

Abstract
Major Depressive Disorder (MDD) is considered a “circuitopathy”, and brain stimulation therapies hold promise for ameliorating MDD symptoms, including hippocampal dysfunction. It is unknown if stimulation of upstream hippocampal circuitry, such as the entorhinal cortex (Ent), is antidepressive, although Ent stimulation improves learning and memory in lab animals and humans. Here we show molecular targeting (Ent-specific knockdown of a psychosocial stress-induced protein) and chemogenetic stimulation of Ent neurons induce antidepressive-like effects in mice. Mechanistically, we show that Ent stimulation-induced antidepressive-like behavior relies on the generation of new hippocampal neurons. Thus, controlled stimulation of Ent hippocampal afferents is antidepressive via increased hippocampal neurogenesis. These findings emphasize the power and potential of Ent glutamatergic afferent stimulation - previously well known for the ability to influence learning and memory - for MDD treatment.
Introduction
Antidepressant medications are widely used to treat major depressive disorder (MDD), but 50% of depressed patients receiving these medications relapse, and 30% are drug-resistant1,2. MDD is marked by disordered activity of neuronal circuits that regulate mood, cognition, perception, reward, and motor function. However, both deep brain stimulation (DBS) and more superficial stimulation approaches (i.e. transcranial direct current stimulation [tDCS], transmagnetic stimulation [TMS] and electroconvulsive treatment [ECT]) have drawbacks including cognitive side effects, seizures, and high relapse rate after remission3. Therefore, more treatments for MDD needed, particularly stimulation therapies that more precisely target depression-linked dysfunctional circuitry.
Notable hallmarks of MDD are the structural and functional deficits apparent in the hippocampus, a brain region linked to mood regulation and memory. For example, depressed humans and chronically-stressed animals have reduced hippocampal activity and volume, and decreased expression of activity-dependent genes and processes, including reduced adult neurogenesis in the hippocampal dentate gyrus (DG)4,5. These hippocampal deficits have long been the target of depression treatments in an effort to ameliorate depression symptoms5,6. Interestingly, DBS of a major hippocampal input, the entorhinal cortex (Ent), enhances hippocampal-dependent memory in humans and rodents and increases DG neurogenesis in rodents7,8. While Ent volume is decreased in late onset and remitted MDD9, it is unknown if specific stimulation of Ent-DG afferents combats the symptoms of depression.
Here we used molecular and chemogenetic approaches to test whether Ent-DG stimulation is antidepressive in rodents. For the molecular approach, we targeted TRIP8b (tetratricopeptide repeat-containing Rab8b interacting protein, also called PEX5R), a brain-specific auxiliary subunit of the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel10. TRIP8b splice variants differentially regulate cell surface expression of HCN channels and their current (Ih)11–14. Notably, disruption of TRIP8b (germline knockout) or HCN1 function (hippocampal specific knockdown) increases the excitability of hippocampal neurons and results in antidepressive-like behavior11,14. Using viral-mediated Ent neuron-specific knockdown of TRIP8b, we found that disinhibited Ent afferents to the DG drive activity-dependent DG processes and facilitate antidepressive-like behavior under both basal and stressful condition. Mechanistically, we found this antidepressive-like behavior to be dependent on adult hippocampal neurogenesis. Since our viral-mediated TRIP8b knockdown targeted all Ent neurons, we then used viral-mediated gene transfer, transgenic mice, and chemogenetics15 to parse whether stimulation specifically of glutamatergic Ent-DG circuitry drives antidepressive-like behavior. We found that chronic stimulation of glutamatergic entorhinal cortical afferents to the DG ameliorates behavioral symptoms of depression under both acute and chronic stressful conditions.
Results
Trip8b germline knockout mice have increased dentate gyrus neurogenesis and dendritic morphology relative to wild type mice
Hippocampal neurons fire more frequently in Trip8b germline knockout mice due to lack of Ih currents11, and Trip8b germline knockout mice display antidepressive-like behavior11. Given links among antidepressive-like behavior, hippocampal activity, and DG neurogenesis5, we hypothesized Trip8b germline knockout mice would have more DG neurogenesis. Indeed, 8-week-old Trip8b germline male and female knockout mice had more DG neurogenesis relative to control littermates (Supplementary Fig. 1), particularly in the temporal DG, a DG subregion related to stress response and emotion16,17.
Psychosocial stress increases isoform-specific TRIP8b levels in the dentate gyrus
Epifluorescent immunohistochemical analysis in 8-week-old wild type mice revealed TRIP8b protein in Ent layer II/III cell bodies and their terminals in the DG molecular layer, but extremely sparse TRIP8b-immunoreactive (+) cells or fibers in the DG granule cell layer (Supplementary Fig. 2). This suggested that TRIP8b from Ent-DG projections - not from DG intrinsic cell bodies or their fibers - was poised to play a role in activity-dependent processes of the hippocampus. To test this, we first considered that stress precipitates MDD in humans and depression-like symptoms in laboratory rodents. The antidepressive behavior and increased neurogenesis in Trip8b KO mice and the presence of TRIP8b in the Ent-DG pathway led us to hypothesize that stress would increase TRIP8b levels in Ent-DG projections. Using a well-established chronic social defeat stress (CSDS) model of depression18, susceptible and resilient mice were identified after a 10-day social defeat paradigm (Fig. 1a, Supplementary Fig. 3). We then analyzed the Ent-DG circuit in stressed vs. control mice for levels of the most predominant alternatively-spliced isoforms of TRIP8b12,13: IsoA4 (exon 1a-4 or 1a-2-4) which upregulates surface expression of HCN channel and enhances Ih amplitude; IsoA5 (containing exon 1a, without exon 2 and 4), which reduces Ih current; and IsoB containing exon 1b (exon 1b-2 or 1b-2-4) which also abolishes Ih current. Western blot analysis revealed 27% more TRIP8b isoform IsoA4 in the Ent-DG circuit of stressed (both susceptible and resilient) vs. control mice (Fig. 1b–c), with no change in total TRIP8b, IsoA5, or IsoB (Fig. 1d–e, Supplementary Fig. 3i, l, o). Also, in resilient - but not susceptible - mice, IsoA4 level was negatively correlated with a social interaction ratio (SI ratio, Supplementary Fig. 3k). As IsoA4 maintains HCN channel surface expression and enhances Ih amplitude12,13, higher levels of IsoA4 may be related to our hypothesis that there is decreased Ent-DG circuit activity after stress.
Figure 1. Chronic social defeat stress (CSDS) increases levels of TRIP8b isoform IsoA4 in the dentate gyrus (DG).
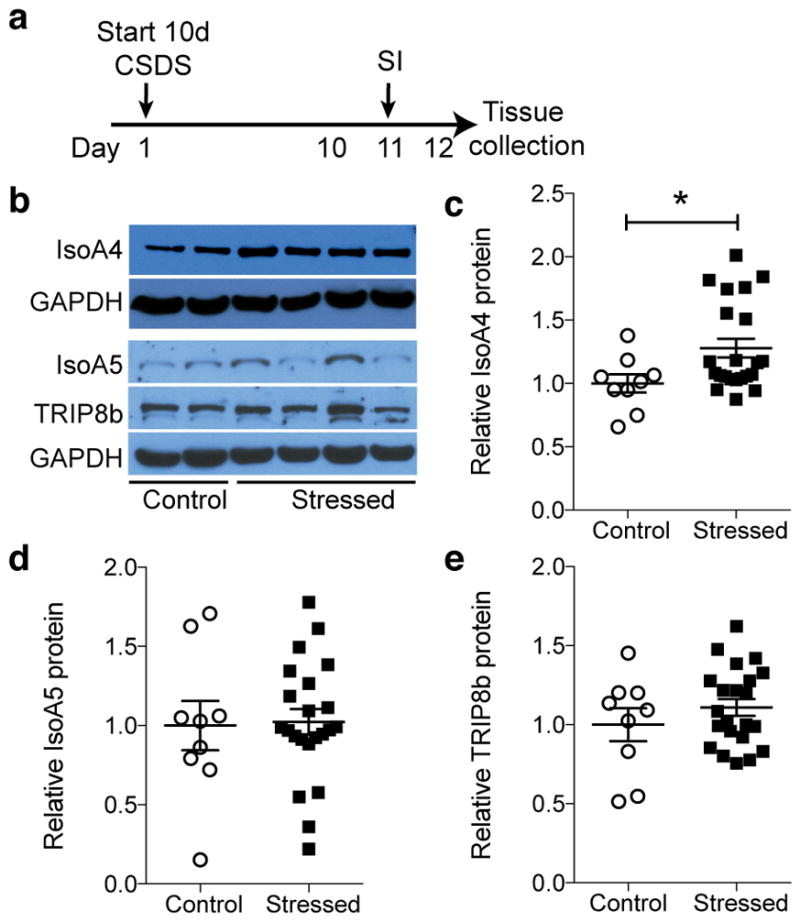
(a) CSDS and social interaction (SI) timeline. (b) Western blot of DG TRIP8b and major hippocampal TRIP8b isoforms, IsoA4 and IsoA5, in control and stressed mice. Uncropped image of blots provided in Supplementary Fig. 3a–e. (c–e) Relative to control and normalized to GAPDH, IsoA4 level is greater after stress (c, unpaired t-test, *p<0.05), with no change in IsoA5 (d) or TRIP8b (e) levels (unpaired t-test, p’s>0.05). n=9 control, 22 stressed mice. Mean±SEM shown with data points from individual animals overlaid (c–e). See Supplementary Table 1 for detailed statistical information.
Entorhinal cortex TRIP8b knockdown increases intrinsic excitability of stellate cells and enhances neurogenesis but does not induce abnormal brain network activity
Since Trip8b germline KO mice show antidepressive behaviors, TRIP8b is expressed in the Ent-DG circuit, and the major TRIP8b isoform, IsoA4, is increased in Ent-DG circuitry after stress, we next hypothesized that loss of TRIP8b specifically in Ent projections to the DG enhances DG activity-dependent processes and antidepressive-like behavior. To knockdown TRIP8b, we generated two viruses [AAV-TRIP8b shRNA-EGFP (TRIP8b shRNA; Fig. 2a); AAV-scrambled shRNA-EGFP (SCR shRNA)]13 and used viral-mediated gene transfer to verify TRIP8b knockdown efficiency in vitro (Fig. 2b–c, Supplementary Fig. 4a–b) and in vivo (Fig. 2d–e). In vitro, there was 87% less TRIP8b expression in TRIP8b shRNA vs. SCR shRNA transfected cells (Fig. 2b–c). Supporting the in vitro efficiency of the virus, there was 87% less TRIP8b expression in TRIP8b shRNA vs. SCR shRNA transfected cells (Fig. 2d–e). To assess the in vivo functional impact of the virus, whole-cell current-clamp recordings were performed in brain slices 3–5 weeks after stereotaxic Ent infusion of TRIP8b shRNA or SCR shRNA into 7-week-old male mice (Fig. 2f–h)19. Action potentials were more frequent in TRIP8b shRNA vs. SCR shRNA transfected stellate cells and also vs. neighboring untransfected stellate cells, resulting in overall increased neuronal excitability (Fig. 2h, Supplementary Fig. 4c–e). Since brain stimulation approaches can have drawbacks such as seizure induction or abnormal brain activity, in vivo electroencephalography (EEG) recordings were performed in awake and behaving male and female mice 5 weeks after Ent viral infusion (Fig. 2i). In vivo EEG recording revealed no abnormal or epileptiform cortical or hippocampal activity, with power spectral density analysis revealing similar activity in mice that had received either SCR shRNA or TRIP8b shRNA into the Ent (Fig. 2i–j, Supplementary Fig. 4f–g). Neither SCR shRNA- nor TRIP8b shRNA-infused mice showed epileptiform-like cortical or hippocampal network activity seen in pilocarpine-treated mice (Fig. 2j).
Figure 2. Entorhinal cortical (Ent) TRIP8b knockdown (KD) increases intrinsic excitability of Ent stellate cells and enhances DG neurogenesis.

(a) TRIP8b shRNA KD construct packaged into adeno-associated virus (AAV). (b, c) Qualitative (b) and quantitative (c) in vitro efficacy of TRIP8b KD via Western blot (SCR shRNA n=4, TRIP8b shRNA n=4; unpaired t-test, **p<0.01). Uncropped image of blots provided in Supplementary Figure 4a–b. (d, e) Photomicrographs of Ent after immunohistochemistry for TRIP8b (red) and EGFP (green) supporting in vivo efficiency of Ent TRIP8b KD. No green Ent cell bodies were red (d, e), consistent with expression of green terminals and diminished red terminals in DG molecular layer (Mol; see m). Dashed-line box in (d) magnified in (e). Scale bar (d)=200 μm, (e)=40 um. (f) Timeline of electrophysiology whole-cell patch experiments. (g, h) Qualitative and quantitative ex vivo assessment of Ent stellate cells transfected in vivo shows TRIP8b shRNA EGFP+ cells (n=5 cells/3 animals) have more spikes vs. SCR shRNA EGFP+ cells (n=4 cells/4 animals) or non-transfected stellate cells (TRIP8b shRNA EGFP-, n=14 cells/4 animals; SCR shRNA EGFP-, n=13 cells/5 animals; two-way repeated measures [RM] ANOVA, main effects of input [F8, 256 = 188.0, p<0.0001], treatment [F3, 32= 4.143, p<0.05], subject [matching; F32, 256= 13.87, p<0.0001] and input X treatment interaction [F24, 256= 2.326, p<0.001], TRIP8b shRNA EGFP+ vs. TRIP8b shRNA EGFP-: posthoc a p<0.05, a′ p<0.01; TRIP8b shRNA EGFP+ vs. SCR shRNA EGFP-: b p<0.05, b′ p<0.01; TRIP8b shRNA EGFP+ vs. SCR shRNA EGFP+: c p<0.05, c′ p<0.01, c″ p<0.001). Calibration (g)=200 ms, 20 mv. (i) Timeline of in vivo EEG. (j) Qualitative and quantitative (Supplementary Fig. 4d–e) assessment of hippocampal (Hip) and cortical (Ctx) EEG in awake and behaving mice showed normal brain activity and no epileptiform activity in SCR or Trip8b shRNA transfected mice, as seen in positive-control mice which received pilocarpine ~1 mon prior. Calibration (j)=10 sec, 0.5 mV. (k–m) Timeline of neurogenesis studies (k), and schematic (l) and photomicrograph (m) of viral stereotaxic infusion into the lateral and medial Ent (lEnt, mEnt) and subsequent EGFP expression in perforant path (PP) terminals in outer (o) and middle (m) DG Mol. Dotted lines in (m) delineate DG granule cell layer (GCL). Scale bar (m)=200 μm. (n–s) Qualitative (n,p,r) and quantitative (o,q,s) assessment of DCX+ cells (n–o), BrdU+ surviving cells (p–q), and BrdU+NeuN+ neurons (r–s) shows AAV-TRIP8b shRNA Ent infusion led to more DCX+ cells (n=6 SCR shRNA mice, 5 TRIP8b shRNA mice), surviving BrdU+ cells ((p) arrowheads, n=8 SCR shRNA mice, 7 TRIP8b shRNA mice), and BrdU+NeuN+ neurons ((r) arrowheads, n=8 SCR shRNA mice, 7 TRIP8b shRNA mice) vs. AAV-SCR shRNA (unpaired t-test, *p<0.05). Dotted lines in (p) delineate DG GCL. Scale bars (n, p)=200 μm, (r)=20 μm. (t) Dendritic tree reconstruction of DCX+ DG neurons in SCR shRNA vs. TRIP8b shRNA mice. Scale bar (t)=10 um. (u–w) TRIP8b shRNA mice had longer DCX+ dendrites (u), and more nodes (v) and ends (w) vs. control mice (unpaired t-test, **p<0.01, (u–w) n=76 neurons in SCR shRNA mice, 95 neurons in TRIP8b shRNA mice, 4–5 mice/group). Mean±SEM shown (c, h, o, q, s, u–w) with data points from individual animals (o, q, s) or cells (c, u–w) overlaid. See Supplementary Table 1 for detailed statistical information.
Having established in vitro and in vivo efficacy and the lack of abnormal cortical and hippocampal network activity, we next assessed the ability of Ent TRIP8b KD to target the DG and promote an activity-dependent process such as DG neurogenesis. Targeting of the Ent-DG circuitry was supported by EGFP+ DG molecular layer terminals 4 weeks after Ent viral infusion (Fig. 2k–m, Supplementary Fig. 5a–h). Ent TRIP8b knockdown enhanced several DG neurogenesis and new-neuron-maturation indices (Fig. 2n–w), as there were 11% more DCX+ cells (Fig. 2n–o), 55% more surviving BrdU+ cells (Fig. 2p–q), and 64% more BrdU+NeuN+ neurons (Fig. 2r–s) in the DG of mice that received Ent TRIP8b shRNA vs. SCR shRNA, but no change in the number of type-1 neural stem cells (Supplementary Fig. 6). Dendritic reconstruction of DCX+ cells showed 23% longer dendrites, 25% more nodes, and 20% more ends (Fig. 2t–w)20,21 in mice that received Ent TRIP8b shRNA vs. SCR shRNA. Thus, TRIP8b knockdown in Ent-DG afferents enhanced both the generation and dendritic maturation of adult-generated DG neurons.
Entorhinal cortex TRIP8b knockdown promotes antidepressive behavior that is neurogenesis-dependent
Since TRIP8b knockdown in the Ent-DG circuit enhanced DG neurogenesis - an activity-dependent process - and Trip8b germline knockout mice had antidepressive behavior, we next hypothesized that Ent TRIP8b knockdown would promote antidepressive-like behavior. Eight-week-old male mice received bilateral Ent infusion of TRIP8b shRNA or SCR shRNA, and 4 weeks later mice were tested for locomotor behavior, and then examined in the forced swim test or for contextual fear conditioning (Fig. 3a). Compared to control SCR shRNA mice, TRIP8b shRNA mice had normal locomotor behavior activity (Fig. 3b), but displayed antidepressive-like behavior, spending 17% less time immobile in the forced swim test (Fig. 3c). Since this same neuronal circuit is involved in context-dependent memory7, we also hypothesized Ent TRIP8b knockdown would promote hippocampal-dependent memory after contextual fear conditioning. Indeed, mice that received Ent TRIP8b shRNA spent 51% more freezing time in the shock-paired context vs. SCR shRNA mice (Fig. 3d), with no difference in the cue test (Fig. 3e). Thus, Ent TRIP8b knockdown indeed improved hippocampal-dependent memory.
Figure 3. Ent TRIP8b KD-induced promotion of antidepressive behavior is neurogenesis-dependent.
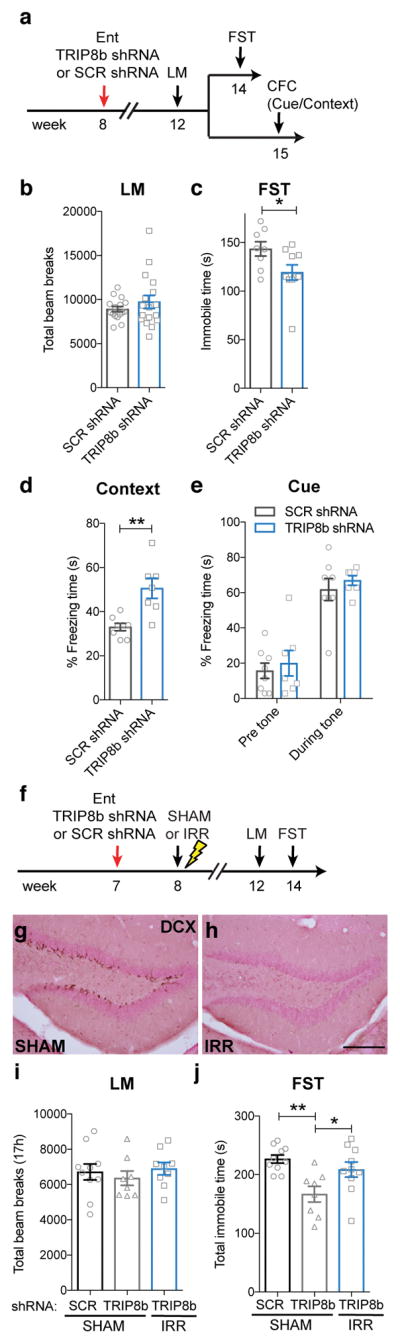
(a) Timeline of viral infusion and behavioral testing. Four weeks post-bilateral Ent AAV-infusion, mice were tested for locomotion (LM), and then for forced swim test (FST) or contextual fear conditioning (CFC). (b–e) AAV-TRIP8b shRNA Ent infusion did not change LM (b, unpaired t-test, p>0.05, n=16 SCR shRNA mice, 17 TRIP8b shRNA mice), but decreased FST immobile time (c, unpaired t-test, *p<0.05, n=8 SCR shRNA mice, 10 TRIP8b shRNA mice) and increased CFC context freezing time (d, unpaired t-test, **p<0.01, n=8 SCR shRNA mice, 7 TRIP8b shRNA mice) without altering cue freezing time (e, two-way RM ANOVA, main effect of tone [F1,13=55.16, p<0.0001], treatment [F1,13=1.117, p>0.05], subjects [matching; F13,13=0.5089, p>0.05], tone X treatment interaction [F1,13=0.004857, p>0.05]). (f–j) Timeline (f) and efficacy (g–h) of neurogenesis ablation study. One week post-bilateral Ent AAV-infusion, mice received sham (SHAM) or image-guided DG-targeted irradiation (IRR) to ablate neurogenesis. Six weeks later, when DCX+ cells were still depleted (g–h), groups did not differ in LM activity (i, one-way ANOVA, F2,24=0.4149, p>0.05, n=10 SCR shRNA/SHAM, 8 TRIP8b shRNA/SHAM, 9 TRIP8b shRNA/IRR). AAV-TRIP8b shRNA Ent infusion decreased FST immobile time in SHAM mice, but not in IRR mice (j, one-way ANOVA, F2,25=7.07, posthoc *p<0.05, **p<0.01, n=10 SCR shRNA/SHAM, 8 TRIP8b shRNA/SHAM, 10 TRIP8b shRNA/IRR). Scale bar (h)=200 μm applies (g–h). Mean±SEM shown with data points from individual animals overlaid (b–e, i–j). See Supplementary Table 1 and the Life Sciences Reporting Summary for detailed statistical information.
Multiple lines of evidence implicate DG adult neurogenesis in antidepressant efficacy, particularly under stressful conditions22–25. Thus, the Ent TRIP8b knockdown-induced increase in neurogenesis may have been a causative factor in driving antidepressive-like behavior. However, it was also possible that our Ent TRIP8b knockdown-induced increase in neurogenesis was merely correlated with the Ent TRIP8b knockdown-induced antidepressive behavior, particularly after stress. To test these two possibilities, 7-week-old male mice received Ent infusion of TRIP8b shRNA or SCR shRNA, followed one week later by sham irradiation or DG-directed image-guided irradiation to ablate neurogenesis (Fig. 3f). Four weeks after irradiation, mice were assessed for locomotor behavior and in the forced swim test after a brief restraint stress (Fig. 3f–j). As expected, ablation of neurogenesis did not change locomotor activity (Fig. 3i, Supplementary Fig. 7a). However, DG-directed irradiation significantly blunted Ent TRIP8b knockdown-induced antidepressive behavior (Fig. 3j, Supplementary Fig. 7b).
Entorhinal cortex TRIP8b knockdown promotes antidepressive-like behaviors under conditions that mimic chronic stress
Since TRIP8b knockdown in the Ent-DG circuit enhances antidepressive behavior under basal and after a brief stress (Fig. 3c, j), we next hypothesized that TRIP8b knockdown in the Ent-DG circuit would enhance antidepressive behavior under conditions that mimic chronic stress, such as long-term exposure to the stress hormone corticosterone26–29. To test this hypothesis, 7-week-old female mice received Ent infusion of TRIP8b shRNA or SCR shRNA, and began to receive chronic administration of corticosterone or vehicle one week later (Fig. 4a). Behavioral testing for locomotor behavior and anxiety- and antidepressive-like behavior, including novelty suppressed feeding as well as the forced swim test, began 4 weeks after viral infusion and 3 weeks into the chronic corticosterone administration (Fig. 4b–g, Supplementary Figs. 8, 9). Neither chronic corticosterone nor Ent TRIP8b knockdown changed locomotor activity (Fig. 4b) or basal anxiety-like behavior in the dark/light test or elevated plus maze (Fig. 4c–d, Supplementary Fig. 9a–d). However, chronic corticosterone increased the latency to feed in the novelty suppressed feeding test vs. vehicle (Supplementary Fig. 8a), and Ent TRIP8b knockdown blocked this corticosterone effect (Fig. 4e, Supplementary Fig. 8a). Ent TRIP8b knockdown did not change the corticosterone-induced effect on food consumption in the home cage post-novelty suppressed feeding (Fig. 4f). Similarly, chronic corticosterone increased the time immobile in the forced swim test, while Ent TRIP8b knockdown blocked this effect (Fig. 4g, Supplementary Fig. 8b).
Figure 4. Ent TRIP8b knockdown (KD) promotes antidepressive-like behaviors under conditions that mimic chronic stress.

(a) Timeline of viral infusion and behavioral testing. (b–d) CORT did not change LM (b, one-way ANOVA, F2,48=0.01135, p>0.05, n=16 SCR shRNA/VEH, 16 SCR shRNA/CORT, 19 TRIP8b shRNA/CORT) or DL (c, time spent in light, one-way ANOVA, F2,48=0.3328, p>0.05, n=16 SCR shRNA/VEH, 16 SCR shRNA/CORT, 19 TRIP8b shRNA/CORT; d, latency to enter light, one-way ANOVA, F2,48=0.249, p>0.05, n=16 SCR shRNA/VEH, 16 SCR shRNA/CORT, 19 TRIP8b shRNA/CORT) in either Ent SCR shRNA or TRIP8b shRNA-infused mice relative to SCR VEH control mice. (e–f) Ent-infused TRIP8b shRNA mice that received CORT had shorter latency to feed in NSF vs. SCR shRNA that received CORT (e, one-way ANOVA, F2,24=2.934, p=0.0725, n=8 SCR shRNA/VEH, 8 SCR shRNA/CORT, 11 TRIP8b shRNA/CORT, posthoc *p<0.05). Food consumption in home cage in both SCR shRNA- and TRIP8b shRNA-infused CORT mice was significantly less than SCR VEH mice (f, Food mass after NSF, one-way ANOVA, F2,24=4.569, p<0.05, posthoc *p<0.05, **p<0.01, n=8 SCR shRNA/VEH, 8 SCR shRNA/CORT, 11 TRIP8b shRNA/CORT). (g) Ent-infused SCR shRNA CORT mice had more time immobile in FST vs. SCR shRNA VEH mice, as well as vs. TRIP8b shRNA CORT mice (g, FST, one-way ANOVA, F2,48=4.03, p<0.05, posthoc *p<0.05, n=16 SCR shRNA/SHAM, 16 SCR shRNA/CORT, 19 TRIP8b shRNA/CORT). Mean±SEM shown with data points from individual animals overlaid (b–g). See Supplementary Table 1 and the Life Sciences Reporting Summary for detailed statistical information.
While one interpretation of these data is that Ent TRIP8b knockdown promotes antidepressive-like behaviors under conditions that mimic chronic stress, it is also possible that these effects are due to the action of Ent projections to other brain and hippocampal regions. However, analysis of behavior data from animals which had additional off-target expression (e.g. terminal expression in DG molecular layer and hippocampal CA1 [Mol+CA1], CA2/3 [Mol+CA2/3] or only CA2/3) regions revealed no antidepressant-like behavior in the forced swim test at baseline or after acute or chronic stress (Supplementary Fig. 5i–k). Thus, these data support the interpretation that TRIP8b knockdown in the Ent-DG circuit - and not in other brain or hippocampal areas - promotes antidepressive behavior.
Chronic stimulation of glutamatergic entorhinal cortical afferents drives dentate gyrus activity-dependent processes but not abnormal network activity
Disinhibition of the Ent-DG circuit by TRIP8b knockdown promotes antidepressive behavior, suggesting that our manipulation was ultimately driving Ent excitatory neurons. To test that stimulation of glutamatergic30 - and not non-glutamatergic31- projections from the Ent to DG are responsible for our effects, we used a chemogenetic approach to control Ent glutamatergic neuronal excitability. Seven-week-old CamKIIα-iCre male and female mice32 received Ent infusion of Cre recombinase-dependent virus containing the construct for either the designer receptor Gq-coupled modified human M3 muscarinic receptor (AAV-hSyn-DIO-hM3Dq-mCherry [hM3Dq]) or a reporter (AAV-hSyn-DIO-mCherry [mCherry])33(Fig. 5a). Supporting appropriate targeting of the Ent-DG circuit (Fig. 5b, Supplementary Fig. 10a–h), CamKIIα-iCre-driven mCherry expression was evident in Ent and DG molecular layer of both hM3Dq and mCherry mice (Fig. 5c). After 4 weeks of daily administration of the designer ligand clozapine-N-oxide (CNO, i.p.), cfos+ cells were more abundant in Ent and DG of hM3Dq vs. mCherry mice (Fig. 5d, e; Supplementary Fig. 10i–j). There was hippocampal subregion specificity, as there were 56% more cfos+ cells in the DG of hM3Dq vs. mCherry mice, but no difference in CA1 or CA3 cfos+ cells in hM3Dq vs. mCherry mice (Fig. 5e). Stimulation of the Ent-DG circuit also increased DG neurogenesis, with 65% more BrdU+ cells and 129% more BrdU+NeuN+ neurons in hM3Dq vs. mCherry mice (Fig. 5f–j).
Figure 5. Chemogenetic stimulation of Ent-DG circuit drives activity-dependent processes in the DG and antidepressive-like behavior.

(a) Timeline of DREADD neurogenesis studies. CamKIIa-iCre mice received BrdU prior to stereotaxic Ent infusion (AAV-hSyn-DIO-mCherry [mCherry, control] or AAV-hSyn-DIO-hM3Dq-mCherry [hM3Dq, CNO-dependent stimulation]). Four weeks later, and at a time point when mCherry-transfected Ent glutamatergic neurons were evident in the DG Mol (b–c), daily i.p. CNO led to more cfos+ cells in Ent (d top row, e) and DG (d bottow row, e) but not in other hippocampal regions (e) in hM3Dq vs. control mice (d, e). Scale bars (c, d)=200 μm (e: two-way ANOVA, effects of subregion [F2,24=6.065, p<0.01], treatment [F1,24=2.442, p>0.05], and Bregma X treatment interaction [F2,24=4.221,p<0.05], treatment posthoc: *p<0.05; n=5, mCherry, 5 hM3Dq). (f–j) hM3Dq mice had more DG BrdU+ cells (f–h) and BrdU+NeuN+ neurons (i–j) vs. controls. Scale bars (f)=200 μm, (i)=20 μm. (g, j: unpaired t-tests, **p<0.01, ***p<0.001; h: two-way ANOVA, effects of Bregma [F13,364=27.90, p<0.0001], treatment [F1,364=51.07,p<0.0001], and Bregma X treatment interaction [F13,364= 2.901, p<0.001], treatment posthoc: a p<0.05, a′ p<0.01, a″ p<0.001; g, h: n=14 mCherry, 14 hM3Dq, j: n=5 mCherry, 5 hM3Dq). (k) Timeline of chronic DREADD behavior studies. (l) Ent-infused hM3Dq mice had similar LM activity vs. controls before and after daily CNO injection (two-way RM ANOVA, effects of time [F79,1580=10.75, p<0.0001], treatment [F1,20=0.7316, p>0.05], subjects [matching; F20,1580=3.971, p<0.0001] and time X treatment interaction [F79,1580= 0.2065, p>0.9999]). (m, n) Ent-infused hM3Dq mice had shorter latency to feed in NSF but similar home cage food consumption vs. controls (unpaired t-test, *p<0.05, n=8 mCherry, 7 hM3Dq), which was not seen with acute DREADD stimulation (o–r) ((p) two-way RM ANOVA, effects of time [F79,1106=14.82, p<0.0001], treatment [F1,14=0.7466, p>0.05], subjects [matching; F14,1106=9.23, p<0.0001] and time X treatment interaction [F79,1106= 0.7006, p>0.05], (q–r) unpaired t-test, p>0.05, n= 7 mCherry, 9 hM3Dq). (s–t) 10 days of CSDS decreased the time of SI in mCherry vs SHAM/mCherry group. Chronic DREADD stimulation of the Ent/PP of mice that received CSDS (s) increased the time in hM3Dq vs. control mice spent in the interaction zone (t). Two-way RM ANOVA, effects of target [F1,36=7.494, p<0.01], virus [F3,36=3.155, p<0.05], and target X virus interaction [F3,36=2.132, p>0.05], Subjects [matching; F36,36=2.242, p<0.01], virus posthoc: *p<0.05, **p<0.01; n=6 mCherry/Control, 6 hM3Dq/Control,16 mCherry/CSDS, 12 hM3Dq/CSDS. Mean±SEM shown (e, g–h, j, l–n, p–r, t) with data points from individual animals overlaid (e, g, j, m–n, q–r, t). See Supplementary Table 1 and the Life Sciences Reporting Summary for detailed statistical information.
As stimulation of Ent-DG glutamatergic afferents might produce abnormal or epileptiform brain activity34, we recorded the cortical and hippocampal EEG of male and female CamKIIa-iCre mice that received hM3Dq or mCherry viral infusion at 7 weeks of age followed 3 weeks later by CNO (1 mg/kg daily i.p., 5 weeks, Supplementary Fig. 11a). In vivo EEG recordings of these awake and behaving mice revealed no aberrant epileptiform or hippocampal network activity seen after 5 weeks of CNO (1 mg/kg, Supplementary Fig. 11a–c), and no measurable difference between hM3Dq and mCherry mice even across the day-night cycle (Supplementary Fig. 11e–g). However, CNO-treated hM3Dq mice given a single high dose of CNO (10 mg/kg) showed epileptiform activity (Supplementary Fig. 11d). These data show that hM3Dq and mCherry mice had similar cortical and hippocampal activity, and support our ability to finely tune the activity of Ent-DG circuit depending on CNO dose.
Chronic stimulation of glutamatergic entorhinal cortical afferents to the dentate gyrus ameliorates behavioral symptoms of stress-induced depression
Having established that stimulation of the glutamatergic Ent-DG circuit enhances DG cellular activity and activity-dependent processes without driving abnormal brain network activity, we examined whether chronic chemogenetic stimulation of glutamatergic Ent cells drove antidepressive-like behaviors24,35. Seven-week-old CamKIIa-iCre male mice received Ent infusion of hM3Dq or mCherry, and then 3 weeks later began to receive daily administration of CNO for 5 weeks (Fig. 5k). Locomotor activity in Ent hM3Dq mice was similar to Ent mCherry mice when examined after 4 weeks of CNO (Fig. 5l). However, in the latency to feed test performed after 5 weeks of CNO, Ent hM3Dq mice displayed a 47% shorter latency to feed vs. Ent mCherry (Fig. 5m), suggesting an antidepressive-like effect. This was a specific effect, as both Ent hM3Dq and mCherry mice consumed the same amount of food in the home cage after the conclusion of the novelty suppressed feeding test (Fig. 5n) and weighed the same after the home cage food intake. In contrast to the antidepressive-like behaviors seen in hM3Dq mice after chronic CNO injections, Ent hM3Dq and mCherry male mice given acute CNO 3 weeks post-viral infusion (Fig. 5o) showed similar locomotor behavior and latency to feed (Fig. 5p–r).
Finally, we hypothesized that chronic stimulation of glutamatergic Ent afferents to DG would enhance antidepressive-like behavior under conditions of an ethologically-relevant chronic social stress, such as CSDS18,36,37. To test this hypothesis, 7-week-old CamKIIa-iCre male mice received Ent infusion of hM3Dq or mCherry, began to receive daily CNO injections 3 weeks after viral infusion, and began 10-days of CSDS or control housing 4 weeks into the CNO regimen (Fig. 5s). SI testing revealed while there were no differences among groups when social target was absent (Fig. 5t), mCherry mice that underwent CSDS had less interaction with a social target than non-stressed mCherry mice, as expected. In addition, hM3Dq mice that underwent CSDS had more interaction time with a social target than mCherry mice that underwent CSDS (Fig. 5t). Taken together, our findings show stimulation of glutamatergic neurons in the Ent-DG circuit is antidepressive, even in an animal model of stress-induced depression.
Discussion
The Ent-hippocampal circuit is well known for its role in processing episodic memory of spatial and object information38,39, and recent research shows that Ent stimulation has physiological benefits for human learning- and memory-related processes. For example, in humans Ent DBS enhances spatial learning, and is accompanied by a resetting of hippocampal theta rhythms8. In contrast, direct DBS of the human hippocampus does not affect hippocampal-dependent memory8, emphasizing the utility of targeting a hippocampal input rather than the hippocampus itself. In addition to this work in humans, preclinical work also supports that stimulation of the Ent via DBS improves spatial learning and memory, specifically driving activity-dependent adult neurogenesis in the rodent hippocampus7. Our present work supports this, as we show stimulation of Ent-DG circuitry in mice improves hippocampal-dependent memory. However, our main goal was to test the novel hypothesis that Ent stimulation would be effective in ameliorating depressive-like behaviors. This hypothesis was inspired by the Ent and hippocampal structural deficits4,5,9 seen in the brains of humans with MDD and animal models of MDD, and by the ability of many antidepressant drugs and even non-pharmacological treatments like ECT to ameliorate these structural deficits40–42. In support of our hypothesis, we show both molecular- and chemogenetic-based stimulation of Ent-DG circuitry in mice combats stress-induced depressive-like behavior. Critical considerations of any novel therapeutic are the intensity and duration of treatment. Interestingly, prior work showed longer-term - but not acute - Ent deep brain stimulation improved hippocampal learning- and memory-related function7,8,43. Our present work also shows chronic - but not acute - Ent stimulation drives antidepressive-like behaviors in mice. It remains to be tested how long the Ent stimulation-induced memory enhancement and antidepressive-like behavior persists. Importantly, it also remains unknown if Ent stimulation will alleviate MDD symptoms in humans. However, our findings in laboratory mice emphasize the power and potential of Ent glutamatergic afferent stimulation - previously known for the ability to positively-influence learning and memory - for the treatment of MDD.
In light of the efficacy of Ent stimulation to both alleviate stress-induced depressive-like behavior in mice (present results) and improve learning- and memory-related processes7,8, it is notable that many psychiatric disorders (MDD, bipolar disorder, schizophrenia, post-traumatic stress disorder, substance-use disorders) share symptoms reminiscent of abnormal DG function: dysregulated memory, mood regulation, pattern separation, and reward processing. Future studies are warranted to examined whether Ent stimulation normalizes DG function and thus ameliorates symptoms in other disorders aside from MDD. In addition, given the functional heterogeneity of the mammalian hippocampus44, it will be important to assess whether targeting of subregions of the Ent result in specific functional outcomes, or whether the entire perforant path needs to be engaged. For example, direct stimulation of only the ventral DG mature granule neurons suppresses innate anxiety, which may be a potential intervention for these common symptoms44. Also, it remains unknown whether the functions of the DG closely relate to each other, such that improvement in mood regulation, for example, also results in improve memory. A corollary of this is whether or not Ent stimulation will improve DG functions that are far less studied, such as contextual pattern separation and reward processing. For example, disruption of Ent also disrupts pattern separation45, and inducible increase of adult DG neurogenesis improves pattern separation23, but it remains to be seen whether stimulation of Ent-DG circuit improves pattern separation.
Mechanistically, we show that Ent stimulation-induced antidepressive-like behavior relies on intact DG neurogenesis. These data are in line with causative studies in rodents showing newly-generated DG neurons are required for antidepressant efficacy, particularly under stressful conditions24,35,46. While “increased hippocampal neurogenesis” has been proposed to even underlie the effectiveness of certain antidepressants47,48, it has thus far been unclear how this action was achieved. Thus our results showing stimulation of Ent-DG circuitry spurs adult hippocampal neurogenesis and drives antidepressant behavior raise the possibility that these actions could be harnessed to improve mood more effectively and/or enhance resilience in the face of stressful situations.
Our findings provide the first evidence that the Ent-DG circuit may be targeted for depression treatment, with chronic - but not acute - stimulation driving antidepressive-like behaviors. The ability to produce antidepressive behaviors by stimulating the Ent-DG circuitry fits well with the modern framing of depression as a “circuitopathy”49. As stimulation of the Ent-DG enhances hippocampal function from mood to memory, targeting of this novel circuit for MDD treatment may preclude side effects of current brain stimulation therapies (i.e. memory loss and cognitive impairment), and will address the great demand for novel avenues of treatment for humans who suffer from MDD.
ONLINE METHODS
for Yun et al., Stimulation of entorhinal cortex-dentate gyrus circuitry is antidepressive
Animals and ethics statement
Experiments were approved by the Institutional Animal Use and Care Committee at the University of Texas Southwestern Medical Center (UTSW; APN 0960-07-02-1 and 2015-100851) and at the Children’s Hospital of Philadelphia (PennMed/CHOP; APN IAC-17-001245). Mice were group-housed in a UTSW or PennMed/CHOP vivarium accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), and were kept on a 12 hr light/dark cycle with ad libitum access to food and water. 7- or 8-week old C57BL/6J male or female mice were purchased from Jackson Laboratory (stock number: 000664), and nestin-GFP mice were bred at UTSW and CamKIIα-icre mice were bred at UTSW and PennMed/CHOP32,50, resulting in hemizygous transgenic mice. Trip8b germline knock-out (KO) mice were generated by the Chetkovich Laboratory11 by breeding heterozygous Trip8b+/− mice. Mice were genotyped by PCR using genomic DNA and previously published primers11,32. All experiments were performed in compliance with local and national guidelines on the care and use of animals.
AAV vectors, virus production, and purification
Short hairpin RNAs were designed to target the TRIP8b C-terminus region (exon 11) as previously published13. The following oligonucleotides with overhanging ends identical to those created by Sap I and Xba I restriction enzymes were used: For TRIP8b shRNA, 5′-TTTGAGCATTTGAAGAAGGCTTAATTCAAGAGATTAAGCCTTCTTCAAAT GCTATTTTT-3′; Scramble shRNA (SCR shRNA), 5′-TTTGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTT-3′. Hairpin oligonucleotides were phosphorylated by T4 polynucleotide kinase (New England Biolabs, Beverly, MA) followed by annealing at 100°C for 5 min and cooling in the heat block for 3 hr. Each annealed oligonucleotide was ligated into the adeno-associated virus (AAV2) plasmid (pAAV-EGFP-shRNA; Stratagene, La Jolla, CA). Virus production was achieved either using helper-free triple transfection in HEK293 cells (American Type Culture Collection, VA), with purification as previously described51,52 in the lab or the virus were purchased from University of Pennsylvania Vector core. AAV8-hSyn-DIO-hM3Dq-mCherry and AAV8-hSyn-DIO-mCherry virus were purchased from the University of North Carolina Vector Core Facilities (Chapel Hill, NC). Virus titer was approximately 1.9x10E12.
Stereotaxic injection
Mice were anesthetized with a mixture of ketamine (120 mg/kg) and xylazine (16 mg/kg) in saline (0.9% NaCl, i.p.). Bilateral stereotaxic injection of 0.4–0.5 μl of purified high titer AAV (TRIP8b shRNA, SCR shRNA, hM3Dq, or mCherry) was directed into the lateral entorhinal cortex (lEnt; from Bregma: A/P −3.7 mm, M/L +4.4 mm, angle 4°; from Lambda: D/V −4.5 mm) and medial Ent (mEnt; from Bregma: A/P −3.9 mm, M/L +3.9 mm, angle 4°; from Lambda: D/V −4.5 mm) using 33 gauge Hamilton syringes (Hamilton, Reno, NV). Injection rate was 0.1 μl/min, with needles kept in place for an additional 5 min to enable diffusion. Animals were randomly allocated to experimental vs. control groups. Accurate virus targeting (EGFP+ or mCherry+ soma in Ent II/III and EGFP+ or mCherry+ terminals in perforant path, middle and/or outer DG molecular layer, Mol; Supplementary Fig. 5a–h, Supplementary Fig. 10a–h) was verified after brain collection. Data from animals in which labeled terminals were seen in CA1 or CA3 (stratum oriens, SO; stratum pyramidalis, SP; or stratum radiatum, SR), subiculum (Sub), or amygdala (Amyg) were excluded from the cellular and behavioral analyses in the main text. In animals with strong terminal expression in the DG Mol, immunohistochemistry with amplification and high magnification revealed fine projections in the stratum lacunosum-moleculare (SLM). Those animals’ cellular and behavioral data are also included in the main text if GFP+ or mCherry+ terminals were not detected in other CA1 regions (SO, SP, SR). Data from mistargeted mice were analyzed to confirm lack of antidepressive-like behavior with off-target TRIP8b shRNA animals (Supplementary Fig. 5i–k).
Drug administration
Mice were injected twice (6 hr inter-injection interval) with 5′-bromo-2′-deoxyuridine (BrdU, Roche Applied Sciences Indianapolis, IN, made as 10 mg/ml in 0.9% saline, injected i.p. as 15 ml/kg) at a dose of 150 mg/kg53 24 hr prior to stereotaxic surgery to label dividing cells and eventually quantify surviving BrdU immunoreactive (+) cells. Female C57BL/6J mice that received virus intra-Ent (AAV2-TRIP8b shRNA or AAV2-SCR shRNA) were given oral corticosterone (Sigma-Aldrich, Cat #C2505, 7.5 mg/kg/day, stock solution 52.5 ug/ml) dissolved in 0.3% EtOH in 0.45% 2-hydroxypropyl-β-cyclodextrin (Vehicle [Veh], Sigma-Aldrich, Cat. # H107)23,26 in opaque bottles to protect them from light, available ad libitum in the drinking water for 7 weeks. Male CamKIIα-icre mice that received virus intra-Ent (AAV8-hSyn-DIO-hM3Dq-mCherry or AAV8-hSyn-DIO-mCherry) were injected with clozapine-N-oxide (CNO; NIMH Chemical Synthesis and Drug Supply Program) dissolved in 0.5% DMSO (Calbiochem, Cat. #317275) in 0.9% saline33. The dose of CNO was as follows: CSDS experiments, 1 mg/kg of 0.2 mg/ml solution, i.p. daily for 6 weeks; non-CSDS chronic and acute CNO behavior and cellular experiments, 2 mg/kg of 0.2 mg/ml solution, i.p. daily for 4–5 weeks (chronic) or 1 injection prior to each behavioral test (acute); EEG experiments, 1 mg/kg of 0.2 mg/ml solution i.p daily for 5 weeks and then either 1 mg/kg of 0.2 mg/ml solution or 10 mg/kg (high dose) of 1mg/ml solution during EEG monitoring).
Electrophysiology
Horizontal slices (300 μm) to be used for whole-cell current clamp patch recording of Ent Layer II neurons were prepared as described previously19 and according to timeline shown in main figures (Fig. 2f). Slices recovered in a holding chamber for ≳1 hr before use. During recording, slices were superfused with artificial cerebrospinal fluid (ACSF; 31.5–32.5°C) saturated with 95% O2/5% CO2 and containing (in mM) 119 NaCl, 2.5 KCl, 1.0 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose. During recordings, ACSF-containing picrotoxin (100 mM) was used to block GABAA receptor-mediated inhibitory postsynaptic potentials and kynurenic acid (2 mM) to block glutamate receptors. All recordings were performed on stellate neurons in Ent layer II. Layer II neurons were classified as stellate or non-stellate neurons based on electrophysiological characteristics described previously54. Briefly, stellate neurons were characterized by the presence of low-frequency, subthreshold membrane potential oscillations, a depolarizing afterpotential following spikes, and prominent inward rectification in response to hyperpolarizing current pulses. To quantify firing properties, whole-cell current-clamp recordings were performed with electrodes (3–5 MΩ) containing (in mM) 120 K-gluconate, 20 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, and 0.3 NaGTP at a pH of 7.20–7.25. Data were filtered at 5 KHz, digitized at 10 KHz, and collected and analyzed via Clampex 10.3.0.2 (Molecular Devices, Inc.). Membrane potentials were maintained at −70 mV. Series resistances (10–18 MΩ) and input resistances were monitored on-line with a 40 pA current injection (150 msec) given before each 700 msec current injection stimulus. Only cells with a stable Ri (Δ<10%) for the duration of the recording were kept for analysis. Firing rate for a given cell was the average value measured from 2 to 4 cycles (duration: 700 ms at 0.1 Hz, range: −320 to +320 pA, increment: 40 pA step).
Electroencephalogram (EEG) recordings
EEG monitoring was performed as previously described55 and according to timeline shown in main figures (Fig. 2i). Mice were stereotaxically implanted with an electrode assembly under continuous isoflurane anesthesia. The electrode assembly consisted of 6 channels: 2 surface electrodes attached to miniature skull screws placed over the left and right frontal cortices (from Bregma: A/P −1.2 mm, M/L ±1.1 mm); a double-depth electrode in right hippocampus (A/P −2.2 mm, M/L +1.2 mm, D/V −1.3 mm); and finally a reference and a ground electrode directly behind Lambda on either side of midline. Teflon-coated silver wires (0.13 mm diameter) were attached to each electrode and connected to a 6-pin pedestal (Plastics One, Roanoke, VA). The entire assembly was secured on the skull with dental cement (Ortho-Jet, LangDental, Wheeling, IL). A flexible cable connected each animal’s headcap assembly to a commutator thus allowing the mouse to move freely during the recordings. Mice were given ≥72 hr post-surgery to recover prior to the being placed in recording cage, and were acclimatized for ≥24 hr in EEG recording cage before recording initiation. Video-monitored EEG recordings of awake and behaving mice were performed in custom-made Plexiglas cages using a Stellate Harmonie acquisition interface (Natus Medical, Pleasanton, CA) and sampled at 2 kHz.
EEG signals were processed offline for the detection of seizures and for the purpose of power spectral density analyses. Seizures were detected manually through inspection of EEG traces. Power spectra analyses were performed using NeuroExplorer (ver. 5, Nex Technologies, Madison, AL) in conjunction with a custom-made Matlab code (developed by RC Ahrens-Nicklas). Frequency bands were separated into delta (0.1–4 Hz), theta (4.1–8 Hz), alpha (8.1–13 Hz), beta (13.1–25 Hz) and gamma (25.1–50 Hz) ranges. The relative power for each frequency band was calculated by dividing the absolute power for each frequency by the total power and then normalizing it with a log transformation to allow comparisons between mice.
Data was analyzed separately for the day and night cycles (lights on, 6:15 AM; lights off, 6:15 PM). Furthermore, for the effects of CNO, data were sampled and analyzed between 90 and 360 min following CNO administration. Given that CNO was injected at 3:15 PM on days of administration, post-CNO data were further segregated into daytime and nighttime segments. In summary, data were analyzed for TRIP8b shRNA and SCR shRNA mice during daytime and nighttime, and for hM3Dq and mCherry mice during daytime pre-CNO, daytime post-CNO, and nighttime post-CNO. For the pilocarpine-induced epilepsy model, 6–8 week old C57BL/6 mice were injected with scopolamine (1 mg/kg, i.p.), and 30 min later with pilocarpine (315 mg/kg i.p.), which typically trigger status epilepticus within <30 min56. Sixty min after status epilepticus onset, diazepam (5 mg/kg, i.p.) was administered to quell seizure activity. Animals were left to recover for at least 2 weeks before they were surgically implanted with EEG electrode assemblies as described above. In pilocarpine-treated mice, spontaneous electrographic and/or behavioral seizures typically start manifesting within 8–15 days post status epilepticus56, and recordings presented here were collected ~1 mon post-pilocarpine. Data for power analyses were sampled in the form of 10 pseudo-randomly selected, noise- and artifact-free, 2-min epochs for each designated time period (see above) for each day of recording. The relative power values for these 10 epochs were subsequently averaged, resulting in a single relative power value for each time period for each day. Where data were collected across multiple days, data from multiple days were averaged for each designated time period. Experimenters remained blind throughout quantification and analysis of data.
Code availability
MatLab code for spike and frequency analysis of in vivo electroelectroencephalogram experiments can be accessed via contacting Rebecca Ahrens-Nicklas at AhrensNicklasR@email.chop.edu.
Image-guided cranial irradiation
Image-guided cranial irradiation was delivered via the X-RAD 225Cx self-contained irradiation system (Precision X-Ray) as previously published25,57. Briefly, 8 week-old male C57BL/6J mice were anesthetized with 1.5–2.5% isoflurane. Mice received diagnostic imaging to align the body and Bregma coordinates with the collimator (6 × 14 mm rectangular collimator) to ensure hippocampal targeting. Mice then received image-guided cranial irradiation (10 Gy, 225 Kv, 13 mA, 207 sec). Control mice (Sham) were handled the same way as irradiated mice (e.g. exposed to isoflurane for the same period of time) except were not placed in the irradiator.
Tissue collection and western blot
Brains and adrenal glands from decapitated mice were immediately dissected and frozen on dry ice (Fig. 1). Animals with only successful bilateral extraction of adrenal glands were selected for Supplementary Fig. 3h, and were used for cellular and behavioral data analyses. For western blot quantification of TRIP8b isoforms, brains were sectioned coronally at 300 μm using a cryostat (Leica) through the anterior-posterior length of the hippocampus58. The hippocampal dentate gyrus (DG; distance range from Bregma: −1.34 to −2.54 mm) was sampled (0.8 mm diameter punch, Fine Science Tools, Foster City, CA) and tissues were homogenized in RIPA Buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, and 50 mM TRIS, pH 8.0) with protease inhibitors (Roche, Indianapolis, IN). Protein concentration was determined using the BCA Protein Assay Reagent (Pierce, Rockford, IL). 20 μg of the protein was electrophoretically separated on a 7.5% Tris-acetate gel and transferred to nitrocellulose membranes (Millipore, Billerica, MA). After transferring proteins from gel to nitrocellulose membrane, membranes were horizontally cut to enable immunoblotting for proteins on subsections of membrane. Membranes were incubated with primary antibodies against rabbit anti-TRIP8b (total, from DM Chetkovich), Guinea pig anti-TRIP8b (1a5, from DM Chetkovich), mouse anti-TRIP8b (exon 4, Neuromab, Cat. #73-208). mouse anti-TRIP8b (1b, Neuromab, Cat. #73-245) or mouse anti-GAPDH (Millipore, Cat. #MAB374). Appropriate donkey anti-rabbit IgG-HRP (Calbiochem, Cat. #401393), donkey anti-guinea pig IgG-HRP (Jackson ImmunoResearch Laboratories, Cat. #106-035-003) and donkey anti-mouse IgG-HRP secondary antibodies (Calbiochem, Cat. #401253) were used. Immunoreactive proteins were detected via chemiluminescent substrate (Thermofisher Scientific, Cat. #34077) using X-ray film (Phenix Research Product, Cat. #F-BX810). Stripping and reprobing was used only for the membrane subsection blotted first for IsoA5 and subsequently for TRIP8b. Stripping of IsoA5 was performed using Restore™ Western Blot Stripping Buffer (Thermo Fisher Scientific, Cat. #21059) following manufacturer’s instructions, and the blot was then incubated with TRIP8b antibody. Images of immunoreactive bands were captured using 3CCD color video camera (Sony, Cat. #DCX-3900), and the intensities were analyzed by densitometry using Scion image software (Scion Corporation). The measurements of immunoreactivity for each protein of interest were normalized to GAPDH in each sample.
For in vitro knockdown efficiency test of pAAV-TRIP8b shRNA, HEK293 cells were co-transfected with TRIP8b including exon 1a-4 expressing plasmids (from DM Chetkovich) with either pAAV-SCR shRNA-GFP or pAAV-TRIP8bshRNA-GFP using lipofectamin 2000 (ThermoFisher Scientific) according to the manufacturer’s protocol. Cells were harvested 72 hr post-transfection. 30 ug cell lysates was electrophoretically separated on a Mini-Protean TGX Gel, 4–15% (Bio-Rad) and transferred to nitrocellulose membranes (LI-COR Bioscience) for 1 hr. For quantification of loading control, total protein stain was performed using REVERT™ Total Protein Stain (LI-COR Bioscience, Cat. #926-11010) following manufacturer’s instructions. Nitrocellulose membranes were blocked for >60 min at room temperature with blocking buffer (Licor Odyssey Blocking Buffer), then probed overnight at 4°C with primary antibodies against rabbit anti-TRIP8b antibody (from DM Chetkovich) and GAPDH (Millipore, Cat. #MAB374) in blocking buffer (Licor Odyssey Blocking Buffer). The following day, blots were rinsed and incubated in IRDye® 680LT Goat anti-Mouse IgG (H + L; LI-COR Bioscience, Cat. #P/N 925-68020) and IRDye® 800CW Donkey anti-Rabbit IgG (H + L; LI-COR Bioscience, Cat. #P/N 926-32213) in blocking buffer (Licor Odyssey Blocking Buffer). Immunoreactive bands were visualized using an Odyssey CLx chemifluorescence detector (LI-COR) with quantitation performed using LI-COR Image studio 5.x. Signal intensity of TRIP8b was normalized by signal intensity of each lane of total protein staining59.
Tissue collection and immunohistochemistry (IHC)
Mice were anesthetized with chloral hydrate (Sigma-Aldrich, Cat. #C8383, 400 mg/kg, stock solution 400 mg/ml made in 0.9% NaCl solution, i.p.) and underwent intracardial exsanguination with cold 0.1M PBS (7 ml/min, 6 min) followed by perfusion with 4% paraformaldehyde in 0.1 M PBS (15 min). After subsequent cryoprotection, brains were sectioned coronally on a freezing microtome (Leica), collecting 30 μm sections through the entire anterior-posterior length of the hippocampus and entorhinal cortex (distance range from Bregma: −0.82 to −4.24 μm). Serial sets of sections were stored in 0.1% NaN3 in 1xPBS at 4°C until processing for slide-mounted IHC36,60.
For Ki67, doublecortin (DCX), BrdU, GFP, TRIP8b, NeuN, and mCherry IHC, one series of sections was mounted on glass slides (Superfrost/Plus, Fisher) that were coded to ensure experimenters remained blind throughout quantification and analysis of data. Sections were processed for antigen retrieval (0.01 M citric acid, pH 6.0, 95°C, 15 min) and nonspecific staining was blocked by incubating in blocking solution (3% normal donkey serum [NDS], vol/vol in 0.1% Triton X-100 in 1xPBS) for 30 min. For BrdU IHC, permeabilization was performed using 0.1% Trypsin in 0.1 M Tris and 0.1% CaCl2, and denaturation was performed using 2 N HCl in 1xPBS before blocking step. After blocking, sections were then incubated in one of the following primary antibodies: rabbit anti-Ki67 (1:500; Thermo Scientific, Cat. #RM-9106), goat anti-DCX (1:500; Santa Cruz, Cat. #SC-8066), rat anti-BrdU (1:800; Accurate Chemicals Cat. #Obt0030), chicken anti-GFP (1:3000; Aves Cat. #GFP-1020), guinea pig anti -TRIP8b, mouse anti-TRIP8b1a5 (both from D. M. Chetkovich), rabbit anti-mCherry (1:1000; Clontech, Cat. #632496) in 0.1% Tween-20 in 1xPBS overnight. For BrdUNeuN double IHC, sections were incubated in mouse-anti-NeuN (1:500; Millipore Cat. #MAB377, Billerica, MA) and rat anti-BrdU (1:800; Accurate Chemicals Cat. #Obt0030). After primary incubation, sections for single labeling IHC were rinsed and incubated in the appropriate secondary antibody: biotinylated-donkey anti-rabbit IgG (Cat. #711-065-162), biotinylated-donkey-anti-goat IgG antibody (Cat. #705-065-003), biotinylated-donkey-anti-rat-IgG (Cat. #712-065-153), biotinylated-donkey-anti-chicken-IgY (Cat. #703-065-155), biotinylated-donkey-anti-guinea pig IgG (Cat. #706-065-148) or biotinylated-donkey-anti-mouse-IgG (Cat. #715-065-150), all 1:200 (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) in 1.5% NDS in 1xPBS for 1 hr. After rinses and 30 min in 0.3% hydrogen peroxide in 1xPBS, sections were incubated in avidin-biotin complex (ABC Elite, Vector Laboratories) for 60 min. After rinsing, staining was visualized using DAB/metal concentration (Thermo Scientific, Cat. #1856090), or Fluorescein-, Cyanine 3-, or Cyanine 5-labeled Tyramide Signal Amplification (TSA, PerkinElmer, Cat. #SAT701, #SAT704 or #SAT705). For double labeling IHC (BrdUNeuN), after NeuN primary incubation sections were incubated in Cyanine 5-donkey-anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories Inc., Cat. #715-175-150), fixed with 4% paraformaldehyde for 15 min, then incubated in BrdU primary followed by incubation in biotinylated donkey-anti-rat secondary antibody (Jackson ImmunoResearch Laboratories Inc., Cat. #712-065-153) and then ABC Elite, and finally visualized via incubation in Fluorescein-TSA (TSA, PerkinElmer, Cat. #SAT701). Nuclear Fast Red (Vector Laboratories, Cat. #H-3403), DAPI (Roche, Cat. #236276) or Neurotrace 500/525 green fluorescent Nissl (Invitrogen, Cat. #N-21480) were used as counterstains.
Assessment and quantification of immunoreactive soma and terminals
Unbiased analysis of Ki67+, BrdU+, DCX+, GFP+, and cfos+ cell number in the DG was performed via stereologic quantification on a BX51 System Microscope (Olympus America, Center Valley, PA, USA) by an observer blind to experimental condition as previously described36,61. Ki67+ subgranular zone (SGZ) cells (Trip8b KO tissue), BrdU+ granular cell layer (GCL) cells, GFP+ SGZ type-1 radial glial-like cells (nestin-GFP tissues) and cfos+ GCL cells (AAV-hM3Dq tissue) in the DG were visualized with a 40x, 0.63 NA oil-immersion objective and quantified with the formula:
where ssf is the section sampling fraction (Ki67: 1/9, BrdU, GFP and cfos: 1/8), asf is the area sampling fraction (1 for these rare populations of cells; thus, all cells were counted in 1/9 or 1/8 of the sections), hsf is the height sampling fraction (1 given the minimal effect edge artifacts have in counting soma <10um with ssf 1/9 or 1/8). As both hemispheres were counted for Ki67, the resulting formula was:
One hemisphere was counted for BrdU, GFP or cfos, thus the resulting formula was:
For Bregma graphs of immunoreactive cells, the number of cells was examined per coronal section over the rostrocaudal axis of the DG and the X axis displays the distance from Bregma in mm. For brain regional qualitative assessment of TRIP8b expression level in WT mice and KO mice (Supplementary Fig. 2i), relative expression in the parietal cortex, Ent, and hippocampal subregions was examined via epifluorescence and 400× magnification and was ranked from - (not detected) to +++ (greatest relative expression). For brain regional assessment of GFP or mCherry expression after Ent infusion of virus (Supplementary Figs. 5, 10), stained sections were examined for presence of GFP+ or mCherry+ soma or terminals via epifluorescence and 400× magnification.
For the cfos+ Ent cell density, cells were visualized with a 20× objective and cfos+ cells in lEnt and mEnt images (two images/subject, mCherry n=5; hM3Dq n=4) were taken using DP74 camera with Cellsens software (Olympus America, Center Valley, PA, USA). Cells were quantified via an observer blind to experimental condition using ImageJ software with automated analysis (US National Institutes of Health, Bethesda, Maryland, USA)62. lEnt and mEnt area were measured by Cellsens standard software (ver. 1.16, Olympus America, Center Valley, PA, USA). For the DCX+ population of GCL cells, which were not rare, cells were visualized with a 0.75 NA (DCX) oil-immersion objective, the Optical Fractionator Probe within Stereo Investigator software (ver. 11.03, MBF Bioscience, Williston, VT, USA) was applied, and an unbiased counting frame was superimposed on the region of interest using a MicroFIRE A/R camera (Optronics, Goleta, CA, USA). To account for tissue shrinkage, an optical dissector height of 12 μm was applied. Both hemispheres were counted for Trip8b KO tissue or one hemisphere was counted for AAV-TRIP8b shRNA or AAV-hM3Dq tissue. As the ssf was 1/9 (Trip8b KO tissue) or 1/8 (AAV-TRIP8b shRNA or AAV-hM3Dq tissue), total counts were multiplied by 9 or 16 to obtain the total number of DCX+ GCL or DG cells.
Confocal microscopy and colocalization analyses
Colocalization of immunoreactive cells was assessed by an observer blind to experimental condition with confocal microscopy (LSM510-META, Carl Zeiss, Oberkochen, Germany; emission wavelengths 488 and 633 nm)63. Analyses were performed using a 63x, 1.2 NA water-immersion lens. 50–100 cells BrdU+ cells were analyzed in 5–6 brain sections from the anterior, middle, and posterior hippocampus of each mouse, and were classified as NeuN+ or NeuN- by analysis of adjacent Z-sections, orthogonal sectioning through Z-sections, and three-dimensional (3D) reconstruction with rotation. The percent of BrdU+ cells that was NeuN+ cells in each mouse was multiplied by total BrdU+ cells for each mouse, which was obtained via stereology. The resulting number of BrdU+NeuN+ cells for each mouse were combined for each group and subjected to statistical analysis.
Dendritic tree reconstruction
Dendrite and cell body morphological analyses of DCX+ neurons stained with DAB were performed by an observer blind to experimental condition using NeuroLucida software (ver. 10, MBF Bioscience) on a BX51 System Microscope (Olympus America, Center Valley, PA, USA) and a 40x, 0.75 NA oil-immersion lens with a MicroFIRE A/R camera (Optronics, Goleta, CA, USA). DCX+ cells selected for tracing had three or more nodes, and the cell body and branches were visible within the thickness of the section. The dendritic tree and cell body of each neuron was traced in its entirety with an unbiased systematic random sampling of the visual field20. 6–8 DCX+ neurons from anterior (−1.46 to −2.06 mm from Bregma) and posterior (−2.76 to −3.28 mm from Bregma) DG sections and spaced equally throughout the anterior-posterior axis of the hippocampus were analyzed. The coordinate files generated by 3D reconstruction were analyzed in NeuroLucida Explorer, generating morphologic measurements that included cell body cross-section, dendrite length and surface area, number of nodes and endings, and maximum branch order for each neuron.
Behavioral tests
Brief descriptions of the behavioral tests are provided below. Male or female mice were used for behavioral tests, as described below, and behavior tests were done during light cycle unless otherwise noted. Behavioral scoring was done by a single rater, blind to the treatment condition.
Chronic social defeat stress (CSDS) and Social Interaction (SI) test
Exposure to social defeat and subsequent interaction testing was performed as described previously18,25,36. Retired male CD1 breeders (4–6 months old; Charles River Laboratories, Wilmington, MA) were screened for aggressiveness by ensuring it took <60 sec latency to attack another CD1 mice placed into its home cage. For CSDS, a CD1 aggressor mouse was housed on one side of a cage partitioned with a plastic divider, and a test mouse (C57BL/6J or CamKIIα-icre) on the other side of the divider. Two to three hr before the onset of the dark cycle, the “defeat” bout was triggered by removing the plastic divider for 5 (C57BL/6J experiments) or 10 (CamKIIα-icre experiments) min, which allowed the CD1 aggressor to physically attack the test mouse. The test mouse was moved to a different cage opposite a new aggressor either after the defeat bout (C57BL/6J experiments) or immediately prior to the next defeat (CamKIIα-icre experiments) in order to instill generalization of stress-induced defeat to the CD1 strain, not to just a single aggressor. Control mice were housed in partitioned cages identical to those used for defeat, but they were housed opposite to other control mice. Control mice were handled each day at the same time of day as when the defeat was performed. The defeats or handling continued daily for 10 d.
On day 11 (24 hr after the last day of the 10 d CSDS), social interaction was measured via two trials lasting 150 sec each. Test mice were individually placed in the novel environment of a white open-field chamber (L 45 × W 45 × H 30 cm) with a discrete “interaction zone” against one wall (L 14 × W 26 cm) that encompassed an empty plastic enclosure (L 6.5 × W 10 × H 42 cm, Nationwide Plastics custom order). For the first trial, the mouse was placed randomly into either corner opposite to the interaction zone, and the movements of the mouse were tracked using Ethovision software (Noldus Information Technology). Specifically, the time the mouse spent in the interaction zone or corners opposite to the interaction zone was quantified. For the second trial, which began ~5 min after the first trial, an unfamiliar CD1 aggressor mouse was placed into the plastic enclosure in the interaction zone. The Social Interaction (SI) ratio was calculated as follows:
Mice with SI ratio <1 were categorized as susceptible, while mice with SI ratio ≥1 were categorized as resilient. The distribution of SI ratio is shown in Supplementary Fig. 3f, and the weights of mice on Day 1 and 11 of CSDS is shown in Supplementary Fig. 3g. Immediately after testing for social interaction, mice were singly-housed until they were euthanized 24 hr later.
Locomotor test (LM)
Mice were individually placed in a mouse cage with fresh bedding, and the cage was placed between photocells under dim/red lights during the light cycle and red lights during the dark cycle. A computer-controlled photobeam activity system (San Diego Instruments) recorded total movement of mice in the XY plane, with photocell beam breaks recorded with 5 min bins for 17 or 20 hr. In Fig. 3i, one mouse from the TRIP8b/IRR group was excluded due to a husbandry error (water bottle flooded cage during LM test).
Restraint stress
Except for under basal condition (Fig. 3a–e) or conditions to model stress (CORT, Fig. 4a–g; CSDS experiments, Fig. 5s–t), mice were restrained for 30 min prior to depressive-like behavior tests (i.e. FST and NSF)24. The restraint apparatus (50 ml falcon tube) restricted movement, but did not interfere with normal breathing since it was modified with numerous holes.
Forced swim test (FST)
The FST was performed to evaluate behavioral despair induced by stress, as previously described64,65. Mice were placed in a 5L beaker (Corning Inc. Life Sciences, Lowell, MA, USA) filled with 4L of 25±2°C water, and the entire session was videotaped. For the last 4 min (Fig. 3c) or total 6 min (Fig. 3j, g, Supplementary Fig 7b, Supplementary Fig 8b) of the session, latency to immobility and immobile time were measured.
Novelty suppressed feeding (NSF) test
Group-housed mice were food-deprived for 18 hr. For the NSF test, a single pellet of regular mouse food chow was placed in the center of a novel open field arena (L 44 × W 44× H 30 cm) under dim light24,35. A mouse was placed in a corner of the arena and allowed to explore for up to 12 min. The trial ended when the mouse initially bit and consumed the chow. After the NSF test, the mouse was immediately removed from the test arena and placed individually in the home cage where they were presented with a pre-weighed amount of chow for 5 min. The food was weighed at the end of the session and the amount consumed (difference) was recorded in grams.
Contextual fear conditioning test (CFC) and cue fear conditioning test
Context- and cue-dependent fear conditioning were performed using a contextual conditioning system (Med Associates, St. Albans, VT, USA) as previously described63. Mice were trained and tested in contextual and cued fear condition within the chamber (L 25 × W 29 × H 25 cm) with clear plastic walls and ceilings, and a standard floor made of metal bars. For training, after 2 min habituation a tone was presented (80 dB white noise, 30 sec), which co-terminated with a footshock (0.5 mA, 2 sec). The tone-shock pairing was repeated for a total of 3 shocks over 6 min, with 1-min interval between tone presentations. For the contextual conditioning test, 24 hr after training mice were placed into the training context for 5 min, but the tone and shock were not presented. For the cued conditioning test, 48 hr after training mice were placed in a novel environment (plastic floor over the grid bars, triangle roof, vanilla scent) for 6 min with the tone presented continuously for the last 3 min. Scoring of freezing behavior was automatically performed by the Med Associates software using a threshold of 20 arbitrary units and minimum freeze duration of 0.5 sec.
Dark/Light test (D/L)
The apparatus consisted of a polypropylene cage (L 44 × W 21 × H 21 cm) unequally divided (⅔ and ⅓) into two chambers by a wall with a small vestibule. The large chamber was open, transparent, and brightly-illuminated by two 20W fluorescent lights (1388 lux at cage floor), while the small chamber was closed, painted black, and dark. Initially, the mouse was placed in the dark-lit side for 2 min, and the transitions of the mouse between the two chambers was automatically detected by four photocells located in the vestibule for 10 min. The time spent in brightly-lit side and latency to enter the brightly-lit side were measured by automated system.
Elevated plus maze (EPM)
The apparatus was built (elevation 99 cm, two open and two closed arms each L 67 × W 6 cm, closed arm walls H 17 cm) as previously described66. Mice were placed in the center of the apparatus and allowed to freely explore for 5 min under dim light. Time spent in open or closed arms and frequency in each arm were scored by Ethovision software (Noldus Information Technology).
Splash test
Animals were singly-housed a day prior to test. The day of the test, the nestlet was removed from their home cage at least one hour prior to test, as previously described67. A freshly made 10% sucrose solution was placed into a fine water spray mister and nozzle was depressed a single time to dispense solution onto the back/dorsal region. Grooming activity (frequency and duration) was recorded for 5 min after the application of the solution.
Image presentation
For image presentation, journal guidelines were applied (www.nature.com/authors/policies/image.html). For western blot analyses, images of adjacent immunoreactive bands were captured using 3CCD color video camera (Sony, Cat. #DCX-3900), and intensities were analyzed by densitometry using Scion image software (Scion Corporation). Full western blots images are provided in Supplementary Fig. 3 and Supplementary Fig. 4. Photomicrographs of immunostained tissue sections were collected on either a DP74 camera with Cellsens software (Olympus America, Center Valley, PA, USA) or a MicroFIRE A/R camera (Optronics, Goleta, CA, USA). For orthogonal images captured using confocal microscopy (LSM510-META, Carl Zeiss, Oberkochen, Germany; emission wavelengths 488 and 633 nm)63, analyses were performed using a 63x, 1.2 NA water-immersion lens by analysis of adjacent Z-sections, orthogonal sectioning through Z-sections, and three-dimensional (3D) reconstruction with rotation. Scale bars photomicrographs were collected at the time of image collection. After collection of images, files were opened in Adobe Photoshop and images were cropped and equal adjustments (brightness, contrast) in control and experimental group were made to the entire image; only linear adjustments were made68. Where red and green images were both present in a single image, images were color-shifted in Photoshop to enable visualization by color-blind individuals68. Processed images were then opened in Adobe Illustrator and panel indicators (arrowheads, dotted lines, molecular weight indicators) were added and final figures were assembled.
Statistical analysis
Data are reported as mean±SEM. Prior to statistical analyses, data assumptions (e.g. normal distribution, similar variation between experimental groups, etc.) were verified. Statistical approaches and results are provided in Supplementary Table 1, statistic analysis summaries are provided figure legends, and additional information can be found in the Life Sciences Reporting Summary that accompanies this paper and is available online. Analyses with two groups were performed using an unpaired, two-tailed unpaired t-test. Analyses with more than two groups and one variable were performed with one-way ANOVA and either Bonferroni posthoc test or Fisher’s LSD posthoc test, with posthoc significance indicated by asterisks. Analyses with more than two variables were performed with two-way ANOVA with Bonferroni posthoc test or Fisher’s LSD posthoc test, with posthoc significance indicated by lowercase letters. Repeated measures (RM) were utilized where appropriate, as indicated in figure legends and Supplementary Table 1. For correlation of DG TRIP8b isoforms levels with SI ratio, linear regression was performed and r square and p-value were reported. Statistics were performed using Prism software (ver. 6.0, ver. 7.0). Statistical significance was defined as p<0.05, and exact p values and F value are provided in Supplementary Table 1. For all molecular and behavioral studies, mice were randomly assigned to groups. Many behavioral and cellular results were confirmed via independent replication of the experiments, as shown in Supplementary Table 1. Additionally, investigators were blinded to the treatment group until all data had been collected. Most behavioral and cellular results were confirmed via at least one independent replication (i.e. Fig. 1, 2h, j, n–w, 3i–j, 4b–g, 5g–t; Supplementary Figs. 1b–m, 3a–l, 4a–e, 5i–k, 7a–b, 8a–b, 9a–f, and 11e–g). Samples sizes determined by extensive laboratory experience and verified via power analysis.
Supplementary Material
Acknowledgments
We thank SG Birnbaum, IM Bowen, ED Marsh, AJ McCoy, L Peca, S Stojadinovic, CA Tamminga, Z Zhang, and JM Zigman for sharing experimental reagents and guidance on use of computer code and providing guidance or assistance in experimental techniques, and IM Bowen and SE Latchney for feedback. This work was supported by grants from the National Institutes of Health to AJE (DA023701, DA023555, MH107945), DMC (R01 NS059934, R21 MH104471), the National Aeronautics and Space Administration to AJE (NNX07AP84G, NNX12AB55G, NNX15AE09G), and an Independent Investigator Award from the National Alliance for Research on Schizophrenia and Depression/Brain and Behavior Foundation to AJE. SY was funded by NIMH T32-MH076690, Basic Science Training Program in the Neurobiology of Mental Illness, PI: CA Tamminga. PDR was funded by NIDA T32-DA007290, Basic Science Training Program in the Drug Abuse Research, PI: AJ Eisch.
Footnotes
Data Availability Statement
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contributions
SY conceived the study, performed most experiments, generated the figures, and wrote the manuscript.
RPR assisted with experiments and generated figure schematics.
IP performed electroelectroencephalogram experiments.
AW performed electroelectroencephalogram experiments.
PDR assisted with experiments.
AS performed the electrophysiology experiments.
ADG assisted with experiments.
MS assisted with experiments.
MJD assisted with experiments.
NI assisted with experiments.
SM assisted with experiments.
DRR assisted with experiments.
CEK provided Trip8b KO mouse brains and TRIP8b and isoform antibodies.
RCA-N wrote the code for in vivo electroelectroencephalogram experiment analysis.
IS assisted with experiments.
DMC provided Trip8b KO mouse brains and TRIP8b and isoform antibodies.
SK performed the electrophysiology experiments.
DAC guided electroelectroencephalogram experiments.
AJE conceived the study, assisted with experiments, guided figure preparation, and wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379:1045–1055. doi: 10.1016/S0140-6736(11)60602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trivedi MH. Modeling predictors, moderators and mediators of treatment outcome and resistance in depression. Biol Psychiatry. 2013;74:2–4. doi: 10.1016/j.biopsych.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Rosa MA, Lisanby SH. Somatic treatments for mood disorders. Neuropsychopharmacology. 2012;37:102–116. doi: 10.1038/npp.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kronmüller KT, et al. Hippocampal volume and 2-year outcome in depression. Br J Psychiatry. 2008;192:472–473. doi: 10.1192/bjp.bp.107.040378. [DOI] [PubMed] [Google Scholar]
- 5.Yun S, Reynolds RP, Masiulis I, Eisch AJ. Re-evaluating the link between neuropsychiatric disorders and dysregulated adult neurogenesis. Nat Med. 2016 doi: 10.1038/nm.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller BR, Hen R. The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol. 2015;30:51–58. doi: 10.1016/j.conb.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stone SSD, et al. Stimulation of entorhinal cortex promotes adult neurogenesis and facilitates spatial memory. J Neurosci. 2011;31:13469–13484. doi: 10.1523/JNEUROSCI.3100-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suthana N, et al. Memory enhancement and deep-brain stimulation of the entorhinal area. N Engl J Med. 2012;366:502–510. doi: 10.1056/NEJMoa1107212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerritsen L, et al. Depression, hypothalamic pituitary adrenal axis, and hippocampal and entorhinal cortex volumes--the SMART Medea study. Biol Psychiatry. 2011;70:373–380. doi: 10.1016/j.biopsych.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 10.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- 11.Lewis AS, et al. Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J Neurosci. 2011;31:7424–7440. doi: 10.1523/JNEUROSCI.0936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santoro B, et al. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis AS, et al. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim CS, Chang PY, Johnston D. Enhancement of dorsal hippocampal activity by knockdown of HCN1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron. 2012;75:503–516. doi: 10.1016/j.neuron.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2015;55:399–417. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- 16.Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci. 2007;10:1110–1115. doi: 10.1038/nn1969. [DOI] [PubMed] [Google Scholar]
- 18.Krishnan V, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Kourrich S, Glasgow SD, Caruana DA, Chapman CA. Postsynaptic signals mediating induction of long-term synaptic depression in the entorhinal cortex. Neural Plast. 2008;2008:840374. doi: 10.1155/2008/840374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latchney SE, Jiang Y, Petrik DP, Eisch AJ, Hsieh J. Inducible knockout of Mef2a, -c, and -d from nestin-expressing stem/progenitor cells and their progeny unexpectedly uncouples neurogenesis and dendritogenesis in vivo. FASEB J. 2015 doi: 10.1096/fj.15-275651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo W, et al. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med. 2011;17:559–565. doi: 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrik D, Lagace DC, Eisch AJ. The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology. 2012;62:21–34. doi: 10.1016/j.neuropharm.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill AS, Sahay A, Hen R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology. 2015;40:2368–2378. doi: 10.1038/npp.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476:458–461. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker AK, et al. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry. 2015;20:500–508. doi: 10.1038/mp.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.David DJ, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone EA, Lin Y. An anti-immobility effect of exogenous corticosterone in mice. Eur J Pharmacol. 2008;580:135–142. doi: 10.1016/j.ejphar.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 28.Gourley SL, et al. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008;63:353–359. doi: 10.1016/j.biopsych.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray F, Smith DW, Hutson PH. Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur J Pharmacol. 2008;583:115–127. doi: 10.1016/j.ejphar.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 30.White WF, Nadler JV, Hamberger A, Cotman CW, Cummins JT. Glutamate as transmitter of hippocampal perforant path. Nature. 1977;270:356–357. doi: 10.1038/270356a0. [DOI] [PubMed] [Google Scholar]
- 31.Melzer S, et al. Long-range-projecting GABAergic neurons modulate inhibition in hippocampus and entorhinal cortex. Science. 2012;335:1506–1510. doi: 10.1126/science.1217139. [DOI] [PubMed] [Google Scholar]
- 32.Casanova E, et al. A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis. 2001;31:37–42. doi: 10.1002/gene.1078. [DOI] [PubMed] [Google Scholar]
- 33.Krashes MJ, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507:238–242. doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vismer MS, Forcelli PA, Skopin MD, Gale K, Koubeissi MZ. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front Neural Circuits. 2015;9:27. doi: 10.3389/fncir.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santarelli L, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 36.Lagace DC, et al. Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc Natl Acad Sci U S A. 2010;107:4436–4441. doi: 10.1073/pnas.0910072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15:1475–1484. doi: 10.1038/nn.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eichenbaum H. A cortical-hippocampal system for declarative memory. Nat Rev Neurosci. 2000;1:41–50. doi: 10.1038/35036213. [DOI] [PubMed] [Google Scholar]
- 39.Tulving E. Episodic memory: from mind to brain. Annu Rev Psychol. 2002;53:1–25. doi: 10.1146/annurev.psych.53.100901.135114. [DOI] [PubMed] [Google Scholar]
- 40.Surget A, et al. Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry. 2011;16:1177–1188. doi: 10.1038/mp.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma DK, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boldrini M, et al. Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol Psychiatry. 2012;72:562–571. doi: 10.1016/j.biopsych.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs J, et al. Direct Electrical Stimulation of the Human Entorhinal Region and Hippocampus Impairs Memory. Neuron. 2016;92:983–990. doi: 10.1016/j.neuron.2016.10.062. [DOI] [PubMed] [Google Scholar]
- 44.Kheirbek MA, et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron. 2013;77:955–968. doi: 10.1016/j.neuron.2012.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vivar C, et al. Monosynaptic inputs to new neurons in the dentate gyrus. Nat Commun. 2012;3:1107. doi: 10.1038/ncomms2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Airan RD, et al. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- 47.Crupi R, Marino A, Cuzzocrea S. New therapeutic strategy for mood disorders. Curr Med Chem. 2011;18:4284–4298. doi: 10.2174/092986711797200417. [DOI] [PubMed] [Google Scholar]
- 48.Malberg JE, Schechter LE. Increasing hippocampal neurogenesis: a novel mechanism for antidepressant drugs. Curr Pharm Des. 2005;11:145–155. doi: 10.2174/1381612053382223. [DOI] [PubMed] [Google Scholar]
- 49.Lozano AM, Lipsman N. Probing and regulating dysfunctional circuits using deep brain stimulation. Neuron. 2013;77:406–424. doi: 10.1016/j.neuron.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- 51.Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med. 2003;9:1539–1544. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- 52.Zolotukhin S, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 53.Mandyam CD, Harburg GC, Eisch AJ. Determination of key aspects of precursor cell proliferation, cell cycle length and kinetics in the adult mouse subgranular zone. Neuroscience. 2007;146:108–122. doi: 10.1016/j.neuroscience.2006.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alonso A, Klink R. Differential electroresponsiveness of stellate and pyramidal-like cells of medial entorhinal cortex layer II. J Neurophysiol. 1993;70:128–143. doi: 10.1152/jn.1993.70.1.128. [DOI] [PubMed] [Google Scholar]
- 55.Corbett BF, et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2013;33:7020–7026. doi: 10.1523/JNEUROSCI.2325-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dengler CG, Yue C, Takano H, Coulter DA. Massively augmented hippocampal dentate granule cell activation accompanies epilepsy development. Sci Rep. 2017;7:42090. doi: 10.1038/srep42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clarkson R, et al. Characterization of image quality and image-guidance performance of a preclinical microirradiator. Med Phys. 2011;38:845–856. doi: 10.1118/1.3533947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spencer S, et al. Circadian genes Period 1 and Period 2 in the nucleus accumbens regulate anxiety-related behavior. Eur J Neurosci. 2013;37:242–250. doi: 10.1111/ejn.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eaton SL, et al. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS One. 2013;8:e72457. doi: 10.1371/journal.pone.0072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ables JL, et al. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J Neurosci. 2010;30:10484–10492. doi: 10.1523/JNEUROSCI.4721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeCarolis NA, et al. In vivo contribution of nestin- and GLAST-lineage cells to adult hippocampal neurogenesis. Hippocampus. 2013;23:708–719. doi: 10.1002/hipo.22130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petrik D, et al. Functional and mechanistic exploration of an adult neurogenesis-promoting small molecule. FASEB J. 2012;26:3148–3162. doi: 10.1096/fj.11-201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakasato A, et al. Swim stress exaggerates the hyperactive mesocortical dopamine system in a rodent model of autism. Brain Res. 2008;1193:128–135. doi: 10.1016/j.brainres.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 66.Mulder GB, Pritchett K. The elevated plus-maze. Contemp Top Lab Anim Sci. 2004;43:39–40. [PubMed] [Google Scholar]
- 67.Surget A, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64:293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 68.Johnson J. Not seeing is not believing: improving the visibility of your fluorescence images. Mol Biol Cell. 2012;23:754–757. doi: 10.1091/mbc.E11-09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.