

Abstract
Background
Hepatocellular carcinoma (HCC) is the most common malignancy of the liver and recent studies have revealed that circular RNA (circRNA) plays an important role in the pathogenesis of HCC. Some circRNAs may act as a microRNA (miRNA) sponge to affect miRNA activities in the regulation of messenger RNA (mRNA) expression. However, the circRNA-miRNA-mRNA network in HCC remains largely unknown.
Material/Methods
The circRNA expression profiles (GSE94508 and GSE97332), miRNA and mRNA expression profile (GSE22058) were downloaded from Gene Expression Omnibus microarray data and then a circRNA-miRNA-mRNA regulatory network in HCC was constructed. Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed (DE) genes were performed. The functional circRNA-miRNA-mRNA regulatory modules were constructed using cytoHubba plugin based on Cytoscape and KEGG enrichment analysis.
Results
The network contained 60 circRNA-miRNA pairs and 4982 miRNA-mRNA pairs, including 29 circRNAs, 16 miRNAs, and 1249 mRNAs. GO and KEGG pathway analysis revealed the network might be involved in the procession of carcinogenesis such as cell proliferation, cell cycle, and p53 signaling pathway. In addition, 3 top ranked circRNAs (hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913) related networks were identified to be highly correlated with the pathogenesis of HCC. Furthermore, the functional circRNA-miRNA-mRNA regulatory modules were constructed based on the 3 top-ranked circRNAs and those DE genes enriched in carcinogenesis related pathways.
Conclusions
This study suggests that a specific circRNA-miRNA-mRNA network is associated with the carcinogenesis of HCC, which might aid in the identification of molecular biomarkers and therapeutic targets for HCC.
MeSH Keywords: Biological Markers; Carcinoma, Hepatocellular; Computational Biology; RNA, Untranslated
Background
Hepatocellular carcinoma (HCC) is the most common types of hepatic malignancies and has become one of the leading causes of cancer-related deaths world-wide [1]. With the advancements in treatment modalities, the therapeutic efficacy of HCC has gradually improved. However, high frequency of metastasis and recurrence remains the main problem for clinical practices [2]. Thus, there is an urgent need to better understand the molecular mechanism of HCC development and to identify potential biomarkers and targets for diagnosis and therapy.
Circular RNA (circRNA) is a novel type of endogenous noncoding RNA that is expressed widely in different species and has been demonstrated to be a class of RNA with tissue/developmental stage-specificity, which makes circRNA a potential diagnostic and prognostic biomarkers [3]. Recently, hsa_circ_0004018 was found to harbor HCC-stage-specific expression features in diverse chronic liver diseases [4]. Another study reported that hsa_circ_0001649 expression was significantly downregulated in HCC tissues and correlated with tumor size and occurrence of tumor embolus [5]. In addition, the expression of circZKSCAN1 (hsa_circ_0001727) was found to be significantly lower in HCC and associated with the tumor number, cirrhosis, and vascular invasion [6]. These findings indicate that circRNA might function as a novel potential biomarker for HCC and might be associated with tumorigenesis and progression of HCC. Due to the covalently closed loop structures without the polyadenylated tail, circRNAs show more stable than traditional linear RNAs when exposed to RNase R and RNA exonuclease, which facilitates circRNAs to exert their regulatory function at the transcriptional or post-transcriptional level [7]. Increasing research has revealed that circRNA participates in a variety of biological processes by acting as microRNA (miRNA) sponges, thereby relieving miRNA-mediated target repression. For instance, circMTO1 (hsa_circ_0007874) serves as a sponge of oncogenic miR-9 to promote p21 expression, resulting in the suppression of HCC cell proliferation and invasion [8]. Another report showed that cerebellar-degeneration-related protein 1 antisense RNA (CDR1as), an oncogenic circRNA, may exert its effects on cell proliferation via targeting miR-7 to regulate the expression of EGFR, CCNE1, and PIK3CD in HCC cells [9,10]. Notably, circRNA CDR1as is viewed as the typical miRNA sponge, and even harbors 74 binding sites for miR-7 [11]. Although several circRNAs have been identified as participating in the pathogenesis of HCC, it is still necessary to conduct a more comprehensive analysis of circRNA-miRNA-mRNA regulatory network in HCC, which may help to advance our understanding of the molecular mechanism underlying the development and progression of HCC.
In the present study, we performed an integrated analysis of circRNAs, miRNAs, and messenger RNAs (mRNAs) expression profiles of HCC from the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO). The flowchart for this procedure is shown in Figure 1. Combining the identified differentially expressed (DE) RNAs, the circRNA-miRNA-mRNA regulatory network was constructed. Furthermore, analysis of the regulatory network identified some important circRNAs, miRNAs, and mRNAs and their corresponding regulatory networks that were highly related to the progression of HCC. In an attempt to better understand the pathogenesis of HCC, the functional circRNA-miRNA-mRNA regulatory modules were also constructed.
Figure 1.
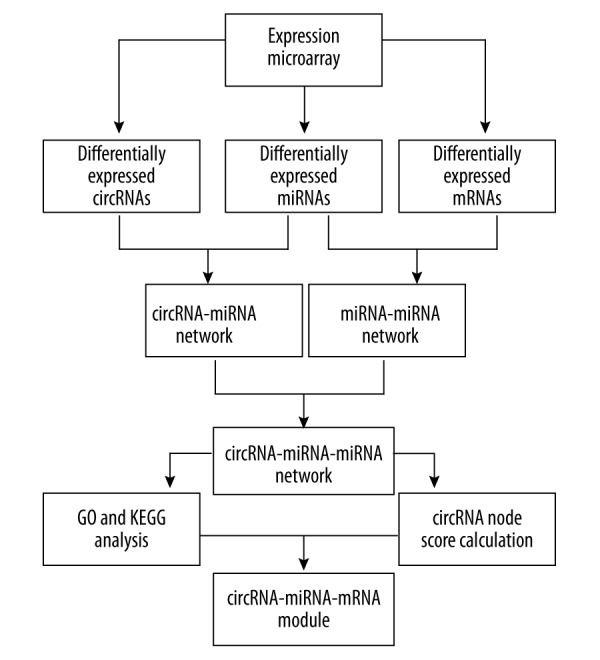
Flow-chart of data analysis. First, we extracted the differential expressed circRNAs, miRNAs, and mRNA in HCC from GEO microarray data. Then the circRNA-miRNA and miRNA-mRNA network were constructed according to Circular RNA Interactome and Targetscan database, respectively. Combining these 2 networks, a circRNA-miRNA-mRNA regulatory network were constructed. Based on the results of GO and KEGG analysis and the node scores of circRNAs, we further constructed the circRNA-miRNA-mRNA regulatory modules. CircRNA – circular RNA; miRNA – microRNA; mRNA – messenger RNA; GEO – Gene Expression Omnibus; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Material and Methods
Data collection
The microarray data used in this study were retrieved from GEO database (http://www.ncbi.nlm.nih.gov/gds/). We only selected the original experimental microarray studies that analyzed circRNA, miRNA, and mRNA expression profiling between HCC and matched non-tumor tissues in human. The circRNA expression data were obtained from GSE94508 (5 pairs of HCC and paracancerous liver tissues) and GSE97332 (7 pairs of HCC and matched non-tumor liver tissues). The miRNA and mRNA expression data were derived from GSE22058, including 96 pairs of HCC and adjacent non-tumor liver tissues.
Differential expression analysis of circRNAs, miRNAs and mRNAs
The raw data of microarray datasets were preprocessed via background correction and normalization. We used the Bioconductor Limma package to identify DE RNAs for each dataset. The adjusted P value (false discovery rate, FDR) of each gene was calculated using the Benjamini Hochberg method. Additionally, robust rank aggregation method was used to calculate the fold change (FC) and significance score (P value) of all DE circRNAs from multiple studies [12]. The DE circRNAs were selected according to the significance score <0.01 and |log2FC| >1. Because the |log2FC| of most miRNAs is less than 1, we set FDR <0.01 for the identification of differential miRNA expression. Moreover, DE mRNAs were selected with the cutoff criteria of FDR <0.01 and |log2FC| >1.
Construction of the circRNA-miRNA-mRNA regulatory network
According to the results of the differential expression analysis, the regulatory relationships between circRNAs and miRNAs were predicted using Circular RNA Interactome (https://circinteractome.nia.nih.gov/). As the sponge of miRNAs, the expression level of circRNAs usually do not affect the expression of miRNAs. Thus, we selected the circRNA-miRNA pairs without the constraint that the expression patterns between circRNAs and miRNAs must be different. In addition, we extracted significant DE miRNAs and DE mRNAs to construct the miRNA-mRNA regulatory network. Among the DE mRNAs, the potential targets of DE miRNAs were predicted by TargetScan database (http://www.targetscan.org). Those miRNA–mRNA pairs with inverse expression relationships were included for network construction. The circRNA-miRNA-mRNA regulatory network was constructed using a combination of circRNA-miRNA pairs and miRNA-mRNA pairs. Finally, the regulatory network was visualized using Cytoscape 3.4.0 (http://cytoscape.org/).
Functional enrichment analysis
Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DE mRNAs were carried out by Database for Annotation, Visualization and Integrated Discovery (DAVID; http://www.david.abcc.ncifcrf.gov/). A GO term or KEGG pathway with FDR <0.05 was considered statistically significant. The top 10 enriched GO terms and pathways of DE mRNAs were ranked by enrichment score (−log10 (P value)).
Generation of functional circRNA-miRNA-mRNA regulatory modules
In order to detect the functional circRNA-miRNA-mRNA regulatory modules of HCC, the degree, betweenness centrality, and closeness centrality of circRNAs were calculated using cytoHubba plugin based on Cytoscape 3.4.0 [13]. The overlapped top-ranked circRNAs among 3 calculation methods were extracted. The functional enrichment analysis of the DE mRNAs involved in the overlapped top-ranked circRNAs related network were performed by DAVID. We then created functional modules based on the overlapped top-ranked circRNAs related regulatory network and enriched KEGG pathways. Then, visualizations of functional modules were performed using Cytoscape 3.4.0.
Results
DE circRNAs, miRNAs, and mRNAs in HCC
The integrated analysis of GSE94508 and GSE97332 identified 50 DE circRNAs, in which 31 circRNAs were found to be upregulated and 19 circRNAs were downregulated in HCC. Based on the dataset GSE22058, 134 DE miRNAs (76 upregulated and 58 downregulated miRNAs) and 1592 DE mRNAs (590 upregulated and 1002 downregulated mRNAs) were obtained in HCC.
CircRNA-miRNA-mRNA network in HCC
According to the differentially expressed results, circRNA-miRNA pairs and miRNA-mRNA pairs were predicted by Circular RNA Interactome and Targetscan database, respectively. Then, circRNA-miRNA-mRNA network was generated using a combination of data obtained from circRNA-miRNA pairs and miRNA-mRNA pairs. This network contained 60 circRNA-miRNA pairs and 4982 miRNA-mRNA pairs, including 29 circRNAs, 16 miRNAs, and 1249 mRNAs (Figure 2). Next, all of the DE mRNAs were performed to analyze their potential function. GO analysis revealed that there were 596 enriched GO terms with the statistical significance in the „Biological Process (BP)”. The top10 enriched BP terms are shown in Figure 3A, among which some genes were involved in cell proliferation and regulation of cell proliferation. KEGG analysis showed the top 10 pathways associated with DE mRNAs, including cell cycle, PPAR signaling pathway, chemical carcinogenesis, and p53 signaling pathway (Figure 3B).
Figure 2.

The circRNA-miRNA-mRNA regulatory network. CircRNA, miRNA, and mRNA were indicated to deformed triangle, diamond, and circle, respectively. The red color represents high expression and blue color represents low expression. CircRNA – circular RNA; miRNA – microRNA; mRNA – messenger RNA.
Figure 3.

Functional analysis of differentially expressed mRNAs. The top 10 significant enriched biological process terms by GO analysis (A). The top 10 significant enriched pathways by KEGG pathway analysis (B). GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Subnetwork analysis of circRNA
It is well acknowledged that hub nodes play important roles in biological networks. Based on the results of degree calculation using cytoHubba plugin of Cytoscape, 5 circRNAs were identified to have relative high degrees in the regulatory network, including hsa_circ_0078279 (degree=5), hsa_circ_0007456 (degree=4), hsa_circ_0004913 (degree=4), hsa_circ_0000098 (degree=4), and hsa_circ_0004018 (degree=4). The circRNA-miRNA-mRNA network of hsa_circ_0078279 is shown in Figure 4A. This subnetwork was composed of 5 circRNA-miRNA pairs and 1567 miRNA-mRNA pairs, including 1 circRNA, 5 miRNAs, and 934 mRNAs. GO analysis showed that genes in the subnetwork of hsa_circ_0078279 were also involved in cell proliferation and regulation of cell proliferation in the top 10 enriched BP term (Figure 4B). Moreover, PPAR signaling pathway and chemical carcinogenesis were also shown in the top 10 enriched KEGG pathways for the genes in the subnetwork of hsa_circ_0078279 (Figure 4C).
Figure 4.

The subnetwork of hsa_circ_0078279 and functional analysis. The hsa_circ_0078279-miRNA-mRNA regulatory network (A). The top 10 significant enriched biological process terms by GO analysis (B). The top 10 significant enriched pathways by KEGG pathway analysis (C). CircRNA, miRNA, and mRNA were indicated to deformed triangle, diamond, and circle, respectively. The red color represents high expression and blue color represents low expression. GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes; circRNA – circular RNA; miRNA – microRNA; mRNA – messenger RNA.
Functional circRNA-miRNA-mRNA regulatory modules
In order to specify the key circRNA-miRNA-mRNA regulatory modules in the process of HCC carcinogenesis, the degree, betweenness centrality, and closeness centrality of all circRNAs were calculated using cytoHubba plugin based on Cytoscape. The results of top 5 circRNAs calculated by 3 methods are listed in Table 1. Interestingly, 3 circRNAs, namely hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913, ranked top among all 3 calculation methods. This suggests that the 3 circRNAs may play crucial roles in the carcinogenesis and development of HCC, and could be considered as the key circRNAs. Thus, we used the 3 circRNAs to construct the circRNA-miRNA-mRNA regulatory subnetwork. This subnetwork was composed of 13 circRNA-miRNA pairs and 3053 miRNA-mRNA pairs, including 3 circRNAs, 10 miRNAs, and 1138 mRNAs (Figure 5A). GO and KEGG analysis were performed to evaluate the function of the DE mRNAs in the subnetwork. As shown in Figure 5B, the results of the top 10 enriched BP terms showed that the DE mRNAs were associated with cell proliferation, cell adhesion, and metabolic process. The top 10 KEGG pathways showed the DE mRNAs might be involved in cell cycle, PPAR signaling pathway, and p53 signaling pathway and that these 3 pathways were also indicated in the top 10 significant KEGG pathways of all DE mRNAs in the whole network (Figure 5C). Thus, hsa_circ_0078279, hsa_circ_0007456 and hsa_circ_0004913, and those DE mRNAs enriched in the aforementioned 3 pathways of the subnetwork were selected to generate the functional circRNA-miRNA-mRNA regulatory modules. As shown in Figure 6, the functional modules contained 13 circRNA-miRNA pairs and 119 miRNA-mRNA pairs, including 3 circRNAs, 10 miRNAs, and 47mRNAs. In particular, 3 circRNAs all harbored the binding sites for hsa-mir-182. Both hsa_circ_0004913 and hsa_circ_0007456 could act as the sponge for hsa-mir-346.
Table 1.
The node scores of circRNAs calculated by cytoHubba plugin based on Cytoscape.
| Name | Degree | Name | Betweenness | Name | Closeness |
|---|---|---|---|---|---|
| hsa_circ_0078279 | 5 | hsa_circ_0078279 | 566.42 | hsa_circ_0078279 | 165324.29 |
| hsa_circ_0007456 | 4 | hsa_circ_0007456 | 558.75 | hsa_circ_0007456 | 150036.96 |
| hsa_circ_0004913 | 4 | hsa_circ_0004913 | 543.25 | hsa_circ_0004913 | 103999.27 |
| hsa_circ_0000098 | 4 | hsa_circ_0000098 | 521.00 | hsa_circ_0004720 | 87057.26 |
| hsa_circ_0004018 | 4 | hsa_circ_0004720 | 514.33 | hsa_circ_0003570 | 59501.01 |
Figure 5.

The subnetwork of hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913 and functional analysis. The hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913 regulatory subnetwork (A). The top 10 significant enriched biological process terms by GO analysis (B). The top 10 significant enriched pathways by KEGG pathway analysis (C). CircRNA, miRNA and mRNA were indicated to deformed triangle, diamond, and circle, respectively. The red color represents high expression and blue color represents low expression. GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes; circRNA – circular RNA; miRNA – microRNA; mRNA – messenger RNA.
Figure 6.
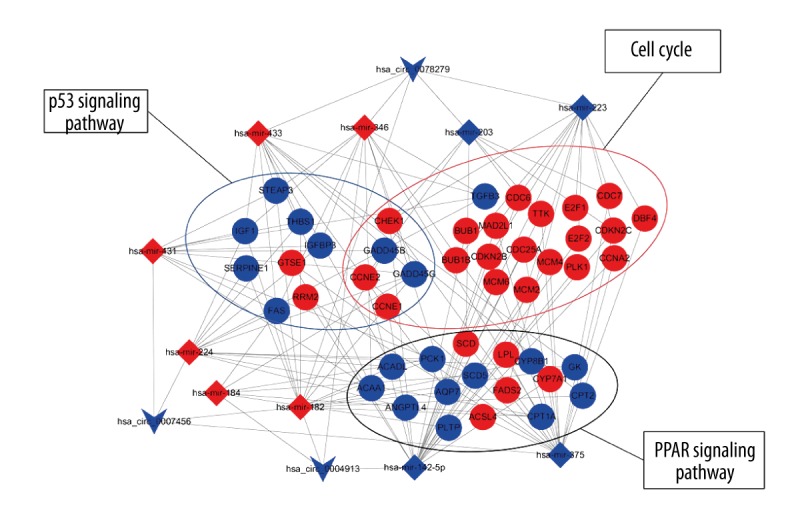
Functional circRNA-miRNA-mRNA regulatory modules. CircRNA, miRNA, and mRNA were indicated to deformed triangle, diamond, and circle, respectively. The red color represents high expression and blue color represents low expression. CircRNA – circular RNA; miRNA – microRNA; mRNA – messenger RNA.
Discussion
In this study, we first systematically analyze the regulatory network of circRNAs, miRNAs, and mRNAs in HCC. Functional analysis by GO and KEGG enrichment analysis reveal the potential role of noncoding RNAs and coding RNAs in the carcinogenesis and development of HCC. Based on the results of the KEGG pathway analysis and the node scores of circRNA, we constructed the circRNA-miRNA-mRNA regulatory modules containing 3 circRNAs, 10 miRNAs, and 47 mRNAs, in an attempt to better understand the pathogenesis of HCC.
Accumulating evidence has revealed that circRNAs could act as miRNA sponges to counteract miRNA mediated repression of mRNA [14]. Lastly, the regulatory axes of circRNAs, miRNAs, and mRNAs are gradually demonstrated in different kinds of diseases [15,16]. In this study, we generated a global triple network according to predicted circRNA-miRNA and miRNA-mRNA pairs. The circRNA-miRNA-mRNA network was composed of 29 circRNAs, 16 miRNAs, and 1249 mRNAs. In this network, the circRNAs mainly bound to hsa-mir-346, hsa-mir-203, and hsa-mir-182. Additionally, hsa-mir-182 was the largest node that interacted with a considerable number of coding and noncoding RNAs in the network, which suggested that circRNA-hsa-mir-182 mediated network might play a critical role in the pathogenesis of HCC. The significantly enriched GO terms for the DE mRNAs are highly related to the carcinogenesis, such as cell proliferation, cell adhesion and cell migration, which are consistent with the previous bioinformatics analysis of key genes in HCC [17]. Pathway analysis also revealed that cell cycle, PPAR signaling pathway, chemical carcinogenesis, and p53 signaling pathway are significant enriched. And the aforementioned pathways have been shown to play pivotal roles in the development of HCC [18–21].
Generally, a circRNA harbors more binding sites for different miRNAs, indicating that it owns more potentiality to be involved in the regulation of miRNA target gene expression. In order to identify the key circRNAs participating in the regulatory network, the degree, betweenness centrality, and closeness centrality of all circRNAs were calculated using cytoHubba plugin based on Cytoscape. The results showed that hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913 were the 3 top-ranked circRNAs among 3 calculation methods. It suggests that the 3 circRNAs with the largest node scores may play important roles in the carcinogenesis and development of HCC. Although the 3 circRNAs have not been investigated in HCC, further regulatory network construction and functional analysis revealed that 3 circRNAs related network might be highly associated with the pathogenesis of HCC. Particularly, KEGG pathway analysis showed that the 3 circRNAs regulatory network was involved in the cell cycle, PPAR signaling pathway, and p53 signaling pathway, which have been commonly reported in the carcinogenesis of HCC. Notably, these pathways of the 3 circRNAs related subnetwork are also indicated in the top 10 significant enriched KEGG pathways of all DE mRNAs in the whole circRNA-miRNA-mRNA network. Actually, compared with the 1249 DE mRNAs involved in the whole circRNA regulatory network, most of DE mRNAs (1138) were included in the 3 circRNA related subnetwork, which suggests that the 3 circRNAs related subnetwork have profound effect on HCC development. Although KEGG pathway analysis showed that several pathways, such as Prion diseases, Malaria, and Amoebiasis, might be independent of HCC, some DE mRNAs enriched in those pathways were correlated with the carcinogenesis of HCC. For instance, both IL1B and IL6 are enriched in Malaria, Prion diseases, Amoebiasis. Moreover, TLR2, TLR4, and TGFB3 are enriched in Malaria and Amoebiasis. It was known that the activation of TLR signaling leads to the release of inflammatory factors, such as IL1B and IL6, which are involved in the process of hepatic inflammation [22,23]. And the sustained hepatic inflammatory status contributes to the pathogenesis of HCC [24]. In this study, we selected the 3 key circRNAs in combination with DE mRNAs involved in cell cycle, PPAR signaling pathway, and p53 signaling pathway to reconstruct the functional circRNA-miRNA-mRNA regulatory modules. In the regulatory modules, 3 circRNAs could interact with 10 DE miRNAs, including 6 upregulated miRNAs (hsa-mir-182, hsa-mir-184, hsa-mir-224, hsa-mir-431, hsa-mir-433, and hsa-mir-346) and 4 downregulated miRNAs (hsa-mir-203, hsa-mir-223, hsa-mir-375, and hsa-mir-142-5p). Particularly, hsa-mir-182 could interact with 3 circRNAs and hsa-mir-346 could bind to hsa_circ_0004913 and hsa_circ_0007456. This indicated that these circRNAs may competitively bind to these 2 miRNAs and thereby affect their respective regulatory networks. Several miRNAs have been demonstrated to be involved in the pathogenesis of HCC [25]. For instance, upregulated miR-182 significantly increases the viability of HCC cells by inhibiting the expression of tumor suppressor gene tumor protein 53-induced nuclear protein 1 [26]. A previous study showed that miR-184 was upregulated in HCC tissues, and overexpression of miR-184 in HCC cells increased cell cycle progression and cell proliferation [27]. It was also reported that miR-224 induced G1/S checkpoint release and increased the growth rate of HCC cells by targeting p21, p15, and CCNE1 [28]. In contrast, some miRNAs, such as miR-203 and miR-375, are considered as tumor-suppressive miRNAs in HCC [29]. These miRNAs may also act as regulatory factors and play pivotal roles in the regulatory network.
In future studies, we will collect HCC specimens for verification and try to assess the relationship between these 3 circRNAs and clinical characteristics, such as age, sex, serum alpha-fetoprotein level, TNM staging, and prognosis. After screening of circRNA with clinical diagnostic and prognostic predictive value, we will explore the function of circRNA by in vitro and in vivo experiments.
Conclusions
In summary, we conducted an integrated analysis of differentially expressed circRNAs, miRNAs, and mRNAs in HCC and constructed a circRNA-miRNA-mRNA regulatory network. The potentially functional circRNA-miRNA-mRNA regulatory modules generated in the present study uncovered 3 important circRNAs (hsa_circ_0078279, hsa_circ_0007456, and hsa_circ_0004913) that might be involved in the carcinogenesis related pathways, which provides new insight for mechanistic investigation and may offer potential therapeutic targets for HCC. Future studies are needed to validate the sponge effect of these 3 specific circRNAs and to better understand the role of these regulatory modules in the carcinogenesis of HCC.
Acknowledgements
We thank Dr. Genwen Chen for the technical advice.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Lee JI, Kim JK, Kim DY, et al. Prognosis of hepatocellular carcinoma patients with extrahepatic metastasis and the controllability of intrahepatic lesions. Clin Exp Metastasis. 2014;31:475–82. doi: 10.1007/s10585-014-9641-x. [DOI] [PubMed] [Google Scholar]
- 3.Rybak-Wolf A, Stottmeister C, Glazar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 4.Fu L, Yao T, Chen Q, et al. Screening differential circular RNA expression profiles reveals hsa_circ_0004018 is associated with hepatocellular carcinoma. Oncotarget. 2017;8:58405–16. doi: 10.18632/oncotarget.16881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin M, Liu G, Huo X, et al. Hsa_circ_0001649: A circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016;16:161–69. doi: 10.3233/CBM-150552. [DOI] [PubMed] [Google Scholar]
- 6.Yao Z, Luo J, Hu K, et al. ZKSCAN1 gene and its related circular RNA (circZKSCAN1) both inhibit hepatocellular carcinoma cell growth, migration, and invasion but through different signaling pathways. Mol Oncol. 2017;11:422–37. doi: 10.1002/1878-0261.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki H, Tsukahara T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci. 2014;15:9331–42. doi: 10.3390/ijms15069331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology (Baltimore, Md) 2017;66:1151–64. doi: 10.1002/hep.29270. [DOI] [PubMed] [Google Scholar]
- 9.Yang X, Xiong Q, Wu Y, et al. Quantitative proteomics reveals the regulatory networks of circular RNA CDR1as in hepatocellular carcinoma cells. J Proteome Res. 2017;16:3891–902. doi: 10.1021/acs.jproteome.7b00519. [DOI] [PubMed] [Google Scholar]
- 10.Yu L, Gong X, Sun L, et al. The Circular RNA Cdr1as act as an oncogene in hepatocellular carcinoma through targeting miR-7 expression. PLoS One. 2016;11:e0158347. doi: 10.1371/journal.pone.0158347. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–38. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 12.Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics (Oxford, England) 2012;28:573–80. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin CH, Chen SH, Wu HH, et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–88. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 15.Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Yuan B, Wu Z, et al. Microarray profiling of circular RNAs and the potential regulatory role of hsa_circ_0071410 in the activated human hepatic stellate cell induced by irradiation. Gene. 2017;629:35–42. doi: 10.1016/j.gene.2017.07.078. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Peng L, Zhang Y, et al. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med Oncol. 2017;34:101. doi: 10.1007/s12032-017-0963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dituri F, Mazzocca A, Lupo L, et al. PI3K class IB controls the cell cycle checkpoint promoting cell proliferation in hepatocellular carcinoma. Int J Cancer. 2012;130:2505–13. doi: 10.1002/ijc.26319. [DOI] [PubMed] [Google Scholar]
- 19.Cao LQ, Shao ZL, Liang HH, et al. Activation of peroxisome proliferator-activated receptor-gamma (PPARgamma) inhibits hepatoma cell growth via downregulation of SEPT2 expression. Cancer Lett. 2015;359:127–35. doi: 10.1016/j.canlet.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Fukuda T, Isaji T, et al. Loss of alpha1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 2015;29:3217–27. doi: 10.1096/fj.15-270710. [DOI] [PubMed] [Google Scholar]
- 21.Hao PP, Li H, Lee MJ, et al. Disruption of a regulatory loop between DUSP1 and p53 contributes to hepatocellular carcinoma development and progression. J Hepatol. 2015;62:1278–86. doi: 10.1016/j.jhep.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 22.Liu QS, Cheng ZW, Xiong JG, et al. Erythropoietin pretreatment exerts anti-inflammatory effects in hepatic ischemia/reperfusion-injured rats via suppression of the TLR2/NF-kappaB pathway. Transplant Proc. 2015;47:283–89. doi: 10.1016/j.transproceed.2014.10.045. [DOI] [PubMed] [Google Scholar]
- 23.Inokuchi S, Tsukamoto H, Park E, et al. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol Clin Exp Res. 2011;35:1509–18. doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan HX, Wu HP, Zhang HL, et al. p53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J Hepatol. 2013;59:762–68. doi: 10.1016/j.jhep.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Chen J, Liu Y, et al. Plasma miR-15b-5p, miR-338-5p, and miR-764 as biomarkers for hepatocellular carcinoma. Med Sci Monit. 2015;21:1864–71. doi: 10.12659/MSM.893082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin J, Luo M, Qian H, Chen W. Upregulated miR-182 increases drug resistance in cisplatin-treated HCC cell by regulating TP53INP1. Gene. 2014;538:342–47. doi: 10.1016/j.gene.2013.12.043. [DOI] [PubMed] [Google Scholar]
- 27.Wu GG, Li WH, He WG, et al. Mir-184 post-transcriptionally regulates SOX7 expression and promotes cell proliferation in human hepatocellular carcinoma. PLoS One. 2014;9:e88796. doi: 10.1371/journal.pone.0088796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An F, Olaru AV, Mezey E, et al. MicroRNA-224 Induces G1/S checkpoint release in liver cancer. J Clin Med. 2015;4:1713–28. doi: 10.3390/jcm4091713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuta M, Kozaki KI, Tanaka S, et al. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–76. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]