

Abstract
Background
Hurler syndrome (MPS IH), the severe, neurodegenerative form of type one mucopolysaccharidosis, is associated with rapid neurocognitive decline during toddlerhood and multi-system dysfunction. It is now standardly treated with hematopoietic cell transplantation (HCT), which halts accumulating disease pathology and prevents early death. While norm-based data on developmental functioning in untreated children have previously demonstrated neurocognitive decline, advances in methodology for understanding the cognitive functioning of children with neurodegenerative diseases have highlighted that the previous choice of scores to report results was not ideal. Specifically, the lowest possible norm-based score is 50, which obscures the complete range of cognitive functioning at more advanced stages of neurodeterioration. To a set of cognitive data collected on a sample of untreated children, we applied a modern method of score analysis, calculating a developmental quotient based on age equivalent scores, to reveal the full range of cognitive functioning beneath this cutoff of 50, uncovering new information about the rapidity of decline and the profound impairment in these children.
Results
Among 39 observations for 32 patients with untreated Hurler syndrome, the full array of cognitive functioning below 50 includes many children in the severely to profoundly impaired range. The loss of skills per time unit was 14 points between age 1 and 2. There was a very large range of developmental quotients corresponding to the norm-based cutoff of 50.
Conclusions
This report enables clarification of functioning at levels that extend beneath the floor of 50 in previous work. At the dawn of newborn screening and amidst a proliferation of new therapies for MPS I, these data can provide crucial benchmark information for developing treatments, particularly for areas of the world where transplant may not be available.
Keywords: Mucopolysaccharidosis type I, Developmental quotient, Neurodegenerative disease, Age equivalent, Cognitive decline, Newborn screening, Natural history
Background
Hurler syndrome, the severe, neurodegenerative form of type one mucopolysaccharidosis (MPS I), is now standardly treated with hematopoietic cell transplantation (HCT), which halts accumulating disease pathology and prevents early death. However, the vast majority of affected children are left with less than normal cognitive and adaptive skills [1–4] which interferes with their long-term quality of life. Earlier intervention owing to newborn screening, as well as a surge in innovative treatments, may allow better cognitive and adaptive development [5–7]. Yet comprehensive cognitive information about the untreated natural history is lacking. We have previously published norm-based data on untreated children from our center [1, 8, 9] but advances in methodology for understanding the cognitive functioning of children with MPS IH have highlighted that the previous choice of scores used to report results was not ideal. Specifically, the first and second editions of the Bayley Scales of Infant Development (BSID) [10, 11] that were used with these children had a lowest possible score of 50 (i.e., “floor”) and thus the lowest end of the developmental trajectory was necessarily truncated at that level: It did not reveal the full range of how profoundly impaired children may be, thus obscuring the complete natural history of this disorder [12]. Furthermore, it was not until the results of early HCT were published in 1996 and 1998 [13, 14] that age equivalent scores were first used, and since then, this methodology has become the gold standard for demonstrating cognitive functioning in highly impaired patients [3, 7, 9, 15–19]. However, these valuable data were never published for those children who were not transplanted, such that the literature is lacking detailed information about declining cognitive age equivalent scores and developmental quotients related to age.
This paper presents detailed cognitive information on untreated children with MPS IH, diagnosed before age 3 years. Most of them were part of an NIH study (NS 29099) from 1991 to 1997 exploring the effectiveness of bone marrow transplant (BMT) treatment. Because initial treatment with BMT was only possible with sibling donors, the majority of these children lacked available donors and were not transplanted. That changed when unrelated donors and cord blood (now called hematopoietic cell transplant, HCT, referring to all sources of donor cells) became part of the therapeutic armamentarium, enabling treatment of nearly all patients. Further, emergence of enzyme replacement therapy has enabled treatment, albeit inferior to HCT, for all children with MPS IH, even in parts of the world where there is insufficient access to transplant [20–23]. As such, assessment of completely untreated children is essentially impossible in the modern era; therefore, the current study is not repeatable but offers crucial information about cognitive age equivalents, described comprehensively.
Methods
Participants
Children who were diagnosed with MPS IH, mostly by urinary glycosaminoglycan (GAG) and enzyme determination, were sent to the University of Minnesota for consideration for BMT. This study comprises patients seen between 1985 and 1995. None were genotyped as this was previous to the era where this was standard procedure. All patients were seen by one of the authors (CW and/or WK1) and referred for developmental examination.2 All patients completed a consent form as required by the IRB. All patients in this sample are now deceased.
Most of the patients were untreated due to lack of available donors in the pre-ERT era (Group 1, N = 23), and a smaller proportion eventually proceeded to transplant but died, such that all data gathered represent their untreated status (Group 2, N = 9). Due to likely differences in health status between the two groups, a conservative analytic approach was adopted to provide additional perspective on the variability in this cohort, in which the entire group was analyzed and Group 1 and Group 2 were also examined separately. Two patients from this time period who did not have BMT were eliminated because they were likely patients with attenuated MPS I (Hurler-Scheie syndrome), as they were diagnosed later and were more cognitively intact. Furthermore, as we have no follow-up, we have no indication that either are deceased.
Procedures
Children who were seen prior to 1993 were administered the original BSID [10], and those after 1993 were administered the second edition [11]. While both the Mental and Physical scales of the BSID were administered in most cases, the Mental Scale results are reported here. We report 1) age at testing; 2) year of testing; 3) age equivalent score for the mental scale; 4) developmental quotient (DQ; defined as mental age divided by chronological age times 100 [15]); 5) MDI (standardized score of cognitive development) for comparison to developmental quotient; and 6) age at death. Information regarding vital status was gathered from two sources 1) communication to WK who stayed in contact with many of these patients; and 2) if we did not have the exact date, internet sources such as social security death index.
Results
Findings are summarized by group in Table 1. All patient-level results are presented in Table 2. There were 32 patients who had at least one visit, did not have BMT at the time of visit, and had a BSID I or II. Seven patients had 2 visits; thus a total of 39 observations were made.
Table 1.
Summary data for the full sample and each group separately. Ages are in months unless otherwise indicated
| Age at test | Mental age | DQ | SS/MDI | Age at death (Years) | |
|---|---|---|---|---|---|
| Group 1 (N = 23) | |||||
| Mean | 27.00 | 15.06 | 61.04 | 63.93 | 7.41 |
| SD | 16.60 | 5.76 | 21.89 | 17.55 | 2.81 |
| Median | 22.80 | 16.80 | 58.90 | 56.00 | 6.90 range: 3.3–11.47 |
| Group 2 (N = 9) | |||||
| Mean | 21.41 | 16.75 | 83.33 | 82.00 | 3.38 |
| SD | 10.80 | 5.70 | 12.92 | 15.43 | 2.35 |
| Median | 18.50 | 14.60 | 85.00 | 82.00 | 1.85 range: 1.22–7.71 |
| Total Sample (N = 32) | |||||
| Mean | 25.19 | 15.61 | 68.29 | 69.80 | 6.24 |
| SD | 15.05 | 5.73 | 21.97 | 18.76 | 3.23 |
| Median | 21.40 | 15.70 | 70.30 | 66.50 | 5.85 range: 1.22–11.47 |
Group 1 = Untreated
Group 2 = Eventually proceeded to BMT but died
Age at Test = age at time of neurocognitive testing
Mental Age = BSID Age Equivalent Score
DQ = Per Delaney et al. [15], Mental Age divided by Age at Test, times 100
SS/MDI = norm-based Bayley standard score, where mean = 100, SD = 15, and floor is 50
Table 2.
Raw data for all patients. Ages are presented in months unless otherwise specified
| ID | HCT? | Sex | Year seen | Visit # | Bayley edition | Age at test | Mental age | DQ | SS/MDI | Age at death (Years) |
|---|---|---|---|---|---|---|---|---|---|---|
| H201 | No | female | 1991 | 1 | 1 | 13.8 | 12.5 | 90.4 | 90 | 11.13 |
| 1992 | 2 | 1 | 22.8 | 14.0 | 61.5 | 50 | ||||
| H203 | No | male | 1990 | 1 | 1 | 7.1 | 2.2 | 31.0 | 50 | 8.17 |
| H205 | No | male | 1992 | 1 | 1 | 21.9 | 16.8 | 76.8 | 64 | 8.74 |
| H207 | No | male | 1992 | 1 | 1 | 13.3 | 12.5 | 93.9 | 100 | N.A. |
| H210 | No | male | 1990 | 1 | 1 | 42.7 | 19.0 | 44.5 | 67 | 10.48 |
| H217 | No | female | 1995 | 1 | 2 | 40.4 | 20.6 | 50.9 | 50 | N.A. |
| 1995 | 2 | 2 | 41.1 | 19.7 | 48.0 | 50 | ||||
| H218 | No | male | 1989 | 1 | 1 | 12.8 | 11.3 | 88.5 | 82 | 7.12 |
| H220 | No | male | 1990 | 1 | 1 | 36.8 | 24.4 | 66.4 | 50 | 11.00 |
| H223 | No | male | 1992 | 1 | 1 | 17.3 | 14.2 | 81.9 | 78 | 6.21 |
| H225 | No | female | 1992 | 1 | 1 | 25.7 | 20.0 | 77.7 | 81 | 11.47 |
| H226 | No | male | 1991 | 1 | 1 | 11.0 | 7.1 | 64.5 | 56 | 4.12 |
| 1992 | 2 | 1 | 14.2 | 7.0 | 49.2 | 50 | ||||
| H228 | No | female | 1994 | 1 | 2 | 36.2 | 27.0 | 74.6 | 74 | 13.36 |
| H232 | No | female | 1988 | 1 | 1 | 30.2 | 17.8 | 58.9 | 50 | 6.68 |
| H238 | No | male | 1987 | 1 | 1 | 91.9 | 12.2 | 13.3 | 50 | 8.77 |
| H239 | No | female | 1988 | 1 | 1 | 22.0 | 17.0 | 77.4 | 68 | 5.13 |
| H240 | No | male | 1990 | 1 | 1 | 18.8 | 10.0 | 53.3 | 50 | 4.05 |
| H247 | No | female | 1987 | 1 | 1 | 36.1 | 19.3 | 53.5 | 50 | 5.85 |
| H249 | No | female | 1991 | 1 | 1 | 80.5 | 17.8 | 22.1 | 50 | 4.67 |
| H291 | No | female | 1985 | 1 | 1 | 32.1 | 14.0 | 43.7 | 50 | 4.81 |
| 1985 | 2 | 1 | 35.3 | 17.8 | 50.4 | 50 | ||||
| H328 | No | female | 1994 | 1 | 2 | 16.9 | 17.0 | 100.6 | 103 | 8.97 |
| H341 | No | male | 1994 | 1 | 2 | 16.8 | 7.6 | 45.2 | 56 | 3.30 |
| H357 | No | male | 1995 | 1 | 2 | 24.5 | 20.0 | 81.5 | 94 | N.A. |
| H375 | No | female | 1995 | 1 | 2 | 16.1 | 7.8 | 48.4 | 63 | 8.14 |
| H227 | Yes | female | 1994 | 1 | 2 | 15.8 | 14.6 | 92.4 | 94 | 7.71 |
| 1994 | 2 | 2 | 20.9 | 17.8 | 85.0 | 85 | ||||
| H234 | Yes | male | 1988 | 1 | 1 | 26.8 | 19.3 | 71.9 | 66 | 6.38 |
| H243 | Yes | male | 1989 | 1 | 1 | 14.4 | 12.0 | 83.3 | 82 | 1.22 |
| H244 | Yes | female | 1993 | 1 | 1 | 20.4 | 17.6 | 86.3 | 81 | 1.64 |
| H292 | Yes | male | 1986 | 1 | 1 | 11.0 | 11.8 | 107.3 | 106 | 3.70 |
| H297 | Yes | female | 1989 | 1 | 1 | 15.2 | 14.2 | 93.2 | 98 | 1.83 |
| H299 | Yes | male | 1993 | 1 | 1 | 13.5 | 12.4 | 91.9 | 97 | 1.72 |
| H302 | Yes | female | 1988 | 1 | 1 | 25.6 | 17.6 | 68.7 | 57 | 4.38 |
| 1988 | 2 | 1 | 32.7 | 20.6 | 63.1 | 63 | ||||
| H336 | Yes | male | 1992 | 1 | 1 | 12.9 | 12.2 | 94.4 | 94 | 1.85 |
| 1992 | 2 | 1 | 18.5 | 14.6 | 78.8 | 76 |
Age at Test = age at time of neurocognitive testing
Mental Age = Bayley Age Equivalent Score
DQ = Per Delaney et al. [15], Mental Age divided by Age at Test, times 100
SS/MDI = norm-based Bayley standard score, where mean = 100, SD = 15, and floor is 50
The developmental growth curve in Fig. 1 elucidates the early growth and plateauing in this group, with evidence of very low functioning after three years of age.
Fig. 1.
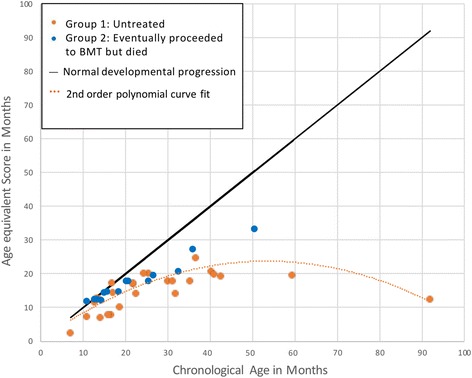
Developmental Growth Curves for Untreated Hurler Syndrome; Seen between 1983 and 1995. Developmental growth curves depict the patients’ mental ages as compared with their chronological ages at the time of testing. At younger chronological ages, most patients are measuring closer to the curve of normal development. With age, their mental functions depart from the normal developmental trajectory, revealing a plateauing in development and eventual decline, illustrated with a second order polynomial curve fit to the data
The mean DQ for patients seen between 1 and 2 years of age was 78.1 (S.D. = 17.7, n = 19); between 2 and 3 years of age was 64.5(13.1, 8); and between 3 and 4 years was 56.3 (19.4, 6). Thus, the change per year respectively was 14 and 8 DQ points. Median DQs were similar (83, 66, and 52 respectively) with a change per year of 17 and 14 points respectively.
For those patients who had an MDI over 50, the correlation with DQ was 0.87. For those who had an MDI of 50 or less (n = 13), the range of DQs was 13.3 to 66.4.
Figure 2 compares the MDI to the developmental quotients for all 32 patients; the slope of cognitive decline is steeper for the DQ than for the MDI for the same patients.
Fig. 2.
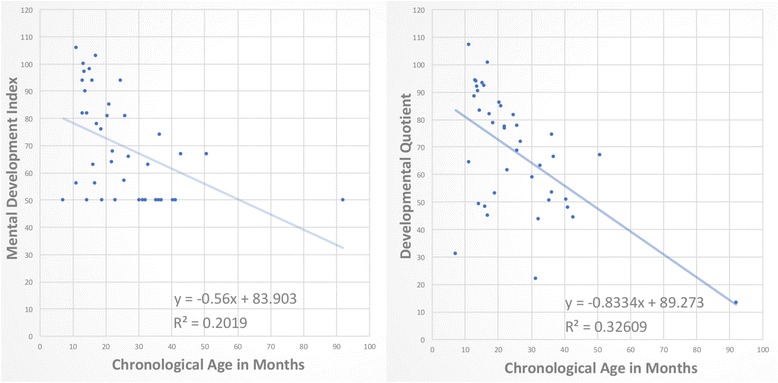
Comparison of slopes of decline using Standard Score versus Developmental Quotient (DQ). When plotted against chronological age, Standard Scores reveal the floor effect, with many data points clustered at a score of 50 (left), which affects calculation of slope of decline. By contrast, use of DQ scores eliminates the floor effect and shows many data points lower than 50 (right). With more comprehensive representation of DQs, the slope of decline is more accurately calculated as steeper
Discussion
This study presents the natural history of children with MPS IH in using modern analytic methods, to provide detailed clarification of the trajectory of neurocognitive decline using age equivalent scores. In the modern era, several works have characterized neurocognitive functioning in MPS IH using age equivalent scores [2, 3], particularly to shed light on treatment outcomes, yet none have quantified the loss in developmental quotient points per year on an untreated population. Previous reports of this decline have used standard scores, which highlights the limitation of past methodology. A graph in Krivit, Peters & Shapiro [8] shows the floor of 50 on the BSID mental scale, and some of these patients overlap with our patients depicted in Fig. 2. Common to both the original graph [8] and the current graph depicting the Mental Development Index (MDI) in Fig. 2, children falling below this floor of 50 could not be sensitively monitored. This contrasts with the present study’s new graph in Fig. 2, depicting the developmental quotient of these same children, where many data points are beneath the floor.
Unlike the historical graph [8], we did not include patients who were transplanted after 1995 or those who survived in our new analyses, in order to focus on the untreated natural history. This cohort comprises patients who lacked available donors (Group 1) as well as patients who eventually proceeded to BMT but died (Group 2), such that all cognitive data represent the untreated natural history. The groups were believed possibly to be different, which is reflected in higher scores in Group 2, for a few reasons. First, for those who did not have an HLA identical sibling, the search for a matched donor may have delayed the assessments, leading to accumulating disease burden, including lower DQ scores and poorer health status in Group 1. Second, the mean age at testing was younger in Group 2, which may indicate less accumulated disease effects. However, the variability is greater in Group 1 than Group 2, while the medians are closer together, which assures some overlap of ages in the two groups. Last, physicians may have been more reluctant to perform a BMT on a patient with more cognitive impairment. As these patients lived in an era when comparatively less was known about MPS IH, much less data on health status, functioning, and treatment planning were routinely collected in standard fashion. Thus, these explanations for differences are speculated based on current knowledge.
This report uncovers the degree of neurodeterioration that extends beneath the floor of 50 in previous work. The full array of cognitive functioning in untreated MPS IH includes many children in the severely to profoundly impaired range, below 50 (33%) with a preponderance of those over 30 months of age (9 of 13). This cohort displayed a loss of skills per time unit (14 points in the 1–2 year olds) similar to that of Sanfilippo syndrome type A, another neurodegenerative type of MPS (i.e., MPS III A) [16, 24] with a decline of 14.6 points per year. Because of the few MPS IH patients over age 4 the decline cannot be as clearly defined beyond that chronological age. Similar to MPS III A patients who do not exceed an age equivalent score much over 30 months [16, 24], the highest age equivalent attained in this sample was 24.4 months.
There is a good correspondence between the developmental quotients and the standard scores in those with an MDI over 50. However, the 13 children with a 50 MDI had a very large range of DQ scores from 13.3 to 66.4, highlighting the arguments of previous work that standard scores do not sensitively reflect the true status of cognitive functioning in neurodegenerative disorders of childhood [15].
Raw scores, i.e., the number of items correct or points earned on the test, would have been another way to present these data; however, raw scores are less meaningful because they differ depending on the edition of the test or the age bracket of the child, and therefore fewer points could simply reflect a young age or different test. Age equivalent scores are the easiest and most appropriate approach to level the artificial differences. Delaney et al. [15] note that in severely impaired children, age equivalent scores enable monitoring of children with cognitive decline to clarify the effects of the disease process prospectively on the gaining of milestones, halting in developmental progress, and losing skills already acquired. A consensus conference on cognitive endpoints in clinical trials in MPS disorders also recommended the use of age equivalent scores to track the inflection points in the developmental curve of developmental arrest and subsequent decline [18]. The sensitivity of this approach has been demonstrated in MPS III [16].
The age of this data set is the most serious limitation. Medical science has progressed and particularly with the use of enzyme replacement [22, 23], survival is much better than it was during the time period that this data set was collected. In addition, this data set may not be fully representative of modern patients with MPS IH. First, cognitive impairment may have been more severe in this data set than currently for several reasons. Specifically, diagnoses were not made as early, as physicians were less informed about rare diseases and diagnoses were often delayed. Further, cognitive impairment can be tied to somatic difficulties which are now better managed [22, 25, 26]. Second, given that these children were all seen before the era of genotyping, the lack of information on the array of MPS IH genotypes represented may limit the generalizability. Third, this data set covers a narrow age range, with few patients over age 42 months, which limits characterization of cognitive status at older ages. It is speculated that accumulating disease burden limited travel, or that families may not have been motivated to pursue additional neurocognitive assessments in the absence of any feasible treatment options. Last, it should be noted that most of these children were seen at the University of Minnesota. Initially it was the first center in the United States to offer transplant as well as to provide opinions regarding its feasibility. Nevertheless, these are the most unalloyed data available on the cognitive course of untreated MPS IH. Historical data offer a unique perspective on the natural history of MPS IH, and may be closer to an authentic demonstration of the raw, untreated course of the disease, than can be obtained with current data on untreated children. That is, current diagnostic (e.g., MRI, SSEPs), interventional (e.g., shunting, antibiotics, hearing aids), supportive (e.g., physical therapy, speech/language) and palliative techniques are now more sophisticated and better tailored to the needs of people with MPS IH, which likely leads to better functioning even in children who do not have access to transplant, than this historical cohort.
The present study aims to increase understanding of the rate of neurocognitive decline in untreated MPS IH, which is needed for comparison when measuring the efficacy of therapies. Due to strong evidence that earlier intervention leads to more favorable outcomes in MPS I [1–3, 6], it was added to the Recommended Uniform Screening Panel (RUSP) for newborn screening. However, it is accepted that currently approved therapies are not perfect, and that people with MPS IH experience significant persisting disease burden [1, 2, 20, 23, 27–29]. This foundation of research and the addition of MPS I to the RUSP have energized the proliferation of new therapies for MPS I, many of which are currently in trial. The loss of DQ points quantified in the present study, and the refined calculation of the slope of decline, shows that neurodeterioration in MPS IH is even steeper and more devastating than previously understood. With the potential for earlier treatment increasing with newborn screening, the present study further argues for expeditious therapeutic intervention, ideally in the first few months of life.
Conclusions
The natural history of MPS IH involves deterioration to profound intellectual impairment, which we have been able to quantify for the first time by using modern analytic methods to revisit data that were previously obscured by a norm-based cutoff of 50. Due to the array of therapies currently available as well as those on the horizon, this study will never be repeatable yet offers crucial enhanced understanding of the untreated course of this disease. Therefore, at the dawn of newborn screening and amidst a proliferation of new therapies for MPS I, these data can provide important benchmark information for developing treatments particularly for areas of the world where transplant may not be available.
Acknowledgements
We are indebted to the courageous families who enabled this research. Pi-Nian Chang, Ph.D., is the Founder and Director Emeritus of Pediatric Psychology, and evaluated some of these children.
Funding
Original data collection supported by NIH grant # NS 29099, 1991–1996. “Value of Bone Marrow Transplant for Storage Diseases.” Analysis supported by NIH U54NS065768. The Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NCATS. This consortium is funded through a collaboration between NCATS, NINDS, and NIDDK.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- DQ
Developmental quotient
- GAG
Glycosaminoglycan
- HCT
Hematopoietic cell transplantation
- MPS I
Mucopolysaccharidosis type I
- MPS IIIA
Mucopolysaccharidosis type IIIA, Sanfilippo syndrome
Authors’ contributions
ES was a co-PI on the original NIH grant # NS 29099, collected all of the original data, conceptualized the present study and re-analysis, composed the first draft and conducted revisions, and approved the final draft; CBW medically evaluated many of the original patients, interpreted data, critically reviewed study design; reviewed and edited drafts of the manuscript and approved the final manuscript as submitted; JBE provided critical review of data analysis and presentation, drafted substantial portions of the original manuscript, interpreted the data, substantially revised multiple drafts of the manuscript, and approved the final manuscript as submitted.
Ethics approval and consent to participate
This study was approved by the University of Minnesota IRB and patients signed a consent form, approved by the IRB, at the time of enrollment.
Competing interests
ES is a Partner in Shapiro Neuropsychology Consulting, LLC; CBW has received consulting fees and research support from ArmaGen, Sangamo, BioMarin and Sanofi Genzyme; JBE has received honoraria, consulting fees, and/or research support from ArmaGen, Regenxbio, Sangamo, Sanofi Genzyme and Shire, and has done contract work for Shapiro Neuropsychology Consulting, LLC.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
William Krivit, MD PhD; Deceased, December 8, 2005. Principal investigator of the NIH grant (#NS 29099) that recruited many of these patients; he and Dr. Whitley screened for and performed the initial bone marrow transplants at the University of Minnesota.
Either by author (ES) or by Pi-Nian Chang, Ph.D., now retired.
Contributor Information
Elsa G. Shapiro, Email: shapi004@umn.edu
Chester B. Whitley, Email: whitley@umn.edu
Julie B. Eisengart, Email: eisen139@umn.edu
References
- 1.Shapiro EG, Nestrasil I, Rudser K, Delaney K, Kovac V, Ahmed A, Yund B, Orchard PJ, Eisengart J, Niklason GR. Neurocognition across the spectrum of mucopolysaccharidosis type I: age, severity, and treatment. Mol Genet Metab. 2015;116:61–68. doi: 10.1016/j.ymgme.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldenhoven M, Wynn RF, Orchard PJ, O’Meara A, Veys P, Fischer A, Valayannopoulos V, Neven B, Rovelli A, Prasad VK. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. doi: 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 3.Poe MD, Chagnon SL, Escolar ML. Early treatment is associated with improved cognition in Hurler syndrome. Ann Neurol. 2014;76:747–753. doi: 10.1002/ana.24246. [DOI] [PubMed] [Google Scholar]
- 4.Kunin-Batson A, Shapiro E, Rudser K, Lavery C, Bjoraker K, Jones S, Wynn R, Vellodi A, Tolar J, Orchard P. Long-term cognitive and functional outcomes in children with Mucopolysaccharidosis (MPS)-IH (Hurler syndrome) treated with hematopoietic cell transplantation J Inherit Metab Dis Rep. 2016;29:1–8. [DOI] [PMC free article] [PubMed]
- 5.Clarke LA, Atherton AM, Burton BK, Day-Salvatore DL, Kaplan P, Leslie ND, Scott CR, Stockton DW, Thomas JA, Muenzer J. Mucopolysaccharidosis type I newborn screening: best practices for diagnosis and management. J Pediatr. 2017;182:363–370. doi: 10.1016/j.jpeds.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 6.Grosse SD, Lam WK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet Med. 2017;19:975–82. [DOI] [PMC free article] [PubMed]
- 7.Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of hurler syndrome. Orphanet J Rare Dis. 2017;12:32. doi: 10.1186/s13023-017-0583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krivit W, Peters C, Shapiro EG. Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Curr Opin Neurol. 1999;12:167–176. doi: 10.1097/00019052-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Janzen D, Delaney K, Shapiro E. Cognitive and adaptive measurement endpoints for clinical trials in mucopolysaccharidoses types I, II, and III: a review of the literature. Mol Genet Metab. 2017;121:57-69. [DOI] [PubMed]
- 10.Bayley N. Manual for the Bayley scales of infant development. New York: Psychological Corporation; 1969.
- 11.Bayley N. Manual for the Bayley scales of infant development (2nd Ed.). San Antonio, TX: Psychological Corporation; 1993.
- 12.Ziegler R, Shapiro E. Metabolic and neurodegenerative diseases across the life span principles and practice of lifespan developmental neuropsychology. New York: Cambridge University Press; 2007. pp. 427–448. [Google Scholar]
- 13.Peters C, Balthazor M, Shapiro E, King R, Kollman C, Hegland J, Henslee-Downey J, Trigg M, Cowan M, Sanders J. Outcome of unrelated donor bone marrow transplantation in 40 children with hurler syndrome. Blood. 1996;87:4894. [PubMed] [Google Scholar]
- 14.Peters C, Shapiro E, Anderson J, Henslee-Downey P, Klemperer M, Cowan M, Saunders E, deAlarcon P, Twist C, Nachman J. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood. 1998;91:2601. [PubMed] [Google Scholar]
- 15.Delaney KA, Rudser KR, Yund BD, Whitley CB, Haslett PA, Shapiro EG. Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome, JIMD reports. 2014;13:129–37. [DOI] [PMC free article] [PubMed]
- 16.Shapiro EG, Nestrasil I, Delaney KA, Rudser K, Kovac V, Nair N, Richard CW, Haslett P, Whitley CB. A prospective natural history study of mucopolysaccharidosis type IIIA. J Pediatr. 2016;170:278–287. doi: 10.1016/j.jpeds.2015.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shapiro E, Bernstein J, Adams HR, Barbier AJ, Buracchio T, Como P, Delaney KA, Eichler F, Goldsmith JC, Hogan M. Neurocognitive clinical outcome assessments for inborn errors of metabolism and other rare conditions. Mol Genet Metab. 2016;118:65–69. doi: 10.1016/j.ymgme.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Lee JH, Morton J, Adams HR, Clarke L, Ebbink BJ, Escolar ML, Giugliani R, Harmatz P, Hogan M, Jones S, Kearney S, Muenzer J, Rust S, Semrud-Clikeman M, Wijburg F, Yu Z, Janzen D, Shapiro E. Cognitive endpoints for therapy development for neuronopathic mucopolysaccharidoses: results of a consensus procedure. Mol Genet Metab. 2017;121:70–79. [DOI] [PubMed]
- 19.Martin HR, Poe MD, Reinhartsen D, Pretzel RE, Roush J, Rosenberg A, Dusing SC, Escolar ML. Methods for assessing neurodevelopment in lysosomal storage diseases and related disorders: a multidisciplinary perspective. Acta Paediatr. 2008;97:69–75. doi: 10.1111/j.1651-2227.2008.00651.x. [DOI] [PubMed] [Google Scholar]
- 20.Dornelles AD, de Camargo Pinto LL, de Paula AC, Steiner CE, Lourenço CM, Kim C, Horovitz DDG, Ribeiro EM, Valadares ER, Goulart I. Enzyme replacement therapy for Mucopolysaccharidosis type I among patients followed within the MPS Brazil network. Genet Mol Biol. 2014;37:23–29. doi: 10.1590/S1415-47572014000100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Silva Franco JF, El Dib R, Agarwal A, Soares D, Milhan NVM, Albano LMJ, Kim CA. Mucopolysaccharidosis type I, II and VI and response to enzyme replacement therapy: results from a single-center case series study. Intractable Rare Dis Res. 2017;6:183–190. doi: 10.5582/irdr.2017.01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisengart JB, Jarnes J, Ahmed A, Nestrasil I, Ziegler R, Delaney K, Shapiro E, Whitley C. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol Genet Metab Rep. 2017;13:64–68. doi: 10.1016/j.ymgmr.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisengart JB, Rudser KD, Xue Y, Orchard P, Miller W, Lund T, Van der Ploeg A, Mercer J, Jones S, Mengel KE, Gökce S, Guffon N, Giugliani R, de Souza CFM, Shapiro EG, Whitley CB. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. 2018. 10.1038/gim.2018.29. [DOI] [PMC free article] [PubMed]
- 24.Buhrman D, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type a. J Inherit Metab Dis. 2014;37:431–437. doi: 10.1007/s10545-013-9661-8. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed A, Rudser K, Kunin-Batson A, Delaney K, Whitley C, Shapiro E. Mucopolysaccharidosis (MPS) physical symptom score: development, reliability, and validity, JIMD reports. 2015;26:61–8. [DOI] [PMC free article] [PubMed]
- 26.Ahmed A, Shapiro E, Rudser K, Kunin-Batson A, King K, Whitley CB. Association of somatic burden of disease with age and neuropsychological measures in attenuated mucopolysaccharidosis types I, II and VI. Mol Genet Metab Rep. 2016;7:27–31. [DOI] [PMC free article] [PubMed]
- 27.Escolar ML, Aldenhoven M. Leukodystrophies and lysosomal storage disorders. In: Boelens JJ, Wynn R, editors. Stem cell biology and regenerative medicine. New York: Springer; 2013. pp. 63–125. [Google Scholar]
- 28.Polgreen L, Tolar J, Plog M, Himes J, Orchard P, Whitley C, Miller B, Petryk A. Growth and endocrine function in patients with hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008;41:1005–1011. doi: 10.1038/bmt.2008.20. [DOI] [PubMed] [Google Scholar]
- 29.Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183–1197. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.