 6D). Notably, however, treatment with antimycin A had no effect on OCR (Fig. 6A–C), suggesting that mitochondrial OCR was unaffected by CA inhibition (Fig. 6E). In addition, we confirmed that mitochondrial function was not altered with CA inhibition by measuring mitochondrial oxidoreductase activity (Fig. 6F). In summary, inhibition of CA catalytic activity decreased extracellular H+ accumulation and oxygen consumption rates of NP cells, independent of mitochondrial oxidative respiration.
Figure 6. Inhibition of CA9 and CA12 in rat NP cells leads to reduced OCR without affecting mitochondrial respiration or functional activity.
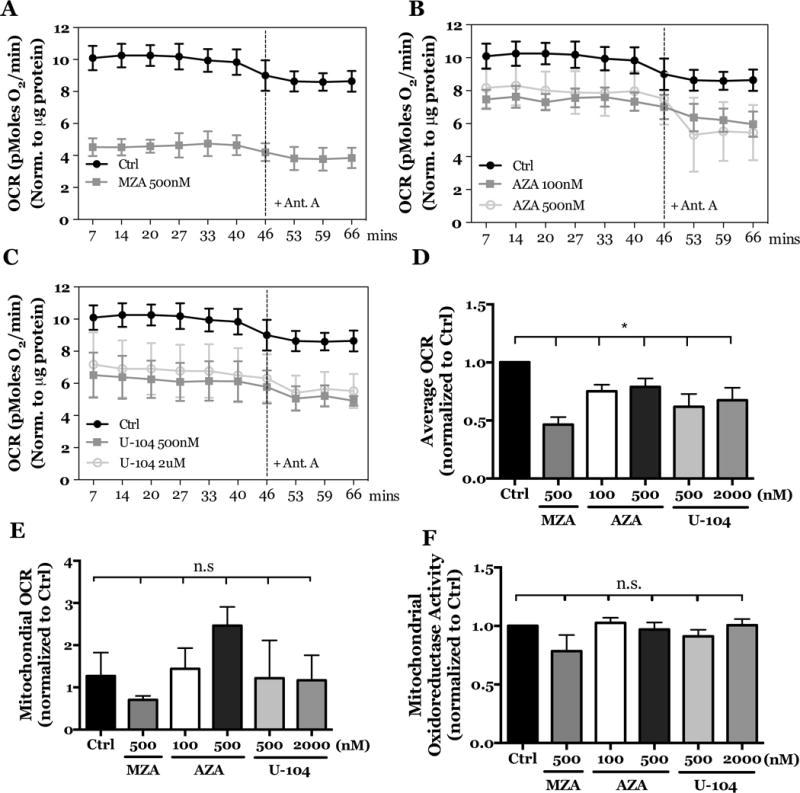
A–C) OCR was reduced in rat NP cells treated with MZA (500 nM) (A), AZA (100 nM, 500 nM) (B) and U-104 (500 nM, 2 μM) (C) before and after treatment with antimycin A. D) Treatment of rat NP cells with MZA, AZA, and U-104 significantly decreased average OCR. E) Treatment of rat NP cells with MZA, AZA, and U-104 has no effect on mitochondrial OCR. F) Rat NP cells treated with MZA, AZA, and U-104 showed no change in their mitochondrial oxidoreductase activity, implying preservation of mitochondrial function. All quantitative data is represented as mean ± SE, n=3 independent biological replicates with 4 technical replicates per biological replicate, p<0.05.
Inhibition of CAs causes a significant decrease to the total extracellular proton production rate without affecting glycolysis in NP cells
To investigate the functional effect of CA inhibition on NP cells we delineated the source of extracellular proton production that contributed to decreased ECAR- i.e. H+ produced via glycolysis or CO2 hydration. Since we determined that ~66% of total proton production in NP cells is from extracellular CO2 hydration, it is expected that part of the decrease in ECAR after CA inhibition is directly due to abrogating proton production from the CA reaction. However, the other potential source of extracellular proton production measured by the analyzer is those transported out of the cells with lactate via monocarboxylate transporter 4 (MCT4). This measurement is often considered a surrogate measure of glycolytic flux as MCT4 exports equal units of lactate and H+, the end products of anaerobic glycolysis (Fig. 7A) (54).
Figure 7. Inhibition of CA9 and CA12 in rat NP cells has no effect on glycolytic flux.
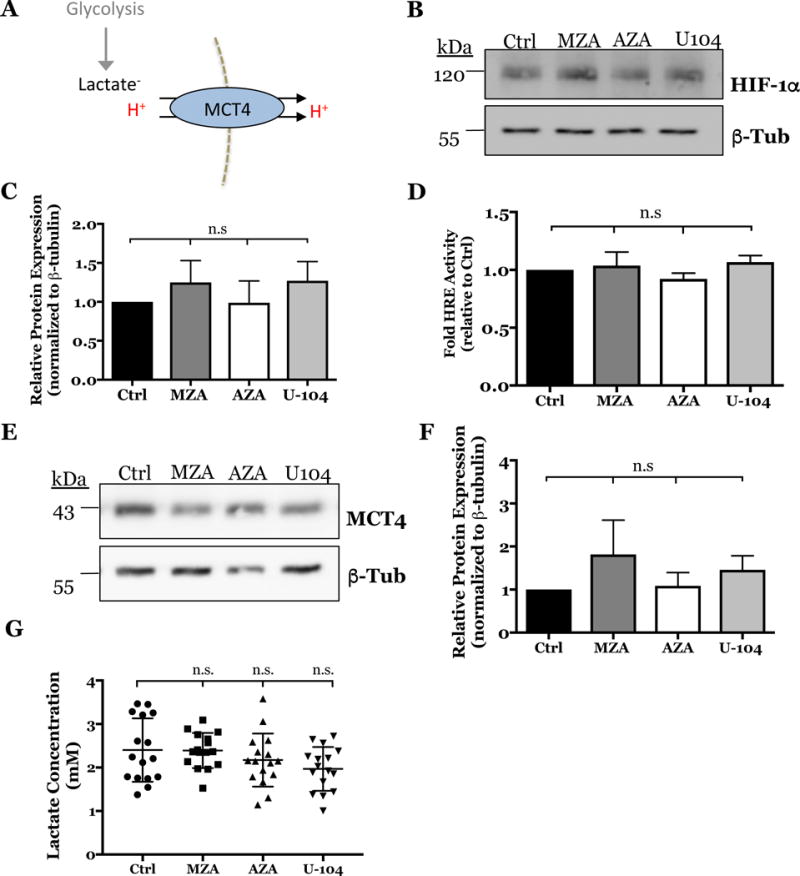
A) Schematic of the glycolytic production of lactate/H+ exported into the extracellular space via MCT4. B) Representative Western blot analysis of HIF-1α after inhibiting CA activity with MZA (500 nM), AZA (500 nM), and U-104 (2 μM) for 18 h in hypoxia. C) Densitometric analysis of Western blot experiment shown in (B) (n=4). D) HRE luciferase reporter activity was unaffected by CA inhibition (n=4, 3 technical replicates each). E) Representative Western blot analysis of MCT4 after inhibiting CA activity with MZA, AZA, and U-104. F) Densitometric analysis of Western blot experiment shown in (E) (n=4). G) Extracellular lactate concentrations were unaffected by CA inhibition with MZA, AZA and U-104 (n=5, 3 technical replicates each). Data is represented as mean ± SE, n≥ 4 independent biological replicates, * p<0.05, ** p<0.01, *** p<0.001.
To investigate if glycolytic flux was perturbed, we measured the effect of CA inhibition on HIF-1α, a major regulator of glycolysis. We showed that levels of HIF-1α protein (Fig. 7B, C) and HIF-1α transcriptional activity measured by HRE luciferase reporter is unaltered (Fig. 7D). We also confirmed that MCT4 protein levels did not change after inhibiting CAs (Figs. 7E, F) and that extracellular lactate concentrations remained unaffected by CA inhibitors (Fig. 7G). Taken together, these results suggested that the decrease in extracellular proton production contributing to decreased ECAR was not due to a change in glycolytic flux or lactate transport. Rather, the data confirm that catalytic activity of extracellularly facing CAs favors the hydration of CO2 generating H+ and HCO3− ions, and inhibition of this reaction causes a measurable decrease in extracellular H+ and HCO3− units.
Unlike CA inhibition, silencing of HIF-1α causes a shift from glycolytic to oxidative metabolism in NP cells
Since our results clearly showed that CA9 and CA12 are HIF-1α targets, we determined if the metabolic profile of NP cells following CA inhibition is like that of cells lacking HIF-1α. Similar to effect of CA inhibition on ECAR, HIF-1α silencing significantly decreased ECAR compared to cells transduced with shCtrl, importantly however, this reduction was rescued by blocking mitochondrial function with antimycin A (Fig. 8A, B). These results suggest that the reduction in ECAR by HIF-1α silencing is likely caused by a rewiring of substrate flux, such that pyruvate is now diverted into the mitochondria and away from reduction to lactate. Moreover, the subsequent increase in ECAR upon antimycin treatment indicated that the HIF-1α silenced cells retain reserved glycolytic capacity.
Figure 8. HIF-1α silencing in rat NP cells leads to metabolic switch from glycolytic to oxidative metabolism.
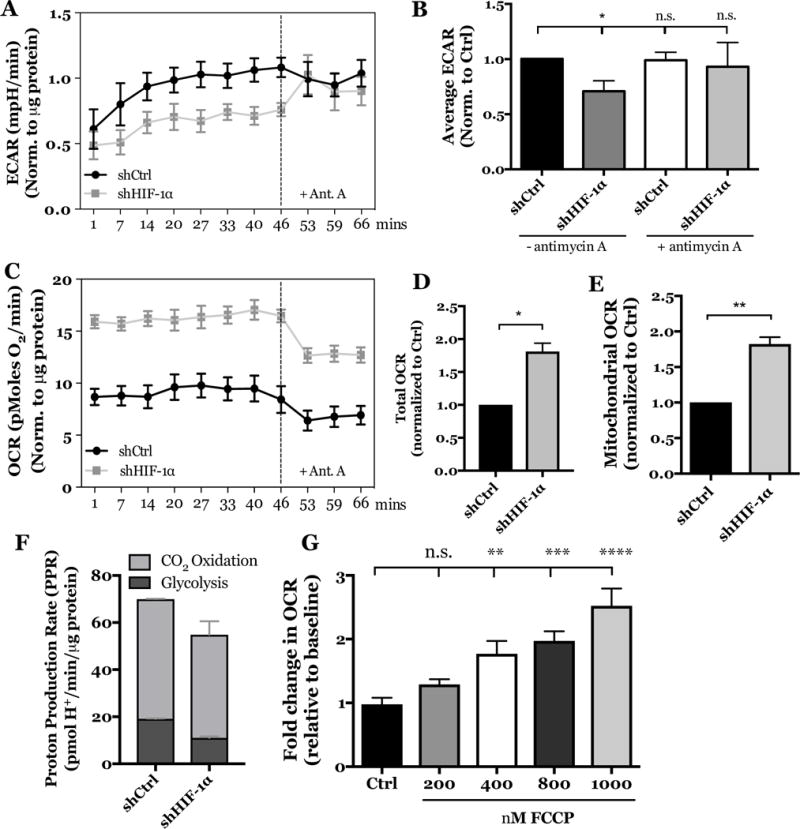
A) Effect of stable HIF-1α silencing on ECAR of rat NP cells over time, before and after treatment with antimycin A. B) Stable HIF-1α knockdown lead to decreased average ECAR compared to cells transduced with shCtrl and treatment with antimycin A rescued ECAR in HIF-1α silenced cells. C) Effect of stable HIF-1α silencing on OCR of rat NP cells over time, before and after treatment with antimycin A. D) Stable HIF-1α knockdown significantly increased total OCR (mitochondrial + non-mitochondrial) compared to cells transduced with shCtrl. E) Stable HIF-1α knockdown significantly increased mitochondrial OCR. F) Graphical representation of glycolytic and CO2-derived proton production rates in the control and HIF-1α knock down cells. G) FCCP treatment at 400, 800, and 1000 nM significantly increased the total OCR by NP cells. All quantitative data is represented as mean ± SE, n=3 independent biological replicates with 4 technical replicates per biological replicate, * p<0.05, ** p<0.01.
HIF-1α silencing also caused a significant increase in total OCR (mitochondrial + non-mitochondrial). Interestingly, this increase was substantially suppressed by antimycin A (Fig. 8C). We determined the mitochondrial fraction of total OCR and found that HIF-1α silencing increased not only the total OCR but also the mitochondrial OCR fraction (Figs. 8D, E). Furthermore, we calculated rates of glycolytic and CO2-dependent proton production and found that HIF-1α knockdown decreased the relative contribution from glycolysis and increased the contribution from CO2 hydration (total proton production rate was ~65 and ~55 pmol H+/min/μg protein for ShCtr and Sh-HIF-1α group, respectively), suggesting an increase in TCA-cycle derived CO2 (Fig. 8F). Given that the results of HIF-1α knockdown assays implied that NP cells are capable of oxidative metabolism, we determined whether NP cells possess reserve electron transport chain capacity by uncoupling mitochondrial ATP generation from electron transport chain activity using the chemical uncoupler, FCCP (Fig. 8G). Interestingly, the fold change in OCR increased in a dose-dependent manner with FCCP treatment from 400 to 1000 nM concentrations. These data suggest that NP cells do, in fact, have reserve electron transport chain capacity that can be tapped into under specific circumstances, such as when HIF-1 signaling is compromised. Taken together, these results clearly showed that HIF-1α actively suppresses electron transport chain function and oxidative respiration in NP cells, and silencing this crucial transcription factor caused a shift in cellular metabolism beyond that evidenced by dysregulation of CAs.
Inhibition of CAs causes a significant decrease in intracellular pH by blocking the generation of recycled bicarbonate
Given that CA inhibition caused a significant decrease in extracellular H+ and HCO3− production, we investigated the fate of this extracellular HCO3−. In some hypoxic cancer cells CA9/12 produced extracellular HCO3− is retrieved to buffer cytosolic pH (19, 55) (Fig. 9A). We measured pHi of rat NP cells after CA inhibition using fluorescent pH-sensitive probes. In line with previous studies of bovine NP cells, the baseline pHi in rat NP cells was found to be acidic (pH~6.8) (11). Importantly, our results clearly showed that the pHi became more acidic when CA9/12 were inhibited (Fig. 9B). These results confirmed that the CA9/12 reaction indeed favors production of extracellular HCO3− which is recycled to buffer intracellular pH. Noteworthy, we performed these experiments in HCO3− buffered medium to verify that the equilibrium of the CA reaction was not shifted to favor H+ and HCO3− production as a reflection of the non-HCO3− buffered conditions in the Seahorse assay.
Figure 9. Inhibition of CA9/12 causes a significant decrease in intracellular pH by blocking the generation of recycled bicarbonate.
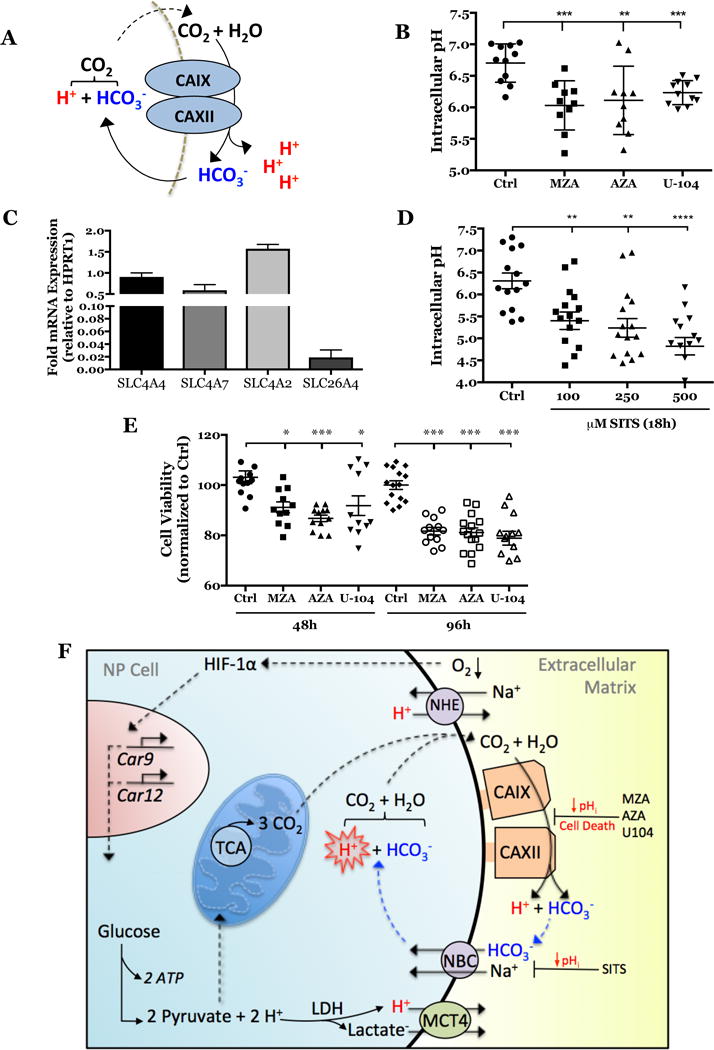
A) Schematic of extracellular HCO3− generation and its intracellular recycling. B) Intracellular pH significantly decreased after CA inhibition (n=3, 3-4 technical replicates each). C) qRT-PCR results showing expression of bicarbonate transporters in rat NP cells. D) Intracellular pH significantly decreased after SITS treatment for 18 hours under hypoxia (n=4, 3 technical replicates each). E) Treatment of rat NP cells with MZA (500 nM), AZA (500 nM), and U-104 (2 μM) for 48 and 96 hours in hypoxia significantly decreased cell viability. Data is represented as mean ± SE, n ≥3 biological replicates, 3-6 technical replicates per biological replicate, * p<0.05, ** p<0.01, *** p<0.001. (F) Schematic of the proposed model of intracellular pH regulation through HCO3− recycling by the HIF-1α-Carbonic anhydrase axis in NP cells.
HCO3− recycling is dependent on the expression and function of bicarbonate transporters in the NP. Analysis of our previous RNA-Seq studies indicated that NP cells express various plasma membrane bicarbonate transporters of the SLC4 and SLC26 families (GEO Accession #GSE86552) (25). Of the bicarbonate transporters expressed, SLC4A2 (AE2), SLC4A4 (NBCe1), SLC4A7 (NBCn1), and SLC26A4 (Pendrin) had the highest levels. qRT-PCR results confirmed the expression of these four isoforms in rat NP cells (Fig. 9C). Na+-coupled/HCO3− exchangers, NBCn1 and NBCe1, are known to function as HCO3− importers and can be catalytically inhibited by the stilbene derivative, SITS (36). We measured pHi after inhibiting bicarbonate transport with SITS. The results recapitulated the significant decrease in pHi seen after CA9/12 inhibition (Fig. 9D). This result indicated that HCO3− produced pericellularly by CA9 and CA12 activity is recycled by NP cells to buffer cytosolic pH.
Chronic inhibition of CA9 and CA12 in hypoxia results in NP cell death
Although short-term inhibition of CA9 and CA12 had no measurable effect on cell viability, we hypothesized that longer-term inhibition and resultant intracellular acidification will lead to pathological changes in NP cell viability. To test this, we measured NP cell viability after inhibiting CA9 and CA12 for 48 and 96 hours. Our results clearly demonstrate that NP cell viability decreased by 15% and 20% after 48 and 96 hours of CA inhibition, respectively (Fig. 9E).
DISCUSSION
The avascular NP tissue relies on a predominantly glycolytic metabolism for energy generation (10). Pathologically acidic pH (~6.5) exacerbates the breakdown of extracellular matrix proteins and decreases cellular glycolytic flux by NP cells (7, 14–16); as a result cells tightly control metabolite concentrations to maintain a physiological pHi. Carbonic anhydrases are the major regulators of acid-base metabolism in other cell types (22). Despite their critical contribution in buffering intracellular and extracellular pH, the detailed molecular mechanisms that control expression of carbonic anhydrases and their function in hypoxic NP cells is not yet fully elucidated (42, 43). In this study, we showed for the first time that CO2 hydration contributes to the majority of extracellular H+ and HCO3− production in the NP cell matrix through actions of HIF-1 dependent membrane-associated and extracellularly facing carbonic anhydrase isoforms 9 and 12. Notably, our data supports the contribution of CA9 and 12 to a HCO3− transport metabolon and pHi regulation, underscoring their importance in NP physiology.
Given that we determined a large portion of extracellular proton production is from CO2 hydration, we hypothesize that NP cells do in fact utilize the TCA cycle to generate metabolic precursors, NADH, and generate CO2 from this pathway. Our previous studies have shown that although NP cells do not rely on mitochondria for ATP production, they are capable of using the TCA cycle for fatty acid oxidation (10). Moreover, they are not limited by availability of intermediates like α-ketoglutarate that are generated through TCA cycle (5, 41). In addition, CO2 (and NADPH) could be generated from decarboxylation reactions in pentose phosphate pathway known to generate five-carbon sugars glucose-6-phosphate in the cytosol.
Given such prominent contribution of CO2 hydration to extracellular proton generation it was logical to investigate if membrane-associated and extracellularly-facing carbonic anhydrases CA9 and 12 are expressed in NP cells. These are the only isoforms of the CA family that have been shown to be hypoxia inducible in other cell types (29–31). Although their in vivo expression is restricted to few adult tissues (32, 33, 56), our results clearly showed robust expression of both isoforms in adult rat NP tissue. Our results also highlighted a positive relationship between hypoxia and their mRNA stability and expression. Since HIF-1 is a major regulator of hypoxic response in NP cells, and due to the known dependency of CA9 and CA12 on HIF-1 pathway, we determined if such a relationship exists in NP cells. Bioinformatic analyses demonstrated that the proximal promoters of Car9 and Car12 contain three putative HRE regions, of which one HRE region in Car9 and two HRE regions in Car12 are conserved between rat and human promoters, suggesting that these regions could be important in regulation. Confirmation for this hypothesis was forthcoming from results of ChIP analysis, which clearly showed HIF-1α binding to the conserved HRE region (+1/+8 bp) at the Car9 transcription start site in rat NP cells. These results are in agreement with an earlier study that demonstrated HIF-1α binding to a corresponding HRE in the human CA9 promoter (57). Additionally, hypoxic enrichment of HIF-1α binding to the conserved HRE region (−91/−123 bp) closest to the transcription start site in the rat Car12 promoter suggested that these binding sites may play an important role in HIF-1α-dependent regulation of CA9 and CA12 in the NP. Taken together, our results suggested that, under hypoxia, both post-transcriptional regulation and HIF-1α binding to HREs contributed to Car9/12 mRNA stability and directly or indirectly to their expression in NP cells.
Notably, dependency of CA9 and CA12 on HIF-1α was evident from both in vitro and in vivo gain and loss of function approaches. First, we demonstrated that stabilization of HIF-1α in normoxia up-regulated Car12 mRNA expression, however the inductive effect on Car9 was smaller. Considering that ChIP results showed similar HIF-1α binding to the Car9 promoter in normoxia and hypoxia, it is plausible that the expression of CA9 under normoxia is saturated. Alternatively, this may suggest requirement of hypoxia specific co-activator/s for induction of CA9. Supporting gain of function experiments, stable silencing of HIF-1α in vitro resulted in a significant decrease in CA9 and CA12 mRNA and protein expression irrespective of oxemic tension establishing a positive relationship between HIF-1α signaling and CA9/12 expression. Importantly, decreased expression of CAIX and CAXII in NP of mice with NP-specific deletion of HIF-1α provided, for the first time, conclusive in vivo evidence that CA9 and CA12 are transcriptional targets of HIF-1α in this hypoxic tissue.
Relevant to NP cell function, our studies showed that inhibition of CA9 and CA12 led to decreased ECAR and OCR that was not associated with changes in glycolytic flux or mitochondrial function. Our results suggest that CA inhibition has no effect on select pathways regulating or regulated by glycolytic flux, regardless of the HCO3− buffering capacity of the experimental setup. For example, HIF-1α transcriptional activity, expression of membrane-associated lactate exporters (MCT4), and extracellular lactate concentrations were not significantly altered upon CA inhibition. Similarly, catalytic inhibition of CA9 and CA12 lead to decreased oxygen consumption rate that was not correlated with any decrease in mitochondrial respiration or functional dysregulation as measured by oxidoreductase activity. Together, these results demonstrated that effect of CA inhibition on ECAR was independent of glycolysis but relied on abrogation of extracellular proton production from carbonic-anhydrase mediated CO2 hydration, likewise, decreases in OCR were due to changes in non-mitochondrial pathways of O2 consumption. Noteworthy this mode of action was different than when HIF-1α was silenced, which resulted in decreased glycolytic proton production and increased CO2-dependent proton production, likely by increasing rates of TCA cycle production. This result further implicates HIF-1α as the gate-keeper of mitochondrial function and activity in the NP cell. Although it is important to note that healthy NP cells have non-mitochondrial oxygen consumption, the mechanism or purpose of this utilization has not been elucidated in this study and requires further investigation.
Our functional assays confirm that CO2 hydration catalyzed by CA9 and CA12 produces HCO3− and H+ ions in the pericellular space of NP cells. These HCO3− ions are then shuttled into the cytosol by NBCs to buffer intracellular pH, in a reaction that regenerates CO2 and H2O inside the NP cell. This model of HCO3− recycling is supported by our findings that clearly showed decreased intracellular pH in NP cells following both CA9/12 or NBC inhibition using specific pharmacological agents.
It must be appreciated that the exact molecular players of the HCO3− transport metabolon in NP cells are unknown. This term was coined to describe the coordinated efforts of common pH-regulating proteins to transport HCO3− across the cell membrane, and was first detected by the interactions between CAII and EA1, a Cl−/HCO3− exchanger in erythrocytes (58). Investigations of membrane associated HCO3− transporters in the disc, however, is seldom. In the present study, however, we confirmed the expression of four membrane-associated bicarbonate transporters, namely NBCe1, NBCn1, AE2, and Pendrin, a rare enzyme commonly associated with thyroid pathology (35). While these results are interesting, further research into the coordinated HCO3− transporters that work in tandem with CA9/12 is essential for a thorough understanding of pH regulation in the NP. Concerning the fate of extracellular H+ units produced along with HCO3− by CA9 and CA12 catalytic reaction, we hypothesize that they will only be transiently available in the pericellular space and will be secreted out of the disc or, alternatively, could be used as a form of energy currency. In this sense, extracellularly facing CA9 and CA12 may act as H+ donors for nearby membrane associated co-transporters (such as MCTs) that also function in the complex network of pH sensors (59, 60).
Our results suggested that in NP cells, extracellularly-facing and membrane-associated CA9 and CA12 isoforms serve as a critical line of defense against accumulation of intracellular H+. Since plasma-membrane H+ extrusion systems alone may not be sufficient in maintaining physiological pHi in NP cells (11) and the otherwise ubiquitous CA2 isozyme is not as highly expressed (GEO Accession #GSE86552) (25), generation of pericellular HCO3− ions by CA9/12 assumes high significance. This mechanism of pH regulation is both essential and unique to NP cells which are prone to high intracellular lactate and H+ concentrations. Without the essential activity of CA9 and CA12, NP cells will have compromised survival in the hypoxic disc environment, a notion supported by our viability measurements following long-term CA inhibition. In summary, the HIF-1α-Carbonic anhydrase axis is critical for NP cell survival and alteration in their expression and activity will compromise tissue function and exacerbate the diseased state.
Supplementary Material
Acknowledgments
This work is supported by grants from the National Institutes of Health AR055655 and AR064733 (MVR). Elizabeth Silagi is supported by T32 AR052273.
Footnotes
DISCLOSURES: None for all authors.
Conflict of Interest: The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions: ESS, ES, IMS, and MVR conceived the study. ESS, ZRS, ELS, and CM conducted the experiments, analyzed data, and wrote the manuscript. IMS designed the study, wrote the manuscript, and secured funding. MVR designed experiments, interpreted results, secured funding, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
References
- 1.Katz JN. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J Bone Joint Surg Am. 2006;88(Suppl 2):21–24. doi: 10.2106/JBJS.E.01273. [DOI] [PubMed] [Google Scholar]
- 2.Murray CJL. The State of US Health, 1990–2010. JAMA. 2013;310:591. doi: 10.1001/jama.2013.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson ZI, Shapiro IM, Risbud MV. Extracellular osmolarity regulates matrix homeostasis in the intervertebral disc and articular cartilage: evolving role of TonEBP. Matrix Biol. 2014;40:10–6. doi: 10.1016/j.matbio.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Risbud MV, Guttapalli A, Stokes DG, Hawkins D, Danielson KG, Schaer TP, Albert TJ, Shapiro IM. Nucleus pulposus cells express HIF-1 alpha under normoxic culture conditions: a metabolic adaptation to the intervertebral disc microenvironment. J Cell Biochem. 2006;98:152–159. doi: 10.1002/jcb.20765. [DOI] [PubMed] [Google Scholar]
- 5.Fujita N, Chiba K, Shapiro IM, Risbud MV. HIF-1alpha and HIF-2alpha degradation is differentially regulated in nucleus pulposus cells of the intervertebral disc. J Bone Min Res. 2012;27:401–412. doi: 10.1002/jbmr.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruber HE, Ashraf N, Kilburn J, Williams C, Norton HJ, Gordon BE, Hanley EN. Vertebral endplate architecture and vascularization: application of micro-computerized tomography, a vascular tracer, and immunocytochemistry in analyses of disc degeneration in the aging sand rat. Spine (Phila Pa 1976) 2005;30:2593–600. doi: 10.1097/01.brs.0000187877.30149.83. [DOI] [PubMed] [Google Scholar]
- 7.Bibby SRS, Jones DA, Ripley RM, Urban JPG. Metabolism of the intervertebral disc: effects of low levels of oxygen, glucose, and pH on rates of energy metabolism of bovine nucleus pulposus cells. Spine (Phila Pa 1976) 2005;30:487–496. doi: 10.1097/01.brs.0000154619.38122.47. [DOI] [PubMed] [Google Scholar]
- 8.Urban JPG, Smith S, Fairbank JCT. Nutrition of the intervertebral disc. Spine (Phila Pa 1976) 2004;29:2700–9. doi: 10.1097/01.brs.0000146499.97948.52. [DOI] [PubMed] [Google Scholar]
- 9.Bartels EM, Fairbank JC, Winlove CP, Urban JP. Oxygen and lactate concentrations measured in vivo in the intervertebral discs of patients with scoliosis and back pain. Spine (Phila Pa 1976) 1998;23:1–7. doi: 10.1097/00007632-199801010-00001. discussion 8. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal A, Guttapalli A, Narayan S, Albert TJ, Shapiro IM, Risbud MV. Normoxic stabilization of HIF-1alpha drives glycolytic metabolism and regulates aggrecan gene expression in nucleus pulposus cells of the rat intervertebral disk. Am J Physiol Cell Physiol. 2007;293:C621–C631. doi: 10.1152/ajpcell.00538.2006. [DOI] [PubMed] [Google Scholar]
- 11.Razaq S, Urban JP, Wilkins RJ. Regulation of intracellular pH by bovine intervertebral disc cells. Cell Physiol Biochem. 2000;10:109–15. doi: 10.1159/000016340. [DOI] [PubMed] [Google Scholar]
- 12.Diamant B, Karlsson J, Nachemson A. Correlation between lactate levels and pH in discs of patients with lumbar rhizopathies. Experientia. 1968;24:1195–1196. doi: 10.1007/BF02146615. [DOI] [PubMed] [Google Scholar]
- 13.Nachemson A. Intradiscal Measurements of pH in Patients with Lumbar Rhizopathies. Acta Orthop Scand. 1969;40:23–42. doi: 10.3109/17453676908989482. [DOI] [PubMed] [Google Scholar]
- 14.Ohshima H, Urban JP. The effect of lactate and pH on proteoglycan and protein synthesis rates in the intervertebral disc. Spine (Phila Pa 1976) 1992;17:1079–82. doi: 10.1097/00007632-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Razaq S, Wilkins RJ, Urban JPG. The effect of extracellular pH on matrix turnover by cells of the bovine nucleus pulposus. Eur Spine J. 2003;12:341–349. doi: 10.1007/s00586-003-0582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilbert HTJ, Hodson N, Baird P, Richardson SM, Hoyland JA. Acidic pH promotes intervertebral disc degeneration: Acid-sensing ion channel -3 as a potential therapeutic target. Sci Rep. 2016;6:37360. doi: 10.1038/srep37360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007;26:299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- 18.Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene. 2010;29:6509–6521. doi: 10.1038/onc.2010.455. [DOI] [PubMed] [Google Scholar]
- 19.McIntyre A, Hulikova A, Ledaki I, Snell C, Singleton D, Steers G, Seden P, Jones D, Bridges E, Wigfield S, Li J-L, Russell A, Swietach P, Harris AL. Disrupting Hypoxia-Induced Bicarbonate Transport Acidifies Tumor Cells and Suppresses Tumor Growth. Cancer Res. 2016;76:3744–3755. doi: 10.1158/0008-5472.CAN-15-1862. [DOI] [PubMed] [Google Scholar]
- 20.McMurtrie HL, Cleary HJ, Alvarez BV, Loiselle FB, Sterling D, Morgan PE, Johnson DE, Casey JR. The bicarbonate transport metabolon. J Enzyme Inhib Med Chem. 2004;19:231–6. doi: 10.1080/14756360410001704443. [DOI] [PubMed] [Google Scholar]
- 21.Parks SK, Chiche J, Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13:611–23. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- 22.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 23.Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev. 1967;47:595–781. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- 24.Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta - Bioenerg. 2015;1847:171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Johnson ZI, Shapiro IM, Risbud MV. RNA sequencing reveals a role of TonEBP in regulation of pro-inflammatory genes in response to hyperosmolarity in healthy nucleus pulposus cells: A homeostatic response? J Biol Chem. 2016 doi: 10.1074/jbc.M116.757732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markova DZ, Kepler CK, Addya S, Murray HB, Vaccaro AR, Shapiro IM, Greg Anderson D, Albert TJ, Risbud MV. An organ culture system to model early degenerative changes of the intervertebral disc II: profiling global gene expression changes. Arthritis Res Ther. 2013;15:R121. doi: 10.1186/ar4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao T-H, Peng Y-J, Tsou H-K, Salter DM, Lee H-S. Nerve growth factor promotes expression of novel genes in intervertebral disc cells that regulate tissue degradation. J Neurosurg Spine. 2014;21:653–661. doi: 10.3171/2014.6.SPINE13756. [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto T, Muneta T, Tabuchi T, Matsumoto K, Saito H, Tsuji K, Sekiya I. Intradiscal transplantation of synovial mesenchymal stem cells prevents intervertebral disc degeneration through suppression of matrix metalloproteinase-related genes in nucleus pulposus cells in rabbits. Arthritis Res Ther. 2010;12:R206. doi: 10.1186/ar3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009;69:358–368. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- 30.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83. [PubMed] [Google Scholar]
- 31.Švastová E, Hulíková A, Rafajová M, Zat’Ovičová M, Gibadulinová A, Casini A, Cecchi A, Scozzafava A, Supuran CT, Pastorek J, Pastoreková S. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004;577:439–445. doi: 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 32.Kallio H, Pastorekova S, Pastorek J, Waheed A, Sly WS, Mannisto S, Heikinheimo M, Parkkila S. Expression of carbonic anhydrases IX and XII during mouse embryonic development. BMC Dev Biol. 2006;6:22. doi: 10.1186/1471-213X-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kivelä A, Parkkila S, Saarnio J, Karttunen TJ, Kivelä J, Parkkila A-K, Waheed A, Sly WS, Grubb JH, Shah G, Türeci Ö, Rajaniemi H. Expression of a Novel Transmembrane Carbonic Anhydrase Isozyme XII in Normal Human Gut and Colorectal Tumors. Am J Pathol. 2000;156:577–584. doi: 10.1016/S0002-9440(10)64762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMurtrie HL, Cleary HJ, Alvarez BV, Loiselle FB, Sterling D, Morgan PE, Johnson DE, Casey JR. The bicarbonate transport metabolon. J Enzyme Inhib Med Chem. 2004;19:231–6. doi: 10.1080/14756360410001704443. [DOI] [PubMed] [Google Scholar]
- 35.Cordat E, Casey JR. Bicarbonate transport in cell physiology and disease. Biochem J. 2009;417:423–439. doi: 10.1042/BJ20081634. [DOI] [PubMed] [Google Scholar]
- 36.Boron WF, Chen L, Parker MD. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol. 2009;212:1697–706. doi: 10.1242/jeb.028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schoepflin ZR, Shapiro IM, Risbud MV. Class I and IIa HDACs Mediate HIF-1α Stability through PHD2-Dependent Mechanism while HDAC6, a Class IIb Member, Promotes HIF-1α Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. J Bone Miner Res. 2016 doi: 10.1002/jbmr.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gogate SS, Fujita N, Skubutyte R, Shapiro IM, Risbud MV. Tonicity enhancer binding protein (TonEBP) and hypoxia-inducible factor (HIF) coordinate heat shock protein 70 (Hsp70) expression in hypoxic nucleus pulposus cells: Role of Hsp70 in HIF-1?? degradation. J Bone Miner Res. 2012;27:1106–1117. doi: 10.1002/jbmr.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran CM, Fujita N, Huang BL, Ong JR, Lyons KM, Shapiro IM, Risbud MV. Hypoxia-inducible factor (HIF)-1alpha and CCN2 form a regulatory circuit in hypoxic nucleus pulposus cells: CCN2 suppresses HIF-1alpha level and transcriptional activity. J Biol Chem. 2013;288:12654–12666. doi: 10.1074/jbc.M112.448860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suyama K, Silagi ES, Choi H, Sakabe K, Mochida J, Shapiro IM, Risbud MV, Suyama K, Silagi ES, Choi H, Sakabe K, Mochida J, Shapiro IM, Risbud MV. Circadian factors BMAL1 and RORα control HIF-1α transcriptional activity in nucleus pulposus cells: implications in maintenance of intervertebral disc health. Oncotarget. 2016;7:23056–23071. doi: 10.18632/oncotarget.8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita N, Gogate SS, Chiba K, Toyama Y, Shapiro IM, Risbud MV. Prolyl hydroxylase 3 (PHD3) modulates catabolic effects of tumor necrosis factor-alpha (TNF-α) on cells of the nucleus pulposus through co-activation of nuclear factor kappa-B (NF-|B)/p65 signaling. J Biol Chem. 2012;287:39942–39953. doi: 10.1074/jbc.M112.375964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Power KA, Grad S, Rutges JPHJ, Creemers LB, van Rijen MHP, O’Gaora P, Wall JG, Alini M, Pandit A, Gallagher WM. Identification of cell surface-specific markers to target human nucleus pulposus cells: expression of carbonic anhydrase XII varies with age and degeneration. Arthritis Rheum. 2011;63:3876–86. doi: 10.1002/art.30607. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Fang XQ, Wang Q, Wang SW, Hu ZJ, Zhou ZJ, Xu WB, Wang JY, Qin A, Fan SW. PHD/HIF-1 upregulates CA12 to protect against degenerative disc disease: a human sample, in vitro and ex vivo study. Lab Invest. 2016 doi: 10.1038/labinvest.2016.32. [DOI] [PubMed] [Google Scholar]
- 44.Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J Med Chem. 2011;54:1896–1902. doi: 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- 45.Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Perez SF, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnóczky G. MICU1 Controls Both the Threshold and Cooperrative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013;17:976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Risbud MV, Schoepflin ZR, Mwale F, Kandel RA, Grad S, Iatridis JC, Sakai D, Hoyland JA. Defining the phenotype of young healthy nucleus pulposus cells: Recommendations of the spine research interest group at the 2014 annual ORS meeting. J Orthop Res. 2015;33:283–293. doi: 10.1002/jor.22789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merceron C, Mangiavini L, Robling A, Wilson TL, Giaccia AJ, Shapiro IM, Schipani E, Risbud MV. Loss of HIF-1?? in the notochord results in cell death and complete disappearance of the nucleus pulposus. PLoS One. 2014 doi: 10.1371/journal.pone.0110768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathelier A, Fornes O, Arenillas DJ, Chen C, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, Zhang AW, Parcy F, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Windt GJW, Chang CH, Pearce EL. Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr Protoc Immunol. 2016;113(3.16B.1) doi: 10.1002/0471142735.im0316bs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan B, Xiao H, Li F, Zeng L, Yin Y. The profiles of mitochondrial respiration and glycolysis using extracellular flux analysis in porcine enterocyte IPEC-J2. Anim Nutr. 2015;1:239–243. doi: 10.1016/j.aninu.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett. 2010;20:3467–74. doi: 10.1016/j.bmcl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J Med Chem. 2011;54:1896–1902. doi: 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- 53.Harju A-K, Bootorabi F, Kuuslahti M, Supuran CT, Parkkila S. Carbonic anhydrase III: A neglected isozyme is stepping into the limelight. J Enzyme Inhib Med Chem. 2012;28:1–9. doi: 10.3109/14756366.2012.700640. [DOI] [PubMed] [Google Scholar]
- 54.Dimmer KS, Friedrich B, Lang F, Deitmer JW, Bröer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350(Pt 1):219–27. [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan PE, Pastorekova S, Stuart-Tilley AK, Alper SL, Casey JR. Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. AJP Cell Physiol. 2007;293:C738–C748. doi: 10.1152/ajpcell.00157.2007. [DOI] [PubMed] [Google Scholar]
- 56.Türeci O, Sahin U, Vollmar E, Siemer S, Göttert E, Seitz G, Parkkila AK, Shah GN, Grubb JH, Pfreundschuh M, Sly WS. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci U S A. 1998;95:7608–13. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaluz S, Kaluzová M, Liao SY, Lerman M, Stanbridge EJ. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochim Biophys Acta - Rev Cancer. 2009;1795:162–172. doi: 10.1016/j.bbcan.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vince JW, Reithmeier RA. Carbonic anhydrase II binds to the carboxyl terminus of human band 3, the erythrocyte C1-/HCO3- exchanger. J Biol Chem. 1998;273:28430–7. doi: 10.1074/jbc.273.43.28430. [DOI] [PubMed] [Google Scholar]
- 59.Jamali S, Klier M, Ames S, Barros LF, McKenna R, Deitmer JW, Becker HM. Hypoxia-induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non-catalytic function. Sci Rep. 2015;5:13605. doi: 10.1038/srep13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stridh MH, Alt MD, Wittmann S, Heidtmann H, Aggarwal M, Riederer B, Seidler U, Wennemuth G, McKenna R, Deitmer JW, Becker HM. Lactate flux in astrocytes is enhanced by a non-catalytic action of carbonic anhydrase II. J Physiol. 2012;590:2333–51. doi: 10.1113/jphysiol.2011.220152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.