

Abstract
Heat shock protein (Hsp) 90 is a key component of the super-chaperone complex that maintains functionally active conformation of various client proteins. Many of these client proteins regulate important nodal points in multiple signalling pathways that promote cancer cell growth and survival. Inhibitors of Hsp90, therefore, have the potential of functioning as anti-cancer agents with pleiotropic effects. Identification of novel Hsp90 inhibitors with more favourable pharmacological properties is a priority in cancer therapy. To achieve this goal, we screened a compound library using a biochemical assay based on refolding of denatured firefly luciferase. The assay revealed high sensitivity, reliability and reproducibility with a Z-factor of 0.81 ± 0.17. Six Hsp90 inhibitory compounds identified by this screening with IC50 values between 1.0 and 6 μM were further characterised for anti-proliferative activity by Cell Titer-Blue Cell Viability Assay using multiple tumour cell lines. Of particular interest was ONO4140 with lowest GI50 values in three different cancer cell lines viz; DU-145, BT-474 and K562 cell lines. This study also revealed that short-term exposure of tumour cells with ONO4140 is sufficient to inhibit the catalytic activity of Hsp90, evaluated through disruption of Hsp90-p23 association by immunoprecipitation. This short term exposure appears to initiate events like degradation of Hsp90 client proteins such as ErbB2/Her-2 and Akt with concomitant inhibition of survival signalling leading to the apoptotic death of tumour cells as seen by western blotting and Caspase Glow-3,7 assay. The study also reveals that apoptosis following Hsp90 inhibition with ONO4140 occurs via Caspase9–Caspase3 intrinsic apoptotic pathway, a process that is likely triggered by inactivation of Akt. In conclusion, we have identified a novel class of synthetic compounds which show potent Hsp90 inhibitory action in preclinical studies. The discovery of this novel class of synthetic Hsp90 inhibitors with simple chemical backbone allows us to conduct further structural modifications to improve their potency and pharmacokinetic properties for use in cancer therapy.
Keywords: Hsp90 inhibitors, Heat shock factor 1, Rabbit reticulolysate, Firefly luciferase, Renaturation
1. Introduction
Molecular chaperones such as heat shock protein (Hsp) 90 have multiple functions, the most important being their ability to mediate protein holding and protein folding activities. While hsp90 is required by all cells, tumour cells especially require Hsp90 in higher levels as they are ‘oncogene addicted’ and therefore play a critical role in the maintenance of malignant phenotype by facilitating folded and functionally active conformation of several mutant and aberrant oncoproteins. Unlike conventional anticancer drug targets, Hsp90 is an especially promising target for anticancer drugs as many of its client proteins regulate nodal points of multiple signalling pathways that govern cell growth, cell survival, angiogenesis and metastasis [1–3]. There are over 200 known Hsp90 client proteins (for an updated list see: htpp://www.picard.ch/downloads/HSP90interactors [4]). These include many kinases such as ERBB2, EGFR, CDK4, CRAF, BRAF, AKT, MET and BCR-ABL, as well as transcription factors such as oestrogen and androgen receptors, HIF-1 and p53; MMP2 and chromatin remodelling proteins like histone deacteylases (HDACs) and SMYD3. Inhibition of HSP90, therefore, leads to simultaneous combinatorial depletion (via the ubiquitin–proteasome pathway) of a wide range of its client proteins thereby providing a mechanism for the simultaneous derailment of multiple signalling pathways that favour transformation [5–7]. Furthermore tumour cells are especially sensitive to hsp90 inhibitors as they are present in high levels in tumour cells compared to normal cells.
Hsp90 is made up of three functional domains: an NH2-terminal Adenosine triphosphate or adenosine diphosphate (ATP/ADP) binding domain, a middle domain involved in client protein binding and a COOH-terminal dimerisation domain [8–10]. In vivo, Hsp90 exists as a homodimer and the chaperone activity of Hsp90 depends on an orchestrated series of conformational changes and interactions with client proteins and accessory proteins or co-chaperones, operating in a cycle that is driven by nucleotide binding and ATP hydrolysis [10]. The complex structure and mechanism of action of the Hsp90 super-chaperone complex suggest numerous alternative opportunities for pharmacological intervention.
A number of natural products have been found to exhibit Hsp90 inhibitory activity which includes the ansamycin antibiotic, Geldanamycin (GA) [9,11,12], the macrocyclic antifungal antibiotic, Radicicol [13] and the amino-coumarin containing antibiotic, Novobiocin [14]. However, they suffer from several disadvantages like Geldanamycin which exhibits sever toxicity due to the electrophilic nature of the quinone moiety and the redox-active behaviour of this ring [15,16]. Further, a derivative of geldanamycin, tanespimycin (17-AAG) although exhibited greater therapeutic index but proved to be a substrate for P450, CYP3A4 and the P-glycoprotein efflux pump and was reported to be metabolised by the oxidoreductaseNQO1/DT-diaphorase making its formulation for clinical delivery a challenge [17]. This issue has been addressed, in part, through the identification of the water-soluble analogue alvespimycin (17-DMAG) and the hydroquinone derivative known as IPI-504 [18]. Both these compounds were successful in preclinical and Phase I clinical trials [22]. Different formulations and combination therapies that employ hsp90 inhibitors (like 17-AAG) and other anticancer drugs (like proteasome inhibitor, bortezomib) are currently being evaluated in clinical Phase II/III and Phase III trials in various cancers (www.clinicaltrials.gov). Radicicol, was found to be a potent inhibitor in vitro, but was found to be inactive in vivo [19] while Novobiocin, although less potent than Radicicol, has shown selectivity for HSP90 both in vivo and in vitro [20]. A series of synthetic small molecule HSP90 inhibitors have been recently developed and include purine scaffold drugs like PU3, PU24FCl, CNF2024 [21,22]; histone deacteylase (HDAC) and anti-HSP90 [23,24].
Because of tremendous potential of Hsp90 inhibitors for treatment of Cancer and other disorders characterised by accumulation of toxic protein aggregate like Huntington and Parkinson’s diseases [25–27], identification of novel Hsp90 inhibitors with more favourable pharmaceutical properties is still desired. In this paper, using a sensitive and simple screening assay based on Hsp90-dependent refolding of firefly luciferase, we describe the identification and characterisation of a novel class of synthetic compounds which act as potent Hsp90 inhibitors in vitro and in vivo.
2. Materials and methods
2.1. Materials
Rabbit reticulocyte lysate (L4960), luciferase assay system (E1501), Creatine phosphokinase (C-3755) and molecular biology grade acetylated bovine serum albumin (B2518) were purchased from Promega. Firefly luciferase (L-9506), ATP (A76999), creatine phosphate, Novobiocin (N6160) and haemoglobin (H7255) were purchased from Sigma. Hsp90 (SPP-770) and Geldanamycin (HPK-102J) were purchased from Stressgen. Coster 96-well black clear bottom plates (07-200-567) were purchased from fisher scientific.
Stability buffer
25 mM Tricine–HCl (pH 7.8), 8 mM MgSO4, 0.1 mM ethylene diaminetetraaceticacid (EDTA), 10% glycerol, 1% Triton X-100 and 10 mg/mL acetylated Bovine serum albumin (BSA).
Cold mix
100 mM Tris–HCl (pH 7.7), 10 mM Mg(OAc)2, 375 mM KCl, 15 mM ATP and 25 mM creatine phosphate.
Creatine phosphokinase (CPK)
Creatine phosphokinase (CPK) was prepared as a 10 mg/mL stock in 50% glycerol.
2.2. Preparation of denatured luciferase
Luciferase at a concentration of 0.5 mg/mL was dissolved in stability buffer lacking glycerol and Triton X-100. After luciferase was completely dissolved, 10% glycerol and 1% Triton X-100 were added and luciferase was denatured by heating at 41.8 °C for 10 min. The activity of luciferase was continuously monitored at one min. intervals during this procedure to avoid its irreversible aggregation that occurs upon over-heating or prolonged incubation at elevated temperatures [28].
2.3. Preparation of denatured luciferase reagent and reticulocyte lysate for 96 well assay
Luciferase reagent (10 mL) was prepared by mixing 8 mL of cold mix, 0.8 mL of 10 mg/mL CPK, 1.075 mL of deionised water; and 0.125 mL of denatured luciferase. After preparation, the luciferase reagent was aliquoted, snap frozen in liquid nitrogen and stored at ←80 °C.
Commercially available Rabbit reticulocyte lysate (L4960) was stored in liquid nitrogen. After thawing, immediately before use, one volume of rabbit reticulocyte lysate was diluted with three volumes of 20 mM Tris–HCl (pH 7.4) containing 75 mM KCl. and the resulting 1:3 diluted reticulocyte was centrifuged at 25,000g for 20 min to remove any particulates.
2.4. Luciferase renaturation assay
Black 96-well clear bottom plates containing 20 μL of 2.5% dimethylsulphoxide (DMSO) in water in the first two columns within the plate (negative and positive controls), and screening compounds at 5 and 10 μM (in duplicates) dissolved in 2.5% DMSO in the remaining lanes were utilised. Tris-buffered saline (10 μL; 20 mM Tris–HCl (pH 7.5), 150 mM NaCl) containing 1% haemoglobin and 4% bovine serum albumin (tris buffered saline (TBS)/HbBSA) were added to the first column as the negative control. Diluted reticulocyte lysate (10 μL) was dispensed to the remainder of the plate. The refolding reaction was initiated by the injection of 10 μL of the denatured luciferase mix to each well to give the following final concentrations: 20 mM Tris (pH 7.7), 3 mM ATP, 5 mM creatine phosphate, 2.0 mM Mg(OAc)2, 75 mM KCl, 0.2 mg/mL CPK and 1.5 μg/mL denatured luciferase. Plates were centrifuged for one min to assure complete mixing of the components. After incubation at room temperature for 2 h, luciferase activity was measured by injection of 40 μL of assay buffer (E1501). Light emission from each well was read with a 1 s integration time using a Perkin-Elmer Victor V 1420 plate reader three minutes after addition of the injection of assay buffer [29].
2.5. Control assay to identify direct inhibitors of native luciferase
After the initial screen, duplicate plates were prepared containing compounds that were found to inhibit luciferase renaturation by ~70% at 5 or 10 μM. Serial dilutions of each compound in 2.5% DMSO (20 μL) were dispensed in duplicates into adjacent wells. The effect of each compound on the activity of luciferase was determined by addition of 10 μL of native luciferase mix (prepared as described above, but not denatured) in modified stability buffer (25 mM Tricine–HCl pH 7.8, 8 mM MgSO4, 0.1 mM EDTA, 30% glycerol, 3% Triton X-100, and 10 mg/mL BSA with 1 ng of native luciferase). Luciferase activity was measured after addition of 30 μL of assay buffer containing 4 mg/mL BSA. Plates were read immediately after dispensing the assay buffer as described above.
2.6. Initial estimate of the IC50 of compounds ability to inhibit Hsp90-dependent luciferase renaturation
The second set of plates were prepared with serial dilutions of each drug loaded into adjacent wells in duplicates and assayed for inhibition of luciferase renaturation as described above in Section 2.4. The concentration-dependent inhibition of luciferase refolding was used to estimate the IC50 of each compound. The positive control (2.5% DMSO) and negative control (TBS/HbBSA) were used as the limits for 0% and 100% inhibition, respectively.
2.7. IC50 determination for inhibitory activity of lead compounds
Hsp90-dependent refolding of firefly luciferase in rabbit reticulocyte lysate was carried out as previously described with minor modifications [29]. Denatured luciferase (0.5 mg/mL) was prepared as described above. Reactions were carried out in triplicate at room temperature in 96 well microtitre plates and experiments were repeated at least twice. Each well contained 1 μL of DMSO or experimental compound dissolved in DMSO to give the desired final concentration: 4 μL of cold mix, 0.4 μL of 10 mg/mL creatine phosphokinase, 5.5 μL of deionised water and 0.1 μL of denatured luciferase. The refolding reaction was initiated by the injection of 10 μL of reticulocyte lysate [one volume of reticulocyte diluted with six volumes of 20 mM Tris–HCl (pH 7.4) containing 75 mM KCl]. After a 30 min incubation at 25 °C, luciferase activity was measured upon injection of 100 μL of assay buffer (E1501) followed by a 10 s integration time to measure relative light unit (RLU) production using a Perkin-Elmer Victor 3V1420 micro plate reader. The IC50 value was calculated as the concentration required inhibiting the recovery of luciferase activity by 50% relative to the DMSO control. The effect of experimental compound on the activity of native luciferase was also assayed as a control.
2.8. Anti-proliferative effect of Hsp90 inhibitors
Anti-proliferative effect of lead compounds with Hsp90 inhibition properties was tested for GI50 determination in three different cancer cell lines viz: BT-474 (invasive ductal breast cancer cell line), DU-145 (a prostate cancer cell line) and K562 (bcr-abl positive CML cell line) [30–32]. BT-474, DU-145 and K562 cell lines were maintained, respectively, in Dulbecco’s Modified Eagle’s Medium, Eagle’s Minimum Essential Medium and Iscove’s Modified Dulbecco’s Medium, each supplemented with non-essential amino acids, L-glutamine (2 mM), streptomycin (500 μg/mL), penicillin (100 units/mL) and 10% foetal bovine serum. Dose response curve for GI50 determination of hit compounds was done by Cell Titer-Blue Cell Viability Assay kit (Promega Corporation #G8080). Briefly, exponentially growing BT-474 cells were seeded into 96-well plates (Corning #3603) at a density of 11,000 cells/100 μL of medium. Cells were treated 24 h later with either vehicle control (DMSO) or compounds for 72 h at 37 °C. After exposure of cells for indicated time, plates were equilibrated to room temperature (20–25 °C) and 20 μL of Cell Titer-Blue reagent was added to each well. Plates were shaken for 10 s on an orbital shaker and then incubated under standard cell culture conditions for 4 h. The plates were shaken for 10 more seconds before recording fluorescence at 490/570 nm from each well in a Glo-max Multi-detection system (Promega) with an integration time of 0.5 s well−1. The percentage cell growth inhibition was calculated by comparison of the fluorescence reading obtained from treated versus control cells.
2.9. Caspase Glow-3,7 assay
To elucidate whether cell demise following exposure to ONO4140 compound was attributable to apoptosis, we first determined effector Caspase-3,7 activities in BT-474 cells, using Caspase Glow-3,7 assay. Cells were plated in 96-well plates and treated with ONO4140 as described in the Cell Titer-Blue Growth Inhibition Assay (see Section 2.8) (for 72 h). Following exposure, plates were taken from incubator and equal volume (100 μL) of Caspase Glow 3/7 reagent was added to each well. Plates were shaken at 300–500 rpm for 30 s and incubated at room temperature for 2–3 h. The luminescence from each well was measured in a Glo-max Multi-detection system (Promega) with an integration time of 0.5 s well–1. The percentage increase in apoptotic cells was calculated by comparison of the luminescence reading obtained from treated versus control cells.
2.10. Western blot analyses of hit compounds
BT-474 breast cancer cells were plated at a density of 1.0 ← 106 cells/plate in culture dishes and allowed to attach overnight. The cells were treated with different concentrations of each compound for indicated periods. Treated cells were harvested and protein lysates were electrophoresed in SDS–polyacrylamide gel and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore Corporation, Billerica, MA, United States of America (USA)). The following antibodies were used for western blotting: Her-2/Neu (CB11) (sc-52349 Santa Cruz Biotechnology, Inc) and phospho Her2 (Tyr1248) (sc-12352-R Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), phosphor-Akt (Ser473) (#9271; Cell Signalling Technology, Inc.), Akt (#9272, Cell Signalling Technology, Inc.), HSP70 (#4873, Cell Signalling Technology, Inc), HSP90#4874, Cell Signalling Technology, Inc; anti actin (A2066, SIGMA).
To assess total levels of Her-2, pHer-2, AKT, phosphorylated AKT and β-actin in the cell extracts, 30–50 μg of total protein was resolved by the appropriate-percentage SDS–PAGE. The above antibodies were used for blotting proteins on the PVDF membrane. Bound antibodies on PVDF immunoblots were detected by appropriate IR Dye 680 tagged secondary Odysseys antibodies by infrared fluorescence detector (Li-COR Biosciences, Lincoln, NE, USA).
To delineate the mechanism of cell death, the lysate obtained following exposure of BT-474 cells to ONO4140 was subjected to western blot analysis using the following antibodies at appropriate concentrations: anti-cleaved caspase-3 rabbit polyclonal antibody (9661, Cell Signalling), anti-cleaved caspase-9 rabbit polyclonal antibody (9502, Cell Signalling), anticleaved caspase-9 mouse monoclonal antibody (MAB4609, Chemicon), anti-PARP p85fragment rabbit polyclonal antibody (G7341, Promega) and anti-caspase-2 mouse monoclonal antibody (611022, BD Biosciences).
2.11. Immunoprecipitation
For immunoprecipitation BT-474 cells were treated with ONO4140 (at 5, 10 and 20 μM concentrations), Geldanamycin (500 nM) and DMSO for 16 h and then lysed in 20 mM Tris–HCl (pH 7.4), 25 mM NaCl, 2 mM DDT, 20 mM Na2MoO4, 0.1% NP-40 and protein inhibitors for 2 h at 4 °C, rotating, and then centrifuged at 13,000g for 10 min [33]. 500 μg of total protein was immunoprecipitated with 8 μL of anti-p23 antibody (cat. No. ALX-804–023, mouse monoclonal, clone JJ3, isotype: IgG1;ALEXIS Corporation, Lausen, Switzer-land Inc.) for 2 h at 4 °C, rotating. Protein A agarose (30 μL; Upstate) was added to each sample, and samples were then incubated for 1 h at 4 °C, rotating. The beads were washed five times with 1 mL lysis buffer. Bound proteins were isolated by boiling in sample buffer and resolved by 12% SDS–PAGE and transferred to PVDF membranes. The levels of co-immunoprecipitating hsp90a proteins were analysed by western blot using anti-HSP90a antibody (cat. No. SPS-771, rabbit polyclonal; Assay Designs, Inc.).
3. Results
3.1. Optimisation and utilisation of the luciferase refolding assay for screening of compounds with hsp90 inhibition function
Luciferase has been used as model substrate to study folding and renaturation of proteins because of its very high bioluminescence activity [28]. It also has been previously demonstrated that refolding of heat denatured luciferase is Hsp90-dependent, making it a very sensitive assay for studying Hsp90-dependent protein refolding [29]. Since reticulocyte lysate is a rich source of Hsp90 and contains the full complement of co-chaperones, it represents a model quasi-physiological system to screen for Hsp90 inhibitors. Geldanamycin (GA), an N-terminal Hsp90 inhibitor has been shown to prevent refolding of luciferase at submicromolar concentrations (IC50 ~0.2 μM) [34] while Novobiocin exhibits the same activity at concentration, IC50 ~500 μM [29].
Five hundred compounds (description of compound library available from Dr. M.V. Ramana: Head of chemistry division, Onconova Therapeutics) in Black 96 well clear bottomed plates were screened for Hsp90 inhibition at two different concentrations of 5 and 10 μM using the assay described in Materials and Methods (Fig. 1). The assay produced an average Z value of 0.81 ± 0.17 indicating statistically reliable and reproducible results. From this library of compounds, we could identify 55 compounds that inhibited luciferase activity by >70% at 5/10 μM concentration. These compounds were re-screened to identify false positives (direct inhibitors of luciferase) and to obtain an initial estimate of the IC50 value for each compound. Of the 55 compounds, 11 were found to inhibit the Hsp90-dependent luciferase refolding activity in reticulocyte lysate by >70% at a concentration of less than 5 μM. The rest of the compounds (44) were found to inhibit luciferase refolding activity at greater than 5 μM. Among these, 28 compounds that displayed potent inhibition were selected for IC50 determination. All of the 28 compounds showed concentration dependent inhibition of Hsp90 and were used for the initial estimation of IC50 of compounds.
Fig. 1.
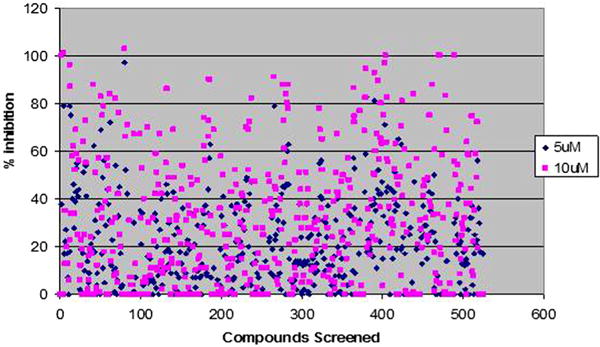
Scatter plot showing percentage control inhibition of 500 screened compounds. Compounds at two different concentrations 5 and 10 μM were screened for percentage inhibition relative to DMSO control (100% activity or 0% inhibition) minus background [(TBS/HbBSA) (for any auto refolding)]. The Z-factor (z = 0.81 ± 0.17) of the optimised assay reveals its high reliability, sensitivity and reproducibility.
3.2. IC50 and GI50 determination
Lead compounds (n = 28) identified from this screen were dissolved in DMSO and IC50 values for inhibitory activity were determined by the method described in Section 2.7. A wide-range of IC50 values were obtained (1–12 μM). Of which, six most active compounds (IC50 between 1 and 6 μM) (Fig. 2A) were selected for further characterisation using enzymatic and cell biological assays.
Fig. 2.
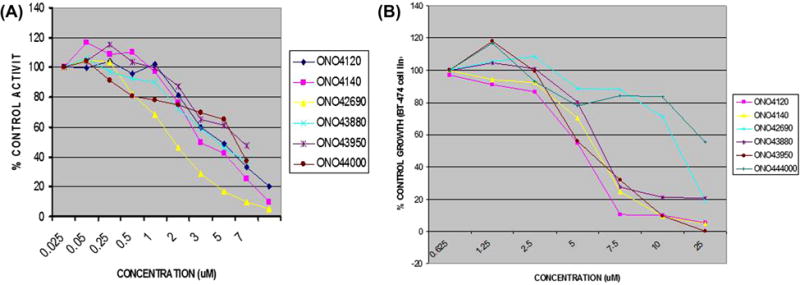
(A) IC50 curve of lead compounds (ONO4120, ONO4140, ONO42690, ONO43880, ONO43950 and ONO44000) as determined by IC50 luciferase refolding assays performed as described in Section 2.7. The assay measures the ability of the compounds to inhibit Hsp90 dependent refolding of denatured luciferase compared to the DMSO control (0% inhibition/100% luciferase refolding) The experiments were performed in triplicates (μM) and repeated at least twice. (B) Growth inhibition curve for GI50 determination of lead compounds viz: ONO4120, ONO4140, ONO42690, ONO43880, ONO43950 and ONO44000 in BT-474 cell lines as determined by Cell Titer-Blue Anti-proliferative Assay performed as described in Section 2.8. BT-474 cells were incubated with increasing concentrations of different compounds. Growth over 72 h was assessed by a method that determines intracellular ATP levels. GI50 value is taken as the concentration that decreases the growth by 50% relative to DMSO control compounds were assayed in triplicate. All compounds were used as DMSO stocks.
We next tested these compounds in tumour cell growth inhibition assays in BT-474, a human invasive ductal breast carcinoma cell line. The results of this study showed that these lead compounds exhibited GI50 values that ranged between 4.0 and 15 μM (Fig. 2B). When these compounds were tested for their tumour cell killing activities using two other cell lines, DU-145, a prostate carcinoma cell line and K562, a bcr-abl positive CML cell line, two lead compounds, ONO4140 and ONO4120, induced cell death in these cell lines at similar concentrations seen with BT-474 (table).
3.3. Molecular validation of hsp90 inhibition of the hit compound, ONO4140
Of the six compounds identified in this study, we chose to further characterise one compound, ONO4140 (structure shown in Fig. 3), which showed the best biological activity in cell growth inhibition assays. This lead compound was further examined for molecular hallmark features of Hsp90 inhibition in vivo, which include the proteasome-dependent degradation of the Hsp90 client proteins and the up regulation of hsp70. BT-474, breast carcinoma cell line was chosen to conduct these studies since this cell line has been found to overexpress Her-2 and AKT, two client proteins of Hsp90. Treatment of BT-474 cells with ONO4140 for 24 h resulted in a concentration-dependent degradation of both phosphorylated and un-phosphorylated forms of both client proteins (Fig. 4A). Complete degradation of Her-2 and AKT was seen at a concentration of 10–20 μM. We also examined the concentration dependent induction of hsp70, another hallmark feature of hsp90 inhibition following the treatment of BT-474 cells for 24 h with ON4140 (Fig. 4B). Maximum induction of Hsp70 was seen between 10 and 20 μM, the same concentration at which the maximum degradation of client proteins is seen. However, no effect of this compound on total hsp90 levels was observed (Fig. 4B).
Fig. 3.
Structure of ONO4140 with highest biological activity of HSP90 inhibition.
Fig. 4.
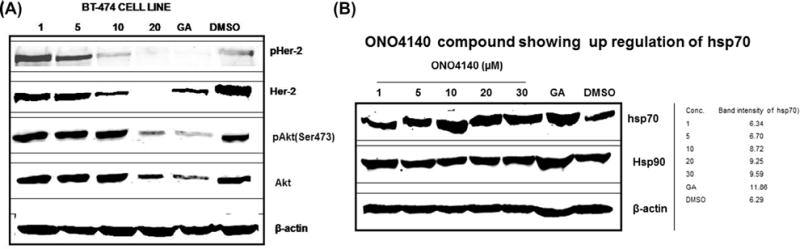
Protein expression analysis of Her-2 overexpressing invasive ductal carcinoma breast cancer cell line, BT-474, upon treatment with lead compounds for hsp90 client protein degradation and hsp70 upregulation, two hallmark features of hsp90 inhibition. (A) Representative picture showing concentration dependent degradation of Hsp90 client proteins, Her-2, Akt and their phospho forms upon treatment with ONO4140. Geldanamycin (GA, 500 nM) and DMSO (1%) were used as positive and negative controls. (B) Representative picture showing concentration dependent upregulation of hsp70 (inducible) with no effect on total hsp90 levels upon treatment with ONO4140 in BT-474 cell line. Geldanamycin (GA, 500 nM) and DMSO (1%) were used as positive and negative controls.
We next examined the effect of time of exposure to ONO4140 on the phosphorylation status of Her-2 and AKT in BT-474 cells. Results of this study (Fig. 5) show that phosphorylation of Her-2 was lost within 2 h following the treatment of cells with 10 μM concentration of ONO4140 while the degradation of this client protein is seen 10 h after the treatment. When the same study was conducted for AKT, loss of AKT phosphorylation as well as degradation of this protein was seen between 2 and 16 h of exposure at 10 μM concentration of ONO4140 (Fig. 5). The induction of hsp70 was also evident in a time dependent manner (Fig. 5).
Fig. 5.
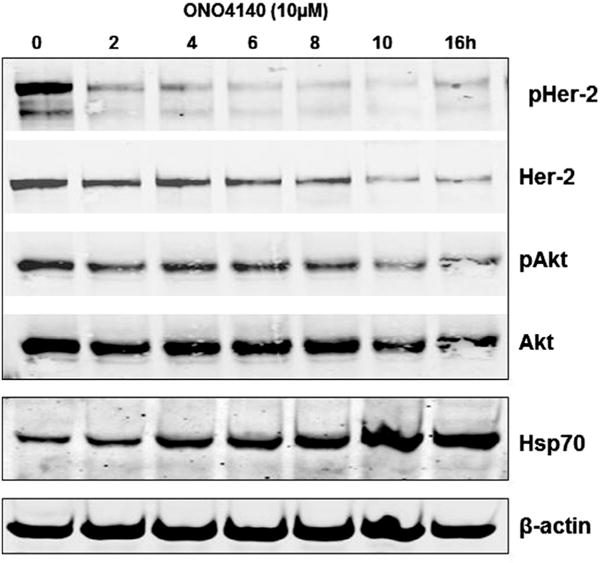
The kinetics of ONO4140 in BT-474 cell lines on Hsp90 client protein degradation and Hsp70 upregulation, two hallmark features of hsp90 inhibition.
3.4. Disruption of HSP90-p23 association
The hsp90 inhibition property of ONO4140 was further validated by studying its cellular effects on the catalytic activity of HSP90 which can be monitored through the evaluation of disruption of the HSP90-p23 complex [35]. HSP90 recruits various co-chaperones at different points in its catalytic cycle, and the association of HSP90 with p23 is essential for HSP90 activity and client protein stability. HSP90-p23 interaction requires ATP binding but not ATP hydrolysis in biochemical assays, and p23 specifically recognises HSP90-ATP complexes but not HSP90 alone [35]. Destabilisation of the HSP90-p23 interaction in tumour cells and the subsequent measurement using immunoprecipitation, therefore, can be used to monitor the effect of HSP90 inhibitors on the HSP90 catalytic cycle. Treatment of BT-474 with ONO4140 caused a concentration-dependent decrease in the amount of HSP90a co-immunoprecipitating with p23 (Fig. 6). The maximum loss of association of Hsp90 with p23 was observed at 10 μM, the same concentration at which the maximum degradation of client proteins occurs. The immunoprecipitation experiment was repeated thrice. This observation substantiates the inhibitory effect of ONO4140 on the catalytic activity of hsp90.
Fig. 6.
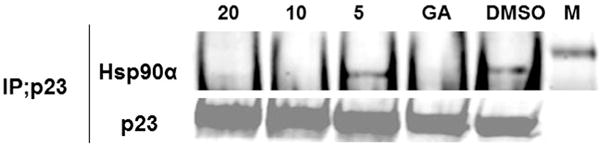
The effect of ONO4140 on the catalytic activity of hsp90 as evaluated through disruption of HSP90-P23 association. Geldanamycin (GA, 500 nM) and DMSO were used as positive and negative controls. M is molecular wt marker (100 KDa).
3.5. Mechanism of cell death
To determine whether ONO4140-induced cell death was attributable to apoptosis, we treated BT-74cells with ONO4140 and analysed its effects on several effectors and mediators of apoptosis. Given the absence of a functional death receptor pathway, apoptosis in Her-2 overexpressing breast cancer cell line, BT-474, can be expected to be via the intrinsic/mitochondrial pathway mediated [36]. To substantiate this hypothesis, we first examined the effect of ONO4140 on effector caspases 3/7 activities as described in Materials and Methods using Caspase Glow assay. These studies showed a substantial increase in apoptotic population of BT-474 cells (as evident from increase in effector caspases 3,7 activities) upon treatment with ONO4140 (72 h). A two fold increase in caspases 3/7 activity was observed between 5 and 10 μM concentration which corresponds to decrease in cell viability (Fig. 7A) Activation of the effector caspases 3/7 upon treatment was further demonstrated by their cleavage following treatment with ONO4140 (Fig. 7B). Several caspases may be responsible for effector caspase-3 activation. Here, caspase-3 activation by ONO4140 was associated with cleavage of both caspase-9 and caspase-2, and further paralleled by PARP cleavage (Fig. 7B)) suggesting the activation of caspase9–caspase3 intrinsic apoptotic pathway. Activation/cleavage of caspases was evident at doses that correlate well with the Hsp90 inactivation (10–20 μM) (Fig. 7B). Furthermore, maximum activation of caspases was seen 16 h following the treatment of cells with ONO4140 (Fig. 7C). Interestingly, it has been reported that Caspase-9 activity is regulated through phosphorylation by AKT which phosphorylates Pro-Caspase9 at Ser196 [37]. This phosphorylation event is known to inhibit the proteolytic processing of pro-Caspase-9. Since, maximum de-phosphorylation and/degradation of Akt and activation/cleavage of caspase 9, are evident at the same time-point i.e. 16 h post treatment (at 10 μM), our results suggest that inhibition of hsp90 by ONO4140, inhibits HER2/PI3 K/Akt pathway which results in the activation of caspase 9–caspase 3 intrinsic apoptotic pathway, at least, in the breast cancer cell line, BT-474.
Fig. 7.
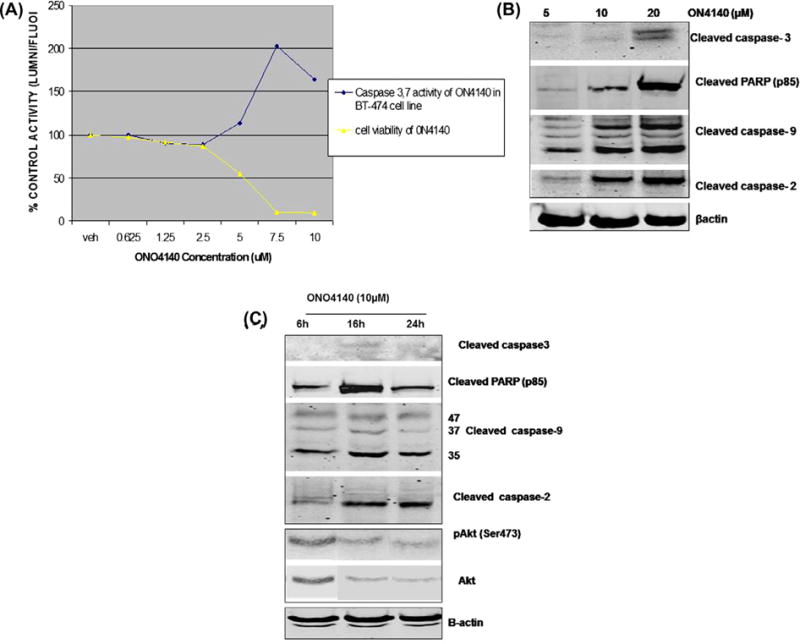
The PI(3)K-Akt pathway and the intrinsic apoptotic pathway are targets of Hsp90 inhibitor-mediated cell death. (A) Caspase 3,7 activity and cell viability of ONO4140 in BT-474 cell line. BT-474 cells were treated with the indicated doses of ONO4140 for 72 h to measure apoptosis by Caspase Glow assay. The assay measures effector Caspase-3,7 activity as the potency of ONO4140 in cleaving the luminogenic Caspase 3/7 substrate containing tetrapeptide sequence-DEVD and generating glow type luminescent signal. The percentage increase in apoptotic cells was calculated by comparing the luminescence readings obtained from ONO4140 treated cells with those obtained from vehicle-treated cells. Caspase-3/7 activity in vehicle-treated cells was normalised to 100%. The increase in Caspase activity from ONO4140 treated cells also corresponds to decrease in cell viability (as measured by Cell Titer-Blue assay described in Section 2.8). (B) Activation of Caspase 9–Caspase 3 intrinsic apoptotic pathway upon exposure to ONO4140. BT-474 cells were treated for 24 h with indicated concentrations of ONO4140 compound and cells were lysed for western blot analysis for various Caspases. (C) The kinetics of Caspase 9–Caspase 3 intrinsic apoptotic pathway upon exposure to ONO4140 (10 μM). BT-474 cells were treated with ONO4140 compound (10 μM) for different time periods and cells were lysed for western blot analysis. β-actin was used as loading control for western blots. Maximum activation of caspase 9, and/degradation of Akt occur at the same time point i.e. 16 h post treatment (at 10 μM), suggesting that inhibition of hsp90 by ONO4140 results in the inhibition of HER2/PI3 K/Akt pathway which in turn results in the activation of Caspase 9–Caspase 3 intrinsic apoptotic pathway.
4. Discussion
The molecular chaperone, hsp90 is a ubiquitously expressed protein that interacts with a plethora of proteins in a low-affinity dynamic fashion to help them in proper folding and conformational maturation. However, in malignant cells this protein appears to play a more important role by maintaining functionally active conformation of various mutant and growth promoting proteins that regulate important nodal points in multiple signalling pathways [1,3]. Given the important role of hsp90, inhibitors of this chaperone have been found to exhibit a debilitating effect on the networks that maintain cancer cell survival and hence are envisaged to have an important application for the treatment of various cancers [4–6]. Although a number of hsp90 inhibitors have been developed some of which are in clinical trials [15,16,18], they appear to suffer from low bioavailability, variable potencies and hepatotoxicity suggesting an important need for identification and development of new inhibitors with more favourable pharmacological properties.
In the present study, we report the utilisation of a highly sensitive assay for high throughput screening of a library of compounds for the identification of novel hsp90 inhibitors. Our results suggest that the assay is statistically reliable, sensitive and reproducible (Z-factor of 0.81 ± 0.17). The rationale of the assay is refolding of denatured luciferase in rabbit reticulocyte lysates (RRL) which serves as a good source of HSP90 and co-chaperones needed for active chaperoning [28]. The assay has the capability of identifying inhibitors that obstruct the chaperone activity of Hsp90 either by direct binding to its N-terminal or C-terminal nucleotide binding sites or by interference with the ability of the chaperone to switch conformations [29]. In our screen, we identified six possible hits which showed IC50 values that ranged between 1.0 and 6 μM.
Interestingly, two lead compounds ONO4120 and ONO4140 appear to be growth inhibitory in three different cancer cell lines (DU-145, BT-474 and K562) at similar lowest concentrations. The variable growth inhibition potencies shown by these six lead compounds (some with IC50 1 μM) in different cancer cell lines could be, possibly, due to differential diffusion through lipid bilayer in different cell lines, or rapid metabolism of drug under cell culture conditions.
We next studied the biochemical and biological activities of ONO4140 to gain an understanding of the mechanism of action of this class of compounds. Our studies show that ONO4140 disrupts the catalytic activity of hsp90 as evidenced by the destabilisation of hsp90-p23 complexes, a feature that has been recently used as an alternative method of monitoring the efficacy of hsp90 inhibitors on the catalytic activity of HSP90. HSP90 recruits various co-chaperones at different points in its catalytic cycle, and the association of ATP bound HSP90 with p23 is essential for HSP90 activity and client protein stability. Since p23 specifically recognises HSP90-ATP complexes, disruption of Hsp90-p23 association, therefore, has been used to measure efficacy of hsp90 inhibitors on the catalytic activity of Hsp90 in human cell lines and xenografts [35,38]. Furthermore, the ability of ONO4140 to mediate hsp90 client protein degradation and HSP70 induction in BT-474 cell line proves its HSP90 inhibitory action in a classical way. The two experiments, therefore, confirm HSP90 specific inhibition properties of ONO4140.
The present study reveals that treatment of BT-474 cell line with ONO4140 results in the degradation and inactivation of growth factor receptors such as Her2 and subsequent inactivation of the PI3K/AKT signalling pathway. In addition, inhibition of HSP90 by ONO4140 results in decreased conformational stability of AKT leading to loss of cellular AKT levels. These observations suggest that ONO4140, as an HSP90 inhibitor, has the potential for acting as an anti-tumourigenic agent, at least, in tumours that are characterised by oncogenic activation of PI3/AKT signalling pathway [39,40]. Our data also suggest that short-term exposure of tumour cells with high drug concentrations results in a potent inhibition of Hsp90activity as evidenced by hsp90-p23 dissociation. This in turn appears to result in the initiation of downstream events such as client protein degradation leading to cessation of the proliferative activity of tumour cells.
Interestingly, our data also suggest that Hsp90 could act as a major inhibitor of apoptosis in BT-474 cells, and pharmacologic inactivation of Hsp90 results in activation of apoptosis in these cells. The significant activation of Caspase9–Caspase3 after Hsp90 inactivation by ONO4140 (16 h post treatment) suggests that cell death is probably mediated by the Caspase9–Caspase3 intrinsic apoptotic pathway, an event most probably triggered by the inactivation of the AKT pathway.
In the present study the molecular mechanism of action of ONO4140 revealed that the drug acts by promoting degradation of Hsp90 client proteins and, therefore, would be useful against cancer cell types that are characterised by overexpression of Hsp90 client proteins like HER-2, RB, AKT, etc. The more potent derivative of this compound has, therefore, the potential of using alone in cancers that develop resistant to conventional cytotoxic drugs or alternately achieve synergistic action when used in combination with conventional chemotherapy and radiation therapy [41]. The present study reveals that treatment of cancer cell lines with ONO4140 results in down regulation of AKT survival signalling. The compound can be, therefore, used in cancers that develop resistance to protein kinase inhibitors like in midostaurin (FLT3 tyrosine kinase inhibitor) resistant leukaemias or in Imatinib (BCR-ABL tyrosine kinase inhibitor) resistant CML patients. Further the present study reveals that the cell demise following treatment with ONO4140 occurs via Caspase 9–caspase 3 intrinsic apoptotic pathways. The compound, therefore, can be used against cancers that are resistance to drugs that target extrinsic apoptotic pathways (TNF and TRIAL resistant tumours) or alternatively synergistic action can be achieved, when used in combination, by inducing both apoptotic pathways. ONO4140, as an HSP90 inhibitor, has the potential of achieving highest antitumour activity when used in combination with proteasome inhibitors. The marked antitumour activity might arise from their complementing abilities to simultaneously trigger intracellular accumulation of unfolded proteins and prevent their cellular protection action.
In conclusion, we have identified a novel class of synthetic compounds which act as potent Hsp90 inhibitors in vitro and mediate the disruption of Hsp90-p23 complexes in vivo, thus leading to the catalytic inactivation of Hsp90. This event appears to result in the degradation of client proteins, which include growth factor receptors such as Her2 and cell survival proteins such as AKT1. The degradation of these proteins appears to occur concomitantly with transcriptional induction of HSP70 via HSF-1(as has been seen with pharmacological inhibition of Hsp90) and initiation of cell demise via activation of caspase9–caspase3 intrinsic apoptotic pathways. The discovery of this novel class of synthetic Hsp90 inhibitors with simple chemical backbone allows us to conduct further structural modifications to improve their potency and pharmacokinetic properties for use in cancer therapy after testing the modified compound in xenograft mouse models and eventually in clinical trials. Further the mechanism of action of ONO4140 reveals that the drug has antitumour activity in cancers that overexpress hsp90 client proteins. In future, the more potent derivative of ONO4140 as an HSP90 inhibitor has the potential of using alone in tumours that are resistant to conventional anticancer drugs or in combination with other anticancer drugs.
Supplementary Material
Acknowledgments
The authors also gratefully acknowledge Dr. M.V. Ramana: Head of Chemistry division, Onconova Ther apeutics, for providing a library of compounds for screening, the Fels Institute of Cancer Research and Molecular Biology, Temple University, for providing equipments for conducting experiments and the technical staff, Venkat R. Pallela, Revathi Patti, of Fels Institute for providing technical help.
Role of funding source
The authors gratefully acknowledge the Onconova Therapeutics for providing funds for research work.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ejca.2014.04.017.
Footnotes
Conflict of interest statement
None declared.
References
- 1.Sharp SY, Jones K, Workman P. HSP90 inhibitors: targeting the cancer chaperone for combinatorial blockade of oncogenic pathways. Cancer Drug Des Discov. 2008:305–35. [Google Scholar]
- 2.Calderwood SK, Khaleque MdA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 4.htpp://www.picard.ch/downloads/HSP90interactors.
- 5.Chiosis G. Targeting chaperones in transformed systems—a focus on Hsp90 and cancer. Expert Opin Ther Targets. 2006;10:37–50. doi: 10.1517/14728222.10.1.37. [DOI] [PubMed] [Google Scholar]
- 6.Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–57. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med. 2004;82:488–99. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 8.Pearl LH, Prodromou C. Structure and mechanism of Hsp90 molecular chaperone machinery. Ann Rev Biochem. 2006;75:271–94. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 9.Neckers L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb Exp Pharmacol. 2006;172:259–77. doi: 10.1007/3-540-29717-0_11. [DOI] [PubMed] [Google Scholar]
- 10.Workman P, Maloney A. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Ochel HJ, Eichhorn K, Gademann G. Geldanamycin: the prototype of a class of antitumor drugs targeting the heat shock protein 90 family of molecular chaperones. Cell Stress Chaperones. 2001;6(2):105–12. doi: 10.1379/1466-1268(2001)006<0105:gtpoac>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitesell L, Mimnaugh EG, Costa BD, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6(24):4459–62. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 14.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 15.Banerji U, Donnell AO, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygel-danamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–61. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 16.Solit DB, Ivy SP, Kopil C, Sikorski R, Morris MJ, Slovin SF, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–82. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxy-geldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91:1940–9. doi: 10.1093/jnci/91.22.1940. [DOI] [PubMed] [Google Scholar]
- 18.Sydor JR, Normant E, Pien CS, Porter JR, Grenier L, Pak RH, et al. Development of 17-allylamino-17-demethoxy geldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A. 2006;103:17408–13. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soga S, Shiotsu Y, Akinaga S, Sharma SV. Development of radicicol analogues. Curr Cancer Drug Targets. 2003;3:359–69. doi: 10.2174/1568009033481859. [DOI] [PubMed] [Google Scholar]
- 20.Burlison JA, Neckers L, Smith AB, Maxwel A, Blagg BSJ. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of hsp90. J Am Chem Soc. 2006;128:15529–36. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 21.Chiosis G. Discovery and development of purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2006;6:1183–91. doi: 10.2174/156802606777812013. [DOI] [PubMed] [Google Scholar]
- 22.Wright L, Barril X, Dymock B, Seridan L, Surgenor A, Beswick M, et al. Structure–activity relationships in purine based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;1:5–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 23.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 24.Matthews RC, Rigg G, Hodgetts S, Carter T, Chapman C, Gregory C, et al. Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrob Agents Chemother. 2003;47:2208–16. doi: 10.1128/AAC.47.7.2208-2216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, et al. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 27.Shen HY, He JC, Wang Y, Huang QY, Chen JF. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280:39962. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 28.Thulasiraman V, Matts RL. Bioluminescent protocols. In: LaRossa RA, editor. Methods in molecular biology. Totowa, NJ: Humana Press Inc; 1998. pp. 129–41. [DOI] [PubMed] [Google Scholar]
- 29.Galam L, Hadden MK, Ma Z, Ye QZ, Yun BG, Blagg BSJ, et al. High-throughput assay for the identification of Hsp90 Inhibitors Based on Hsp90-dependent refolding of firefly luciferase. Bioorg Med Chem. 2007;15(5):1939–46. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10:R33. doi: 10.1186/bcr1996. http://dx.doi.org/10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams C, Tabios R, Linehan W, Neckers L. Intratumor injection of the Hsp90 inhibitor 17AAG decreases tumor growth and induces apoptosis in a prostate cancer xenograft model. J Urol. 2007;178:1528–32. doi: 10.1016/j.juro.2007.05.120. [DOI] [PubMed] [Google Scholar]
- 32.Blagosklonny MV, Fojo T, Bhalla KN, Kim J-S, Trepel JB, Figg WD, et al. The Hsp90 inhibitor geldanamycin selectively sensitizes Bcr-Abl-expressing leukemia cells to cytotoxic chemotherapy. Leukemia. 2001;15:1537–43. doi: 10.1038/sj.leu.2402257. [DOI] [PubMed] [Google Scholar]
- 33.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–30. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Thulasiraman V, Matts RL. Effect of geldanamycin on the kinetics of chaperone-mediated renaturation of firefly luciferase in rabbit reticulocyte lysate. Biochemistry. 1996;35:13443–50. doi: 10.1021/bi9615396. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan WP, Owen BA, Toft DO. The influence of ATP and p23on the conformation of hsp90. J Biol Chem. 2002;277:45942–8. doi: 10.1074/jbc.M207754200. [DOI] [PubMed] [Google Scholar]
- 36.Dubská L, Anděra L, Sheard MA. HER2 signaling downregu-lation by trastuzumab and suppression of the PI3K/Akt pathway: an unexpected effect on TRAIL-induced apoptosis. FEBS Lett. 2005;579(19):4149–58. doi: 10.1016/j.febslet.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 37.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 38.Chan CT, Paulmurugan R, Gheysens OS, Kim J, Chiosis G, Gambhir SS. Molecular imaging of the efficacy of heat shock protein 90 inhibitors in living subjects. Cancer Res. 2008;68:216–26. doi: 10.1158/0008-5472.CAN-07-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sain N, Krishnan B, Ormerod MG, De Rienzo A, Liu WM, Kaye SB, et al. Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther. 2006;5:1197–208. doi: 10.1158/1535-7163.MCT-05-0445. [DOI] [PubMed] [Google Scholar]
- 40.Maira SM, Voliva C, Garcia-Echeverria C. Class IAphosphat-idylinositol3-kinase: from their biologic implication in human cancers to drug discovery. Expert Opin Ther Targets. 2008;12:223–38. doi: 10.1517/14728222.12.2.223. [DOI] [PubMed] [Google Scholar]
- 41.Lu X, Xiao L, Wang L, Ruden DM. Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem Pharmacol. 2012;83(8):995–1004. doi: 10.1016/j.bcp.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.