

Abstract
Background
Compelling evidence indicates that metformin, a low-cost and safe orally administered biguanide prescribed to millions of Type 2 diabetics worldwide, induces the osteoblastic differentiation of mesenchymal stromal cells (MSCs) through the AMP-activated protein kinase (AMPK) pathway. As a highly hydrophilic cationic compound, metformin uptake is facilitated by cell membrane organic cation transporters (OCTs) of the solute carrier 22A gene family. We hypothesized that to effectively enhance osteogenic differentiation, and ultimately bone regeneration, metformin must gain access into functional OCT-expressing MSCs.
Methods
Data was obtained through immunoblotting, cellular uptake, mineralization and gene expression assays.
Results
We demonstrate for the first time that functional OCTs are expressed in human-derived MSCs from umbilical cord Wharton’s jelly, an inexhaustible source of non-embryonic MSCs with proven osteogenic potential. A clinically relevant concentration of metformin led to AMPK activation, enhanced mineralized nodule formation and increased expression of the osteogenic transcription factor Runt-related transcription factor 2 (RUNX2). Indeed, targeting OCT function through pharmacological and genetic approaches markedly blunted these responses.
Conclusions
Our findings indicate that functional OCT expression in UC-MSCs is a biological pre-requisite that facilitate the intracellular uptake of metformin to induce an osteogenic effect. Future preclinical studies are warranted to investigate whether the expression of functional OCTs may serve as a potential biomarker to predict osteogenic responses to metformin.
Keywords: Bone regeneration, mesenchymal stromal cells, metformin, organic cation transporters, osteogenesis, stem cells, umbilical cord
INTRODUCTION
Compelling evidence suggest that a number of common drugs that were originally developed for specific chronic diseases can also be beneficial and potentially repurposed for other unrelated conditions. This seems to be the case with the anti-diabetic drug metformin [1]. As a member of the biguanide family, metformin was approved by the United States Food and Drug Administration in 1995 for treating type 2 diabetes, and is currently the most widely used oral anti-diabetic worldwide [2, 3]. Intriguingly, recent studies indicate that metformin can also impact skeletal homeostasis and reduce the risk of bone fractures [4-7]. To this end, in vitro studies have reported that metformin enhances the proliferation and osteoblastic differentiation of bone marrow mesenchymal stromal cells (MSCs) and pre-osteoblastic cell lines [8-12].
Proposed mechanisms for the anti-diabetic effects of metformin have mainly focused on the role of the AMP-activated protein kinase (AMPK) pathway, a conserved signaling cascade that acts as master sensor of cellular energetics [13, 14]. Most studies performed in bone marrow MSCs and pre-osteoblasts have reported that metformin enhances osteoblastic differentiation via AMPK signaling in doses ranging from 0.5-500 μM. These responses include a dose-dependent effect on cell proliferation together with increases in extracellular mineral nodule formation and well-known osteoblastic markers such as type I collagen, osteocalcin, alkaline phosphatase and the transcription factor runt-related transcription factor 2 (RUNX2) [11, 15, 16]. Recently, our group reported similar responses to metformin in induced pluripotent stem cell-MSCs (iPSC-MSCs). We found that metformin when used at a clinically relevant concentration of 10 μM significantly stimulated alkaline phosphatase activity, enhanced mineralized nodule formation and increased expression of the osteogenic markers RUNX2 and Osterix. Furthermore, inhibition of LKB1 activity, a key upstream serine/threonine kinase responsible for activating AMPK, markedly reversed metformin-induced AMPK activation as well as the expression and nuclear localization of RUNX2 [17, 18]. These findings underscore the significance of using therapeutically relevant doses when trying to extrapolate in vitro results to the clinical setting in humans. Plasma concentrations of metformin found in patients within 2-4 hours after an oral dose of 500-1500 mg ranges between 2.7-20 μM [11, 19].
Preclinical animal studies designed to evaluate the effects of metformin on bone mass and quality following traumatic bone fracture or bone loss associated with estrogen deficiency have been overall encouraging, but some conveyed ambiguous results. In general, metformin significantly increases total body bone mineral density, bone trabecular volume, osteocyte density, osteoblast number and exerts protective effects against bone loss [6, 15, 20-22]. In contrast, others report that metformin has no effect on bone mass or fracture healing [23]. Although discrepancies in experimental findings may reflect variability in experimental design and methodology, responses to metformin may also differ relative to its effects in endochondral or intramembranous bones, rodent strain, gender, age, treatment dose and duration, or glycemic status. Yet, a major gap in knowledge that remains elusive is the role played by organic cation transporters (OCTs) on the intracellular uptake of metformin and their contribution to the osteogenic action in MSCs.
As a highly hydrophilic cationic drug, metformin uptake relies on tissue-specific mechanisms facilitated by a group of polyspecific cell membrane transporters of the solute carrier 22A (SLC22A) gene family. OCT-1, OCT-2 and OCT-3 mediate transport of structurally diverse, small hydrophilic organic cationic endogenous compounds, toxins and drugs, including metformin [24-28]. In spite of the critical involvement of OCTs in hepatic and renal cellular transport [29, 30], little is known about the impact of these transporters in metformin-induced osteoblastic differentiation. By elucidating OCT expression and function in MSCs we would afford a better understanding of the intervening cellular determinants mediating the osteogenic action of metformin. Furthermore, as a low-cost drug with well-accepted tolerance after long-term use metformin could emerge as an attractive systemic or locally delivered option to pharmacologically potentiate MSC-based bone regeneration. To this end, we hypothesized that to effectively enhance osteogenic differentiation and ultimately bone regeneration metformin must gain access into functional OCT-expressing MSC cells.
As multipotent cells capable of differentiating into osteoblasts, human bone marrow MSCs are regarded as the “gold standard” for autogenous MSC-based bone tissue engineering [31]. However, bone marrow MSCs are harvested through an invasive procedure, have lower self-renewal ability, and aging and underlying chronic conditions negatively impact their proliferative and differentiating capabilities [32-35]. Other alternative MSC sources are being studied to regenerate skeletal tissues in a more predictable fashion. Recent reports have pointed to the human Wharton’s jelly of the umbilical cord (UC) as an attractive extra-embryonic, inexpensive and inexhaustible source of perinatal MSCs for regenerative medicine, with neither donor site morbidity nor ethical issues associated with their use [36-38]. UC-MSC osteogenic capacity has been demonstrated both in vitro and in experimental preclinical models when seeded in three-dimensional scaffolds [39-44]. Notably, UC-MSCs are able to match the bone regenerative efficacy of bone marrow MSCs in critical size cranial defects in athymic rats [45]. Moreover, to mimic their regenerative potential in congenital oral and craniofacial bone defects in pediatric patients novel preclinical tissue engineering strategies have been recently proposed to repair maxillary alveolar bone defects with nanomicrofiber scaffolds seeded with UC-MSCs in juvenile swines [46, 47].
The present in vitro study was conducted to investigate (a) whether OCTs are expressed and functional in UC-MSCs, and (b) whether they play a key role in mediating the osteogenic action of metformin. This work may not only offer new insight into critical cellular determinants driving the osteogenic capacity of metformin, but also provide a significant step forward in the area of perinatal MSC research and bone regeneration, especially if further translated into novel autogenous, locally delivered therapeutic options to safely and affordably enhance skeletal regeneration in patients with congenital malformations that compromise the oral and craniofacial region.
MATERIALS AND METHODS
Reagents
Cell growth complete medium consisted of low glucose Dulbecco’s modified Eagle’s media (DMEM, Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco), and 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). Primary rabbit polyclonal antibodies against phospho-AMPKα1 Thr172 (1:1,000) and AMPKα1 (1:1,000) were purchased from Cell Signaling Technologies (Danvers, MA), and OCT-1 (1:400), OCT-2 (1:400), β-actin (1:20,000) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:10,000) from Sigma. Rabbit monoclonal antibody against OCT-3 (1:10,000) was obtained from Epitomics (Burlingame, CA) and Runx2 (1:1,000) from Cell Signaling Technologies. Metformin was obtained from Calbiochem (Gibbstown, NJ), and quinidine hydrochloride monohydrate from Sigma-Aldrich. Alizarin Red S (ARS) was purchased from Lifeline Cell Technology (Walkersville, MD). Cell Titer 96® Aqueous One Solution (MTS) was obtained from Promega (Madison, WI).
Cell Culture
Human-derived Wharton’s jelly UC-MSCs harvested from four different cord tissues were purchased from PromoCell (named UC-MSC-1 and UC-MSC-2; Heidelberg, Germany) as well as from ScienCell (named UC-MSC-3 and UC-MSC-4; Carlsbad, CA). All UC-MSCs were grown in complete growth medium consisting of low-glucose DMEM containing 10% FBS, 1% antibiotic/antimycotic solution (Gibco) at 37°C and 5% CO2 atmosphere. Cells were seeded on 75-cm2 flasks, and subcultured using trypsin-EDTA and PBS solution. All cells were plated at passages 4-6.
Western blot analysis
After extraction of whole UC-MSC lysates, equal amounts of protein were separated by SDS-PAGE, electrophoretically transferred onto polyvinylidene difluoride membranes and immunodetection was performed by incubating membranes overnight at 4°C with indicated antibodies as previously described [48]. Blots were stripped and re-probed with either β-actin or GAPDH antibodies as sample loading control.
UC-MSC viability assays
The effect of metformin on cell viability was evaluated through a colorimetric assay by using the CellTiter 96® AQueous One Solution (MTS) reagent following the manufacturer’s protocol as previously reported [48]. UC-MSCs were plated in triplicate overnight in complete growth medium at a density of 15,000-20,000 cells/cm2 in a 96-well plate. The following day, cells were treated with vehicle control or metformin (10 μM) in DMEM containing 0.5% FBS plus 1% antibiotic/antimycotic solution. Treatment was repeated at days 3 and 5. After 7 days, MTS reagent was added for 1-2 hours and absorbance at 490 nm was measured in a microplate reader.
[14C]-metformin uptake assay
Metformin cellular uptake was analyzed as previously described [49]. UC-MSCs were washed once with pre-warmed KRH buffer (125 mM NaCl, 4.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM HEPES, 5.6 mM glucose, pH 7.4). After 10-minute incubation with 10 μM [14C]-metformin plus 40 μM unlabeled metformin with or without 10 μM quinidine the uptake was halted by removing the buffer and washing the cells with ice-cold KRH buffer 3 times. Cells were lysed in 300 μl 1% Triton X-100, and 250 μl cell lysates were transferred to scintillation tube containing 3 ml Econo-Safe™ economical biodegradable cocktail (Research Products International Corp, Mount Prospect, IL). Radioactivity was counted in a Tri-Carb 2910TR liquid scintillation analyzer (Perkin Elmer, Boston, MA).
OCT-1 RNA interference
Short interfering RNA (siRNA)-based experiments were performed as previously published [50]. Briefly, UC-MSCs were plated overnight at 15,000 cells/cm2 in complete growth medium. The following day, transfections were performed following the dilution of commercially available siRNA duplexes targeting human OCT-1 with Hiperfect transfection reagent and serum-free, antibiotic-free DMEM according to manufacturer’s recommendations (Qiagen, Valencia, CA). Cells transfected with a non-targeted control siRNA (Qiagen) were included in all experiments. The next day following transfections, cells were serum-starved and used in the indicated assays following treatment with vehicle control or metformin.
Mineralization assay
UC-MSCs were plated in complete DMEM growth medium in triplicate in 24-well plates at a density of 30,000 cells/cm2. The following day, cells were highly confluent and treatments with either complete growth medium alone or supplemented with metformin were initiated. Media were changed with added treatments every three days. After 21 days, cells were fixed in 10% buffered formalin and stained with 2% ARS at room temperature for 45 minutes to quantify the formation of calcium-containing mineral deposits. Red-stained calcium deposits were eluted with 10% cetylpyridinium chloride solution, and 100 μL from each well was transferred to a 96-well plate. Optical density was read at 550 nM in a microplate reader.
RUNX2 gene expression
To measure RUNX2 gene expression levels in OCT-1 or control siRNA-transfected UC-MSCs exposed to metformin quantitative real-time reverse transcription polymerase chain reaction (qPCR) was used. Cells were plated on 6-well plates at a density of 0.3×106 cells per well. The next day cells were incubated with 1% FBS low glucose DMEM overnight, and the following day treated with metformin. Total cellular RNA was extracted after 7 days with the PureLink RNA Mini Kit (Invitrogen, Waltham, MA) plus TRizol reagent (Invitrogen), and then reverse-transcribed into cDNA by a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). RUNX2 and GAPDH gene expression levels were quantified by qPCR using SYBR Green PCR Master Mix (Applied Biosystems). Commercially synthesized sequences of human specific primers were utilized (Sigma): RUNX2 (forward: gactgtggttaccgtcatggc; reverse: acttggtttttcataacagcgga, and GAPDH (forward: tcaacgaccccttcattgac; reverse: atgcagggatgatgttctgg). Relative expression was normalized by the Ct of the housekeeping gene GAPDH, and calculated using the 2-ΔΔCt methods. The qPCR analyses were performed using Graph Pad software.
Statistical analyses
Statistical analyses were performed using Prism 6.0 biostatistics program (GraphPad Software). All data were expressed as the mean value ± standard error of the mean (S.E.M). Statistical significance was analyzed by using Student’s t-test or one-way analyses of variance (ANOVA) followed by Tukey’s multiple comparison test. P values <0.05 were considered statistically significant.
RESULTS
Metformin induces AMPK activation in OCT-expressing UC-MSCs
To identify whether any of the three OCT isoforms (OCT-1, OCT-2, OCT-3) were expressed in UC-MSCs western blot analyses were performed (Figure 1A). We found that OCT-1 (molecular weight [MW]: 61 kDa) was expressed in all of the analyzed UC-MSCs (UC-MSC-1, UC-MSC-2, UC-MSC-3 and UC-MSC-4). OCT-2 (MW: 62 kDa) was also expressed in UC-MSC-1, UC-MSC-2 and UC-MSC-3, while OCT-3 (MW: 60 kDa) expression was only detected in UC-MSC-1 cells. Regardless of the isoform, we found that OCTs were differentially expressed in this type of MSCs.
Figure 1. OCT protein expression in UC-MSCs.
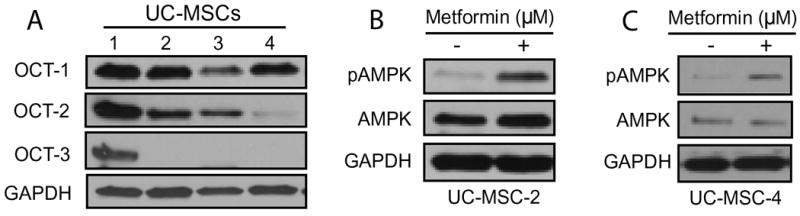
(A) Whole cell lysates extracted from commercially available, human-derived UC-MSCs obtained from four different donors were analyzed by Western blotting to determine the expression levels of OCT-1, OCT-2 and OCT-3. Whole cell lysates obtained from UC-MSC-2 (B) and UC-MSC-4 (C) following a 3-hour treatment with metformin (10 μM) demonstrate an increase in the phosphorylating status of AMPKα1 at Thr172 (pAMPK) as analyzed by Western blotting. In all immunoblots GAPDH served as loading control.
Next, we tested the functionality of OCTs by exposing UC-MSC-2 cells to increasing doses of metformin to determine whether this treatment triggered AMPK activation (Figure S1). Indeed, we found that following a 3-hour treatment metformin in doses ranging from 5-50 μM triggered the activation of the LKB1/AMPK signaling pathway, a well-known and commonly used biochemical end-point signal of metformin intracellular action [51]. We confirmed these results in UC-MSC2 as well as in UC-MSC-4 cells by exposing them to a clinically relevant dose of metformin (10 μM). Our findings demonstrate that when compared to untreated cells OCT-expressing UC-MSCs were responsive to metformin treatment as evidenced by AMPK activation (phosphorylated AMPK or pAMPK) (Figures 1B and 1C).
UC-MSC viability is unaffected by metformin treatment
To investigate the effect of metformin on UC-MSC viability, MTS assays were performed by using a colorimetric assay as described in the Material and Methods section. Our data show that following a 7-day treatment with metformin (10 μM), UC-MSC-2 cell viability remained unaffected and similar to untreated cells (Figure 2A). However, metformin treatment slightly increased the relative number of UC-MSC-4 viable cells without reaching statistical significance (p=0.053; Figure 2B). Overall, these results provide evidence that with the indicated dose, prolonged metformin treatment was not detrimental to UC-MSC viability.
Figure 2. Metformin supports UC-MSC viability.
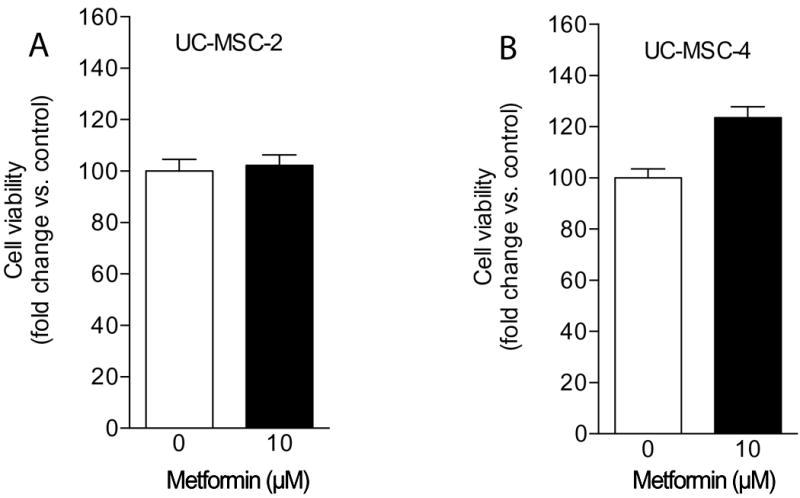
UC-MSC-2 (A) and UC-MSC-4 (B) cell viability in response to metformin was evaluated through an MTS colorimetric assay after 7 days of treatment. No statistically significant differences were observed when compared to untreated controls.
OCT-dependent cellular uptake of metformin leads to AMPK activation in UC-MSCs
To test whether the transport function of OCTs in UC-MSCs plays a key role in the intracellular accumulation of metformin, we carried out uptake assays with radiolabeled metformin in the absence or presence of the commonly used pan-OCT inhibitor quinidine [52]. Based on the sole expression of the OCT-1 isoform in UC-MSC-4 cells as evidenced by immunoblotting (Figure 1A), we used these cells in all of the following experiments to more specifically examine the contribution of OCT function relative to the osteogenic action of metformin. We found that the intracellular accumulation of radiolabeled metformin was significantly reduced in cells pre-treated with quinidine when compared to metformin only (Figure 3A). These data support the notion that in order for metformin to gain substantial intracellular access the presence of functional OCTs in UC-MSCs is critical.
Figure 3. OCT inhibition impairs UC-MSC-4 cellular uptake of metformin and downregulates metformin-induced AMPK activation.

(A) Metformin uptake assay performed in UC-MSC-4 cells to analyze OCT-dependent metformin uptake in the presence or absence of the pan-OCT inhibitor quinidine. OCT inhibition significantly impaired the uptake of 10 μM [14C]-metformin. (B) Whole cell extracts from UC-MSC-4 cells exposed to metformin for 3 hours in the presence or absence of quinidine were subjected to Western blotting. β-actin served as loading control.
Based on the cellular uptake assays, we then performed experiments to elucidate whether the OCT inhibitory action of quinidine in UC-MSCs would impair metformin-induced AMPK activation. As shown in Figure 3B, the phosphorylated status of AMPK was markedly downregulated when cells were pre-treated with quinidine. Overall, these results highlight the essential role of OCTs as facilitators of metformin uptake and intracellular activity.
Metformin significantly increases mineralized nodule formation in UC-MSCs
To further investigate whether the formation of calcium-containing nodules, a commonly used end-point assay of in vitro osteogenic induction, could be enhanced by metformin in UC-MSCs we performed ARS staining assays. Following a 21-day treatment of UC-MSCs with metformin we found a significant increase in ARS staining in comparison to untreated cells (Figure 4A). Moreover, UC-MSCs pre-treated with quinidine exhibited a significantly reduced effect of metformin on mineralized nodule formation underscoring the contribution of OCT function to this response. Although it was clearly evident that quinidine exerted an inhibitory effect on the formation of calcium-rich deposits induced by metformin, our data suggest that quinidine alone was able to induce a small, but statistically significant increase in mineralization (Figure 4B).
Figure 4. Metformin significantly increases mineralized nodule formation in UC-MSCs.
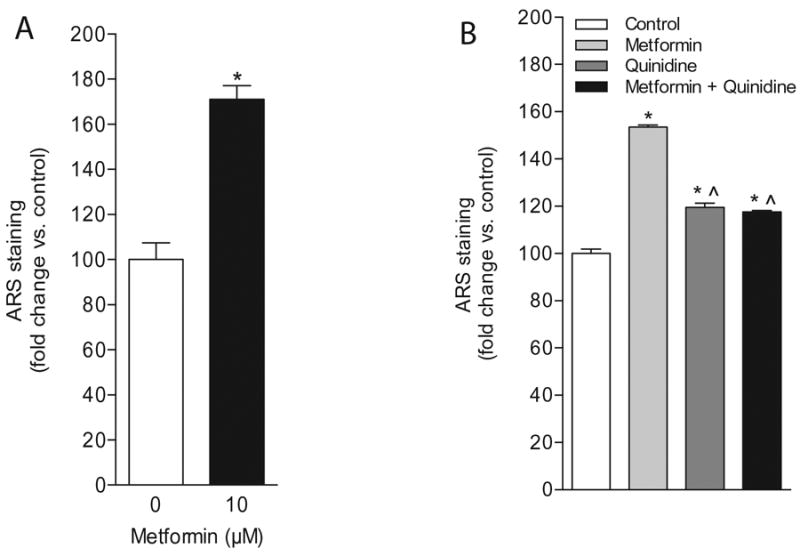
(A) Quantification of ARS-stained, calcium-containing nodules in UC-MSC-4 cells in response to metformin following a 21-day treatment. Data represent mean ± S.E.M. *p<0.05, when compared to untreated control. (B) Treatment of UC-MSC-4 cells with metformin (10 μM) and quinidine (10 μM) for 21 days resulted in a significant decrease in the formation of calcium-rich nodules as determined by ARS staining. Data represent mean ± S.E.M. *p<0.05, when compared to untreated control; ˆp<0.05, when compared to metformin-treated cells.
OCT-1-mediated metformin uptake facilitates RUNX2 expression and mineralized nodule formation
Next, we examined whether OCT-1 targeted genetic disruption would impact metformin uptake and downstream osteogenic activity. To this end, UC-MSC-4 cells were transiently transfected with either a control (CTL) or OCT-1 siRNA duplex. As shown in Figure 5A, increasing doses of OCT-1 siRNA (5-25 nM) markedly knocked down OCT-1 protein expression when compared to untransfected or CTL siRNA-transfected cells. As shown in Figure 5B, when uptake assays were performed to determine whether the intracellular accumulation of radiolabeled metformin was affected in CTL siRNA- versus OCT-1 siRNA-transfected cells, we found that genetically interfering with OCT-1 expression significantly decreased the intracellular accumulation of metformin by approximately 40%.
Figure 5. OCT-1 knock down impairs metformin-induced RUNX2 expression and mineralized nodule formation in UC-MSC-4 cells.
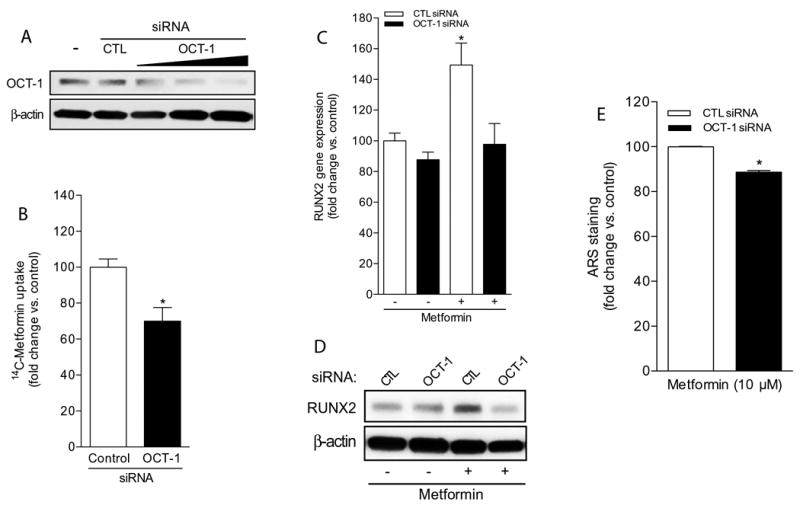
(A) Western blotting to determine OCT-1 expression levels in UC-MSC-4 untransfected cells (-), and in cells transfected with either 25 nM control siRNA (CTL) or with OCT-1 siRNA (5-25 nM). β-actin served as loading control. (B) Metformin uptake assay to analyze OCT-1-dependence in UC-MSC-4 cells transfected with either 25 nM control or OCT-1 siRNA. Data represent mean ± S.E.M. *p<0.05, when compared to control siRNA-transfected cells. (C) Runx2 gene expression analysis by qPCR in UC-MSC-4 cells transfected with either 25 nM control siRNA (CTL siRNA) or OCT-1 siRNA, and then left untreated or exposed to metformin (10 μM) for 7 days. Data represent mean ± S.E.M. *p<0.05, when comparing metformin-treated, CTL siRNA-transfected cells versus other treatment groups. (D) Whole cell extracts from UC-MSC-4 cells transfected with either 25 nM control siRNA (CTL) or OCT-1 siRNA, and then left untreated or treated with metformin (10 μM) for 7 days were subjected to RUNX2 immunoblotting. β-actin served as loading control. (E) Quantification of ARS-stained, calcium-rich mineralized nodules in UC-MSC-4 cells transfected with either 25 nM control siRNA (CTL siRNA) or OCT-1 siRNA following treatment with 10 μM metformin for 21 days. Data represent mean ± S.E.M. *p<0.05, when comparing OCT-1 siRNA-transfected cells versus CTL siRNA-transfected cells.
Furthermore, when CTL siRNA- and OCT-1 siRNA-transfected UC-MSCs were treated for 7 days with metformin, a statistically significant upregulatiom in RUNX2 gene expression was observed in CTL siRNA-transfected cells; however, RUNX2 was markedly inhibited in metformin-treated, OCT-1 siRNA-transfected cells to expression levels similar to untreated controls (Figure 5C). In fact, immunoblotting data obtained from whole cell lysates extracted following a similar treatment protocol show that RUNX2 protein expression validated the gene expression findings (Figure 5D). We also observed a significant decrease in calcium-rich nodule formation as evidenced by ARS staining quantification in metformin-treated, OCT-1 siRNA-transfected cells (Figure 5E). Altogether, these findings emphasize the concept that for metformin to act as a predictable osteogenic factor, UC-MSCs must express functional OCTs.
DISCUSSION
Recent studies indicate that metformin, a biguanide compound used worldwide by millions of Type 2 diabetics induces osteoblastic differentiation in MSCs and pre-osteoblasts. Based on this evidence, the potential repurposing of metformin as a systemic osteoanabolic agent or as a locally delivered drug to particularly enhance oral and craniofacial bone regeneration is very attractive considering its low cost and safety record in type 2 diabetic patients after long-term use. Though therapeutically attractive, pending questions with significant clinical implications remain unanswered. In particular, it is still unclear what role OCTs play on the intracellular uptake of metformin in MSCs. Therefore, we carried out this in vitro study to investigate whether OCTs act as major cellular determinants underlying metformin-induced osteoblastic differentiation. Here, we provide the first evidence supporting a potential osteogenic action of metformin in UC-MSCs. Furthermore, this study demonstrates that UC-MSCs derived from different donors variably express OCT-1, OCT-2 and OCT-3, and that the function of these cell membrane transporters is critical to facilitate the osteogenic effect triggered by metformin likely through the activation of the AMPK pathway.
In general, OCT-1, OCT-2 and OCT-3 mediate transport of structurally diverse, small hydrophilic organic cationic endogenous compounds, toxins and drugs, including metformin [24-28, 53]. Our literature search has found only one report where high OCT-1 gene and protein expression were characterized in osteoblasts derived from rat mandible [54]. In the liver, though, highly expressed OCT-1 mediates the therapeutic anti-diabetic action of metformin as demonstrated by Shu et al., who showed that metformin glucose-lowering effect in OCT-1-deficient mice is completely abolished [25]. In fact, a range of polymorphic genetic OCT variants with reduced uptake function has also been reported [55-58]. Collectively, these studies emphasize the key role of functional OCTs in the response to metformin, and imply that MSCs with different OCT expression levels and/or genotypes may respond differently to the osteogenic effects of metformin.
In agreement with several reports studying the effects of metformin in other cell types, we provide evidence demonstrating that metformin treatment leads to the activation of the LKB1/AMPK signaling pathway, and that this effect may be central to the enhancing action of metformin on UC-MSC osteoblastic differentiation [6]. As a mild and reversible inhibitor of mitochondrial oxidative phosphorylation at complex I of the electron transport chain, metformin reduces the intracellular energy status resulting in an increase of the AMP/ATP ratio [13, 59]. AMPK is bound by AMP, which makes AMPK a better substrate for a phosphorylating activation by its upstream serine/threonine kinase LKB1 [60]. Noteworthy, metformin does not activate AMPK directly, but contributes to AMPK activation as a result of decreasing intracellular energy following the inhibition of mitochondrial oxidative phosphorylation [61].
Through the use of chemical and genetic approaches to inhibit OCT expression and function in UC-MSCs, we were able to significantly impaired metformin uptake and triggered a marked decrease in metformin-induced AMPK phosphorylation. Particularly, these findings underscore the critical functional role of OCTs as facilitators of metformin intracellular access into UC-MSCs. Moreover, in support of its role as a key osteogenic transcription factor, we found that OCT-1 inhibition through RNA interference resulted in a significant downregulation in metformin-induced RUNX2 gene and protein expression.
Similar inhibitory effects on osteogenic marker expression have been previously reported in other osteoprogenitor cells and preosteoblasts when chemical AMPK inhibitors were used prior to metformin treatment [10, 11, 20]. To the best of our knowledge, this study represents the first one to highlight the key role of OCTs on the osteogenic action of metformin in UC-MSCs, and provide a clear indication that functional OCT expression is a biological pre-requisite to facilitate these responses.
Collectively, the results of this study may serve as the basis to considering OCT expression and function as cellular biomarkers to predict whether metformin could be used as an affordable and safe osteogenic compound to enhance MSC-based skeletal regeneration. In addition to enhancing bone regeneration, metformin may be also useful in the repair of other mineralized tissues via different types of MSCs, including those obtained from the bone marrow, dental pulp, and induced pluripotent sources. Future in vitro and in vivo studies are warranted to investigate these promising drug repurposing properties of metformin with the ultimate goal of potentiating mineralized tissue regeneration.
Supplementary Material
Whole cell lysates extracted from UC-MSC-2 cells following a 3-hour treatment with increasing doses of metformin were analyzed by Western blotting to determine the expression levels of phosphorylated AMPKα1 at Thr172 (pAMPK) and total AMPK.
Acknowledgments
This work was supported by the National Institutes of Health grants R01DE023578 (to A.S.) and R01GM099742 (to Y.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J. 2015;471(3):307–22. doi: 10.1042/BJ20150497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey CJ. Metformin: a multitasking medication. Diab Vasc Dis Res. 2008;5(3):156. doi: 10.3132/dvdr.2008.026. [DOI] [PubMed] [Google Scholar]
- 3.Hampp C, Borders-Hemphill V, Moeny DG, Wysowski DK. Use of antidiabetic drugs in the U.S., 2003-2012. Diabetes Care. 2014;37(5):1367–74. doi: 10.2337/dc13-2289. [DOI] [PubMed] [Google Scholar]
- 4.Vestergaard P, Rejnmark L, Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia. 2005;48(7):1292–9. doi: 10.1007/s00125-005-1786-3. [DOI] [PubMed] [Google Scholar]
- 5.Melton LJ, 3rd, Leibson CL, Achenbach SJ, Therneau TM, Khosla S. Fracture risk in type 2 diabetes: update of a population-based study. J Bone Miner Res. 2008;23(8):1334–42. doi: 10.1359/JBMR.080323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarthy AD, Cortizo AM, Sedlinsky C. Metformin revisited: Does this regulator of AMP-activated protein kinase secondarily affect bone metabolism and prevent diabetic osteopathy. World J Diabetes. 2016;7(6):122–33. doi: 10.4239/wjd.v7.i6.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen SC, Brooks R, Houskeeper J, Bremner SK, Dunlop J, Viollet B, Logan PJ, Salt IP, Ahmed SF, Yarwood SJ. Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. Mol Cell Endocrinol. 2017;440:57–68. doi: 10.1016/j.mce.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortizo AM, Sedlinsky C, McCarthy AD, Blanco A, Schurman L. Osteogenic actions of the anti-diabetic drug metformin on osteoblasts in culture. Eur J Pharmacol. 2006;536(1-2):38–46. doi: 10.1016/j.ejphar.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Xue J, Li X, Jia Y, Hu J. Metformin regulates osteoblast and adipocyte differentiation of rat mesenchymal stem cells. J Pharm Pharmacol. 2008;60(12):1695–700. doi: 10.1211/jpp.60/12.0017. [DOI] [PubMed] [Google Scholar]
- 10.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun. 2008;375(3):414–9. doi: 10.1016/j.bbrc.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 11.Jang WG, Kim EJ, Bae IH, Lee KN, Kim YD, Kim DK, Kim SH, Lee CH, Franceschi RT, Choi HS, Koh JT. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone. 2011;48(4):885–93. doi: 10.1016/j.bone.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Yan W, Li X. Impact of diabetes and its treatments on skeletal diseases. Front Med. 2013;7(1):81–90. doi: 10.1007/s11684-013-0243-9. [DOI] [PubMed] [Google Scholar]
- 13.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider A. Mouse Models to Study Metformin Effects in Carcinogenesis. In: Berger NA, editor. Murine Models, Energy Balance and Cancer. Springer International Publishing; Switzerland: 2015. pp. 271–292. [Google Scholar]
- 15.Sedlinsky C, Molinuevo MS, Cortizo AM, Tolosa MJ, Felice JI, Sbaraglini ML, Schurman L, McCarthy AD. Metformin prevents anti-osteogenic in vivo and ex vivo effects of rosiglitazone in rats. Eur J Pharmacol. 2011;668(3):477– 85. doi: 10.1016/j.ejphar.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 16.Jeyabalan J, Shah M, Viollet B, Chenu C. AMP-activated protein kinase pathway and bone metabolism. J Endocrinol. 2012;212(3):277–90. doi: 10.1530/JOE-11-0306. [DOI] [PubMed] [Google Scholar]
- 17.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(10):3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Ma T, Guo D, Hu K, Shu Y, Xu HHK, Schneider A. Metformin induces osteoblastic differentiation of human induced pluripotent stem cell-derived mesenchymal stem cells. J Tissue Eng Regen Med. 2017 doi: 10.1002/term.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334(9):574–9. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 20.Molinuevo MS, Schurman L, McCarthy AD, Cortizo AM, Tolosa MJ, Gangoiti MV, Arnol V, Sedlinsky C. Effect of metformin on bone marrow progenitor cell differentiation: in vivo and in vitro studies. J Bone Miner Res. 2010;25(2):211– 21. doi: 10.1359/jbmr.090732. [DOI] [PubMed] [Google Scholar]
- 21.Gao Y, Li Y, Xue J, Jia Y, Hu J. Effect of the anti-diabetic drug metformin on bone mass in ovariectomized rats. Eur J Pharmacol. 2010;635(1-3):231–6. doi: 10.1016/j.ejphar.2010.02.051. [DOI] [PubMed] [Google Scholar]
- 22.Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP, Zhao L, Jia CH, Wen ZH, Jin DD, Bai XC. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J Cell Biochem. 2011;112(10):2902–9. doi: 10.1002/jcb.23206. [DOI] [PubMed] [Google Scholar]
- 23.Jeyabalan J, Viollet B, Smitham P, Ellis SA, Zaman G, Bardin C, Goodship A, Roux JP, Pierre M, Chenu C. The anti-diabetic drug metformin does not affect bone mass in vivo or fracture healing. Osteoporos Int. 2013;24(10):2659–70. doi: 10.1007/s00198-013-2371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu Y, Leabman MK, Feng B, Mangravite LM, Huang CC, Stryke D, Kawamoto M, Johns SJ, DeYoung J, Carlson E, Ferrin TE, Herskowitz I, Giacomini KM. Evolutionary conservation predicts function of variants of the human organic cation transporter. OCT1 Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):5902–7. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117(5):1422–31. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–80. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakata T, Anzai N, Kimura T, Miura D, Fukutomi T, Takeda M, Sakurai H, Endou H. Functional analysis of human organic cation transporter OCT3 (SLC22A3) polymorphisms. J Pharmacol Sci. 2010;113(3):263–6. doi: 10.1254/jphs.09331sc. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Pawlikowski B, Schlessinger A, More SS, Stryke D, Johns SJ, Portman MA, Chen E, Ferrin TE, Sali A, Giacomini KM. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet Genomics. 2010;20(11):687–99. doi: 10.1097/FPC.0b013e32833fe789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;(201):105–67. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 30.Liang X, Giacomini KM. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J Pharm Sci. 2017;106(9):2245–2250. doi: 10.1016/j.xphs.2017.04.078. [DOI] [PubMed] [Google Scholar]
- 31.Grayson WL, Bunnell BA, Martin E, Frazier T, Hung BP, Gimble JM. Stromal cells and stem cells in clinical bone regeneration. Nat Rev Endocrinol. 2015;11(3):140–50. doi: 10.1038/nrendo.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller SM, Glowacki J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional collagen sponges. J Cell Biochem. 2001;82(4):583–90. doi: 10.1002/jcb.1174. [DOI] [PubMed] [Google Scholar]
- 33.Lee SY, Miwa M, Sakai Y, Kuroda R, Matsumoto T, Iwakura T, Fujioka H, Doita M, Kurosaka M. In vitro multipotentiality and characterization of human unfractured traumatic hemarthrosis-derived progenitor cells: A potential cell source for tissue repair. J Cell Physiol. 2007;210(3):561–6. doi: 10.1002/jcp.20890. [DOI] [PubMed] [Google Scholar]
- 34.Yan J, Tie G, Wang S, Messina KE, DiDato S, Guo S, Messina LM. Type 2 diabetes restricts multipotency of mesenchymal stem cells and impairs their capacity to augment postischemic neovascularization in db/db mice. J Am Heart Assoc. 2012;1(6):e002238. doi: 10.1161/JAHA.112.002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Januszyk M, Sorkin M, Glotzbach JP, Vial IN, Maan ZN, Rennert RC, Duscher D, Thangarajah H, Longaker MT, Butte AJ, Gurtner GC. Diabetes irreversibly depletes bone marrow-derived mesenchymal progenitor cell subpopulations. Diabetes. 2014;63(9):3047–56. doi: 10.2337/db13-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HS, Hung SC, Peng ST, Huang CC, Wei HM, Guo YJ, Fu YS, Lai MC, Chen CC. Mesenchymal stem cells in the Wharton’s jelly of the human umbilical cord. Stem cells. 2004;22(7):1330–7. doi: 10.1634/stemcells.2004-0013. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Ott L, Seshareddy K, Weiss ML, Detamore MS. Musculoskeletal tissue engineering with human umbilical cord mesenchymal stromal cells. Regen Med. 2011;6(1):95–109. doi: 10.2217/rme.10.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding DC, Chang YH, Shyu WC, Lin SZ. Human umbilical cord mesenchymal stem cells: a new era for stem cell therapy. Cell Transplant. 2015;24(3):339–47. doi: 10.3727/096368915X686841. [DOI] [PubMed] [Google Scholar]
- 39.Diao Y, Ma Q, Cui F, Zhong Y. Human umbilical cord mesenchymal stem cells: osteogenesis in vivo as seed cells for bone tissue engineering. J Biomed Mater Res A. 2009;91(1):123–31. doi: 10.1002/jbm.a.32186. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Singh M, Bonewald LF, Detamore MS. Signalling strategies for osteogenic differentiation of human umbilical cord mesenchymal stromal cells for 3D bone tissue engineering. J Tissue Eng Regen Med. 2009;3(5):398–404. doi: 10.1002/term.176. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Dormer NH, Bonewald LF, Detamore MS. Osteogenic differentiation of human umbilical cord mesenchymal stromal cells in polyglycolic acid scaffolds. Tissue engineering Part A. 2010;16(6):1937–48. doi: 10.1089/ten.TEA.2009.0706. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Zhao L, Detamore MS. Human umbilical cord mesenchymal stromal cells in a sandwich approach for osteochondral tissue engineering. J Tissue Eng Regen Med. 2011;5(9):712–21. doi: 10.1002/term.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen Y, Jiang B, Cui J, Li G, Yu M, Wang F, Zhang G, Nan X, Yue W, Xu X, Pei X. Superior osteogenic capacity of different mesenchymal stem cells for bone tissue engineering. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116(5):e324–32. doi: 10.1016/j.oooo.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 44.Mueller AA, Forraz N, Gueven S, Atzeni G, Degoul O, Pagnon-Minot A, Hartmann D, Martin I, Scherberich A, McGuckin C. Osteoblastic differentiation of Wharton jelly biopsy specimens and their mesenchymal stromal cells after serum-free culture. Plast Reconstr Surg. 2014;134(1):59e–69e. doi: 10.1097/PRS.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 45.Chen W, Liu J, Manuchehrabadi N, Weir MD, Zhu Z, Xu HH. Umbilical cord and bone marrow mesenchymal stem cell seeding on macroporous calcium phosphate for bone regeneration in rat cranial defects. Biomaterials. 2013;34(38):9917–25. doi: 10.1016/j.biomaterials.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caballero M, Morse JC, Halevi AE, Emodi O, Pharaon MR, Wood JS, van Aalst JA. Juvenile Swine Surgical Alveolar Cleft Model to Test Novel Autologous Stem Cell Therapies. Tissue Eng Part C Methods. 2015;21(9):898–908. doi: 10.1089/ten.tec.2014.0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caballero M, Jones DC, Shan Z, Soleimani S, van Aalst JA. Tissue Engineering Strategies to Improve Osteogenesis in the Juvenile Swine Alveolar Cleft Model. Tissue Eng Part C Methods. 2017 doi: 10.1089/ten.TEC.2017.0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel H, Younis RH, Ord RA, Basile JR, Schneider A. Differential expression of organic cation transporter OCT-3 in oral premalignant and malignant lesions: potential implications in the antineoplastic effects of metformin. J Oral Pathol Med. 2013;42(3):250–6. doi: 10.1111/j.1600-0714.2012.01196.x. [DOI] [PubMed] [Google Scholar]
- 49.Li Q, Peng X, Yang H, Rodriguez JA, Shu Y. Contribution of organic cation transporter 3 to cisplatin cytotoxicity in human cervical cancer cells. J Pharm Sci. 2012;101(1):394–404. doi: 10.1002/jps.22752. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Schneider A. HIF-2alpha-mediated activation of the epidermal growth factor receptor potentiates head and neck cancer cell migration in response to hypoxia. Carcinogenesis. 2010;31(7):1202–10. doi: 10.1093/carcin/bgq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider A, Gartenhaus RB. AMPK signaling: a targetable tumor suppressor pathway? Cancer Biol Ther. 2010;10(11):1178–81. doi: 10.4161/cbt.10.11.13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L, Schaner ME, Giacomini KM. Functional characterization of an organic cation transporter (hOCT1) in a transiently transfected human cell line (HeLa) J Pharmacol Exp Ther. 1998;286(1):354–61. [PubMed] [Google Scholar]
- 53.Li Q, Shu Y. Role of solute carriers in response to anticancer drugs. Mol Cell Ther. 2014;2:15. doi: 10.1186/2052-8426-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma L, Wu X, Ling-Ling E, Wang DS, Liu HC. The transmembrane transport of metformin by osteoblasts from rat mandible. Arch Oral Biol. 2009;54(10):951–62. doi: 10.1016/j.archoralbio.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Leabman MK, Huang CC, Kawamoto M, Johns SJ, Stryke D, Ferrin TE, DeYoung J, Taylor T, Clark AG, Herskowitz I, Giacomini KM. Polymorphisms in a human kidney xenobiotic transporter, OCT2, exhibit altered function. Pharmacogenetics. 2002;12(5):395–405. doi: 10.1097/00008571-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3) J Pharmacol Exp Ther. 2004;308(1):2–9. doi: 10.1124/jpet.103.053298. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, Urban TJ, Chen L, Yee SW, Choi JH, Huang Y, Brett CM, Burchard EG, Giacomini KM. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19(7):497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta S, Burckhardt G, Hagos Y. SLC22 transporter family proteins as targets for cytostatic uptake into tumor cells. Biol Chem. 2011;392(1-2):117–24. doi: 10.1515/BC.2011.014. [DOI] [PubMed] [Google Scholar]
- 59.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 60.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, Guigas B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54(12):3101–10. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole cell lysates extracted from UC-MSC-2 cells following a 3-hour treatment with increasing doses of metformin were analyzed by Western blotting to determine the expression levels of phosphorylated AMPKα1 at Thr172 (pAMPK) and total AMPK.