

Abstract
It is critical to understand the molecular mechanisms of hepatocarcinogenesis in order to prevent or treat hepatocellular carcinoma (HCC). The development of HCC is commonly associated with hepatocyte death and compensatory proliferation. However, the role of Caspase-3, a key apoptotic executor, in hepatocarcinogenesis is unknown. In this study, we used Caspase-3-deficient mice to examine the role of Caspase-3 in hepatocarcinogenesis in a chemical (diethylnitrosamine, DEN)-induced HCC model. We found that Caspase-3 deficiency significantly increased DEN-induced HCC. Unexpectedly, Caspase-3 deficiency increased apoptosis induced by DEN and the subsequent compensatory proliferation. Intriguingly, we discovered that Caspase-3 deficiency increased the activation of p38 with and without DEN treatment. Moreover, we demonstrated that TNFα and IL1α stimulated increased activation of p38 in Caspase-3 KO hepatocytes compared with wild-type hepatocytes. Finally, we found that inhibition of p38 by SB202190 abrogated enhanced hepatocyte death, compensatory proliferation and HCC induced by DEN in Caspase-3-deficient mice. Overall, our data suggest that Caspase-3 inhibits chemical-induced hepatocarcinogenesis by suppressing p38 activation and hepatocyte death.
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer deaths worldwide1. The overall survival of patients with HCC is <12%, and most patients with HCC have limited treatment options2. Currently, the most effective targeted therapeutic agent for advanced HCC, sorafenib, only increases survival in patients with advanced HCC from 7.9 months to 10.7 months3. There is an urgent need to develop more effective therapeutic strategies and agents to treat HCC. To achieve this goal, the molecular signaling pathways that drive or mediate the development of HCC must be better understood.
HCC is considered a chronic inflammation-related disorder, the development of which is commonly associated with hepatocyte death and compensatory proliferation1,4–10. Several classes of chemicals promote HCC in rodents, including diethylnitrosamine (DEN), which has been extensively studied. In the DEN-induced HCC model, DEN stimulates DNA damage and causes reactive oxygen species to accumulate; it also induces hepatocyte death through the c-Jun N-terminal kinase (JNK) pathway11–14. The dying cells release interleukin-1 alpha (IL1α), which activates Kupffer cells6,15. Activated Kupffer cells produce cytokines and growth factors such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα)5,16–18, which promote expansion of hepatocytes with DNA mutations thus enhancing HCC development5,16,17. We previously reported that deficiency of PUMA (p53 upregulated modulator of apoptosis) significantly decreased DEN-induced liver cancer by blocking acute apoptotic responses and the subsequent compensatory proliferation19. These data provide evidence for the correlation of specific mediators of hepatocyte apoptosis and the development of HCC. Caspase-3 is a member of the cysteine-aspartic acid protease family (caspases) and plays a central role in the execution-phase of cellular apoptosis20. Given the strong association of apoptosis and compensatory proliferation in HCC development, and the specific apoptotic executive function of Caspase-3, we hypothesized that inhibition of Caspase-3 would suppress DNA damage-induced hepatocyte death, and thereby inhibit hepatic carcinogenesis.
Unexpectedly, we found that Caspase-3 deficiency significantly promoted DEN-induced HCC development. Intriguingly, Caspase-3 deficiency increased hepatocyte death and the subsequent compensatory proliferation induced by DEN. In addition, we discovered that Caspase-3 deficiency increased the activation of p38 MAP kinase. Moreover, we demonstrated that deletion of Caspase-3 increased p38 activation by IL1α and TNFα in primary mouse hepatocytes. Furthermore, we found that inhibition of p38 abrogated enhanced hepatocyte death, the compensatory proliferation and HCCs induced by DEN in Caspase-3-deficient mice. Overall, our data suggest that Caspase-3 inhibits chemical-induced hepatocarcinogenesis by suppressing p38 activation and hepatocyte death.
Results
DEN induced Caspase-3 cleavage in mouse livers
To determine the role of Caspase-3 in hepatocarcinogenesis, we used the DEN-induced HCC model because Caspase-3 is cleaved and activated in response to DNA damage or non-genotoxic stimuli21 and DEN is known to induce DNA damage22. We analyzed the activation of Caspase-3 in the livers of WT mice after DEN treatment. Cleaved Caspase-3 but not total Caspase-3 was significantly elevated by day 3 after treatment compared with untreated mice (Fig. 1a). Immunostaining indicated that cleaved Caspase-3 protein was selectively induced in hepatocytes around the centrilobular regions 24 h after DEN treatment, whereas the basal level was undetectable (Fig. 1b).
Fig. 1. Caspase-3 deficiency promoted DEN-induced liver cancer.

a CASPASE-3, active CASPASE-3 and β-actin protein expression in the livers of WT mice 3 days following injection with either saline (Un) or 100 mg/kg of DEN was analyzed by western blotting. Values are means ± SD, n = 3 mice in each group. b Active CASPAPS-3 protein (red) in the livers of WT and Caspase-3 KO mice following indicated treatment was detected by IF with nuclei counterstained with DAPI (magnification, ×400). c Photographs of livers of WT and Caspase-3 KO mice 9 months after DEN injection. d Quantification of liver tumor numbers (n = 6). e Quantification of liver tumor sizes (n = 6). f Histological analysis (H&E staining) and staining of AFP of the livers of WT and Caspase-3 KO mice 9 months after DEN injection. Bars: 20 µM. Values in (d) and (e) are means ± SDs
Caspase-3 deficiency promoted DEN-induced liver cancer
A single injection of DEN to 15-day-old male mice results in efficient HCC induction19. To determine a potential role for Caspase-3 in hepatocarcinogenesis, we compared tumor incidence and size in WT and Caspase-3 knockout (Casp3 KO) littermates 9 months after DEN treatment. All of the mice developed tumors by 9 months of age (Fig. 1c). Interestingly, tumor incidence in Casp3 KO mice increased by about twofold compared with WT mice (8.8 ± 2.2 vs. 3.5 ± 0.8) (Figs. 1c, d). Tumor size was also significantly increased in Casp3 KO mice compared with WT mice (Figs. 1e, f). In addition, immunohistochemical (IHC) staining signals for alpha-fetoprotein (AFP), a common HCC marker, were significantly lower in the livers of WT mice compared with those of Casp3 KO mice (Fig. 1f). These results indicate that Casp3 deficiency promotes DEN-induced hepatocarcinogenesis.
Caspase-3 deficiency increased DEN-induced hepatocyte death
Caspase-3 is a critical apoptotic executor21. Promotion of HCC development by Casp3 deficiency could be due to decreased apoptosis of tumor cells. To determine whether a deficiency of Casp3 affects apoptosis in DEN-induced HCC, we performed Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in liver tumors from WT and Casp3 KO mice treated with DEN. We did not find significant differences in apoptosis in tumors between these two groups of mice (Fig. S1A and S1B). We also analyzed proliferation in the DEN-treated livers from WT and Casp3 KO mice by Ki67 staining. The number of Ki67+ cells was comparable in Casp3-deficient and WT liver tumors (Fig. S1A and S1B). These results show that Casp3 deficiency affects neither apoptosis nor proliferation in DEN-induced HCC.
The development of DEN-induced HCC is associated with hepatocyte death and compensatory proliferation1,4–10. DEN induces significant hepatocyte death in WT mice within 3 days (Figs. 2a, b), notably, in the centrilobular regions where Caspase-3 is activated (Figs. 1b, 2a). Interestingly, there was dramatically enhanced hepatocyte death in Casp3 KO mice compared with WT mice after DEN treatment (Figs. 2a, b). Hepatocyte death decreased by 10 days after DEN treatment in both WT and Casp3 KO mice, but was greater in Casp3 KO mice than in WT mice (Figs. 2a, b). DEN treatment increased serum levels of the liver enzyme alanine aminotransferase (ALT) by day 3, such increase being similar in both Casp3 KO and WT mice (Fig. 2c). As DEN can induce oxidative DNA damage in the liver5, we evaluated DNA double-strand breaks in livers by p-H2AX staining. No difference in DNA damage was found between WT and Casp3 KO mice 3 or 10 days after DEN treatment (Figs. 2d, e).
Fig. 2. Caspase-3 deficiency increased DEN-induced hepatocyte apoptosis.

Liver tissue or blood was harvested from WT and Caspase-3 KO mice 0, 3 or 10 days after a single injection of 100 mg/kg DEN. a Apoptosis in the livers of WT and Caspase-3 KO mice after treatment with vehicle or DEN was examined by TUNEL staining. b Quantification of TUNEL staining for (a) (n = 5). c Serum alanine aminotransferase (ALT) levels were determined at the indicated time points after DEN treatment. Values are means ± SD, n = 4 mice in each group. d Expression of p-H2AX protein in the livers of WT and Caspase-3 KO mice after treatment with vehicle or DEN was examined by IF. e Quantification of p-H2AX staining for (d) (n = 5). Bars: 20 µM. Values in (b), (c) and (e) are means ± SDs
Caspase-6 and Caspase-7 may play similar functions as Caspase-3 in the execution of apoptosis23. To determine if deletion of Caspase-3 increases the activation of Caspase-6 and Caspase-7 after DEN treatment in mouse livers, we examined the expression of total and cleaved Caspase-6 and Caspase-7 in DEN-treated mouse livers. We found a slight decrease in cleavage of Caspase-6 or Caspase-7 in Casp3 KO mice compared with WT mice (Fig. S2), suggesting that increased DEN-induced hepatocyte apoptosis resulting from Caspase-3 deficiency is not the result of enhanced activation of Caspase-6 or Caspase-7 in DEN-treated mouse livers.
Caspase-3 deficiency enhanced DEN-induced compensatory proliferation
DEN-induced hepatocyte death is associated with compensatory proliferation and HCC development19. As expected, elevated proliferation was found in the livers of WT mice 3 and 10 days following DEN treatment as evidenced by Ki67 and PCNA staining (Figs. 3a–d). The proliferation centered on the centrilobular regions where apoptotic cells were detected (Figs. 2a, 3a). The degree of proliferation was significantly increased in Casp3 KO mice compared with WT mice (Figs. 3a–d). IL-6 and its downstream target Stat3 have been shown to play important roles in DEN-induced compensatory proliferation5,16–18. We therefore examined the expression of IL-6 mRNA and phosphorylation of Stat3 in WT and Casp3 KO mouse livers. We found that IL-6 mRNA was increased in Caspase-3 KO mouse livers compared with WT mouse livers after DEN treatment both on day 3 and day 10 (Fig. S3). Phosphorylation of Stat3 was also increased in Caspase-3 KO mouse livers compared with WT mouse livers after DEN treatment on day 3 while the difference was diminished by day 10 (Fig. S3). These results showed that Caspase3 deficiency promotes DEN-induced compensatory proliferation in hepatocytes.
Fig. 3. Caspase-3 deficiency enhanced DEN-induced compensatory proliferation.

Liver tissue was harvested from WT and Caspase-3 KO mice 0, 3 and 10 days after injection of 100 mg/kg DEN. a Ki67 was measured at indicated time points by IHC. b Quantification of Ki67 staining for (a) (n = 5). c Proliferating cell nuclear antigen (PCNA) was measured at indicated time points by IF. d Quantification of Ki67 staining for (c) (n = 5). Bars: 20 µM. Values in (b) and (d) are means ± SDs
Caspase-3 deficiency enhanced p38 activation in mouse livers
Activation of JNK, nuclear factor-κB (NF-κB) and p38 by cytokines, including TNFα and IL1α, plays a critical role in hepatocyte death and compensatory proliferation after DEN treatment24,25. To determine whether deletion of Caspase-3 affects the activation of JNK, NF-κB and p38 in the liver after DEN treatment, we compared the phosphorylation of these three proteins in the livers of WT and Casp3 KO mice. There was no significant difference in the phosphorylation status of JNK in Casp3 KO mouse livers compared with WT mouse livers prior to or after DEN treatment (Fig. 4a). NF-κB was slightly increased by Caspase-3 deficiency in response to DEN treatment by 3 days but not after 10 days (Fig. 4a). Accordingly, phosphorylation of I-kappa-B-alpha (IκBα) on Ser 32 and Ser 36, which results in release and nuclear translocation of active NF-κB26, was also slightly increased by Caspase-3 deficiency in response to DEN treatment on day 3 after treatment (Fig. S4A). We also examined levels of IκBα, Vcam1 and Cxcl10 mRNA, which have been reported to be direct target genes of NF-κB27,28. We found that the expression of Vcam1 and Cxcl10 but not IκBα was increased by Caspase-3 deficiency in response to DEN treatment in mouse livers (Fig. S4B). NF-κB has been shown to protect against DEN-induced hepatocyte death and HCC5,24. Therefore, it is unlikely that an increase in NF-κB activation led to enhanced DEN-induced hepatocyte death, compensatory proliferation and HCC caused by Caspase-3 deficiency. Interestingly, higher p-p38 was observed in Casp3 KO mouse livers compared with WT mouse livers with or without DEN treatment (Fig. 4a), suggesting that deletion of Caspase-3 increases the activation of p38 in mouse livers. Consistently, phosphorylation of MK2, a downstream direct target of p38, was also increased in Casp3 KO mouse livers (Fig. S5A). MMK6 and MMK3 have been shown to phosphorylate and activate p38. We also found that phosphorylation of MAP kinase kinases 3/6 (MKK3/6) was increased in Casp3 KO mouse livers (Fig. S5A).
Fig. 4. Caspase-3 deficiency enhanced TNFα- or IL1α-induced p38 activation in mouse hepatocytes.

a Expression of p-p38, p38, p-JNK, JNK, p-NF-κB, NF-κB, CASPASE-3 and GAPDH proteins in the livers of WT and Caspase-3 KO mice 0, 3 and 10 days following injection with either saline (Un) or 100 mg/kg of DEN was analyzed by western blotting. b mRNA expression of Tnfα and IL1α in the livers of WT and Caspase-3 KO mice 0, 3 and 10 days following injection with either saline (Un) or 100 mg/kg of DEN was analyzed by western blotting. Values are means ± SDs, n = 3 mice in each group. c Expression of p-p38, p38, CASPASE-3 and GAPDH proteins in isolated hepatocytes from WT and Caspase-3 KO mice 1 h after treatment with 0, 5 or 15 ng/ml TNFα was analyzed by western blotting. d Expression of p-p38, p38, CASPASE-3 and GAPDH proteins in isolated hepatocytes from WT and Caspase-3 KO mice 1 h after treatment with 0, 20 or 100 ng/ml IL1α was analyzed by western blotting. Values in (b) are means ± SDs
Caspase-3 deficiency did not affect TNFα nor IL1α expression in mouse livers after DEN treatment
TNFα plays a critical role in hepatocyte compensatory proliferation and the activation of JNK, NF-κB and p38 after DEN treatment24,25. It is possible that Caspase-3 deficiency enhances p38 activation in mouse liver by increasing the expression of TNFα after DEN treatment. We therefore compared mRNA and protein levels of TNFα in the livers of WT and Casp3 KO mice. Expression of both TNFα mRNA and protein was increased when examined 3 days following DEN treatment in WT mice, whereas comparable expression of TNFα was observed in Casp3 KO mice (Fig. 4b and Fig. S5A). IL1α has been shown to phosphorylate and activate p38 in liver cells29. We therefore examined the expression of IL1α and found no significant difference in IL1α mRNA expression (Fig. 4b). These data indicate that Caspase-3 deficiency increases the activation of p38 but does not do so through enhanced expression of TNFα or IL1α in mouse livers.
Caspase-3 deficiency enhanced activation of p38 by in response to TNFα or IL1α in hepatocytes
As the expression of neither TNFα nor IL1α was affected in mouse livers by deletion of Caspase-3, we therefore examined whether Caspase-3 deficiency in hepatocytes enhances the phosphorylation of p38 in response to TNFα or IL1α. We isolated primary hepatocytes from WT and Casp3 KO mice and treated them with TNFα or IL1α. There was a significant enhancement of p38 phosphorylation in Casp3 KO hepatocytes compared with WT hepatocytes prior to or after treatment with TNFα (Fig. 4c). Similarly, phosphorylation of p38 was increased in Casp3 KO hepatocytes compared with WT hepatocytes after 1 h of IL1α treatment (Fig. 4d). Consistently, phosphorylation of MK2 and MKK3/6 was also increased in Casp3 KO hepatocytes compared with WT hepatocytes after 1 h of treatment with IL1α or TNFα (Fig. S5B and C). In general, these data demonstrate that Caspase-3 deficiency enhances the activation of p38 in hepatocytes in response to TNFα or IL1α.
Inhibition of p38 abrogated enhanced DEN-induced hepatocyte death, compensatory proliferation and HCC development induced by deletion of Caspase-3
Emerging evidence indicates that activation of p38 promotes cell death. Activation of p38 by MKK6 has been shown to induce apoptosis in Huh7 and HepG2 cells30. A p38 MAPK inhibitor markedly suppressed cell death induced by TNFα31. Therefore, we reasoned that Caspase-3 deficiency increased DEN-induced hepatocyte apoptosis through enhanced p38 activation. To test this hypothesis, we first examined whether the overexpression of p38 increases TNFα-induced cell death in AML12 cells, a mouse hepatocyte cell line. We found that overexpression of p38 did increase TNFα-induced cell death in these cells (Fig. S6). Further, we treated WT and Casp3 KO mice with a specific inhibitor of p38, SB202190, prior to and after DEN treatment. We found that SB202190 decreased the phosphorylation of p38 in both WT and Casp3 KO mice (Fig. 5a) and inhibited DEN-induced hepatocyte death (Figs. 5b, c). Consistently, SB202190 also decreased DEN-induced compensatory proliferation in Casp3 KO mice (Fig. 5d). Importantly, SB202910 inhibited DEN-induced HCC development in Casp3 KO mice (Figs. 5e, f). These data suggest that deletion of Caspase-3 enhances DEN-induced hepatocyte death, compensatory proliferation and the development of HCC by increasing the activation of p38.
Fig. 5. Inhibition of p38 abrogated enhanced DEN-induced hepatocyte death, compensatory proliferation and HCC development induced by deletion of Caspase-3.

a Expression of p-p38 and GAPDH proteins in the livers of Caspase-3 KO mice 3 days following injection with either saline (Un) or 100 mg/kg DEN plus 20 mg/kg SB202190 was analyzed by western blotting. b Apoptosis in the livers of Caspase-3 KO mice 3 days following injection with either saline (Un) or 100 mg/kg DEN plus 20 mg/kg SB202190 was analyzed by TUNEL staining. Bars: 20 µM. c Quantification of TUNEL staining for (b) (n = 3). d Quantification of Ki67 staining in the livers of Caspase-3 KO mice 3 days after injection with either saline (Un) or 100 mg/kg DEN plus 20 mg/kg SB202190. e Photographs of livers of WT and Caspase-3 KO mice 9 months after DEN injection with or without SB202190. f Quantification of liver tumor numbers (n = 5) and liver tumor sizes (n = 5) for (e). Values in (c), (d) and (f) are means ± SDs
Discussion
Treatment options for HCC are limited2. Currently, the most effective targeted therapeutic agent for advanced HCC, sorafenib, an inhibitor of several protein tyrosine kinases (including vascular endothelial growth factor receptor and platelet-deried growth factor receptor) and Raf kinases, increases survival in patients with advanced HCC by only 3 months3. More effective therapeutic strategies and agents to treat HCC are clearly needed. Therefore, it is necessary to elucidate the molecular signaling pathways that drive or mediate the development of HCC. Hepatocyte apoptosis has been shown to play an important role in HCC development. About 3.5% of HCC samples from The Cancer Genome Atlas (TCGA) liver HCC data sets show deletion of the Caspase-3 gene, which is correlated with reduced Caspase-3 mRNA expression (Fig. S7). A recent study indicated that low expression of Caspase-3 is correlated with poor prognosis in HCC patients32, suggesting that Caspase-3 might be involved in HCC pathogenesis. However, the role of Caspase-3 in HCC development in vivo has not been reported. In this study, we found that deletion of Caspase-3 promotes DEN-induced hepatocyte death, compensatory proliferation and carcinogenesis, indicating that Caspase-3 plays an inhibitory role in hepatocarcinogenesis. Therefore, it is likely that Caspase-3 deletion or downregulation in human HCC samples promotes HCC development.
Caspase-3 is a central executor of apoptosis. Therefore, it was surprising to us that Caspase-3 deletion increases apoptosis induced by DEN in mouse livers. Caspase-6 and Caspase-7, the other two apoptotic executors, have been shown to compensate the functions of Caspase-3 in some cell contexts33. However, we observed no compensatory activation of Caspase-6 or Caspase-7 in DEN-treated WT or Caspase-3 KO livers. Instead, we found that loss of Caspase-3 increases the activation of p38 in response to DEN, TNFα or IL1α in hepatocytes. It is notable that p38 hyperactivation in Casp3 KO mice is largely constitutive (Fig. 4a), although treatment with IL1α or TNFα promotes a further increase in phosphorylation (Fig. S5B). It is possible that Caspase-3 deletion increases p38 activation in response to the basal level of TNFα or IL1α in mouse liver. Activation of p38 by MKK6 has been shown to induce apoptosis in HCC cells30. p38 also mediates cell death induced by TNFα in cancer cells31. We also found that overexpression of p38 enhanced TNFα-induced cell death in mouse liver cells (Fig. S6). In addition, we demonstrated that a p38 inhibitor abrogates the enhanced DEN-induced hepatocyte death induced by deletion of Caspase-3. Overall, our data suggest that Casp3 KO hepatocytes are more sensitive to TNFα, which is likely due to p38 hyperactiation (Fig. S6). Therefore, deletion of Caspase-3 enhances DEN-induced hepatocyte death, compensatory proliferation and hepatocarcinogenesis by increasing the activation of p38 (Fig. 6). To our knowledge, this is the first report of inhibition of p38 activation by Casp-3. It is unknown whether this mechanism is a general one that will be found all in cell types or whether it is in cell context specific. It will be interesting for us to test this question in all our future studies.
Fig. 6. A schematic model.
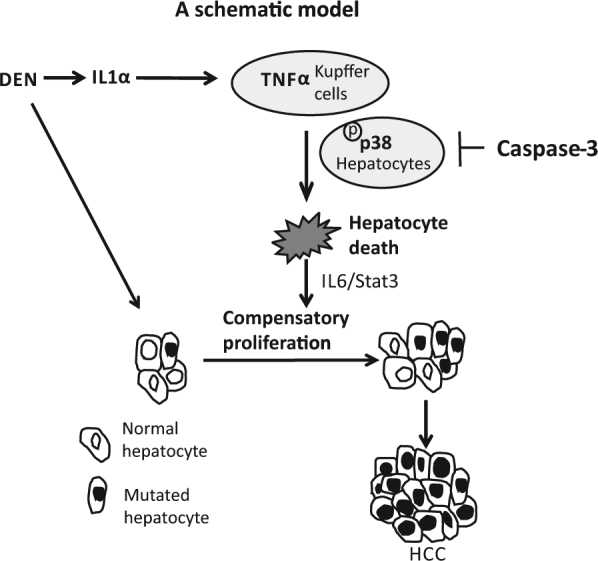
In the DEN-induced HCC model, DEN induces hepatocyte death. The dying cells release IL1α, which activates Kupffer cells. Activated Kupffer cells produce TNFα, which induces hepatocyte death and promotes expansion of the hepatocytes carrying DNA mutations, thus enhancing HCC development. p38 hyperactivation promotes TNFα-mediated hepatocyte death. Caspase-3 suppresses p38 activation in hepatocytes thereby Caspase-3 deletion increases the activation of p38 and enhances hepatocyte death, the subsequent compensatory proliferation and the development of HCC, all induced by DEN.
Increased activation of p38 caused by the deletion of Caspase-3 may be due to the result of multiple actions. MKKs 3 and 6 and have been identified as common activators of p38 in the liver34. Indeed we found that phosphorylation of MKK6/3 is increased in Caspase-3-deficient hepatocytes (Fig. S4), suggesting that Caspase-3 deletion increases the activation of p38 through enhancing MKK6/3 activation in hepatocytes. Several MAP kinase kinase kinases have been shown to trigger MKK6/3 activation; they include apoptosis signal-regulating kinase 1 (ASK1), dual-leucine zipper-bearing kinase 1 (DLK1), thousand-and-one amino acid (TAO) 1 and 2, tumor progression loci 2 (TPL2), mixed lineage kinase 3 (MLK3), MEK kinase 3 (MEKK3), MEKK4, and leucine zipper and sterile-α motif kinase 1 (ZAK1)29. Over three hundred targets of Caspase-3 have been identified35. It is possible that the loss of Caspase-3 increases the activation of MKK6/3 and p38 through regulating its downstream targets. Given the cell context dependence and multiple known targets of Caspase-3, it is not our contention that the increased activation of MKK6/p38 is the only mechanism to explain Caspase-3’s effects. Rather, we posit that this mechanism is a contributor to increased hepatocyte death, compensatory proliferation and hepatic carcinogenesis caused by the loss of Caspase-3, and can account for some, if not all, of the functional changes observed.
In conclusion, our study shows that deficiency of Caspase-3 promotes chemical-induced hepatocyte compensatory proliferation and hepatocarcinogenesis. Recent studies suggest that specific inhibition of Casapse-3 in concert with chemotherapy may be a novel approach for the treatment of cancers because of the role of cell death in promoting tissue regeneration in mammalian cells36,37. Our data suggest that the role of Caspase-3 in cancer development is complex and the inhibition of Caspase-3 might not be suitable for the treatment of HCC patients.
Materials and methods
Mice and treatments
All animals received humane care according to the “Guide for the Care and Use of Laboratory Animals” (http://oacu.od.nih.gov/ac_cbt/guide3.htm). The procedures for all animal experiments were approved by the Institutional Animal Care and Use Committee of Loyola University Chicago. Casp3+/+ and Casp3−/− littermate mice in a C57BL/6 background were generated from heterozygote intercrosses. Genotyping was performed as described in the JAX Lab website (https://www.jax.org/strain/006233). The mice were housed in micro-isolator cages in a room illuminated from 07:00 a.m. to 07:00 p.m. (12:12-h light–dark cycle), and allowed access to water and chow ad libitum.
For the DEN-induced HCC model, DEN (15 mg/kg) was injected intraperitoneally (i.p.) into 15-day-old mice. Mice were sacrificed after 9 months on the standard diet. Surface tumor nodules in each liver lobe were counted and measured with a caliper. For short-term studies of DEN-induced liver injury, 8- to 12-week-old mice were injected with DEN (100 mg/kg body weight) i.p. and sacrificed after 3 or 10 days.
For p38 inhibitor treatments, SB202190 (LC Laboratories, Cat # S-1700) was diluted in 6% captisol (CyDex, Inc., Lenexa, KS). One day prior to DEN treatment, six 2-week-old or 8- to 12-week-old WT and six 2-week-old or 8- to 12-week-old Casp3−/− mice were administered vehicle solution (6% captisol) or 20 mg/kg SB202190 by i.p. daily for 3 days and livers were collected 9 months, 3 or 10 days after DEN treatment.
Isolation and culture of primary mouse hepatocytes
Hepatocytes from Casp3+/+ and Casp3−/− mice were isolated by the non-recirculating two-step perfusion method as previously described38. The hepatocytes were then cultured in Williams’ medium E supplemented with Hepatocyte Maintenance Supplement Pack (Life Technologies, Grand Island, NY) overnight. Cells were treated with 5 ng/ml and 20 ng/ml IL1α or 5 ng/ml and 15 ng/ml TNFα for 1 h; proteins were subsequently collected for western blot analysis.
Culture and treatment of AML12 cells
AML12 cells were purchased from ATCC and cultured as per the manufacturer's recommendations. Cells were transfected with pCNA3 or pCNA3-p38α (Addgene, #20352) plasmids followed by treatment with either saline (vehicle) or 20 ng/ml TNFα for 48 h. Viability of the cells was analyzed by Alamar Blue Assay.
ALT measurement
Venous blood of mice was collected from the tail vein 0, 24, and 72 h after DEN treatment. Blood was kept at 4 °C for overnight and centrifuged at 200 × g for 20 min to isolate serum. ALT was measured using the Infinity™ ALT kit (Thermo Scientific, Middletown, VA) and is reported as mean ± SD. Briefly, 10 μl of serum was added to 100 μl ALT reagent and measured at 37 °C at 340 nm. Each sample was measured in triplicate and three mice were used in each group.
Western blotting
Western blotting was performed as previously described38. Primary antibodies, against Caspase-3 (9662), active Caspase-3 (9661), Caspase-6 (9762), active Caspase-6 (9761), Caspase-7 (9492), active Caspase-7 (8438), JNK (9252), p-JNK (9255), NF-κB (8242), p-NF-κB (3033), p38 (8690), p-p38 (4631), p-MK2 (3007), MKK3 (8535), MKK6 (8550), p-MKK3/6 (12280), were purchased from Cell Signaling Technologies (Danvers, MA).
TUNEL staining
TUNEL staining was performed as previously described38,39. The apoptotic index was scored in at least five fields at ×400 magnification/mouse and reported as mean ± SD. Three mice were used for each group.
IHC staining
IHC was performed as previously described38,40. Cells with positive staining were scored in at least five fields at ×400 or ×200 magnification and reported as mean ± SD. Three mice were used per group.
Total RNA extraction and real-time reverse transcriptase polymerase chain reaction
Approximately 100 mg of fresh tissue was minced and put into 600 μl lysis buffer (Promega). Total RNA was isolated, and complementary DNA was then generated for real-time PCR analysis as described40. Real-time PCR was performed on a Mini Opticon Real-time PCR system (Bio-Rad) with SYBR Green (Invitrogen) and specific primers19. Melting curves and agarose gel electrophoresis of the PCR products were used to verify the specificity of PCR amplification.
Statistical analysis
Statistical analysis was performed using GraphPad Prism V software. Data are presented as means ± standard deviations (SD). Statistical significance was calculated using the Student’s t-test. P < 0.05 was considered to be significant. The means ± SDs are shown in figures where applicable.
Electronic supplementary material
Acknowledgements
We thank Drs. Mitchell F. Denning, Nancy Zeleznik-Le, Jiwang Zhang and Manuel Diaz (all of Loyola University Chicago) for their helpful discussions and advice. This work is supported in part by AASLD Liver Scholar Award (W.Q.), NIH R03CA195183 (W.Q.), R03CA184652 (W.Q.) and R01CA197128 (W.Q.).
Authors' contributions
N.S. performed experiments, analyzed data and wrote the paper. T.B. bred mice and performed experiments. P.B. wrote the paper. X.Z.D. analyzed histology. J.L. performed statistical analysis. B.M.S. designed experiments, analyzed data and wrote the paper. W.Q. designed and performed experiments, analyzed data and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by E. Baehrecke
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41419-018-0617-7).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Baomin Shi, Phone: +8621 66111783, Email: baomingsph@163.com.
Wei Qiu, Phone: +708 327 8191, Email: wqiu@luc.edu.
References
- 1.Bruix J, Sherman M, Practice Guidelines Committee, A.A.f.t.S.o.L.D. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 2.Johnson PJ. Non-surgical treatment of hepatocellular carcinoma. HPB. 2005;7:50–55. doi: 10.1080/13651820410024076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/S1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 5.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulte-Hermann R, Bursch W, Low-Baselli A, Wagner A, Grasl-Kraupp B. Apoptosis in the liver and its role in hepatocarcinogenesis. Cell. Biol. Toxicol. 1997;13:339–348. doi: 10.1023/A:1007495626864. [DOI] [PubMed] [Google Scholar]
- 8.Farber E. Hepatocyte proliferation in stepwise development of experimental liver cell cancer. Dig. Dis. Sci. 1991;36:973–978. doi: 10.1007/BF01297150. [DOI] [PubMed] [Google Scholar]
- 9.Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch. Toxicol. 2012;87:227–247. doi: 10.1007/s00204-012-0931-2. [DOI] [PubMed] [Google Scholar]
- 10.Weber A, Boege Y, Reisinger F, Heikenwalder M. Chronic liver inflammation and hepatocellular carcinoma: persistence matters. Swiss Med. Wkly. 2011;141:w13197. doi: 10.4414/smw.2011.13197. [DOI] [PubMed] [Google Scholar]
- 11.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwabe RF, et al. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 13.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Invest. 2008;118:3943–3953. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakurai T, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 17.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver. Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 18.Johnson C, et al. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl. Gastrointest. Cancer. 2012;1:58–70. doi: 10.3978/j.issn.2224-4778.2011.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu W, et al. PUMA-mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology. 2011;54:1249–1258. doi: 10.1002/hep.24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 21.Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013;5:1–25. doi: 10.1101/cshperspect.a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feinberg A, Zedeck MS. Production of a highly reactive alkylating agent from the organospecific carcinogen methylazoxymethanol by alcohol dehydrogenase. Cancer Res. 1980;40:4446–4450. [PubMed] [Google Scholar]
- 23.Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 2004;13:1979–1987. doi: 10.1110/ps.04789804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breitwieser W, et al. Feedback regulation of p38 activity via ATF2 is essential for survival of embryonic liver cells. Genes Dev. 2007;21:2069–2082. doi: 10.1101/gad.430207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/S0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 27.Hein H, et al. Genomic organization, sequence, and transcriptional regulation of the human eotaxin gene. Biochem. Biophys. Res. Commun. 1997;237:537–542. doi: 10.1006/bbrc.1997.7169. [DOI] [PubMed] [Google Scholar]
- 28.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 29.Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013;4:342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyoda K, et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003;97:3017–3026. doi: 10.1002/cncr.11425. [DOI] [PubMed] [Google Scholar]
- 31.Kang JS, et al. DBM1285 suppresses tumor necrosis factor alpha production by blocking p38 mitogen-activated protein kinase/mitogen-activated protein kinase-activated protein kinase 2 signaling pathway. J. Pharmacol. Exp. Ther. 2010;334:657–664. doi: 10.1124/jpet.109.161687. [DOI] [PubMed] [Google Scholar]
- 32.Huang H, et al. Expression and prognostic significance of osteopontin and caspase-3 in hepatocellular carcinoma patients after curative resection. Cancer Sci. 2010;101:1314–1319. doi: 10.1111/j.1349-7006.2010.01524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 35.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boland K, Flanagan L, Prehn JH. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 2013;4:e725. doi: 10.1038/cddis.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang N, et al. FAK is required for c-Met/beta-catenin-driven hepatocarcinogenesis. Hepatology. 2015;61:214–226. doi: 10.1002/hep.27402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shang N, et al. FAK kinase activity is required for the progression of c-Met/beta-catenin-driven HCC. Gene Exp. 2016;17:79–88. doi: 10.3727/105221616X691604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arteaga M, et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver. Physiol. 2016;310:G1155–G1168. doi: 10.1152/ajpgi.00271.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.