

SUMMARY
Accumulation and aggregation of amyloid-β (Aβ) in the brain is an initiating step in the pathogenesis of Alzheimer’s disease (AD). The ε4 allele of apolipoprotein E (apoE) gene is the strongest genetic risk factor for late-onset AD. Although there is strong evidence showing that apoE4 enhances amyloid pathology, it is not clear what is the critical stage(s) during amyloid development apoE4 has the strongest impact. Using apoE inducible mouse models, we show that increased expression of astrocytic apoE4, but not apoE3, during the seeding stage of amyloid development enhanced amyloid deposition and neuritic dystrophy in amyloid model mice. ApoE4, but not apoE3, significantly increased brain Aβ half-life measured by in vivo microdialysis. Furthermore, apoE4 expression increased whereas apoE3 reduced amyloid-related gliosis in the mouse brains. Together, our results demonstrate that apoE4 has the greatest impact on amyloid during the seeding stage, likely by perturbing Aβ clearance and enhancing Aβ aggregation.
In Brief
Liu et al. have developed cell type-specific and inducible apoE mouse models and demonstrate that astrocytic apoE4 is a potent factor in promoting amyloidosis during the seeding stage but not the rapid growth period of amyloid development. ApoE4 impairs Aβ clearance and accelerates Aβ aggregation leading to enhanced amyloid pathology and neuritic dystrophy.
INTRODUCTION
Alzheimer’s disease (AD), the leading cause of dementia in the elderly, is an age-dependent neurological disorder characterized by the presence of extracellular amyloid plaques composed of amyloid-β (Aβ) and intracellular neurofibrillary tangles (Jucker and Walker, 2011). Mounting evidence supports a role of Aβ accumulation and aggregation as an early trigger of a toxic cascade leading to eventual neurodegeneration in the etiology of AD (Hardy and Selkoe, 2002). Amyloid positivity in cognitive normal individuals was shown to be associated with rates of Aβ deposition and hippocampal atrophy (Villemagne et al., 2013). Emerging findings indicate that Aβ begins to deposit in the brains of dementia patients decades before the onset of clinical symptoms (Bateman et al., 2012; Perrin et al., 2009). Identification of 20–30 year interval between amyloid positivity and dementia implies that there is window of opportunity to intervene as prevention strategies for AD (Jansen et al., 2015). The growth kinetics of amyloid plaques follows a sigmoid-shaped progression with time in amyloid mouse models as well as in humans (Bateman et al., 2012; Burgold et al., 2014). Amyloid pathogenesis was suggested to start with a slow nucleation phase that leads to formation of small aggregates (seeding stage). After that, the extension of these Aβ aggregates occurs more rapidly during amyloid growth period (Eisenberg and Jucker, 2012). Identifying the factors that promote amyloid seeding should help to understand the mechanism underlying the onset of AD and suggest strategies for early intervention.
Apolipoprotein E (apoE), mainly produced by astrocytes in the central nervous system, is a major cholesterol carrier that delivers lipids to neurons for the maintenance synapses, as well as promoting injury repair (Bu, 2009). The ε4 allele of the APOE gene is the strongest genetic risk factor for late-onset AD among its three polymorphic alleles (ε2, ε3 and ε4). APOE4 confers a greater AD risk with an earlier age of disease onset in a gene dose-dependent manner (Corder et al., 1993; Liu et al., 2013). Aβ deposition as senile plaques is more abundant in APOE4 carriers than in non-carriers (Kok et al., 2009). APOE4 gene dose is associated with fibrillar Aβ burden, suggesting that apoE4 might promote Aβ aggregation (Morris et al., 2010; Reiman et al., 2009). Although studies indicate that apoE4 enhances the accumulation and deposition of Aβ in the brain, the critical period when apoE4 has the strongest effects on amyloid pathology remains unclear.
We have developed innovative mouse models, allowing human apoE3 or apoE4 to be expressed specifically in astrocytes in an inducible fashion during different stages of amyloid plaque development. We revealed a crucial pathogenic role of apoE4 in the seeding of amyloids in vivo. Together, our study provides crucial evidence that apoE4 facilitates amyloid development during the early stage, gaining biological insights into apoE-targeted therapies to suppress amyloid pathogenesis to treat AD.
RESULTS
Novel mouse models expressing human apoE3 or apoE4 in a cell type-specific and inducible manner
The apoE-targeted replacement (TR) mice have allowed for studies of human apoE in AD pathogenesis. However, these mice alone provide limited information on how changes in apoE isoform expression in adult mice and in specific cell types impact these events. To address this, we have developed novel mouse models that allow for cell type-specific and inducible expression of apoE3 or apoE4 (termed iE3 and iE4 where “i” means inducible). The apoE3 or apoE4 transgene was targeted into the ROSA locus by homologous recombination (Figure 1A), ensuring controlled expression with no impact on the expression of other genes. To understand how astrocytic apoE isoforms affect amyloid pathogenesis, we first bred the iE3 and iE4 mice with mice expressing glial fibrillary acidic protein (GFAP)-Cre transgene, which drives expression in astrocytes, the predominant cell type that produces apoE in brain parenchyma. Western blotting and ELISA showed that both apoE3 and apoE4 were highly expressed in the brain of Cre+ mice but not in Cre− mice (Figures 1B and 1C). GFP immunostaining also confirmed expression in astrocytes of Cre+ mice (Figure 1D). Two weeks after feeding of doxycycline (Dox) chow, apoE expression in Cre+ mice was completely turned off and returned to the baseline level (Figure 1E), confirming the effectiveness of the Tet-off system and functionality of our iE3 and iE4 mice. To investigate the effects of apoE isoforms on amyloid development, we further bred astrocyte-specific iE3 and iE4 mice into the background of APPSWE/PS1ΔE9 mice (thereafter referred to as APP/PS1) (Jankowsky et al., 2004). Compared with apoE-TR mice, apoE inducible mice exhibited 2–3 fold increases in apoE levels (Figure 1F). In addition, GFP signal which represents the expression pattern of apoE was co-localized with GFAP-positive astrocytes, but not with NeuN-positive neurons (Figure 1G; Figure S1A), confirming the specificity of the Cre driver.
Figure 1. Characterization of Cell Type-specific and Inducible ApoE Mice. See also Figure S1.
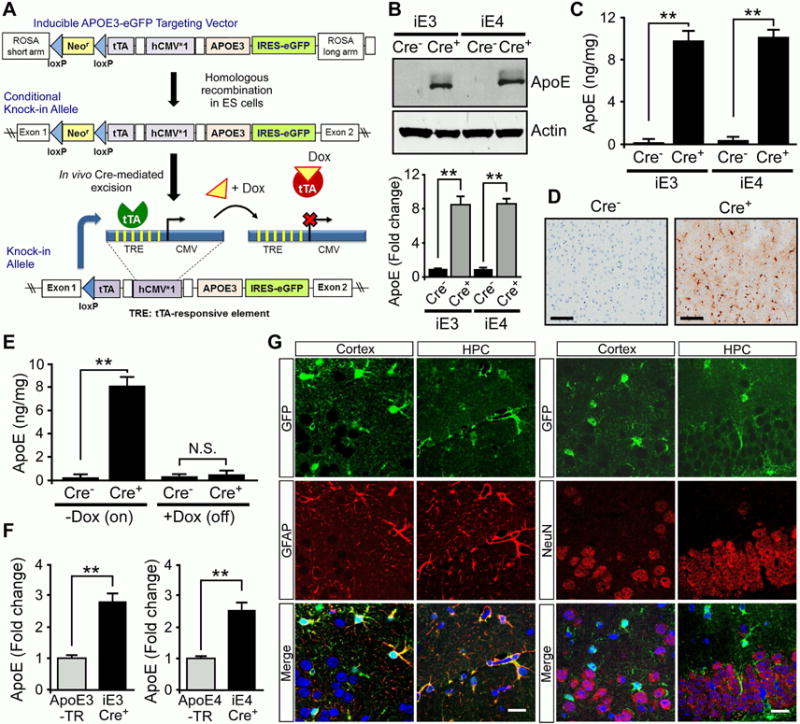
(A) The structure of the ROSA-targeting vector with the Tet-off regulation cassette for APOE and eGFP expression. The resultant mice were named iE3 and iE4. Breeding iE3 or iE4 mice with GFAP-Cre mice which removed the loxP-flanked Neor gene led to expression of apoE3 or apoE4 in astrocytes.
(B, C) ApoE levels in the cortex of iE3 and iE4 mice at 3 months of age with or without GFAP-Cre were analyzed by Western blotting (B) and ELISA (C).
(D) Immunohistochemical analysis of GFP for the cortical region of iE3 mice in the presence or absence of Cre.
(E) The iE3 mice at 3 months of age were fed with regular (−Dox) or Dox-containing chow for 2 weeks. ApoE in the cortex was analyzed by ELISA. Data represent mean ± SEM. **, p<0.01; N.S., not significant.
(F) Comparison of apoE levels in the cortex of apoE inducible amyloid model mice with apoE-TR mice at 3–4 months of age examined by ELISA. Data represent mean ± SEM. **, p<0.01.
(G) Cortex and hippocampus (HPC) from apoE3 inducible (Cre+) amyloid model mice were co-immunostained for GFP which represents apoE distribution (green) and astrocyte-specific (anti-GFAP; red) or neuron-specific (anti-NeuN; red) markers. Scale bar, 100 μm.
Astrocytic expression of apoE4 during the initial seeding stage but not the rapid growth period enhances amyloid pathology and neuritic dystrophy
Previous studies have shown that brain Aβ levels and amyloid plaque loads are apoE isoform-dependent (E4>E3≥E2) (Castellano et al., 2011; Mawuenyega et al., 2010). However, it is not clear at what stage of amyloid development and progression apoE has the highest impact. We chose to address this question using our inducible apoE mouse models bred to APP/PS1 mice which start developing visible amyloid pathology at 4~5 months of age, having the fastest amyloid growth between 6–9 months, and reaching near saturation at ~10 months of age (Garcia-Alloza et al., 2006). To investigate the effects of apoE isoforms on Aβ metabolism and amyloid pathogenesis, we designed three paradigms for controlled apoE expression: 1) the entire 9 months (0–9 m On); 2) during the seeding stage only (0–6 m On); or 3) during the rapid growth period only (6–9 m On) (Figure 2A). These experimental mice grew normally and showed no overt developmental abnormalities in the brain (Figure S1B). As expected, the apoE levels in the brain were dramatically increased in both detergent-soluble (RIPA) and -insoluble (guanidine, GDN) fractions in Cre+ mice with apoE expression for the entire 9 months, and during 6–9 months compared to Cre− mice (Figures 2B and 2C; Figures S1C and S1D). For mice with apoE expression during 0–6 months, the apoE level was low upon Dox treatment which turned off its expression. Interestingly, we found that expression of astrocytic apoE4 for the entire 9 months significantly increased soluble and insoluble Aβ40 and Aβ42 levels, whereas expression of apoE3 did not have such effects (Figure 2D; Figures S1E-G). We next investigated the critical stage by which apoE4 drives the increase of Aβ. Importantly, we found that expression of apoE4 in the Aβ seeding stage (0–6 m On) significantly increased Aβ40 and Aβ42, whereas expression of apoE4 during the plaque rapid growth period (6–9 m On) had no significant effects on Aβ levels (Figures 2E and 2F; Figures S1H and S1I).
Figure 2. Astrocytic ApoE4 Increases Insoluble Aβ and Amyloid Pathology When Expressed Early. See also Figure S1-S3.
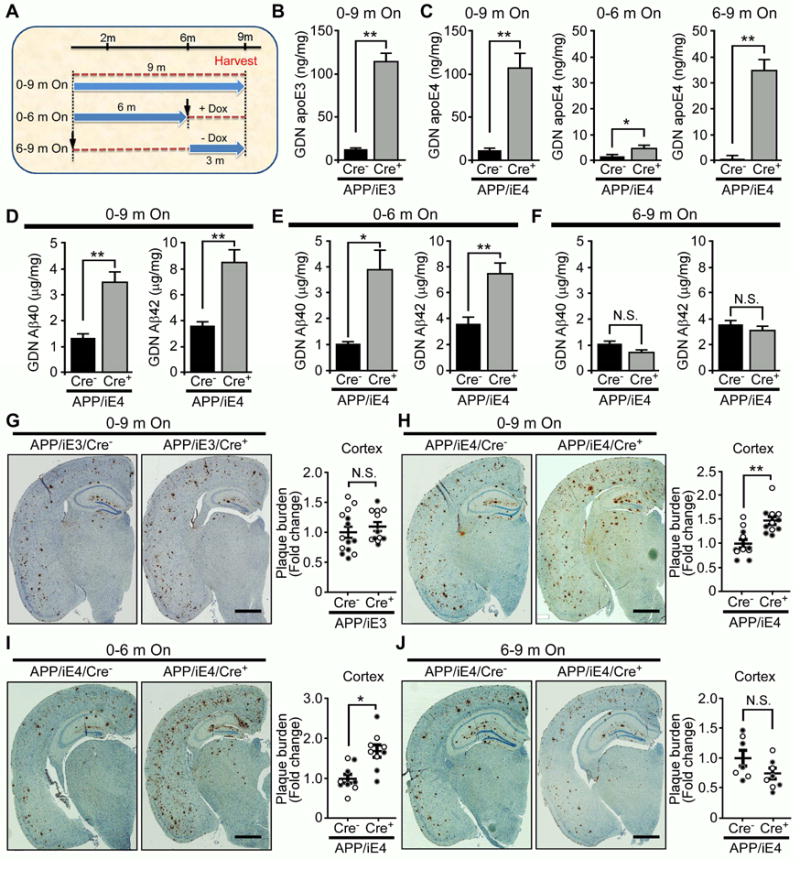
(A) Illustration of apoE induction paradigms at different stages of amyloid pathology. (B, C) Insoluble apoE levels in the cortex of APP/PS1 mice expressing (B) apoE3 (APP/iE3; n=17–18/group) throughout 9 months (0–9 m On) or (C) apoE4 (APP/iE4; n=13–16/group) throughout 9 months, during 0–6 months (0–6 m On; n=6–8/group) or during 6–9 months (6–9 m On; n=8–9/group) were examined by ELISA. Data represent mean ± SEM. *, p<0.05; **, p<0.01. (D–F) Insoluble Aβ40 and Aβ42 levels in the cortex of APP/PS1 mice expressing apoE4 (APP/iE4; n=13–16/group) throughout the entire 9 months, during 0–6 months (n=6–8/group) or during 6–9 months (n=8–9/group) were examined by specific Aβ ELISA. Data represent mean ± SEM. *, p<0.05; **, p<0.01; N.S., not significant.
(G–J) Brain sections from 9-month-old APP/PS1 mice expressing apoE3 (APP/iE3; n=12–14/group), or apoE4 (APP/iE4) throughout the entire 9 months (n=11/group), during 0–6 months (n=9/group) or 6–9 months (n=7–8/group) were immunostained with a pan-Aβ antibody. Representative images of Aβ staining in the cortical and hippocampal regions are shown. Scale bar, 1 mm. Open circles are females; closed circles are males. Data represent mean ± SEM. *, p<0.05; **, p<0.01; N.S., not significant.
We next examined how astrocytic apoE isoforms expressed at different stages impact the development of amyloid pathology. We found that expression of astrocytic apoE4, but not apoE3, for the entire 9 months enhanced amyloid deposition (Figures 2G and 2H; Figures S2A and S2B). More importantly, expression of apoE4 during the seeding stage (0–6 m On), but not the rapid growing period (6–9 m On), was sufficient to promote amyloid pathology (Figures 2I and 2J; Figure S2B). Interestingly, expression of astrocytic apoE3 during the seeding stage did not significantly affect Aβ levels and amyloid deposition in the brain (Figures S2C–S2F). To investigate which Aβ species might be involved in apoE4-mediated effect, we examined whether astrocytic apoE4 affects soluble Aβ oligomer formation in the brains of APP/iE4 mice at different stages. Interesting, we found that apoE4 increased the levels of Aβ oligomers when it was expressed during seeding stage (0–6 m On), but not in the rapid growing period (6–9 m On) (Figure S2G). Together, these results indicate the critical roles of apoE4 in the Aβ seeding, oligomerization and amyloid deposition.
We next examined whether there are changes in the formation of cerebral amyloid angiopathy (CAA) in our mouse models expressing astrocytic apoE isoforms. We found that expression of astrocytic apoE3 or apoE4 did not significantly affect the levels of leptomeningeal CAA in any induction paradigms (Figure S2H). These results indicate that astrocytic apoE plays a critical role in regulating amyloid pathogenesis in the brain parenchyma, but not in cerebrovasculature.
Previous studies suggest a critical role of apoE in Aβ fibrillogenesis (Bales et al., 1997). Thus, we next characterized how astrocytic apoE isoforms impact the formation of fibrillary amyloid plaques in inducible apoE mice by Thioflavin S staining. Consistent with the Aβ immunostaining pattern, expression of astrocytic apoE3 had no effect on the fibrillar plaque load (Figure S3A). In addition, expression of apoE4 for the entire 9 months and during the seeding stage (0–6 m On), but not the rapid growth period (6–9 m On), led to significant increases in fibrillar plaque formation in the cortex and hippocampus (Figures S3B-D). Together, these results support that apoE4 plays a crucial role in driving amyloid pathology during the initial phase of amyloid development.
Neuronal dystrophy is a pathological hallmark of AD and has been shown to be induced by fibrillar Aβ (Grace and Busciglio, 2003). To examine whether astrocytic apoE isoforms affect amyloid-associated neuritic dystrophy, we performed double immunostaining for Aβ and lysosomal-associated membrane protein 1 (LAMP1), a dystrophic neurite marker, in brain sections from the APP/iE3 and APP/iE4 mice. Mice expressing apoE4 for the entire 9 months (0–9 m On) and during seeding stage (0–6 m On) had a significant increase in the dystrophic neurites surrounding amyloid plaques compared with control mice (Figures S3E–H). As expected, mice expressing astrocytic apoE3 for 9 months or apoE4 during plaque growing period did not significantly affect amyloid burden and plaque-associated neuritic dystrophy (Figures S3I and S3J). These results indicate that astrocytic apoE isoforms have differential impacts on amyloid deposition as well as neuritic dystrophy around plaques.
Expression of apoE4 in astrocytes suppresses Aβ clearance
We next examined whether expression of human apoE isoforms in astrocytes differentially regulate Aβ clearance in the interstitial fluid (ISF) of young APP/iE3 and APP/iE4 mice. Using in vivo microdialysis to analyze the elimination kinetics of Aβ after halting Aβ production, we found that expression of apoE3 did not affect the clearance rate of Aβ (Figures 3A and 3B). Intriguingly, the half-life of Aβ in the hippocampus of APP/iE4/Cre+ mice was significantly longer compared to APP/iE4/Cre− mice (Figures 3C and 3D), suggesting an impairment of Aβ clearance by astrocytic apoE4.
Figure 3. Induced ApoE4 Expression in Astrocytes Impairs ISF Aβ Clearance in the Hippocampus of APP/PS1 Mice.
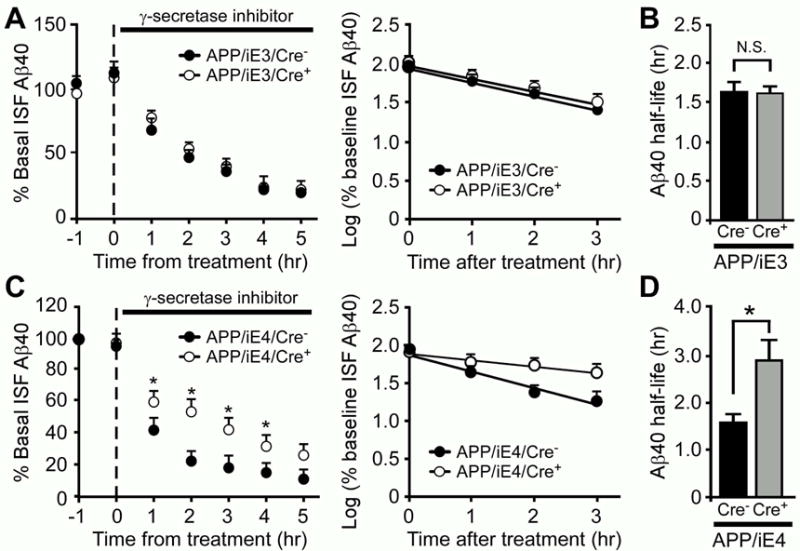
The ISF Aβ40 levels in APP/PS1 mice expressing apoE3 (A-B, APP/iE3; n=8/group) or apoE4 (C–D, APP/iE4; n=4/group) at the age of 3–4 months were analyzed. To assess Aβ40 half-life, the mice were treated with a γ-secretase inhibitor, and the hippocampal ISF Aβ40 levels were monitored. The slope from the individual linear regressions from log (% ISF Aβ40) versus time for each mouse was used to calculate the mean half-life (t1/2) of elimination for Aβ from the ISF (B, D). Data represent mean ± SEM. *, p<0.05; N.S., not significant.
Expression of human apoE isoforms differentially affects Aβ-associated gliosis and synaptic integrity
Prominent activation of astrocytes and microglia along with the presence of amyloid plaques and tangles are observed in AD brains (Heneka et al., 2015). To determine the extent of Aβ-mediated gliosis upon expression of astrocytic apoE, we examined GFAP-positive reactive astrocytes in APP/iE3 and APP/iE4 mice by immunostaining and Western blotting. Interestingly, APP/iE3/Cre+ mice displayed significantly lower GFAP-positive astrocytes than those in APP/iE3/Cre− mice (Figures 4A and 4E), implying an anti-inflammatory function of apoE3. Conversely, GFAP-positive astrogliosis was enhanced in APP/iE4/Cre+ mice compared to APP/iE4/Cre− mice (Figures 4B and 4E). Regression analysis indicated a significant positive correlation between the levels of GFAP and those of Aβ40 and Aβ42 (Figure S4A and S4B), suggesting that astrogliosis is related to Aβ pathology. We further examined how expression of apoE4 in different stages of amyloid development impacts astrogliosis. Consistent with the effects on amyloid, the GFAP levels were significantly enhanced in Cre+ mice with apoE4 expression in the seeding stage, but not the rapid growth period, during amyloid development (Figures S4C-S4E). A significant increase of ionized calcium-binding receptor 1 (Iba1)-positive microglia was also observed in APP/iE4/Cre+ mice but not in APP/iE3/Cre+ mice (Figures 4C and 4D). Furthermore, expression of astrocytic apoE3 decreased the levels of IL-6 and IL-1β, but not TNF-α, in APP/iE3/Cre+ mice (Figure S4F). In contrast, expression of astrocytic apoE4 increased IL-6, IL-1β and TNF-α in APP/iE4/Cre+ mice compared with control mice (Figure S4G). Consistent with the effects of apoE4 on amyloid seeding, the levels of IL-6 were significantly increased in Cre+ mice with apoE4 expression in the seeding stage, but not in the rapid growth period, during amyloid development (Figures S4H). Together, these results indicate that apoE isoforms differentially affect amyloid pathology and associated neuroinflammation.
Figure 4. ApoE Isoform-specific Effects on Aβ-associated Gliosis. See also Figure S4.
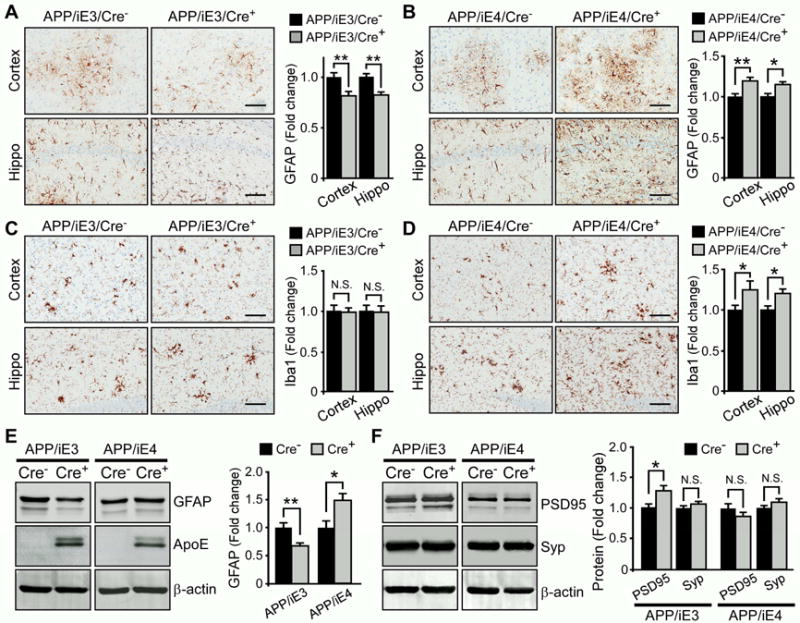
(A–D) Brain sections from 9-month-old APP/PS1 mice expressing apoE3 (APP/iE3) or apoE4 (APP/iE4) in the astrocytes throughout the entire 9 months were immunostained with GFAP antibody (A, B) or Iba1 antibody (C, D). Scale bar, 100 μm. The immunoreactivities of GFAP or Iba1 in cortex (n=11–13/group) and hippocampus (n=9–12/group) were quantified. Data represent mean ± SEM. *, p<0.05; **, p<0.01.
(E, F) The levels of GFAP, presynaptic marker synaptophysin (Syp) and postsynaptic marker PSD-95 in the cortex (n=16–18/group) of APP/PS1 mice expressing apoE3 (APP/iE3) or apoE4 (APP/iE4) in the astrocytes throughout the entire 9 months were examined by Western blotting. Data represent mean ± SEM. *, p<0.05; **, p<0.01; N.S., not significant.
To further understand how apoE isoforms affect the synaptic changes associated with Aβ, we examined the levels of pre- and post-synaptic markers in the brains of APP/iE3 and APP/iE4 mice. We found that the level of postsynaptic density 95 (PSD-95), but not synaptophysin, was slightly increased in APP/iE3/Cre+ mice, whereas no significant changes in the synaptic markers were observed in APP/iE4/Cre+ mice in any induction paradigm (Figure 4F; Figure S4E). This result suggests that apoE3 might ameliorate Aβ-mediated synaptic impairment.
DISCUSSION
APOE4 is the strongest genetic risk factor for late-onset AD likely by driving amyloid pathology; yet, how apoE4 exerts such effects during different stages of pathological development is unclear. Using novel inducible mouse models for apoE isoforms, we showed that expression of apoE4 in astrocytes during the initial seeding stage is sufficient to drive amyloid pathology and plaque-associated neuritic dystrophy. Conversely, if apoE4 is present after the initial seeding stage, it has minimal impact on amyloid pathology. Expression of apoE3 at any stage of the amyloid pathology does not alter amyloid plaque development but benefits synapses and reduces amyloid-associated gliosis. Our findings suggest that therapeutic interventions for asymptomatic APOE4-positive individuals who are predisposed to AD need to start early in the pre-symptomatic phase in terms of targeting the Aβ pathway. With the available genetic testing, as well as brain imaging and CSF-based biomarkers for AD risk assessment, our results support an effort of identifying high-risk individuals and designing prevention or early treatment strategies targeting apoE4-related pathogenic pathways.
Among individuals without dementia, APOE4 is associated with a greater risk and decreased age at onset for amyloid positivity (Jansen et al., 2015). Brain imaging and CSF biomarker studies also indicate that Aβ burden in cognitively normal older people is associated with APOE4 gene dosage (Morris et al., 2010; Reiman et al., 2009). The strong association of APOE4 with amyloid positivity emphasizes apoE as a critical target for both understanding its effects on amyloid and designing anti-Aβ therapy. Increasing studies indicate that Aβ deposition may begin more than two decades before clinically noticeable cognitive decline (Jack and Holtzman, 2013). It is suggested that when the amount of brain Aβ is above a certain critical concentration, it forms aggregates which can serve as nucleating seeds for the development of amyloid plaques. After the slow nucleation phase, the seeds can recruit more Aβ to form larger aggregates at a faster pace during the growth period of fibril formation until the late stage of AD when the deposition slows down (Burgold et al., 2014). Consistent with this notion, Aβ aggregation can be instigated and/or accelerated in young, pre-deposit amyloid model mice through introducing minute amounts of Aβ aggregates as seeds (Meyer-Luehmann et al., 2006). Understanding the seeding factor for Aβ aggregation could furnish clues to early and mechanism-based interventions. ApoE, a component of cerebral amyloid deposits, has been shown to play an important role in amyloid pathogenesis (Bales et al., 2009; Liu et al., 2013). Our findings and studies from others (Castellano et al., 2011; Deane et al., 2008) have demonstrated that apoE4 impairs Aβ clearance, which would lead to increased Aβ concentrations critical for the initiation of Aβ aggregation. A previous study reported that APOE4 carriers exhibited apoE in newly formed Aβ deposits more frequently than non-APOE4 carriers (Thal et al., 2005). In vitro aggregation study showed that apoE4 has stronger effects than apoE3 in enhancing Aβ fibrillogenesis (Wisniewski et al., 1994). Thus, apoE4 may accelerate amyloid development by serving as a potent seeding cofactor to enhance amyloid formation (Figure S5). Consistent with our findings, studies from Huynh et al. (co-submitted) utilizing an apoE antisense oligonucleotide (ASO) to reduce apoE4 expression in the brains of APP/PS1-21/apoE4/4 mice indicated that the timing of apoE4 expression greatly affects its impact on Aβ pathology. Specifically, ASO treatment starting at P0, but not 6 weeks of age (when significant amyloidosis has occurred in this aggressive amyloid model mice), can significantly decrease Aβ deposition. Altogether, results from ours and their studies demonstrate a critical role of apoE4 in the early and seeding stage of plaque formation. Protein aggregation has been implied as one of the common pathogenic mechanisms that impel neurodegenerative diseases, including AD (Jucker and Walker, 2011). ApoE4 is able to form neurotoxic aggregates more readily than apoE3 or apoE2 (Hatters et al., 2006); thus, it is possible that apoE4 could nucleate Aβ aggregation or plaque formation through its self-aggregating propensity. In addition, apoE4 might be potentially more vulnerable to aggregation due to its poor lipidation status (Gong et al., 2002). It will be informative to assess whether reducing the amount and aggregation of apoE4, and/or promoting its lipidation, particularly in the Aβ seeding stage can halt or reduce the development of amyloid pathology.
Brain Aβ clearance is relatively efficient with a half-life of 1–2 hours in mice and ~8 hours in humans (Bateman et al., 2006; Liu et al., 2016). Multiple pathways exist in brain for Aβ clearance, including cellular uptake, proteolytic degradation, and cerebrovasculature-mediated clearance pathways (Bu, 2009). While the specific contribution from each pathway is not clear, increasing evidence indicates that impaired Aβ clearance is a common prelude to late-onset AD (Mawuenyega et al., 2010). Astrocytes represent a major cell type mediating Aβ clearance in the brain. Using in vivo microdialysis, we showed that expression of apoE4 in astrocytes impairs Aβ clearance which is consistent with previous reports (Castellano et al., 2011; Deane et al., 2008). Aβ aggregation and deposition into insoluble plaques is concentration-dependent; thus, the compromised clearance of Aβ in apoE4 increases Aβ concentration which may contribute to Aβ seeding effect. It was shown that apoE4/Aβ complex is cleared at the blood-brain-barrier (BBB) with a substantially slower rate than when Aβ is free or bound to apoE3 (Deane et al., 2008). Another study reported that apoE isoforms differentially block Aβ clearance in astrocytes by competing for the same clearance pathways mediated by the low-density lipoprotein receptor-related protein 1 (LRP1). LRP1 and heparan sulfate proteoglycan (HSPG), two major receptors for both apoE and Aβ, have been shown to mediate Aβ clearance in neurons and various cell types (Bu, 2009; LaDu et al., 2006; Liu et al., 2016). Whether apoE isoforms modulate Aβ metabolism in distinct brain compartments and cell types depending on these receptors warrant further exploration.
Abnormal activation of neuroinflammation contributes to neuronal damage in the pathogenesis of AD (Heneka et al., 2015). Deficiency of apoE in mice led to increased inflammation in response to Aβ, suggesting that apoE may have anti-inflammatory effect (LaDu et al., 2001). Our results showed that the marker for astrocytic reactivity was slightly lower in APP/iE3/Cre+ mice than Cre− mice, indicating expression of apoE3 may reduce astrogliosis. Alternatively, astrogliosis is thought to develop as a response to plaque deposition. Although we did not observe significant differences in plaque burden between APP mice with or without apoE3 expression, it is possible that subtle but not significant changes in amyloid deposition contributed to the changes in astrogliosis. A study revealed an increased inflammatory response in young APOE4 carriers that may relate to AD risk later in life (Ringman et al., 2012). ApoE4 may have pro-inflammatory functions, less effective anti-inflammatory function, or both, which may further exacerbate AD pathogenesis. Consistent with this notion, we showed that astrocytic apoE4 expression enhances plaque-associated inflammatory responses, whereas increased levels of apoE3 suppress Aβ-driven neuroinflammation and ameliorates post-synaptic alteration. In addition, apoE isoforms have been shown to differentially regulate synaptic functions by multiple mechanisms, such as modulating the signaling function of post-synaptic receptors (Chen et al., 2010; Liu et al., 2015b). How expression of astrocytic apoE3 and apoE4 impacts synaptic functions and plasticity in the absence of amyloid pathology requires future investigation.
We examined the human astrocytic apoE3 or apoE4 effects on amyloid development in the context of murine apoE expression. Further studies are required to explore their functions in the background of human apoE. In summary, our findings using cell type-specific and inducible mouse models support that apoE4 has the greatest impact during the initial phase of amyloid plaque development. Thus, an effective disease-modifying therapy targeting Aβ and apoE4 should be introduced before massive amyloid buildup and irreversible neuronal damages occur. Gaining an in-depth knowledge of apoE4-mediated pathological process that instigates amyloid development might prove essential for the design, in particular the timing of apoE4-targeted disease-modifying therapies for AD.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Guojun Bu (bu.guojun@mayo.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
The cell type-specific and inducible apoE mouse models were generated by a knock-in strategy targeting the ROSA-26 locus with a vector system containing a floxed STOP cassette and tetracycline-regulatory elements initially provided by S. Miyazaki (Osaka University, Japan) (Miyazaki et al., 2005). The construct for apoE3 or apoE4 includes: 1) ROSA short and long arms at either ends for targeted integration of ROSA26 locus; 2) a floxed Neor STOP cassette to allow for Cre-mediated, cell type-specific expression; 3) Tet-off regulatory elements to allow for doxycycline-regulated apoE expression, and a CMV promoter with a tetracycline-responsive element (TRE); 4) human APOE3 or APOE4 cDNA; and 5) an enhanced green fluorescent protein (eGFP) cDNA driven by an internal ribosomal entry site (IRES). The constructs are termed inducible APOE3-eGFP and inducible APOE4-eGFP. The vector constructs were confirmed by restriction digestion and sequencing and electroporated into mouse ES cells. The individual ES clones were screened for homologous recombination and the positive clones were confirmed by Southern blotting. Three ES cell lines were chosen, verified by karyotyping, and used to generate apoE knock-in mice by blastocyst injection into C57BL/6J mice. The generation of inducible APOE3-eGFP and inducible APOE4-eGFP constructs, production and screening of the ES clones and the generation of chimeric mice were carried out by Transgenic Vectors Core, Mouse ES Cell Core, and Mouse Genetics Core at the Washington University, respectively. The resulting chimeras were bred with C57BL/6J mice, and the founder mice carrying a Tet-off cassette were identified as inducible apoE3 or apoE4 mice (named in short as iE3 and iE4 mice). To examine the effects of astrocytic apoE isoforms on Aβ pathology and related pathways, iE3 and iE4 mice were crossed with glial fibrillary acidic protein (GFAP)-driven Cre recombinase mice (obtained from NCI Mouse Repository) (Bajenaru et al., 2002), and further bred into the background of APPSWE/PS1ΔE9 (hereafter referred to as APP/PS1) amyloid mouse model (Jankowsky et al., 2004). Littermates (including both male and female mice) of control APP/PS1 (APP/iE3/Cre− or APP/iE4/Cre−) and APP/PS1 mice expressing apoE3 or apoE4 (APP/iE3/Cre+ or APP/iE4/Cre+) were used. We designed three paradigms for controlled apoE expression: 1) the entire 9 months (0–9 m On); 2) during the seeding stage only (0–6 m On); or 3) during the rapid growth period only (6–9 m On). To turn off the expression of apoE at specific periods of time, mice were fed on doxycycline (Dox)-impregnated chow (Bio-Serv). For the group of mice expressing apoE during the seeding stage only (0–6 m On), mice were fed with normal chow at 0–6 months of age and then were fed with Dox chow from 6–9 months of age to turn off the expression of apoE. For the group of mice expressing apoE during the rapid growth period only (6–9 m On), the parents were fed with Dox chow before pregnancy, and their offspring (the experimental mice) were kept on Dox chow until 6 months of age. Mice were housed in a temperature-controlled environment with a 12-h light–dark cycle and free access to food and water. All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Preparation of brain homogenates
Brain tissues were dissected and kept frozen at −80°C until further analysis. Some brain tissues were fixed in 10% neutralized formalin for histological analysis. Mouse brain tissues for biochemical analysis were processed through sequential extraction. Briefly, brain tissues were homogenized with RIPA buffer supplemented with protease inhibitor cocktail (cOmplete; Sigma) and PhosSTOP (Sigma) and incubated with mild agitation for 30 min at 4°C After centrifugation, the supernatant was referred to as RIPA-soluble fraction. The RIPA-insoluble pellet was re-suspended with 5 M guanidine hydrochloride (pH 7.6), incubated with mild agitation for 12–16 hr at room temperature, and centrifuged at 16,000g for 30 min (referred to as GDN-soluble fraction).
In vivo microdialysis
To assess the concentration of interstitial fluid (ISF) Aβ in the hippocampus of awake, freely moving mice, in vivo microdialysis was performed as previously described (Liu et al., 2016). Briefly, the animals were placed in an animal stereotaxic device equipped with dual manipulator arms and an isoflurane anesthetic mask (David Kopf Instruments). Under isoflurane volatile anesthetic, guide cannula (BR style; Bioanalytical Systems) were cemented into the hippocampus (3.1 mm behind bregma, 2.5 mm lateral to midline, and 1.2 mm below dura at a 12° angle). Four to six hours post-surgery, a microdialysis probe (30-kilodalton MWCO membrane, Bioanalytical Systems) was inserted through the guide cannula into the brain. Artificial cerebrospinal fluid (CSF) (mM: 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.4) containing 4% bovine serum albumin (BSA; Sigma) filtered through a 0.1 μm membrane was used as microdialysis perfusion buffer. Flow rate was a constant 1.0 μl/min. Samples were collected every 60–90 min overnight which gets through the 5–6 hr recovery period into a refrigerated fraction collector. The mean concentration of Aβ over the 6 hr preceding treatment was defined as basal levels of ISF Aβ. For each animal, all Aβ levels were normalized to the basal Aβ concentration. To assess Aβ40 half-life, the mice were treated subcutaneously with a γ-secretase inhibitor, LY411575 (5 mg/kg) to rapidly block the production of Aβ, and the hippocampal ISF Aβ40 levels were monitored and measured by ELISA (Liu et al., 2016).
Immunohistochemical and immunofluorescence staining
Paraffin-embedded sections were immunostained with pan-Aβ (Aβ 33.1.1; human Aβ 1-16 specific), anti-GFAP (BioGenex), and anti-ionized calcium-binding adaptor molecule 1 (Iba-1) (Wako) antibodies (Liu et al., 2016). Immunohistochemically stained sections were captured using the ImageScope AT2 image scanner (Aperio Technologies) and analyzed using the ImageScope software. The immunoreactivities of GFAP and Iba-1 staining in the hippocampus were calculated using the Positive Pixel Count Algorithm available with the ImageScope software (Aperio Technologies). Quantification of immunostaining was performed in a blinded manner, For CAA quantification, brain sessions were immunostained with pan-Aβ, and the burden of amyloid deposition in leptomeningeal arteries (13–15 arteries/mouse) was quantified by the Positive Pixel Count program (Aperio Technologies). The fibrillar Aβ was stained with Thioflavin S. The images were captured by Aperio Fluorescent Scanner and the stained areas were quantified by Image J. For double immunostaining, deparaffinized sections were pre-incubated with citrate buffer (10 mM sodium citrate buffer with 0.05% Tween 20, pH 6.0) at 95°C for 20 min. They were incubated at 4°C overnight with a rabbit anti-GFP antibody (Abcam) and a mouse anti-NeuN antibody (Millipore) or a mouse anti-GFAP antibody (Millipore), followed by Alexa488- or Alexa568-conjugated secondary antibodies (Thermo Fisher Scientific) for 2 hr at room temperature. The images were captured by confocal laser-scanning fluorescence microscopy (model LSM510 invert; Carl Zeiss, Germany). To examine amyloid plaque-associated neuritic dystrophy, brain sections were incubated with rat anti-LAMP1 (Abcam) and mouse anti-Aβ antibody MOAB2, a kind gift from Dr. Mary Jo LaDu (University of Illinois at Chicago), overnight at 4°C, followed by incubation with Alexa Fluor-488-conjugated or Alexa Fluor-568-conjugated secondary antibodies for 2 hr at room temperature. After washes, brain sections were mounted using Vectashield containing DAPI (Vector Laboratories). Images were acquired using a Zeiss AxioImager.Z1/ApoTome microscope. The fluorescent intensity of LAMP1 within 15 μm of each plaque was quantified by Image J.
Western blotting
The detailed procedures were performed as previously described (Liu et al., 2015a). Equal amounts of protein from the homogenized lysates were resolved by SDS-PAGE and transferred to PVDF membranes. After the membranes were blocked, proteins were detected with primary antibody. Membrane was probed with LI-COR IRDye secondary antibodies and detected using the Odyssey infrared imaging system (LI-COR). The following antibodies were used in this study: anti-GFAP (Millipore), anti-apoE (WUE4) (Novus Biologicals), anti-PSD-95 (Cell signaling), anti-synaptophysin (Milipore), and anti-β-actin (Sigma) antibodies.
ELISA quantification
ApoE was measured by ELISA as previously described (Zhao et al., 2014). Briefly, 96-well immunoassay plates were coated overnight with a monoclonal antibody WUE4 (Novus Biologicals). After blocking in 1% milk/PBS for 1 hr, apoE standards and samples were diluted in 1% milk/PBS and added to the plates. After 24 hr incubation at 4°C, plates were washed and then detected with biotin-conjugated goat anti-apoE antibody (K74180B; Meridian Life Science). Recombinant human apoE3 and apoE4 (Fitzgerald) were used as standards. After washes, plates were incubated with streptavidin-polyHRP40 (Fitzgerald) for 1 hr, and were developed with Super Slow tetramethylbenzidine (TMB) substrate (Sigma). Colorimetric quantification was performed on a Synergy HT plate reader (BioTek).
Aβ levels in the brain lysates were determined by ELISA with end-specific mAb 2.1.3 (human Aβx-42 specific) and mAb 13.1.1 (human Aβx-40 specific) for capture and HRP-conjugated mAb Ab5 (human Aβ1-16 specific) for detection (Liu et al., 2014). The ELISAs were developed using Super Slow ELISA TMB (Sigma). Murine IL-1β, IL-6 and TNF-α were measured using commercial ELISA kits (R&D; BioLegend Systems) according to manufacturer’s instructions. To detect soluble oligomeric Aβ species, the same antibody was used for both capture and detection as previously described (Liu et al., 2016). Briefly, 96-well immunoassay plates were coated with 3D6 (10 g/ml) antibody overnight at 4°C. After blocking, the samples and standards were added to the plates and incubated overnight at 4°C. After washes, the samples were detected with the biotinylated 3D6 (0.5 g/ml) antibody in assay buffer (0.25% BSA/0.05% Tween 20, pH 7.4, in PBS) for 90 min at 37°C. After washes, avidin-horseradish peroxidase (Vector Laboratories) was then added to the plates and incubated for 90 min at room temperature. The ELISAs were developed using Super Slow ELISA TMB (Sigma).
Statistical analysis
All quantified data represents an average of samples. Unless indicated, the n numbers in the figure legends represent the numbers of animals. Statistical significance was determined by two-tailed Student’s t test, and p < 0.05 was considered significant.
Supplementary Material
Highlights.
ApoE4 drives amyloid pathology during the seeding stage.
ApoE4 has minimal effect on amyloidosis during the plaque rapid growth period.
ApoE isoforms differentially affect amyloid plaque-associated neuroinflammation.
Strategies targeting apoE4 to reduce Aβ pathology should focus on early prevention.
Acknowledgments
This work was supported by NIH grants R01AG046205, R37AG027924, R01AG035355, RF1AG051504, P01NS074969, and P50AG016574 (to G.B.), a grant from the Cure Alzheimer’s Fund (to G.B.), a fellowship from the BrightFocus Foundation A2016346F (to C.-C.L.), a grant from Alzheimer’s Association AARG-17-500335 (to C.-C.L.). We are grateful to Dr. Dennis Dickson, Linda Rousseau, Monica Castanedes Casey, and Virginia Phillips at Mayo Clinic Histology Core for the immunohistochemical analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
C.-C.L. and G.B. conceived and designed the project, and wrote the paper. C.-C.L., N.Z., Y.F. and N.W. executed the experiments and analyzed the data. C.M.L. and C.-W.T. contributed to the animal maintenance and tissue preparation. All authors read and commented on the manuscript.
References
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgold S, Filser S, Dorostkar MM, Schmidt B, Herms J. In vivo imaging reveals sigmoidal growth kinetic of beta-amyloid plaques. Acta Neuropathol Commun. 2014;2:30. doi: 10.1186/2051-5960-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Science translational medicine. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci USA. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Gong JS, Kobayashi M, Hayashi H, Zou K, Sawamura N, Fujita SC, Yanagisawa K, Michikawa M. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J Biol Chem. 2002;277:29919–29926. doi: 10.1074/jbc.M203934200. [DOI] [PubMed] [Google Scholar]
- Grace EA, Busciglio J. Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J Neurosci. 2003;23:493–502. doi: 10.1523/JNEUROSCI.23-02-00493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. 2006;361:932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Aalten P, Aarsland D, Alcolea D, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313:1924–1938. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65:650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, Guo L, Van Eldik LJ. Apolipoprotein E and apolipoprotein E receptors modulate Abeta-induced glial neuroinflammatory responses. Neurochem Int. 2001;39:427–434. doi: 10.1016/s0197-0186(01)00050-x. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Stine WB, Jr, Narita M, Getz GS, Reardon CA, Bu G. Self-assembly of HEK cell-secreted ApoE particles resembles ApoE enrichment of lipoproteins as a ligand for the LDL receptor-related protein. Biochemistry. 2006;45:381–390. doi: 10.1021/bi051765s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Hu J, Tsai CW, Yue M, Melrose HL, Kanekiyo T, Bu G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J Neurosci. 2015a;35:5851–5859. doi: 10.1523/JNEUROSCI.5180-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84:63–77. doi: 10.1016/j.neuron.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci Transl Med. 2016;8:332ra344. doi: 10.1126/scitranslmed.aad3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DS, Pan XD, Zhang J, Shen H, Collins NC, Cole AM, Koster KP, Ben Aissa M, Dai XM, Zhou M, et al. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegener. 2015b;10:7. doi: 10.1186/s13024-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Miyazaki T, Tashiro F, Yamato E, Miyazaki J. Development of a single-cassette system for spatiotemporal gene regulation in mice. Biochem Biophys Res Commun. 2005;338:1083–1088. doi: 10.1016/j.bbrc.2005.10.054. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, Elashoff D, Geschwind DH, Welsh BT, Gylys KH, Lee C, Cummings JL, Cole GM. Plasma signaling proteins in persons at genetic risk for Alzheimer disease: influence of APOE genotype. Arch Neurol. 2012;69:757–764. doi: 10.1001/archneurol.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Schultz C, Rub U, Saido TC, Yamaguchi H, Haass C, Griffin WS, Del Tredici K, Braak H, Ghebremedhin E. Apolipoprotein E co-localizes with newly formed amyloid beta-protein (Abeta) deposits lacking immunoreactivity against N-terminal epitopes of Abeta in a genotype-dependent manner. Acta Neuropathol. 2005;110:459–471. doi: 10.1007/s00401-005-1053-1. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Liu CC, Shinohara M, Nielsen HM, Dong Q, Kanekiyo T, Bu G. Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J Biol Chem. 2014;289:11282–11292. doi: 10.1074/jbc.M113.526095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.