

Abstract
Background
Triple negative breast cancer (TNBC) subtypes are clinically aggressive and are treated with targeted therapeutics commonly used in other BC subtypes. The claudin-low (CL) molecular subtype of TNBC has high rates of metastases, chemoresistance and recurrence. There exists an urgent need to identify novel therapeutic targets in TNBC; however, existing models utilized in target discovery research have limitations. Patient-derived xenograft (PDX) models have emerged as superior models for target discovery experiments because they recapitulate features of patient tumors that are limited by xenograft methods.
Methods
We utilize immunohistochemistry, qRT-PCR and Western Blot to visualize tumor architecture, cellular composition, genomic and protein expressions of a new TNBC PDX model (TU-BcX-2O0). We utilize tissue decellularization techniques to examine extracellular matrix composition of TU-BcX-2O0.
Results
Our laboratory successfully established a TNBC PDX tumor, TU-BCX-2O0, which represents a CL-TNBC subtype and maintains this phenotype throughout subsequent passaging. We dissected TU-BCx-2O0 to examine aspects of this complex tumor that can be targeted by developing therapeutics, including specific cell populations within the tumor, the extracellular matrix, and cancer stem-like cell populations.
Conclusions
Here we characterize a claudin-low TNBC patient-derived xenograft model that can be utilized for therapeutic research studies.
Keywords: Triple-negative breast cancer, claudin-low, patient-derived xenograft, mesenchymal, extracellular matrix, decellularized tumor scaffold, collagen
Background
Triple negative breast cancers (TNBCs) are aggressive, defined by a propensity to metastasize, recur and develop chemoresistance [1,2]. Due to reduced expression of commonly targeted receptors (estrogen receptor (ER), human epidermal growth factor receptor 2 (HER2)), TNBCs are more difficult to treat compared to other breast cancer subtypes, underlining the need to discover novel therapeutic targets in TNBC [3–5]. TNBCs are categorized into four molecular subtypes: Basal-like (BL1 and BL2), mesenchymal (M) and luminal androgen receptor (LAR) [6]. Each subtype has a distinct molecular profile, which dictates responsiveness (or sensitivity) to targeted therapies and immune infiltration [7,8]. Within the basal-like subtypes, there exists an intrinsic tumor cell population that expresses low levels of luminal differentiation markers, high expression of genes associated with epithelial-to-mesenchymal transition (FN1, MMP2, TWIST, SERPINE1, THY1, SPARC) [9] and enriched in genes involved in the immune response and cancer stem cell (CSC) populations [10,11]. This subpopulation of basal-like tumors is known as claudin-low TNBC (CL-TNBC). On a transcriptional level, CL-TNBCs show high genomic instability and are highly undifferentiated. Clinically, CL-TNBCs have high rates of metastasis, recurrence and chemoresistance [8]. The recent classification of the CL subtype revealed unchartered approaches to identify targets that are specific for the different molecular subtypes [12]. Notably, the CL subtype is the least characterized molecular TNBC subtype in the literature.
One obstacle in the field of drug discovery research is the limitation of frequently utilized models. Many of these models, namely immortalized, cancer-derived cell lines, or orthotopic xenografts, are unable to re-create patient-specific features of tumors. More specifically, these models cannot accurately reflect the tumor architecture and surrounding stroma, and cannot reproduce the cellular heterogeneity that is present in the original patient tumor [13–15]. Passaging of immortalized cell lines results in the introduction of irreversible alterations in genetic information and behavioral characteristics [16]. Bypassing these obstacles is crucial to investigating previously unrecognized targets and mechanisms of neoplastic diseases beyond breast cancer.
Patient-derived xenografts (PDXs) are emerging as a novel research models that overcome these limitations [17–19]. Therapeutic discovery in breast cancers can especially benefit from these translational models because of the stability of xenografts that possess the complex stromal architecture and heterogeneous cellular composition of TNBC [20–22]. Furthermore, PDX models facilitate studying mutations and drug interactions that are specific to TNBC subtypes that have a specific molecular signature [23–25]. In fact, PDX models have been found to accurately reflect clinical response to therapeutic agents [26]. One disadvantage to the PDX model is that over time, after multiple passages in mice, murine components invade the tumor and there is evidence of loss of human stromal architecture of the tumors. However, the genomic profiles and clonal dynamics of breast cancer PDX tumors can remain stable throughout sequential in vivo passages [27,28].
Therapeutic discovery research benefits from the preservation of the tumor microenvironment from the patient, including stromal architecture and stromal cells, sub-populations of tumor cells, extracellular matrix components and interactions amongst diverse cell types. Together, investigating how compounds affect various aspects of the tumor strengthens drug discovery experiments to optimize treatment strategies of CL-TNBCs. In this study, we have established and characterized a TNBC PDX model that represents the mesenchymal, claudin-low TNBC subtype based on genomic profiles, composition and tumor architecture. We believe that the use of these translational PDX models will help identify and characterize new molecular targets involved in tumorigenesis and formation of metastases that will ultimately reveal patient-specific therapies.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), Dulbecco’s phosphate-buffered saline (PBS), phenol-red free DMEM, fetal bovine serum (FBS), essential amino acids, non-essential amino acids (NEAA), antibiotic/anti-mitotic, penicillin/streptomycin, sodium pyruvate, and EDTA (0.5 M, pH8) were obtained from GIBCO (Invitrogen; Carlsbad CA). Insulin was purchased from Sigma-Aldrich (St. Louis MO) and charcoal stripped FBS from HyClone (Thermo Scientific; Logan UT). Dimethyl sulfoxide (DMSO) was obtained from Research Organics, Inc (Cleveland OH).
Cell Culture
Human MDA-MB-231 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and are characterized as triple-negative/basal B mammary carcinoma. Cells were maintained in DMEM supplemented with 10% FBS, NEAA, MEM amino acids, anti-anti (100 U/mL), sodium pyruvate and porcine insulin (1 × 10−10 mol/L) at 37°C in humidified 5% CO2. For treatment studies, cells were grown in phenol red-free DMEM supplemented with 5% charcoal-stripped FBS and supplemented with NEAA, MEM amino acids, Gluta-Max and penicillin (100 U/mL).
Patient-derived xenografts
SCID/Beige mice (CB17.Cg-PrkdcscidLystbg/Crl) were purchased from Charles River and are used to prevent rejection of the xenografted human tumors. The autosomal recessive SCID (Prkdcscid) mutation results in severe combined immunodeficiency affecting both the B and T lymphocytes. The autosomal recessive beige (Lystbg) mutation results in defective natural killer (NK) cells. Tumor tissues from each patient were cut into 3 × 3 mm2 pieces under aseptic sterile conditions, coated with full factor Matrigel (Cat No. 354234, Fisher Scientific, Waltham, MA, USA) and implanted bilaterally into the mammary fat pads (mfp) of mice under isoflurane and oxygen. Tumors were measured using a digital caliper after ostensible tumor take was established. Tumors were passaged when tumor volume achieved 750–1000 mm3. To passage, mice were euthanized by CO2 and tumors were removed, dissected, coated in full factor Matrigel, and then implanted bilaterally into new mice that were anesthetized using a mix of isoflurane and oxygen delivered by mask. Before surgery, mice were given Meloxicam (5 mg/kg/day, for 3 days’ post-surgery) for pain.
RNA isolation and quantitative real time PCR
MDA-MB-231 cells were plated in 10% DMEM and grew to 70% confluency. Cells were harvested and total RNA was isolated using RNeasy (Qiagen, Valencia, CA) following manufacturer’s protocol; quantity and quality were determined by absorbance (260, 280 nm). Total RNA (2 μg) was reverse-transcribed (iScript kit, BioRad Laboratories, Hercules, CA) and analyzed by qRT-PCR. All qRT-PCR data were normalized to actin. Primer sequences are as follows (Invitrogen, Carlsbad, CA): β-actin F–5′-GGCACCCAGCACAATGAAGA-3′; β-actin R-5′-ACTCCTGCTTGCTGATCCAC-3′; CDH1 F-5′-AGGTGACAGAGCCTCTGGATAGA-3′, CDH1 R-3′-TGGATGACACAGCGTGAGAGA-3′. CDH2 F-5′-GCCCCTCAAGTGTTACCTCAA-3′; CDH2 R-5′-AGCCGAGTGATGGTCCAATTT-3′, SNAIL F-5′-ACCACTATGCCGCGCTCTT-3′; SNAIL R-5′-GGTCGTAGGGCTGCTGGAA-3′, ZEB1 F-5′-ACACAAGCGAGAGGATCATG-3′; ZEB1 R-5′-CGGAATCTGAATTTGCTTCTACC-3′, FRA1 F-5′-AGCTGCAGAAGCAGAAGGAG-3′; FRA1 R-5′-GGGGAAAGGGAGATACAAGG-3′; PLAU F-5′-GGTGACGCCTTCAGCATGA-3′; PLAU R-5′-CCCACTGCGGTACTGGACAT-3′. qRT-PCR was conducted as previously published [29]. Data were represented as normalized fold expression compared to human actin with biological triplicate samples ± SEM.
Immunohistochemistry staining
Formalin-fixed, paraffin-embedded tumor sections were deparaffinized, rehydrated in a graded solution of Sub-X solutions, stained with hematoxylin and eosin or quenched with 0.3% H2O2 (Sigma-Aldrich, St. Louis MO), rinsed with PBS with Triton X-100 (PBST), blocked with 1% BSA and stained with primary antibodies against Ki-67 (Abcam) overnight at 4°C. Each tumor section was subsequently washed in PBST, incubated with appropriate HRP-conjugated secondary antibody, and washed with PBST. For colorimetric staining, slides were then incubated in 3,3′-Diaminobenzidine (DAB; Vector Laboratories; Burlingame, CA, USA), washed with PBST, counterstained with hematoxylin, and rinsed with deionized water. Slides were sealed with Permount Mounting Medium (Sigma). Images were acquired at 10X, 20X and 40X objectives using an Aperio Scanscope instrument (Aperio Technologies, Inc., Vista, CA, USA) with ImageScope software (Aperio Technologies, Inc).
Immunofluorescence staining
For immunofluorescence staining, paraffin-embedded tissue sections were stained according to the standard protocol of LSUHSC Molecular Histopathology and Analytical Microscopy core. Briefly, tissue sections were deparaffinization in xylene, re-hydration through descending grades of alcohol to water, non-enzymatic antigen retrieval in citrate buffer, followed by PBS wash and then blocking. Sections were then incubated with a first primary antibody overnight at room temperature, rinsed in PBS and a fluorescein-conjugated secondary antibody (1:200 dilution; Invitrogen) was added and incubated for an hour in the dark. After washing with PBS, a second primary antibody was added overnight in the dark, followed by rinsing with PBS and incubation with a second rhodamine-tagged secondary antibody (1:200 dilution; Invitrogen) for an hour. Primary antibodies included mouse monoclonal antibodies against CD11b (1:50 dilution; Abcam), rat monoclonal Gr-1 (1:100 dilution; Abcam), rabbit polyclonal CD68 (1:100 dilution; Abcam), and rabbit polyclonal F4/80 (1:100 dilution; Abcam). Sections were then washed in PBS, mounted with DAPI (Vector Laboratories), and analyzed using a confocal microscope (Olympus FV1000).
Flow Cytometry
Tumors were digested with DNAse and Liberase (Roche) at 37°C for 1 hour, and tumor-infiltrating MDSC were isolated from tumor single-cell suspensions as described [32]. The following fluorochrome-conjugated human antibodies were used to characterize cell subtypes: CD33 (WM53), HLA-DR (G46–6), CD68 (Y1/82A) (all from BD Biosciences) and CD14 (61D3; eBioscence). Fluorochrome-conjugated mouse antibodies specific for CD11b (M1/70), Gr1 (RB6–8C5), Ly6C (AL-21), and Ly6G (1A8) were purchased from BD Biosciences. Live/Dead cell staining kit was from Molecular Probes (Life Technologies).
PDX Decellularization and Histological Analysis
PDX tumors were decellularized through a modified protocol previously described [30,31]. In brief, tumor samples were collected and left in water for 24 hours at 4°C followed by incubation in Triton X-100 (Bio-Rad, Hercules CA, Cat. No. 1610406). Cells were first washed in deionized water and incubated in sodium deoxycholate solution (ThermoFisher Scientific, Waltham MA), then washed in deionized water and incubated in calcium chloride (Sigma-Aldrich, St. Louis MO) and finally washed with deionized water ad treated with DNAse I (1 U/mL, Sigma-Aldrich, St. Louis MO) and antibiotic-antimycotic (100 U/ml; ThermoFisher Scientific, Waltham MA) solutions. Decellularized tumors were then formalin-fixed (10%), paraffin-embedded and sectioned for Picosirius Red staining. Total collagen content was observed through 4X objective on an Olympus BX51 system microscope. Analysis of collagen fibers was performed in Matlab with a custom code. Quantification of collagen I:III and subsequent representative darkfield images were obtained through use of polarized light microscopy, as described previously [32].
Statistical analysis
Studies run in triplicate were analyzed by unpaired Student’s t-test (Graph Pad Prism V.4). p-Values < 0.05 were considered statistically significant. The flow cytometry analysis of the circulating tumor cells was run in duplicate; all other analyses were run in triplicate.
Results
Characterization of TU-BcX-2O0
TU-BcX-2O0 was derived from a 70-year old African American female diagnosed with non-metastatic, Grade 3 invasive ductal carcinoma. Pathology reported the PAM50 subtype was TNBC (Figure 1A). Before implantation in SCID/Beige mice, we covered TU-BcX-2O0 in Matrigel to mimic the extracellular matrix of the tumor microenvironment. After 102 days, the primary tumor grew to a measurable size, and another 49 days for the tumor to grow to our endpoint volume, 750-1000mm3. Tumors were propagated for future ex vivo and in vitro experiments. In gross appearance, both the primary tumor and subsequent passages had nodules on the surface of the tumor that appeared “pustulate-like” (Figure 1B). As expected, both time to tumor palpation after engraftment and tumor growth were highest in the initial passages compared to consecutive passages. (Figure 1C).
Figure 1. TU-BcX-2O0, had a distinct gross appearance and tumor growth pattern and has a consistent cytology throughout passages.
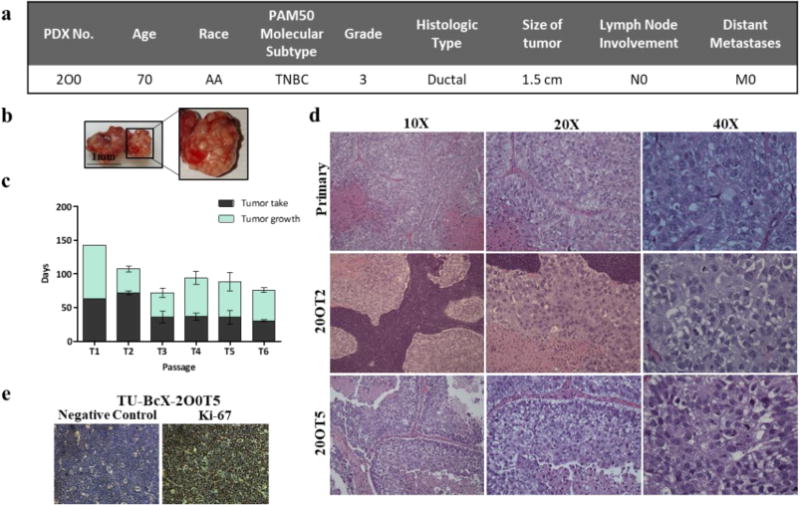
(a) Patient data from TU-BcX-200 shows it is categorized as TNBC based on the PAM50 molecular subtype without lymph node (N0) or distant metastases (M0) involvement at the time of tumor resection. (b) Gross appearance of TU-BcX-2O0 was consistent throughout consecutive passages. The tumor had distinct pockets filled with a pustulate-like substance that was liquid when dissected. (c) Tumor growth patterns of TU-BcX-2O0 after initial engraftment into the mfp of SCID/Beige mice. Tumor growth indicates the number of days from when the tumor first became palpable to when tumors were extracted (at a final volume of 750–1000 mm3). (d) The primary specimen (obtained post-op, not yet engrafted into SCID/Beige mice) histology was compared to lower (T2) and higher passage (T5) PDX tumors. H & E stained images were captured at 10×, 20× and 40×. (e) Proliferation rate determined by Ki-67 immunohistochemistry staining of TU-BcX-2O0T5 PDX tumor compared to negative control.
Formalin-fixed paraffin-embedded sections of primary tumor and subsequent xenograft passages were stained with Hematoxylin and Eosin (H&E) for microscopic evaluation. The histologic appearances of all tumors, primary as well as low and high passages, were similar and revealed high grade histology represented by high grade nuclei, lack of tubule formation and numerous mitoses including atypical mitotic figures, and areas of extensive tumor necrosis (Figure 1D). Furthermore, in concordance with pathology reports, TU-BcX-2O0 tumors revealed high proliferation rate by immunohistochemical staining with Ki-67 antibody (Figure 1E). H&E staining also revealed abundant presence of mixed inflammatory components, namely neutrophils, lymphocytes and eosinophils, although they were restricted to the periphery of the tumor (Supplementary Figure S1). CL-TNBC tumors have an enriched immune response [7], and we found increased mRNA expression of CXCL2 compared to IL-6, IL-8 and CXCR4 (Supplementary Figure S2A). Further, TU-BcX-2O0 had similar mRNA expression of CL-TNBC specific immune genes (CD79A, CD79B, CD14, VAV1 and CXCL12) compared to MDA-MB-231 cells [12,33] (Supplementary Figure S2B). Gr-1+CD11b+ cells, or immune suppressive myeloid derived suppressor cells (MDSCs), are overproduced in tumor-bearing mice, in both the peripheral blood and spleens. A population of these MDSCs were found within the TU-BcX-2O0 tumor (Supplementary Figure 3A). Human macrophages, CD68+ cells, were present both in initial and even in higher passages. Mouse macrophages, identified as F4/80+ cells, were not observed in neither low nor higher passages (Supplementary Figure S3B). These data were confirmed by flow cytometry; the immune composition of the blood and spleen were also analyzed. There was a higher population of mouse MDSCs and macrophages in the blood and spleen compared to the tumor. Further, there were fewer circulating MDSCs (CD11b+Gr1+) and macrophages (CD11b+F4/80+) in tumor-bearing mice than in normal mice, suggestive of the possible migration of these immune subsets from the periphery into the tumor site. It is also of note that the percentages of polymorphonuclear MDSCs (CD11b+Gr1+Ly6ClowLy6G+) were higher than those of monocytic MDSCs (CD11b+Gr1+Ly6ChighLy6G−) in all organs (Supplementary Figure 4 and 5). Finally, the presence of human myeloid cells (CD33 +HLA-DR−) in TU-BcX-2O0 detected by immunohistochemistry was confirmed with flow cytometry data (Supplementary Figure 5). The fact that these cells are HLA-DR− may indicate their immunoregulatory nature.
Molecular characterization of TU-BcX-2O0
TU-BcX-2O0 forms metastatic lesions in the lungs of SCID/Beige mice in both low and high passages, with larger and more frequent lesions in the lower passages (Figure 2A). To evaluate the molecular characteristics of TU-BcX-2O0 over consecutive passages, we analyzed mRNA expression of both luminal and mesenchymal genes in explants using qRT-PCR. While we found evidence of low endogenous levels of the luminal marker E-cadherin (CDH1), there were slightly higher levels of other luminal-associated genes EpCAM and CD24. Of the basal markers analyzed, there were high levels of endogenous VIM expression, and high levels of CDH2, cFOS, TWIST, SNAI1 and SNAI2 mRNA expression in TU-BcX-2O0. These data suggest a “basal-like” phenotype of TU-BcX-2O0 which was consistent throughout passages (Figure 2B). Next, to refine our characterization of the molecular subtype of this TNBC tumor, we analyzed relative claudin (CLDN) expression of the tumors. Specifically, we chose CLDN 3, 4 and 7 because these specific claudins define the “claudin-low” TNBC phenotype. We compared the results to a known CL-TNBC cell line, MDA-MB-231, to observe relative expression. CLDN3 expression was most similar to MDA-MB-231 in both low and high passage TU-BcX-2O0. CLDN4 mRNA expression was lower in TU-BcX-2O0 compared to MDA-MB-231 in both low and high passage. CLDN7 was elevated compared to MDA-MB-231 in low passage, but not higher passage tumors (Figure 2C). Then we evaluated mRNA expression of MUC1, which is also downregulated in CL-TNBC phenotype. We observed similar endogenous mRNA expression as that in MDA-MB-231 cells (Figure 2D). Low expression of MUC1 protein was confirmed with the Alcian Blue stain (Supplementary Figure S7).
Figure 2. TU-BcX-2O0 metastasizes to the lungs and expresses mesenchymal markers.
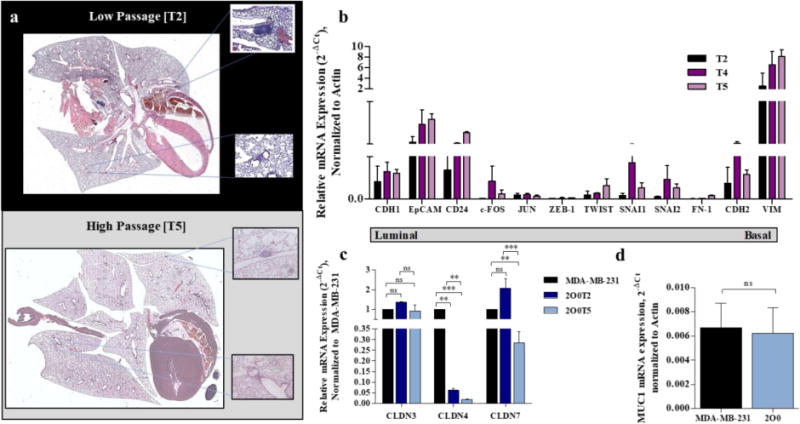
(a) At both lower and higher passages, TU-BcX-2O0 metastasizes to the lungs, although there are fewer lesions in higher passages. Hematoxylin and Eosin stained and imaged. Examples of specific lesions are represented by the boxed areas. (b) qRT-PCR panel of luminal (CDH1, EpCAM, CD24) and basal (c-FOS, JUN, ZEB-1, TWIST, SNAI1, SNAI2, FN-1, CDH2, VIM) gene expressions. Gene expressions in various passages ranging from low (T2) to high (T5) were compared. All data was obtained using qRT-PCR in triplicate and normalized to actin. Error bars represent SEM and significantly different * p < 0.05, *** p < 0.001. (c) The following mRNA baseline expressions of genes enriched or downregulated in the claudin-low TNBC subtype from TU-BcX-2O0 is compared to the claudin-low TNBC cell line, MDA-MB-231. (a) CLDN3, 4 and 7 gene expressions, defining the CL-TNBC subtype, of MDA-MB-231 and 2O0 tumor. (d) 2O0 has comparable MUC1 mRNA expression, a gene associated with the CL subtype, compared to MDA-MB-231. Error bars represent SEM and significantly different * p < 0.05, *** p < 0.001.
Collagen composition of TU-BcX-2O0
To evaluate the relative collagen composition, we stained TU-BcX-2O0 with Picosirius Red to highlight collagen fibers, then use polarized light to examine collagen bundles. Collagen I is represented by red or orange, and collagen III is represented by yellow or green. Using this method, the collagen composition of tumors can be quantified [34]. We show that in both lower and higher passages, TU-BcX-2O0 has a higher expression of collagen I compared to minimal expression of collagen III (Figure 3A). Then, because recent therapeutic discovery research is invested in evaluating roles of novel therapeutics on the extracellular environment, we stripped the tumor of its cellular components using a novel decellurization method to evaluate relative collagen composition without the background of active cell signaling. Decellurization of TU-BcX-2O0 resulted in a similar ratio of collagen I to collagen III, albeit with lower expression levels of collagen type I (Figure 3B). Finally, quantification of these data show significant differences between lower and higher collagen I:III ratio in low versus higher passage, although this is due to less intensity of collagen I density, and not due to an increase in collagen III (Figure 3C). These results are reflected in the decellularized tumor: There was significant changes between passages of decellularized TU-BcX-2O0, although this was also due to reduced intensity of collagen I density and not due to an increase in collagen III (Figure 3D).
Figure 3. Collagen composition of TU-BcX-2O0 was not altered throughout consecutive passages.
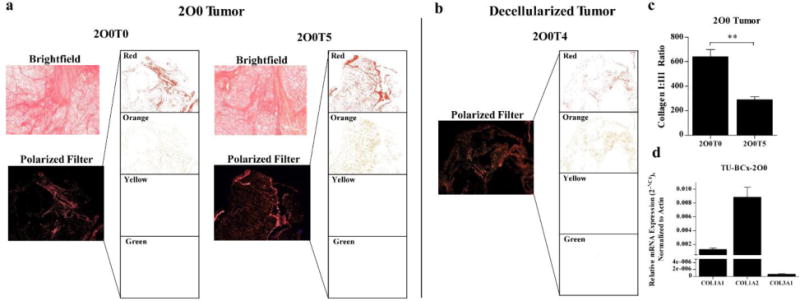
(a) Picosirius Red staining (top panel) was utilized to highlight collagen fibers, and subsequent imaging of the stained slides under filtered light (bottom panels) revealed specific collagen I and collagen III composition of tumors throughout low and higher passages. Also shown are extracted images of the four polarized filters utilized to identify the specific collagens displayed. Red and orange filter indicate collagen I; yellow and green indicate presence of collagen III. (b) Similar architecture is shown when the tumor was decellularized. There is an abundance of collagen I compared to collagen III. (c) Quantification of collagen staining demonstrates collagen I:III ratio was larger in lower passage TU-BcX-2O0 compared to higher passage, although collagen III staining remained low (p=0.0093). (d) Additionally, in decellularized TU-BcX-2O0 tumor, there was a significant variation of collagen I:III ratio between passages (p=0.002), although collagen I remained dominant. Quantification was performed in triplicate and ratio is shown as Red AF+Orange AF/Yellow AF + Green AF; AF stands for Area Fraction (%). n = 3 for TU-BcX-2O0T3 decellularized and TU-BcX-2O0T5; n = 4 for 2O0T0 and n = 5 for TU-BcX-2O0T4 decellularized TU-BcX-2O0.
Discussion
In this study, we describe a novel PDX model that represents the claudin-low TNBC molecular subtype. We demonstrate the value of using translational PDX models in target discovery and therapeutic research, especially in complex tumors such as the claudin-low TNBC subtype. In gross appearance, TU-BcX-2O0 has pustulate-like nodules and areas of liquefactive necrosis. Based on these observations, and our knowledge that the CL-TNBC subtype elicits immune infiltrates more than other subtypes, we investigated TU-BcX-2O0 as a claudin-low TNBC tumor. Claudin-low TNBCs are similar to basal subtypes but have distinctive biological and molecular signatures. They undifferentiated and enriched in epithelial-to-mesenchymal and CSC markers. We first show that TU-BcX-2O0 consistently metastasized to the lungs. Gene expression analyses confirmed endogenous levels of some mesenchymal markers and low expression of epithelial markers. These findings confirm that TU-BcX-2O0 has a mesenchymal phenotype and is capable of spontaneous metastasis. Compared to basal subtypes, CL-TNBC tumors have very low expression of cell-cell adhesion molecules, including E-cadherin, claudins (specifically CLDN3, CLDN4 and CLDN7) and MUC-1, which contributes to the propensity of this tumor subtype to metastasize. We compared claudin gene expression of the TU-BcX-2O0 to a previously established CL-TNBC cell line, MDA-MB-231 [35]. Notably, endogenous mRNA expressions of CLDN3 and CLDN7 in TU-BcX-2O0 resembled MDA-MB-231, while CLDN4 mRNA expression was significantly lower in TU-BcX-2O0. These data were consistent throughout low and high passage TU-BcX-2O0 PDX tumors. We also observed low expression of MUC1 in both MDA-MB-231 cells and TU-BcX-2O0. This was confirmed both with qRT-PCR and immunohistochemistry staining.
The extracellular matrix is another aspect of the tumor microenvironment that has been investigated as a therapeutic target, specifically the collagen networks that surround tumor cells. Although collagens have a diverse role in breast cancer tumorigenesis and metastasis, two collagen subtypes have been identified to predict breast cancer invasiveness. Collagen type I is associated with increased invasiveness and recruitment of stromal cells and other microenvironment components and is increased in metastatic subtypes. Conversely, collagen type III suppresses invasive potential of breast cancer cells. To analyze the collagen composition of the tumor in absence of cell signaling interference we utilized a novel technique to decellularize the tumor tissue, then we used Picosirius Red staining under polarized light to quantify the relative composition of collagen subtypes. We found that in both TU-BcX-2O0 natural tumor and the decellularized tumor there was dramatically higher expression of collagen type I compared to collagen type III, indicating TU-BcX-2O0 has an extracellular matrix that has a higher invasive potential. These data show that TU-BcX-2O0, as well as the associated decellularized model, can be utilized for novel therapeutic target discovery, to evaluate how therapeutics affect collagen composition. We will interrogate this concept in future studies.
Due to the unique gross appearance of TU-BcX-2O0 tumor with the surface areas of pustulate, we evaluated immune-related components of the tumor. Tumor-associated neutrophils and macrophages have been observed in 50% of the total tumor mass in invasive breast carcinomas [36]. Tumor cells drive local neutrophil recruitment and the presence of intratumoral neutrophils has been shown to increase invasive capacity and drive angiogenesis of breast cancer cells [36]. In our data, we did identify a presence of mouse Gr-1+ myeloid cells as well as CD11b+ cells within the tumor. CD11b is involved in recruitment of tumor-associated macrophages (TAMs) to tumors which remodel the microenvironment in to promote tumorigenesis and contribute to formation of the pre-metastatic niche [36]. Interestingly we also discovered a significant human macrophage infiltration that was consistent in both low and high passages. CD11b+GR-1+ mouse MDSCs were present, a cell type that promotes tumorigenesis and metastasis by contributing significantly to tumors’ ability to escape from immune surveillance [37].
Characteristically, CL-TNBC tumors have higher baseline expressions of immune response genes including CD79A, CD79B, VAV1, CD4, CXCL2, CXCR4 and CD14 [12] as well as several cytokines that cytokines promote tumor immune infiltration and angiogenesis (IL-8) and stimulate the growth of TNBC cells [38,39]. The TU-BcX-2O0 model may be useful to investigate the role of various therapeutics on the cross-talk between CL-TNBC and myeloid cells mediated by CXCL2 and other factors. An important limitation of non-humanized PDX models is that immunocompromised mouse strains such as the SCID/Beige strain used in our xenograft experiments lacks T-cells and B-cells, and impairs NK-cell development.
Claudin-low TNBCs remain elusive to small molecule targeted therapies, especially due to the complex microenvironment of the CL-TNBCs and the tumor cell’s interactions with surrounding cellular and stromal components. We demonstrate in our study various applications of utilizing PDX models in therapeutic discovery research. There are many advantages to creating a novel claudin-low cell line derived from PDX tumors. This cell line bypasses common limitations of immortalized cell lines or cell lines that have been used for multiple passages. Over time, cell lines acquire mutations, and are altered by the stress of repeated passaging; this can drastically alter results, especially in drug discovery experiments and cytotoxicity studies. Our establishment of a TU-BcX-2O0 cell line creates a new tool to study the biology of CL-TNBC tumors in a model that has not undergone multiple in vitro passages. Furthermore, the PDX tumor is derived from a post-menopausal African-American patient, a population that has higher rates of TNBC mortality. Louisiana has a high proportion of African-American residents (32.5% in 2015), and the incidence of TNBC in New Orleans is among the highest in the US [40–42]. The application of this model in therapeutic discovery may facilitate more extensive studies on this subset of cancers.
Conclusions
Patient-derived xenografts are being evaluated as models for accelerating target discovery and drug development in a variety of cancers [43,44]. Our findings support the concept that targeted drug discovery research and pre-clinical proof of concept studies of experimental therapeutics can be carried out in PDX models of claudin-low TNBC tumors, which are notoriously difficult to target and exhibit high rates of metastasis.
Supplementary Material
Supplementary Figure S1. Immune components within TU-BcX-2O0. Hematoxylin and Eosin staining shows TU-BcX-2O0 has prominent infiltration of neutrophils, lymphocytes and eosinophils at both lower (T0) and higher (T5) passages in SCID/Beige mice. Representative images are shown in 20X and 60X magnification.
Supplementary Figure S2. TU-BcX-2O0 has an immune system gene signature that is enriched in CL-TNBC. (a) TU-BcX-2O0 has low endogenous levels of IL-6, IL-8 and CXCR4, but higher levels of CXCL2. (b) 2O0 has similar baseline expression of CL-specific immune markers CD79A, CD79B, VAV1, CXCL12 and CD14 as MDA-MB-231 cells. All data was obtained using qRT-PCR in triplicate and normalized to actin. Error bars represent SEM and significantly different * p < 0.05, *** p < 0.001.
Supplementary Figure S3. Immune system components infiltrate TU-BcX-2O0. (a) Both lower and higher passages exhibited presence of mouse-specific markers GR-1, a myeloid differentiation antigen, and CD11b, an integrin expressed on granulocytes, macrophages and myeloid cells. Images were captured using immunofluorescence. Green = Gr-1, Red = CD11b. (b) Human macrophages, represented by CD68 were observed in low and high passage TU-BcX-2O0 tumors. There was no evidence of mouse macrophages, represented by F4/80. Images were captured using immunofluorescence. CD68 = Red; F4/80 = Green.
Supplementary Figure S4. Presence of immune cell populations in blood and spleen of TU-BcX-2O0-bearing and healthy control mice. (a) TU-BcX-2O0T9 tumor has a lower population of circulating murine MDSCs (Gr1+CD11b+) compared to healthy control mice. Within the MDSC subpopulation in both healthy control and tumor-bearing mice, there was a higher presence of granulocytic MDSCs (Ly6G) than monocytic MDSCs (Ly6C). There was also a smaller population of macrophages (F4/80+CD11b+) in circulation in tumor-bearing mice compared to healthy control mice. (b) Similar to data obtained from the blood, within the spleen TU-BcX-2O0T9 tumor-bearing mice had a reduced population of MDSCs (Gr1+CD11b+) compared to healthy control mice and a smaller population of granulocytic MDSCs (Ly6G) than monocytic MDSCs (Ly6C) in both healthy and tumor-bearing mice. We also found a smaller population of macrophages (F4/80+CD11b+) in circulation in tumor-bearing mice compared to healthy control mice.
Supplementary Figure S5. Composition of myeloid cells within TU-BcX-2O0 tumor. (a) Flow cytometry with mouse-specific antibodies shows presence of murine MDSCs (Gr1+CD11b+) within TU-BcX-2O0T9 tumor, and there are both polymorphonuclear and monocytic MDSCs within the tumor. Additionally, there is a small population of mouse macrophages (CD11b+F4/80+) within TU-BcX-2O0T9. (b) Flow cytometry with human-specific antibodies show high population of human myeloid cells (CD33 +HLA-DR -) within the tumor, even at a higher passage (T9). Data was performed with one sample of each group.
Supplementary Figure S6. TU-BcX-2O0 has low protein expression of MUC1. Alcian blue staining of 2O0 reveals low mucin presence in lower passage tumor and less expression in the higher passage tumor. Positive control was from a small intestine; Blue = mucin.
Supplementary Figure S7. TU-BcX-2K1 forms mammospheres in 3D culture. Tumor explant was plated in 3D culture conditions (serum-free media supplemented with B-27, EGF and FGF) in low-attachment plates for 7 days.
Supplementary Figure S8. TU-BcX-2O0 has distinct cell populations in 2D culture. TU-BcX-2O0 was plated in 2D culture and various cell populations were identified. Further cloning and characterization of the individual populations are required. These populations can be utilized.
Acknowledgments
This work was supported by grants from National Institute of Health (NIH) (Grant No. R01-CA125806 (MEB) and R01-CA174785-A1 (BC-B). This work was also supported in part by the Biospecimen Core Laboratory of the Louisiana Cancer Research Consortium. From the LCRC Core we would especially like to acknowledge Alex Alfortish whom played an integral role in coordination and acquisition of TNBC tissue specimen. Additionally, this work was also supported in part by 1 U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds the Louisiana Clinical and Translational Science Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge Sharon Miller for her assistance in coordinating and obtaining the PDX tissue at the time of surgeries. We are also appreciative of Krewe de Pink for their support in this project. Finally, we would also like to acknowledge the patients who consent to donate their tissue to benefit breast cancer research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethical Standards Statement
Experiments performed in this study comply with current laws in the United States of America. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Availability of data and materials
All datasets supporting the conclusion of this article are included within the article.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- 1.Alteri R, Bertaut T, Brinton LA, Fedewa S, Freedman RA, Gansler T, et al. Breast cancer facts and figures 2015–2016. Atlanta: American Cancer Society Inc; 2015. [Google Scholar]
- 2.Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLOS Med. 2010 doi: 10.1371/journal.pmed.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Annals Oncol. 2012;23(S6):vi56–65. doi: 10.1093/annonc/mds196. [DOI] [PubMed] [Google Scholar]
- 4.Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013;73(7):2025–30. doi: 10.1158/0008-5472.CAN-12-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engebraaten O, Vollan HKM, Borresen-Dale A-L. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183(4):1064–74. doi: 10.1016/j.ajpath.2013.05.033. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLOS ONE. 2016;11(6):e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232:142–150. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. Pietenpol Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investigation. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatier R, Finetti P, Guille A, Adelaide J, Chaffanet M, Viens P, Birnbaum D, Bertucci F. Claudin-low breast cancers: clinical, pathological, molecular and prognostic characterization. Mol Cancer. 2014;13:228. doi: 10.1186/1476-4598-13-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pareja F, Geyer FC, Marchio C, Burke KA, Weigelt B, Reis-Filho JS. Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants 2016. Npj Breast Cancer. 2:16036. doi: 10.1038/npjbcancer.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer IA, Abramson VG, Lehmann BD, Pientenpol JA. New strategies for triple-negative breast cancer – deciphering the heterogeneity. Clin Cancer Res. 2014;20(4):782–90. doi: 10.1158/1078-0432.CCR-13-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burdall SE, Hanby AM, Lansdown MRJ, Speirs V. Breast cancer cell lines: friend or foe? Breast Cancer Res. 2003;5(2):89–95. doi: 10.1186/bcr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cifani P, Kirik U, Waldemarson S, James P. Molecular portrait of breast-cancer-derived cell lines reveals poor similarity with tumors. J Proteome Res. 2015;14(7):2819–27. doi: 10.1021/acs.jproteome.5b00375. [DOI] [PubMed] [Google Scholar]
- 15.Manning HC, Buck JR, Cook RS. Mouse models of breast cancer: Platforms for discovering precision imaging diagnostics and future cancer medicine. J Nuclear Med. 2016;57(S1):60S–68S. doi: 10.2967/jnumed.115.157917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillet JP, Clacagno AM, Varma S, Marino M, Green LJ, Vora MI, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA. 2011;108:18709–13. doi: 10.1073/pnas.1111840108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: The best laid plans of mice and men. Clin Cancer Res. 2012;18(19):5160–2. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discovery. 2014;4:998. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G. Patient-derived xenograft models for oncology drug discovery. J Cancer Metastasis Treat. 2015;1:8–15. [Google Scholar]
- 20.DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, Welm AL, Welm BE. Patient-derived models of human breast cancer: Protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol. 2013;14 doi: 10.1002/0471141755.ph1423s60. Unit 14.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18(19):5314–28. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 22.Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumor tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010;12:473. doi: 10.1007/s12094-010-0540-6. [DOI] [PubMed] [Google Scholar]
- 23.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landis MD, Lehmann BD, Pietenpol JA, Chang JC. Patient-derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Research. 2013;15:201. doi: 10.1186/bcr3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertotti A, Migliardi G, Galimi F, Sassi F, Tordi D, Isella C, et al. A molecularly annotated platform of patient-derived xenograft (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 27.Reyal F, Guyader C, Decraene C, Lucchesi C, Auger N, Assayag F, et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Research. 2011;14:R11. doi: 10.1186/bcr3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2013;518:422–26. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhodes LV, Tate CR, Segar HC, Burks HE, Phamduy TB, Hoang V, et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat. 2014;145(3):593–604. doi: 10.1007/s10549-014-2979-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pashos NC, Scarritt ME, Eagle ZR, Gimble JM, Chaffin AE, Bunnell BA. Characterization of an acellular scaffold for a tissue engineering approach to the nipple-areolar complex reconstruction. Cells Tissues Organs. 2017;203:183–193. doi: 10.1159/000455070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonvillain RW, Scarritt ME, Pashos NC, Sullivan DE, Betancourt AM, Tsien F, et al. Characterization of Rhesus Macaque lung-resident multipotent stromal cells. Cell Gene Therapy Insights. 2016;2(1):47–92. [Google Scholar]
- 32.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. OncoImmunology. 2017 doi: 10.1080/2162402X.2017.1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jézéquel P, Loussouarn D, Charbonnel-Guérin C, Campion L, Vanier A, Gouraud W, Lasla H, Guette C, Valo I, Verriéle V, Campone M. Gene-expression molecular subtyping of triple-negative breast cancer tumors: importance of immune response. Breast Cancer Res. 2015;17:43. doi: 10.1186/s13058-015-0550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lattouf R, Younes R, Lutomski D, Naaman N, Godeau G, Senni K, Changotade S. Picosirius red staining: A useful tool to appraise collagen networks in normal and pathological tissues. J Histochem & Cytochem. 2014;62(10):751–758. doi: 10.1369/0022155414545787. [DOI] [PubMed] [Google Scholar]
- 35.Holliday SL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6(3):1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Edwards CM, Mundy GR. Gr-1+CD11b+ Myeloid-derived suppressor cells: Formidable partners in tumor metastasis. J Bone Miner Res. 2010;25(8):1701–06. doi: 10.1002/jbmr.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregory AD, Houghton AM. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011;71(7):2411–6. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 39.Azab B, Bhatt VR, Phookan J, Murukutla S, Kohn N, Terjanian T, Widmann WD. Usefulness of the Neutrophil-to-Lymphocyte ratio in predicting short- and long-term mortality in breast cancer patients. Annals Surg Oncol. 2012;19(1):217–24. doi: 10.1245/s10434-011-1814-0. [DOI] [PubMed] [Google Scholar]
- 40.Loch MM, Estrada J, Reske T, Li X, Chen V, Wu X. Triple negative breast cancer in African American women: Disparity between women in New Orleans versus Louisiana. Abstract: 2013 ASCO Annual Meeting 2013 [Google Scholar]
- 41.Loch MM, et al. New Orleans has the highest incidence rates of triple negative breast cancer. Abstract: SABCS 2016 2016 [Google Scholar]
- 42.Howlader N, Chen VW, Ries LAG, Loch MM, Lee R, DeSantis C, et al. Overview of breast cancer collaborative stage data items – their definitions, quality, usage and clinical implications: A review of SEER data for 2004 – 2010. Cancer. 2014;120(S23):3771–3780. doi: 10.1002/cncr.29059. [DOI] [PubMed] [Google Scholar]
- 43.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14(20):6456–68. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 44.Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272–83. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Immune components within TU-BcX-2O0. Hematoxylin and Eosin staining shows TU-BcX-2O0 has prominent infiltration of neutrophils, lymphocytes and eosinophils at both lower (T0) and higher (T5) passages in SCID/Beige mice. Representative images are shown in 20X and 60X magnification.
Supplementary Figure S2. TU-BcX-2O0 has an immune system gene signature that is enriched in CL-TNBC. (a) TU-BcX-2O0 has low endogenous levels of IL-6, IL-8 and CXCR4, but higher levels of CXCL2. (b) 2O0 has similar baseline expression of CL-specific immune markers CD79A, CD79B, VAV1, CXCL12 and CD14 as MDA-MB-231 cells. All data was obtained using qRT-PCR in triplicate and normalized to actin. Error bars represent SEM and significantly different * p < 0.05, *** p < 0.001.
Supplementary Figure S3. Immune system components infiltrate TU-BcX-2O0. (a) Both lower and higher passages exhibited presence of mouse-specific markers GR-1, a myeloid differentiation antigen, and CD11b, an integrin expressed on granulocytes, macrophages and myeloid cells. Images were captured using immunofluorescence. Green = Gr-1, Red = CD11b. (b) Human macrophages, represented by CD68 were observed in low and high passage TU-BcX-2O0 tumors. There was no evidence of mouse macrophages, represented by F4/80. Images were captured using immunofluorescence. CD68 = Red; F4/80 = Green.
Supplementary Figure S4. Presence of immune cell populations in blood and spleen of TU-BcX-2O0-bearing and healthy control mice. (a) TU-BcX-2O0T9 tumor has a lower population of circulating murine MDSCs (Gr1+CD11b+) compared to healthy control mice. Within the MDSC subpopulation in both healthy control and tumor-bearing mice, there was a higher presence of granulocytic MDSCs (Ly6G) than monocytic MDSCs (Ly6C). There was also a smaller population of macrophages (F4/80+CD11b+) in circulation in tumor-bearing mice compared to healthy control mice. (b) Similar to data obtained from the blood, within the spleen TU-BcX-2O0T9 tumor-bearing mice had a reduced population of MDSCs (Gr1+CD11b+) compared to healthy control mice and a smaller population of granulocytic MDSCs (Ly6G) than monocytic MDSCs (Ly6C) in both healthy and tumor-bearing mice. We also found a smaller population of macrophages (F4/80+CD11b+) in circulation in tumor-bearing mice compared to healthy control mice.
Supplementary Figure S5. Composition of myeloid cells within TU-BcX-2O0 tumor. (a) Flow cytometry with mouse-specific antibodies shows presence of murine MDSCs (Gr1+CD11b+) within TU-BcX-2O0T9 tumor, and there are both polymorphonuclear and monocytic MDSCs within the tumor. Additionally, there is a small population of mouse macrophages (CD11b+F4/80+) within TU-BcX-2O0T9. (b) Flow cytometry with human-specific antibodies show high population of human myeloid cells (CD33 +HLA-DR -) within the tumor, even at a higher passage (T9). Data was performed with one sample of each group.
Supplementary Figure S6. TU-BcX-2O0 has low protein expression of MUC1. Alcian blue staining of 2O0 reveals low mucin presence in lower passage tumor and less expression in the higher passage tumor. Positive control was from a small intestine; Blue = mucin.
Supplementary Figure S7. TU-BcX-2K1 forms mammospheres in 3D culture. Tumor explant was plated in 3D culture conditions (serum-free media supplemented with B-27, EGF and FGF) in low-attachment plates for 7 days.
Supplementary Figure S8. TU-BcX-2O0 has distinct cell populations in 2D culture. TU-BcX-2O0 was plated in 2D culture and various cell populations were identified. Further cloning and characterization of the individual populations are required. These populations can be utilized.