

Abstract
A stereoselective and efficient method for free radical addition of benzyl thiol to aryl acetylene in the presence of Et3B-hexane has been developed for the synthesis of (Z) and (E)-styryl benzyl sulfides where base catalyzed hydrothiolations have failed. The scope of this reaction was successfully extended for the synthesis of (E)-ON 01910·Na, a phase III clinical stage anti-cancer agent and its inactive geometrical isomer (Z)-ON 01910·Na. It is interesting to note that all the E-isomers synthesized have shown better cytotoxicity profile on cancer cells compared to the Z-isomers.
Introduction
ON 01910·Na (Rigosertib®)1 is a styryl benzyl sulfone and it is biologically active only as an (E)-isomer. It is a novel non-ATP competitive anticancer agent that inhibits mitotic progression, promotes G2/M arrest and induces apoptosis in a number of cancer cells while leaving nonmalignant cells virtually unaffected.2 (E)-ON 01910·Na has exhibited both antitumor activity and anti-angiogenic activity with a low toxicity profile in various preclinical tumor xenograft models. Unlike hypomethylating agents, (E)-ON 01910·Na exerts potent antitumor activity against leukemic and pre-leukemic cells by inhibition of the PI/3K/Akt/mTOR pathway and down regulation of cyclin D1 translation.3,4 Cyclin D proteins are elevated in many hematologic malignancies, including the myelodysplastic syndromes (MDS).5 A phase III pivotal trial of (E)-ON 01910·Na in MDS patients has been underway.6 This compound has also shown promising therapeutic effects and drug tolerance in patients with advanced solid tumors including pancreatic (phase II/III) and ovarian (phase II) cancer, both as a single agent and in combination therapy protocols.7,8 The structure activity studies of (E)-ON 01910·Na confirmed that the nature, number, and position of substituents on both aromatic rings of the molecule along with geometry of the styryl group are critical for activity.9
(E)-ON 01910·Na was synthesized by Knoevenagel condensation as described in our earlier publication.9 It exists as a stable E-isomer, which is the preferred form of this molecule. The double bond in (E)-ON 01910·Na is capable of exhibiting geometrical isomerism resulting in both E- and Z-isomers. Stability studies indicate that over a period of time (E)-ON 01910·Na, when exposed to light, accumulates traces of the Z-isomer in both solid and solution form. Similar kinds of photo-induced geometric isomerism changes have been reported previously10 and have resulted in the formation of mixed geometrical isomers11 from a single isomer. Surprisingly, neither thermal exposure of (E)-ON 01910·Na in the absence of light nor storage in amber colored vials resulted in the accumulation of Z-isomer. This indicates that photo isomerization is taking place resulting in the formation of traces of (Z)-ON 01910·Na (Fig. 1), which has the same molecular weight and molecular formula but different retention time in HPLC. The objective of the present study is to synthesize the Z-isomer of ON 01910·Na in sufficient quantities to characterize and compare the pharmacokinetic and biological activity of this compound with that of the E-isomer. As the geometry of a molecule is known to play an important role in SAR, the synthesis of the Z-isomer provides an opportunity to compare the biological and pharmacological properties of both isomers. This study also allows us to design alternate methods for exclusive synthesis of the E-isomer, processes for purification and stabilization, and to minimize or totally eliminate the un-desired product(s).
Fig. 1.
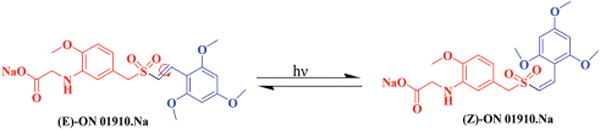
Photoisomerization of ON 01910·Na.
Alkyne hydrothiolation (Fig. 2) is a challenging reaction associated with an atom-economical method for C–S bond formation, Markovnikov and (or) anti-Markovnikov addition, and stereoselectivity. Hydrothiolation is also of great importance as it provides an attractive method for the formation of vinyl sulfides, which can be used as complementary building blocks to carbonyl compounds12 and Michael acceptors13 for synthesis of many polymeric materials,14 natural products,15 and synthetic reagents.16 Conventional approaches to the synthesis of vinyl sulfides include the addition of thiols to alkynes under free-radical17 or transition metal catalyzed conditions,18 Wittig olefination,19 and direct nucleophilic substitution through use of vinyl halides.20 Several metal catalysts have proven effective in hydrothiolation of unsaturated compounds, but the stereo-and regioselectivity control still remains an important challenge.21
Fig. 2.
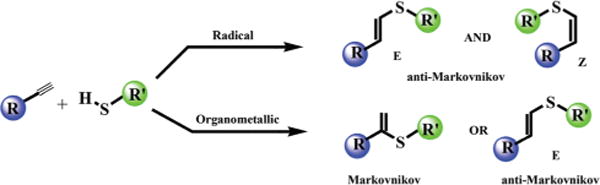
Radical and catalytic alkyne hydrothiolations.
Numerous methods have been developed for the stereo-selective synthesis of (E)-vinyl sulfides.22 In contrast, it has been challenging to prepare Z-isomers stereoselectively.23 The synthesis of (Z)-vinyl sulfides utilizing transition metal catalysts has been limited to neutral and cationic complexes of rhodium and iridium. These methods suffer from limited sub-strate scope and low stereoselectivity.24 Base-mediated hydrothiolations were first reported in 1956 by Truce et al.,25 and, more recently, a cesium carbonate catalyzed process to synthesize (Z)-vinyl sulfides was reported.26 This process requires a radical inhibitor, and the substrate scope was limited to only alkyl thiols. Alkyne hydrothiolation via a radical mechanism was first reported in 1970, whereby a free radical process yielded anti-Markovnikov products with typically low E/Z selec-tivity.27,28,17a,b Synthesis of vinyl sulfides by the addition of thiols to acetylenes in the presence of Et3B was reported29 in 1987, but the scope is limited to alkyl acetylenes only. Although the free-radical addition of aryl and alkyl thiols with various phenyl and alkyl acetylenes in the presence of AIBN has been reported,17b,30 we have noticed no reports concerning the addition of benzyl mercaptans to aryl acetylenes in the presence of Et3B-hexane leading to stereoselective formation of (Z)-styryl benzyl sulfides. Thus, a general and simple method for the synthesis of stereoselective (Z)-styryl benzyl sulfides from alkynes remains a challenge to be addressed. Hence, herein we report the hydrothiolation of aryl acetylenes by benzyl thiols in the presence of Et3B-hexane as a radical initiator, resulting in stereoselective formation of (Z)-styryl benzyl sulfides under mild reaction conditions.
Results and discussion
Synthesis of key starting materials
4-Methoxy-3-nitrobenzyl thiol (3) was synthesized as shown in Scheme 1. 4-Methoxy-3-nitro toluene (1) was brominated with NBS in the presence of a catalytic amount of benzoyl peroxide in CCl4 to obtain 4-methoxy-3-nitrobenzyl bromide (2) which on subsequent reaction with thiourea gave an intermediate isothiouronium salt which on further reduction with ammonia yielded 3 along with its dimer 1,2-bis(4-methoxy-3-nitrobenzyl)-disulfane (4) as a byproduct. 2,4,6-Trimethoxyphenyl acetylene (7) and its propiolic acid derivative 7a were prepared as shown in Scheme 2. Reaction of 2,4,6-trimethoxybenzaldehyde (5) with bromomethyltriphenylphosphonium bromide in the presence of potassium t-butoxide gave 2-(2-bromovinyl)-1,3,5-tri-methoxybenzene (6) which on dehydrobromination with potassium t-butoxide afforded 7 in moderate yields. Com-pound 7a was synthesized starting from 5 and CBr4 in the presence of Ph3P and the resulting 2′,2′-dibromovinyl-1,3,5-trimethoxybenzene (8) was treated with methyl chloroformate and n-BuLi in THF at −78 °C to get methyl ester 9 which on subsequent hydrolysis with lithium hydroxide afforded 7a in low yields.
Scheme 1.
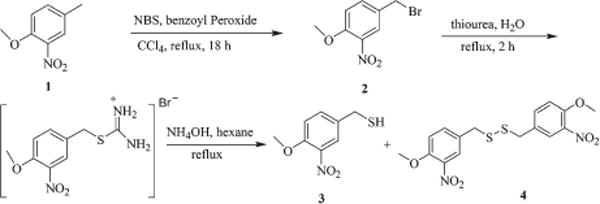
Synthesis of 4-methoxy-3-nitrobenzyl thiol (3).
Scheme 2.
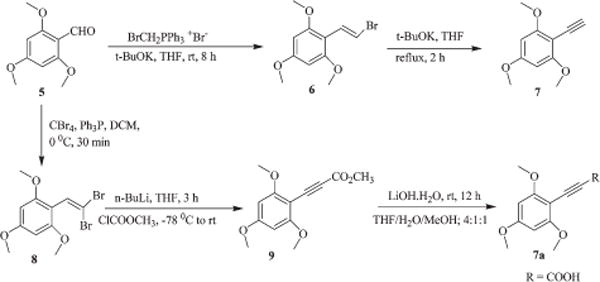
Synthesis of 2,4,6-trimethoxyphenyl acetylene (7) and its propiolic acid derivative (2,4,6-trimethoxyphenyl)propynoic acid (7a).
In our first attempt, we tried different conventional approaches to make 1,3,5-trimethoxy-2-[2-(4-methoxy-3-nitro-benzylsulfanyl)vinyl]benzene (10) by the reaction of 4-methoxy-3-nitro benzyl thiol (3) and 2,4,6-trimethoxyphenyl acetylene (7) in the presence of a number of bases and catalysts (Table 1). Initially, we tested the effect of base on this hydrothiolation reaction. As we have used metallic sodium in ethanol or sodium hydroxide in ethanol in our earlier attempts to make (Z)-vinyl sulfides,31 the same approach was tried for this reaction. Both reactions (entries 1 and 2) did not proceed at reflux temperatures for more than 24 h. Alternatively, with a solid supported Al2O3/KF catalyst at 60 °C, this reaction proceeded to give a mixture of isomers (Z/E = 56 : 44) with yields of less than 10%. We then attempted addition of the thiol with acetylene with a base and catalyst free reaction in the presence of water as the solvent.32 The reaction did not proceed at room temperatures, but at reflux temperatures the reaction produced moderate yields of a mixture of isomers (Z/E = 79 : 21) (entry 4). Reactions with bases such as K2CO3, Et3N and Cs2CO3 in NMP solvent and CuI as catalyst did not yield the addition product (entries 5–7). Recently, coupling reactions initiated by the de-carboxylation of carboxylic acids catalyzed by copper have shown great promise in the synthesis of stereoselective (Z)-vinyl sulfides.33 The reaction of 2,4,6-trimethoxyphenyl acetylene (7) (entry 7) or its propiolic acid derivative (7a) (entry 9) and 3 in the presence of CuI and cesium carbonate did not proceed even after stirring for 24 h at 80–90 °C. Addition of a radical inhibitor such as TEMPO (2,2,6,6-tetramethyl-piperidine-N-oxyl) (entries 8 and 10) resulted only in very poor yields of the product. As hydrothiolation reactions with transition metal catalysts produce predominantly E-isomers, we have not made any attempts to synthesize Z-isomers using these catalysts.
Table 1.
Addition of 4-methoxy-3-nitrobenzyl thiol (3) to 2,4,6-trimethoxyphenyl acetylene (7) or 2,4,6-trimethoxyphenyl propynoic acid (7a) under different conditionsa

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R | Base | Solvent | Catalyst | Temp (°C) | Time (h) | Yieldb (%) | Z/EC |
| 1 | H | Na metal | EtOH | — | 80 | 24 | Traces | Ndd |
| 2 | H | NaOH | EtOH | — | 80 | 12 | 0 | — |
| 3 | H | — | Toluene | Al2O3/KF | 60 | 2 | <10 | 56:44 |
| 4 | H | — | Water | — | 100 | 2 | 40 | 79 : 21 |
| 5 | H | K2CO3 | NMP | CuI | 80 | 24 | Traces | Ndd |
| 6 | H | Et3N | NMP | CuI | 80 | 24 | 0 | — |
| 7 | H | Cs2CO3 | NMP | CuI | 80 | 24 | Traces | Ndd |
| 8 | H | Cs2CO3 + TEMPOe | DMSO | — | 85 | 24 | 10 | 70 : 30 |
| 9 | COOH | Cs2CO3 | NMP | CuI | 90 | 24 | 0 | — |
| 10 | COOH | Cs2CO3 + TEMPOe | DMSO | — | 90 | 4 | <10 | Ndd |
| 11 | H | AIBN | Benzene | — | 80 | 5 | 62 | 79 : 21 |
| 12 | H | AIBN + TEMPOe | Benzene | — | 80 | 5 | 0 | — |
| 13 | H | Et3B-hexanef | Benzene | — | 25 | 2 | 82 | 86 : 14 |
| 14 | H | Et3B-hexanef + TEMPOe | Benzene | — | 25 | 2 | 0 | — |
Unless noted, all the reactions were carried out with 3 (1.0 mmol), 7 or 7a (1.0 mmol), base (1.2 mmol), catalyst (1.2 mmol) in 10 mL of solvent at indicated time and temperature.
Yield of isolated product.
Z/E ratio was determined by the 1H NMR spectrum of crude products.
Not determined.
0.2 mmol was added along with the base.
1.0 M solution from Sigma (Cat # 195030).
As base-catalyzed reactions resulted either in low yields or poor selectivity, we moved to a strategy employing free radical initiators. Addition of 3 to 7 in the presence of AIBN in benzene at 80 °C afforded a mixture of addition products (Z/E = 79 : 21) in 62% yield (entry 11). Encouraged by this result, we attempted the same reaction with Et3B-hexane as the radical initiator which further improved the overall yield and selectivity (Z/E = 86 : 14) (entry 13). This reaction provided very good yields, better stereoselectivity, shorter reaction times and the cleanest product of all the reactions tried. Next, we examined whether this reaction would proceed if free radical formation were suppressed by the addition of 20 mol% of TEMPO. In the presence of TEMPO, the reaction is completely inhibited both in AIBN and in Et3B initiated reactions (entries 12 and 14, respectively), demonstrating that this reaction is mediated by a radical mechanism. We varied the solvents and reaction temperatures in order to establish the optimal conditions for this reaction (Table 2). Among them, non-polar sol-vents such as benzene and toluene yielded addition products with high yields and greater selectivity at room temperatures. Addition of polar solvents such as methanol as an additive to either benzene (entry 3) or toluene (entry 7) completely reversed the stereoselectivity towards the formation of E-isomers with higher overall yields.22d It is known that in polar solvents the proton abstraction from thiol is slow and the formed Z-vinyl radical (10a) (Scheme 3) can easily isomerize to a thermodynamically more stable E-vinyl radical (10b) and then abstracts the hydrogen atom leading to the formation of the E-isomer. This is also supported by the fact that the bond dissociation energy of RS–H would be higher in polar solvents and the formed Z-radical would have enough time to convert to a more stable E-radical.34 The same phenomenon was also observed in reactions with other polar aprotic solvents such as DMSO (entry 15) and DMF (entry 16) where selectivity towards the E-isomer was high with reasonably good yields. Reactions in acetonitrile, 1,4-dioxane, NMP and THF resulted in poor selectivity towards Z-isomer formation, prolonged reaction times and low to moderate yields. When we tried reactions in DCM at 25 °C (entry 10) and at −30 °C (entry 11), the reactions proceeded very rapidly, resulting in very good yields. The selectivity profile of the product formation has changed completely as the reaction temperature changed from 25 °C (Z/E = 58 : 42) to −30 °C (Z/E = 91 : 9) to −78 °C (Z/E = 100 : 0). As hydrothiolation reactions are solvent, temperature and catalyst dependent, a temperature effect was found to be operative in this reaction. The results suggest that the trans addition process giving a cis adduct is presumably a kinetically controlled process in aprotic solvents such as DCM with a high dipole moment leading to the formation of a cis addition product.34b,c,35 Although the reaction proceeded more slowly at lower temperatures, its selectivity improved, resulting in the formation of a single isomer and thereby avoiding the need for separation of isomers. Hence, the addition reaction in DCM at −30 °C or at −78 °C in the presence of Et3B-hexane was chosen as the best method for the preparation of (Z)-styryl benzyl sulfides 10.
Table 2.
Optimization reaction conditions of Et3B-hexane induced radical addition of 4-methoxy-3-nitrobenzyl thiol (3) to 2,4,6-trimethoxyphenyl acetylene (7)a

| |||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yieldb (%) | Z/Ec |
| 1 | Benzene | 25 | 2 | 82 | 86 : 14 |
| 2 | Benzene | 5 | 2 | 75 | 78 : 22 |
| 3 | Benzene + 4 eq. methanold | 25 | 2 | 92 | 0 : 100 |
| 4 | Toluene | 25 | 2 | 85 | 88 : 12 |
| 5 | Toluene | 0 | 3 | 80 | 79 : 21 |
| 6 | Toluene | −20 | 4 | 75 | 86 : 14 |
| 7 | Toluene + 4 eq. methanold | 0 | 2 | 95 | 0 : 100 |
| 8 | Acetonitrile | 25 | 3 | 82 | 72 : 28 |
| 9 | 1,4-Dioxane | 25 | 3 | 60 | 48 : 52 |
| 10 | DCM | 25 | 0.5 | 85 | 58 : 42 |
| 11 | DCM | −30 | 1 | 88 | 91 : 9 |
| 12 | DCM | −78 | 8 | 68 | 100 : 0 |
| 13 | THF | 25 | 4 | 80 | 62 : 38 |
| 14 | NMP | 25 | 2 | 85 | 63 : 37 |
| 15 | DMSO | 25 | 2 | 60 | 8 : 92 |
| 16 | DMF | 25 | 2 | 72 | 28 : 72 |
Unless noted, all the reactions were carried out with 3 (1.2 mmol), 7 (1.0 mmol), base (1.2 mmol) in 10 mL of solvent at indicated time and temperature.
Yield of isolated product.
Z/E ratio was determined by the 1H NMR spectrum of crude products.
Methanol was added to the reaction mixture before Et3B-hexane addition.
Scheme 3.
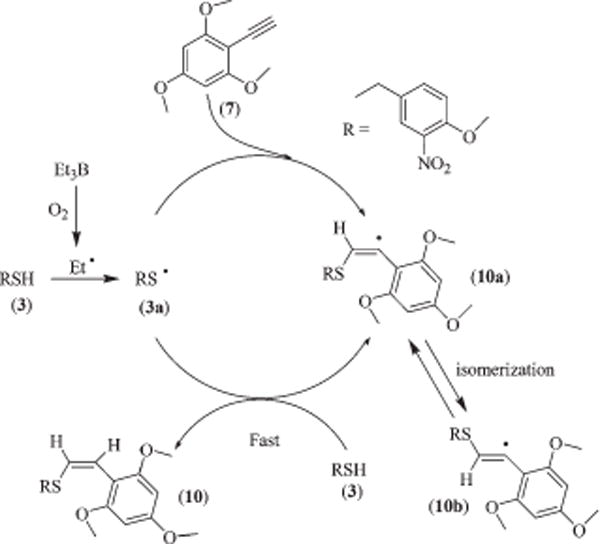
A plausible reaction mechanism of radical addition of the thiol to acetylene.
A possible mechanism for the formation of styryl benzyl sulfides by free radical addition of the thiol to acetylene is out-lined in Scheme 3. At the initiation step, an ethyl radical generated from triethylborane in the presence of molecular oxygen abstracts a hydrogen atom from thiol (3) to form a thiyl radical (3a). As the thiyl radicals are known to behave as electron deficient species, they immediately react with a more electron rich SP-hybridized carbon of the acetylene (7), to furnish a β-sulfanyl radical (10a). Hydrogen abstraction from the starting thiol 3 afforded a styryl benzyl sulfide (10) and a new thiyl radical 3a is regenerated to complete the radical chain process.34b,36 Although, the carbon–sulfur bond formation occurs regioselectively at the β-position of the acetylene where higher electron density resides, there is a possibility of the formation of a mixture of E- and Z-stereoisomers. Though the isomerization between Z- and E-sulfanyl radicals (10a and 10b) is faster than the abstraction of the proton from thiol 3, the rate of interconversion would be largely determined by steric hindrance between their cis-2-substituents (i.e. RS or H) and a radical scavenger rather than by their equilibrium position.30,34c As the cis-2-substituent H of the Z-sulfanyl radical (10a) is smaller than the cis-2 substituent RS of the E-sulfanyl radical (10b), 10a preferentially abstracts hydrogen from 3 leading to the formation of Z-styryl benzyl sulfide (10).30,36 In light of the proposed mechanism and based on the experimental results from Tables 1 and 2, it may be concluded that the addition of 3 to 7 under radical reaction conditions proceeds preferably in trans fashion to afford Z-styryl benzyl sulfides, although the exact stereo chemical course of these addition reactions remains uncertain due to the influence of reaction conditions (e.g. temperature and solvent).
Oxidation of sulfides to sulfoxides and sulfones
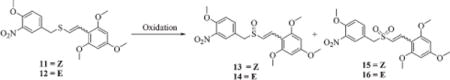
After separating the mixture of Z/E-styryl benzyl sulfide 10 to pure (Z)-styryl benzyl sulfide (11) and (E)-styryl benzyl sulfide (12) by column chromatography, we oxidized them to their cor-responding sulfoxides and sulfones. Several oxidizing agents were tried to optimize the oxidation process to achieve better yields and selectivity without isomerization (Table 3) of the product. Among the reagents used, oxidation of 11 with 30% hydrogen peroxide in glacial acetic acid at room temperature resulted in a decomposed product (entry 1), whereas at 5 °C both 11 and 12 afforded corresponding sulfoxides 13 and 14, respectively, in good yields (entries 3 and 4). Oxidation of 12 with hydrogen peroxide in glacial acetic acid at room temperature for 24 h gave the sulfone (16) in moderate yields (entry 2), whereas when the same reaction was conducted at 60 °C for 6 h sulfone (16) was obtained in much higher yields in shorter reaction times (entry 5). Oxidation of the Z-isomer 11, with m-chloroperoxybenzoic acid (m-CPBA) at room temperature, resulted in decomposition of the product (entry 6) whereas oxidation of the E-isomer 12 under the same conditions gave sulfone 16 in moderate yields (entry 7). Due to poor yields and sensitivity to harsh reaction conditions, oxidation of the Z-isomer was attempted under mild reaction conditions using Oxone in THF/MeOH. With 4 equivalents of Oxone, the Z-sulfide 11 underwent partial oxidation to afford a mixture of sulfoxide (13) and sulfone (15) in a 15/85 ratio (entry 8), whereas the reaction with 6 equivalents of Oxone resulted in complete oxidation to sulfone 15 in relatively good yields (entry 9). When 1,1,1,3,3,3-hexafluoro-2-propanol and 30% H2O2 in THF was used for oxidation of 11, only sulfoxide (13) was formed in moderate yields (entry 10). Thus, from these observations, we conclude that the use of Oxone at room temperature for the oxidation of 11 to Z-sulfones and the use of H2O2 in AcOH at 5 °C for making Z-sulfoxides are the best methods of oxidation for making the sulfones and sulfoxides, respectively. Similarly, oxidations with H2O2 in AcOH at 60 °C and H2O2/AcOH at 5 °C are the best methods for converting (E)-vinyl sulfides (12) into their corresponding sulfones and sulfoxides, respectively. In all these reactions the starting compound geometry was not affected by the oxidation.
Table 3.
Oxidation of (Z) and (E)-1,3,5-trimethoxy-2-[2-(4-methoxy-3-nitrobenzylsulfanyl)vinyl]benzene (11 and 12)
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compd | Oxidizing agent (eq.) | Solvent | Temp (°C) | Time (h) | Producta | Yieldb (%) |
| 1 | 11 | H2O2 | AcOH | rt | 8 | — | —c |
| 2 | 12 | H2O2 | AcOH | rt | 24 | 16 | 40 |
| 3 | 11 | H2O2 | AcOH | 5 | 6 | 13 | 55 |
| 4 | 12 | H2O2 | AcOH | 5 | 6 | 14 | 75 |
| 5 | 12 | H2O2 | AcOH | 60 | 6 | 16 | 85 |
| 6 | 11 | m-CPBA (2.5) | MeOH | rt | 12 | — | —c |
| 7 | 12 | m-CPBA (2.5) | MeOH | rt | 24 | 16 | 50 |
| 8 | 11 | Oxonee (4) | THF/MeOH (2 : 1) | rt | 24 | 13 & 15d | 55 |
| 9 | 11 | Oxonee (6) | THF/MeOH (2 : 1) | rt | 24 | 15 | 65 |
| 10 | 11 | H2O2 | Hexafluoro-2-propanol | rt | 2 | 13 | 40 |
Starting compound geometry was retained in the product.
Yield of isolated product.
Starting compound decomposed.
Partial oxidation leading to mixture of sulfoxide and sulfone (ratio:15/85).
Oxone was dissolved in water (2 mL g−1) and then added to the reaction mixture.
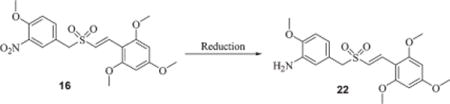
Reduction of nitro styryl benzyl sulfides, sulfoxides and sulfones into their corresponding amino compounds
The reduction of nitro styryl benzyl sulfides, sulfoxides and sulfones to their corresponding amino compounds was tried with various reducing agents to optimize the product yields and to reduce the formation of byproduct impurities (Table 4). Optimization of the reaction conditions for reduction of the nitro compounds was initiated using compound 16. Reduction of the nitro group in 16 with sodium hydrosulfite in acetone/water (2 : 1) at 50 °C gave a clean reduced product 22 with very low yields (entry 1) whereas the reduction with Zn/AcOH at room temperature afforded moderate yields of the amino compound 22 (entry 2). Reduction of 16 with RANEY® Ni/NH2NH2 in MeOH at 40 °C or indium metal/NH4Cl in EtOH at 80 °C or SnCl2 in EtOH at room temperature resulted in the formation 22 in moderate to good yields (entries 3–5). Reduction of the nitro compound 16 with metallic iron powder in AcOH/MeOH (1 : 2) at reflux temperatures gave corresponding amine 22 in very high yields (entry 7). Reduction with 3% Pt/C in acetonitrile at 60 °C in the presence of H2 also resulted in the formation of the corresponding amine compound with yields comparable to that of the iron method (entry 8). Based on the cost and effectiveness of all the reducing agents, the use of iron powder for the reduction of the nitro group in sulfides, sulfoxides and sulfones was considered to be the economically best choice among the reducing agents used. We extended this optimized reduction condition with the iron catalyst for reducing the nitro sulfides 11 and 12 to their corresponding amino sulfides 17 and 18, nitro sulfoxides 13 and 14 to their corresponding amino sulfoxides 19 and 20 and nitro sulfone 15 to amino sulfone 21. The starting compound geometry was not affected by the reduction conditions.
Table 4.
Reduction of 3-nitro (E)-styryl benzyl sulfone (16) to 3-amino (E)-styryl benzyl sulfone (22)
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time (h) | Yielda (%) |
| 1 | Na2S2O4 | Acetone/water (2 : 1) | 50 | 1 | 20 |
| 2 | Zn | AcOH | 25 | 3 | 40 |
| 3 | RANEY® Ni/NH2NH2 | MeOH | 40 | 4 | 50 |
| 4 | Indium/NH4Cl | EtOH | 80 | 5 | 50 |
| 5 | SnCl2 | EtOH | 25 | 4 | 40 |
| 7 | Fe(0) | AcOH/MeOH (1 : 2) | 80 | 3 | 95 |
| 8b | 3% Pt/C (sulfided)/H2 | CH3CN | 60 | 3 | 74 |
Yield of isolated product.
Pt catalyst purchased from Evonik Degussa Corporation, KY, USA.
Synthesis of (Z) and (E)-isomers of ON 01910·Na (25 and 26)
In order to enhance the solubility and bio-availability of these molecules, the (Z) and (E)-amino sulfones 21 and 22 were alkylated to their corresponding glycyl esters 23 and 24 with methyl 2-bromoacetate in the presence of a mild base sodium acetate, which on subsequent hydrolysis afforded the corresponding highly water soluble sodium salt of acids 25 (Z-ON 01910·Na) and 26 (E-ON 01910·Na) (Scheme 4).
Scheme 4.
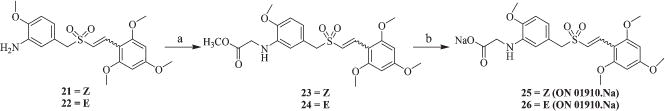
Synthesis of glycine esters and sodium salt of acids 25 and 26: (a) BrCH2COOCH3, MeOH, CH3COONa, 4 h; (b) NaOH, EtOH, 60–65 °C, EtOAc, H2O, rt, 2–3 h.
In vitro cytotoxicity assay
The potency of the (Z) and (E)-isomers synthesized was measured in cell viability studies using two different human tumor cell lines: DU-145 prostate cancer cells and K562 leukemic cells. These cell lines were selected as a benchmark for the initial routine screening of all compounds synthesized in our laboratory. In a recent paper,9 we described in detail the synthesis, structure–activity relationship and biological activity of some (E)-styryl benzyl sulfones. These studies confirmed that the nature, number, and position of substituents on the two aromatic rings of the parent molecule are the determining factors for the biological activity of these compounds. In the present report, we investigated the effect of the geometry on the cytotoxicity of these molecules, as the geometrical isomerism in a molecule plays a crucial role in the biological activity of the compound. The results presented in Table 5 showed that in general all E-isomers showed superior cytotoxic activity compared to Z-isomers. The data from the table show that all of the 3-nitro substituted Z and E-isomers are much less potent than their corresponding 3-amino analogs. Next we examined the contribution of the oxidative state of sulfur in the molecules with regard to biological activity. Analysis of the biological data presented in the table reveals that the sulfides are less active than the sulfoxides and the sulfoxides are, in turn, less active than the sulfones, indicating that the complete oxidative state of sulfur is required for attaining the optimum activity in these molecules. In terms of the role of geometry in the biological activity, E-isomers are more potent than their corresponding Z-isomers.
Table 5.
In vitro cytotoxicity of (Z) and (E)-styryl benzyl sulfides, sulfoxides and sulfones
| Compd | Structure | Isomer | K562b IC50a (μ M) |
DU145c IC50a (μ M) |
|---|---|---|---|---|
| 11 |
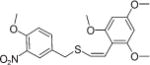
|
Z | 15 ± 0.9 | 35 ± 2.1 |
| 12 |
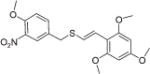
|
E | 5 ± 0.2 | 5 ± 0.2.8 |
| 13 |
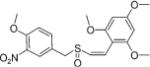
|
Z | 75 ± 5.0 | >100 |
| 14 |
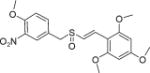
|
E | 15 ± 0.7 | 17 ± 0.6 |
| 15 |
|
Z | 15 ± 0.6 | 15 ± 0.75 |
| 16 |
|
E | 2.5 ± 0.15 | 2.5 ± 0.1 |
| 17 |
|
Z | 0.25 ± 0.01 | 0.75 ± 0.04 |
| 18 |
|
E | 0.03 ± 0.002 | 0.05 ± 0.003 |
| 19 |
|
Z | 5 ± 0.3 | 5 ± 0.2 |
| 20 |
|
E | 0.02 ± 0.0015 | 0.04 ± 0.002 |
| 21 |
|
Z | 0.15 ± 0.006 | 0.15 ± 0.005 |
| 22 |
|
E | 0.003 ± 0.0002 | 0.003 ± 0.0001 |
| 23 |
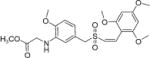
|
Z | 40 ± 2.0 | 38 ± 2.2 |
| 24 |
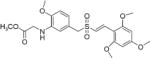
|
E | 0.1 ± 0.004 | 0.1 ± 0.002 |
| 25 |
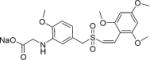
|
Z | 37 ± 1.5 | 35 ± 2.0 |
| 26 |
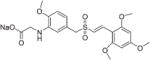
|
E | 0.0075 ± 0.0003 | 0.08 ± 0.004 |
IC50: represents the compound concentration (μM) that is lethal to survival of 50% of tumor cells following 96 h treatment with the tested compound; values represent the average ± SD of two experiments.
K562: myelogenous leukemia.
DU145: human prostate cancer.
The compounds 21 and 22, which have high potency, also have very low solubility in aqueous buffers and solutions. In order to enhance their water solubility and bioavailability, amino groups in 21 and 22 were converted to glycine derivatives 23 and 24. As shown in Table 5, glycyl esters of the Z-isomers are less active than those of the E-isomers. Similarly the sodium salt of (Z)-ON 01910·Na (25) is approximately 5000 times less potent in killing K562 cells and 437 times less potent in killing DU145 cells than the corresponding salt of (E)-ON 01910·Na (26). From this study, it is evident that apart from the nature and position of the substituents in a given molecule, the geometry of the molecule also contributes to its biological activity. As the biological activity of the molecule depends upon its ability to bind to a cellular target, the orientation of the groups within the molecule ultimately determines binding affinity. The trimethoxy styryl group in the trans-configuration may have a better orientation with which to interact with a given target than the cis-isomer. Co-crystal studies of these molecules with their targets would demonstrate these interactions more clearly and also explain the superior biological activity of the E-isomer over the Z-isomer. As the E-configuration of the molecule proved to be essential for biological activity, the inactive Z-isomer could be used as a negative control in the biological assays for determining the mechanism of action of these molecules.
Conclusions
In this communication, we described the Et3B-hexane catalyzed radical hydrothiolation of 2,4,6-trimethoxyphenyl acetylene as a highly efficient method for the synthesis of (Z)-styryl benzyl sulfides where base catalyzed hydrothiolations have failed. Of the various methods tried, only reactions performed in the presence of radical initiators such as AIBN and Et3B offered good yields with better Z-selectivity. Among these, Et3B-hexane in DCM afforded the best yields with very good selectivity towards the Z-isomer. Optimum reaction conditions for the stereoselective synthesis of (Z)-styryl benzyl sulfides in the presence of Et3B-hexane were established with different solvents, temperatures and reaction times. We observed that lower temperatures favor Z-isomer formation. The conditions for oxidation of sulfides to sulfoxides and sulfones and reduction of nitro compounds to amines were optimized by trying various reagents under different reaction conditions. Among the various reducing agents tried, reduction with iron powder was found to be more cost effective producing the product in greater than 90% yield. To increase the solubility and bio-availability of 21 and 22, they were converted to glycine analogs 25 and 26. In the biological assay, all the E-isomers have shown superior biological efficacy compared to Z-isomers, suggesting that the geometry of the molecule also contributes to potency apart from the nature of the substituents on the aromatic ring.
Experimental
General procedures
All reagents and solvents were obtained from commercial suppliers and used without further purification unless otherwise stated. Solvents were dried using standard procedures and reactions requiring anhydrous conditions were performed under a N2 atmosphere. Reactions were monitored by Thin Layer Chromatography (TLC) on pre-coated silica gel F254 plates (Sigma-Aldrich) with a UV indicator. Column chromatography was performed with Merck 70–230 mesh silica gel 60 Å. Yields were of purified product and were not optimized. Melting points were determined using an Electrothermal Mel-Temp® 3.0 micro melting point apparatus and are un-corrected. Nuclear magnetic resonance spectra were recorded on a Bruker Avance III (300 MHz, 1H NMR and 75 MHz, 13C NMR) Spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; dd, doublet of a doublet; t, triplet; m, multiplet; br s, broad singlet. Coupling constants (J values) were measured in hertz (Hz). Compound purity was determined by LC/MS analysis and was confirmed to be >95% for all compounds. All LC/MS data were gathered on an Agilent 1200 LC with Agilent 6410 triple quadrupole mass spectrometer detectors. The compound solution was infused into the electrospray ionization source operating positive and negative modes in methanol/water/trifluoroacetic acid (50 : 50 : 0.1% v/v) at 0.4 mL min−1. The sample cone (declustering) voltage was set at 100 V. The instrument was externally calibrated for the mass range m/z 100 to m/z 1000.
4-Methoxy-3-nitrobenzyl bromide (2)
To a stirred solution of 4-methoxy-3-nitro toluene (1, 10 g, 60 mmol) in carbon tetrachloride (200 mL) were added benzoyl peroxide (1.45 g, 6.0 mmol) and N-bromosuccinimide (12.8 g, 72 mmol). The reaction mixture was heated at reflux for 18 h and after completion of the reaction (TLC monitoring, hexane/ethyl acetate, 9 : 1 on silica gel plate), the contents were cooled to room temperature, water was added, and the product was isolated by extraction with dichloromethane. The organic phase was washed with water, brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the crude product which on purification by silica gel flash column chromatography afforded pure 2. The yield of this reaction was 60%, giving a light yellow solid with a melting point of 106–108 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.99 (s, 3H, OCH3), 4.49 (s, 2H, CH2), 7.09 (d, J = 8.7 Hz, 1H, Ar-H), 7.60 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.91 (d, J = 2.4 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 31.3, 56.7, 113.9, 126.3, 130.2, 134.8, 139.4, 152.9; HRMS (ESI) m/z calcd for C8H8BrNO3 [M + H] 245.9688, found 245.9682.
4-Methoxy-3-nitrobenzyl thiol (3) and 1,2-bis(4-methoxy-3-nitrobenzyl)disulfane (4)
A solution of 4-methoxy-3-nitroben-zyl bromide (2, 10.0 g, 41 mmol) and thiourea (10.0 g, 131 mmol) in 50 mL water was heated under reflux for 2 h. The reaction mixture was cooled and stirred at room temperature for 2 h and the solid was filtered. The resultant isothiouronium salt was decomposed by boiling several times with concentrated ammonia and hexane (100 mL, 15 : 85). Concentration of the combined hexane extracts provided a mixture which on further purification with silica gel flash column chromatography (hexane/ethyl acetate, 4 : 1) yielded pure 3 and 4. The yield of this reaction was 78%.
(3) A yellow solid with a melting point of 48–49 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 1.74 (t, J = 7.8 Hz, SH), 3.67 (d, J = 7.8 Hz, 2H, CH2), 3.89 (s, 3H, OCH3), 6.98 (d, J = 8.7 Hz, 1H, Ar-H), 7.45 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.76 (d, J = 2.1 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 27.7, 56.7, 113.8, 125.2, 133.5, 133.8, 139.4, 152.0; HRMS (ESI) m/z calcd for C8H9NO3S [M − H] 198.0303, found 198.0320.
(4) A yellow solid with a melting point of 108–110 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.64 (s, 4H, (CH2)2), 3.99 (s, 6H, (OCH3)2), 7.08 (d, J = 8.4 Hz, 2H, Ar-H), 7.43 (dd, J = 8.7, 2.1 Hz, 2H, Ar-H), 7.71 (d, J = 2.4 Hz, 2H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 41.6, 56.6, 113.7, 126.4, 129.7, 135.0, 139.2, 152.4; HRMS (ESI) m/z calcd for C16H16N2O6S2 [M − H] 395.0450, found 395.0446.
2-(2-Bromovinyl)-1,3,5-trimethoxybenzene (6)
To a solution of 2,4,6-trimethoxybenzaldehyde (5, 5.0 g, 25.5 mmol) and bromomethyltriphenylphosphonium bromide (13 g, 30.6 mmol) in dry tetrahydrofuran (THF) (100 mL) at 0 °C potassium t-but-oxide (32 mL of 1 M solution in THF, 33 mmol) was added slowly and the resulting mixture was continuously stirred at this temperature for 1 h and at room temperature for 8 h. After completion of the reaction, THF was removed under reduced pressure and the residue was extracted with ethyl acetate. The organic phase was washed with water and brine, dried over anhydrous sodium sulfate, and concentrated under vacuum to obtain the crude product which on purification by silica gel flash column chromatography afforded pure 6. The yield of this reaction was 68%, giving a light yellow solid with a melting point of 104–105 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.84 (s, 3H, OCH3), 3.85 (s, 6H, 2X OCH3), 6.13 (s, 2H, Ar-H), 7.10 (d, J = 13.8 Hz, 1H, ═CH), 7.41 (d, J = 13.8 Hz, 1H, ═CH); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.3, 55.7, 90.6, 106.6, 108.0, 127.4, 158.9, 160.7; HRMS (ESI) m/z calcd for C11H13BrO3 [M + H] 274.1231, found 274.1244.
2,4,6-Trimethoxyphenyl acetylene (7)
To a solution of 2-(2-bromovinyl)-1,3,5-trimethoxy-benzene (6, 4.6 g, 17 mmol) dissolved in THF (100 mL), potassium t-butoxide (35 mL of 1 M solution in THF, 34 mmol) was added slowly and the reaction mixture was maintained at reflux for 2 h. After completion of the reaction, the potassium bromide salts were filtered and washed with an additional 20 mL of THF. All filtrates were then combined, concentrated under reduced pressure, and the crude product was purified by flash column chromatography (hexane/ethyl acetate, 9 : 1) to afford pure 7. The yield of this reaction was 65%, giving a white solid with a melting point of 119–122 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.52 (s, 1H, CH), 3.85 (s, 3H, OCH3), 3.90 (s, 6H, 2×OCH3), 6.12 (s, 2H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 56.1, 83.8, 90.4, 93.1, 161.9, 163.0; HRMS (ESI) m/z calcd for C11H12O3 [M + H] 193.0786, found 193.0764.
2-(2′,2′-Dibromovinyl)-1,3,5-trimethoxybenzene (8)
To a sol-ution of 2,4,6-trimethoxybenzaldehyde (5, 5.0 g, 25.5 mmol) and triphenyl phosphine (13.37 g, 51.0 mmol) in anhydrous dichloromethane (60 mL) was added a solution of tetrabromomethane (9.8 g, 30.0 mmol) in dichloromethane (10 mL), keeping the temperature below 5 °C. The reaction mixture was stirred for an additional 30 min at this temperature. After completion of the reaction, monitored by TLC (hexane/ethyl acetate, 9 : 1 on silica gel plate), the reaction mixture was filtered and concentrated in vacuo. The residue was purified by flash column chromatography (hexane/ethyl acetate, 9 : 1) to afford pure 8. The yield of this reaction was 72%, giving a white solid with a melting point of 128–130 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 6H, 2× OCH3), 3.85 (s, 3H, OCH3), 6.13 (s, 2H, Ar-H), 7.21 (s, 1H, CH═); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 55.7, 90.5, 92.7, 106.8, 130.9, 158.0, 161.7; HRMS (ESI) m/z calcd for C11H12Br2O3 [M + H] 352.9133, found 352.9112.
(2,4,6-Trimethoxyphenyl)propynoic acid methyl ester (9)
To a solution of 2-(2-bromovinyl)-1,3,5-trimethoxybenzene (8, 1 g, 2.8 mmol) in dry THF (10 mL) cooled to −78 °C, n-butyllithium (0.6 mL of 2.5 M solution in hexane, 6.2 mmol) was added drop wise. The resulting mixture was continuously stirred at this temperature for 15 min and then the temperature was raised to room temperature and the solution was stirred for an additional 1 h. Methyl chloroformate (0.32 g, 3.4 mmol) was then added to the reaction mixture at −78 °C drop wise and the mixture was continuously stirred at room temperature for 3 h. After completion of the reaction, the contents were quenched with saturated ammonium chloride solution and extracted with ethyl acetate. The combined organic layers were dried and the solvent was removed under reduced pressure. The crude product obtained was purified by flash chromatography to afford pure 9. The yield of this reaction was 42%, giving a white solid with a melting point of 85–86 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 3H, COOCH3), 3.86 (s, 3H, OCH3), 3.88 (s, 6H, 2× OCH3), 6.08 (s, 2H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 29.7, 52.5, 55.5, 56.1, 56.6, 81.7, 87.8, 90.3, 90.4, 91.0, 155.0, 164.0, 164.3; HRMS (ESI) m/z calcd for C13H14O5 [M + H] 251.0841, found 251.0845.
(2,4,6-Trimethoxyphenyl)propynoic acid (7a)
To a solution of 2,4,6-trimethoxyphenyl-propynoic methyl ester (9, 0.2 g, 0.8 mmol) in 5 mL of THF/water/methanol (4 : 1 : 1), lithium hydroxide monohydrate (50 mg, 1.2 mmol) was added at room temperature and the resulting mixture was continuously stirred for 12 h. After completion of the reaction, 10 mL of ethyl acetate was added and the pH was adjusted to 4 with 1 N hydrochloric acid. The reaction contents were extracted with ethyl acetate, dried over anhydrous sodium sulfate, and filtered and the solvent was removed under reduced pressure. Flash column chromatography of the crude product gave pure 7a. The yield of this reaction was 55%, giving a white solid with a melting point of 128–130 °C; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 3.75 (s, 9H, 3× OCH3), 6.28 (s, 2H, Ar-H), 13.19 (br s, 1H, COOH); 13C NMR (75 MHz, CDCl3) δ (ppm): 32.5, 55.4, 55.5, 55.8, 56.0, 82.1, 90.6, 91.0, 158.4, 159.4, 162.7, 164.3, 201.8; HRMS (ESI) m/z calcd for C12H12O5 [M − H] 235.0685, found 235.0689.
Et3B-hexane mediated addition of 4-methoxy-3-nitrobenzyl thiol (3) to 2,4,6-trimethoxyphenyl acetylene (7): a typical procedure for the synthesis of 10
A hexane solution of triethylborane (0.18 mL, 1.2 mmol) was added to a solution of 2,4,6-trimethoxyphenyl acetylene (7, 0.2 g, 1.0 mmol) and 4-methoxy-3-nitrobenzyl thiol (3, 0.24 g, 1.2 mmol) in dichloromethane (10 mL) at −30 °C. The resulting mixture was stirred for 1 h at −30 °C. After completion of the reaction monitored by TLC (hexane/ethyl acetate, 9 : 1 on a silica gel plate), the reaction mixture was quenched with 1 molar ammonium chloride solution and extracted with dichloromethane. The organic phase was washed with water, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo and the residue was purified by column chromatography (hexane/ethyl acetate, 9 : 1) resulting in a stereo isomeric mixture (10, 0.34 g, Z/E = 91 : 9) with 88% yield as a white solid with m.p.: 98–99 °C; the Z/E ratio was determined by 1H NMR spectroscopy; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.84 (s, 12H, 4× OCH3), 3.96 (s, 2H, CH2S), 6.12 (s, 2H, Ar-H), 6.15 (s, 2H, Ar-H), 6.22 (d, J = 10.2 Hz, 1H, CH═), 6.48 (d, J = 10.5 Hz, 1H,═CH), 6.84 (d, J = 15.6 Hz, CH═), 6.97–7.08 (m, Ar-H and ═CH), 7.51–7.62 (m, Ar-H), 7.83–7.89 (m, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 37.0, 55.3, 55.6, 56.6, 90.4, 106.7, 113.6, 120.7, 126.0, 126.0, 131.0, 134.5, 152.0, 158.2, 161.2; HRMS (ESI) m/z calcd for C19H21NO6S [M + H] 392.1090, found 392.1099.
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (11)
This compound was prepared from mixture 10 by column chromatographic separation as a white solid with a melting point of 102–104 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.84 (s, 12H, 4× OCH3), 3.96 (s, 2H, CH2S), 6.15 (s, 2H, Ar-H), 6.21 (d, J = 10.5 Hz, 1H, CH═), 6.47 (d, J = 10.5 Hz, 1H, ═CH), 7.03 (d, J = 8.4 Hz, 1H, Ar-H), 7.52 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.83 (d, J = 2.4 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 37.0, 55.3, 55.6, 56.6, 90.4, 106.8, 113.6, 120.7, 125.9, 126.1, 131.0, 134.5, 139.5, 152.0, 158.2, 161.2; HRMS (ESI) m/z calcd for C19H21NO6S [M + H] 392.1090, found 392.1084.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (12)
This compound was prepared from mixture 10 by column chromatographic separation as a white solid with a melting point of 121–123 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.82 (s, 9H, 3× OCH3), 3.94 (s, 3H, OCH3), 3.96 (s, 2H, CH2S), 6.12 (s, 2H, Ar-H), 6.84 (d, J = 15.6 Hz, 1H, CH═), 6.99 (d, J = 15.6 Hz, 1H, ═CH), 7.06 (d, J = 8.7 Hz, 1H, Ar-H), 7.59 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.89 (d, J = 2.1 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 36.2, 55.3, 55.7, 56.6, 90.7, 107.3, 113.8, 121.0, 123.1, 125.9, 131.0, 134.7, 139.2, 152.0, 158.8, 160.0; HRMS (ESI) m/z calcd for C19H21NO6S [M + H] 392.1090, found 392.1088.
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfoxide (13)
Method A
A solution of (Z)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (11, 0.5 g, 1.3 mmol) in 40 mL of glacial acetic acid was cooled to 5 °C on an ice bath. Hydro-gen peroxide 30% w/v (0.20 mL, 2.0 mmol) was added and the mixture was stirred at 5 °C for 6 h. After completion of the reaction, 10% sodium hydrogen carbonate solution (25 mL) was added slowly and stirred for 10 min and extracted with ethyl acetate. The organic layer was washed with water (2 × 25 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (hexane/ethyl acetate) to obtain the title compound 13 as a white solid (0.29 g, 55% yield) with m.p.: 121–123 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.76 (s, 6H, 2× OCH3), 3.78 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.90 (d, 2H, CH2S), 5.97 (d, J = 10.8 Hz, 1H, CH═), 6.05 (s, 2H, Ar-H), 6.84 (d, J = 10.8 Hz, 1H, ═CH), 6.98 (d, J = 8.7 Hz, 1H, Ar-H), 7.45 (dd, J = 8.4, 2.1 Hz, 1H, Ar-H), 7.66 (d, J = 2.1 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 55.7, 56.6, 90.5, 104.8, 113.6, 123.0, 127.4, 130.1, 133.7, 136.4, 139.2, 152.9, 158.3, 162.5; HRMS (ESI) m/z calcd for C19H21NO7S [M + H] 408.1039, found 408.1046.
Method B
An aqueous 30% hydrogen peroxide (0.28 mL, 2.6 mmol) was added to a stirred solution of (Z)-2′,4′,6′-tri-methoxystyryl-4-methoxy-3-nitrobenzyl sulfide 11 (0.50 g, 1.3 mmol) in 1,1,1,3,3,3-hexafluoro-2-propanol (5 mL) at rt. The reaction was monitored by TLC. After the complete disappearance of the sulfide (2 h), the excess hydrogen per-oxide was quenched with saturated sodium sulfite (Na2SO3) solution (5.0 mL) and the fluorous organic phase containing the sulfoxide was separated. After removal of the solvent in vacuo, sulfoxide 13 was obtained as a semisolid. Flash column chromatography (chloroform) gave pure 13. The yield of this reaction was 40%. The analytical data of the compound are in accord with the compound synthesized by method A.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfoxide (14)
The title compound was obtained by the oxidation of 12 following the procedure as described in compound 13 (Method A). Yield, 75%; white solid, m.p.: 148–150 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 6H, 2× OCH3), 3.84 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.05 (d, J = 12.9 Hz, 2H, CH2S), 6.10 (s, 2H, Ar-H), 7.04 (d, J = 15.6 Hz, 1H, CH═), 7.09 (s, 1H, Ar-H), 7.41 (d, J = 15.9 Hz, 1H, ═CH), 7.53 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.78 (d, J = 2.4 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 55.7, 56.6, 58.9, 90.4, 104.7, 113.6, 122.4, 127.5, 128.5, 129.4, 136.5, 139.2, 152.8, 160.7, 162.8; HRMS (ESI) m/z calcd for C19H21NO7S [M + H] 408.1039, found 408.1032.
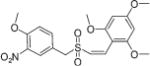
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfone (15)
Compound (Z)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitro-benzyl sulfide (11, 0.4 g, 1.0 mmol) was dissolved in methanol/THF (1 : 1, 20 mL) and cooled to 0 °C. A solution of Oxone® (1.84 g, 6 mmol in 10 mL of water) was added drop wise over 10–15 min at 0 °C. After stirring for 24 h at rt, the reaction mixture was partitioned between dichloromethane (DCM) and water. The aqueous layer was back-extracted, and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product was purified by column chromatography (hexane/ethyl acetate) to obtain the title compound 15 as a white solid (0.27 g, 65% yield) with m.p.: 154–156 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.74 (s, 6H, 2× OCH3), 3.76 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 4.12 (s, 2H, CH2S), 6.05 (s, 2H, Ar-H), 6.09 (d, J = 11.4 Hz, 1H, CH═), 7.00 (d, J = 8.7 Hz, 1H, Ar-H), 7.02 (d, J = 11.7 Hz, 1H, ═CH), 7.54 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.78 (d, J = 2.1 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 55.6, 56.7, 58.9, 90.3, 103.5, 113.9, 120.7, 125.8, 128.1, 135.7, 136.8, 139.4, 153.3, 158.8, 162.9; HRMS (ESI) m/z calcd for C19H21NO8S [M + H] 424.0988, found 424.0994.
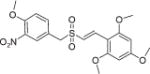
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfone (16)
A solution of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (12, 0.5 g, 1.3 mmol) in glacial acetic acid (40 mL) was taken in a 100 mL round-bottomed flask and hydrogen peroxide 30% w/v (0.20 mL, 2.0 mmol) was added in portions at frequent intervals. Then the reaction mixture was stirred at 60 °C for 6 h. After completion of the reaction monitored by TLC (chloroform/methanol, 9 : 1 on silica gel plate), the reaction mixture was concentrated under vacuum and poured into ice water. The solid formed was filtered, washed with water and dried and the resulting crude product was purified by column chromatography (chloroform/methanol, 9 : 1) to obtain the title compound 16 as a white solid (0.46 g, 85% yield) with m.p.: 184–186 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.84 (s, 6H, 2× OCH3), 3.86 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.23 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 7.03 (d, J = 15.6 Hz, 1H, ═CH), 7.10 (d, J = 8.7 Hz, 1H, Ar-H), 7.63 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 7.80 (d, J = 15.6 Hz, 1H, CH═), 7.85 (d, J = 2.1 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.5, 55.8, 56.7, 60.5, 90.4, 103.6, 113.8, 121.4, 121.4, 128.0, 136.8, 136.9, 139.3, 153.2, 161.6, 164.2; HRMS (ESI) m/z calcd for C19H21NO8S [M + H] 424.0988, found 424.0982.
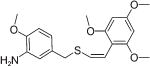
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfide (17)
A solution of (Z)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-nitrobenzyl sulfide (11, 0.5 g, 1.3 mmol) in methanol/acetic acid (2 : 1, 30 mL) was heated to 60 °C and iron powder (0.36 g, 6.5 mmol) was added and the resulting mixture was continuously stirring at 80 °C for 3 h. After completion of the reaction, the contents were cooled to 0 °C, DCM (30 mL) was added and the acetic acid was neutralized with 10% sodium hydroxide solution (10 mL). The contents were stirred for 15 min and the organic phase was separated, washed with water and brine, and dried over anhydrous sodium sulfate. Removal of the solvent under reduced pressure gave the crude product which on purification by silica gel flash column chromatography (ethyl acetate/hexane) afforded pure 17 as a white solid (0.44 g, 95% yield) with m.p.: 90–92 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.67 (s, 2H, CH2), 3.74 (s, 6H, 2× OCH3), 3.75 (s, 6H, 2× OCH3), 6.06 (s, 2H, Ar-H), 6.25 (d, J = 10.5 Hz, 1H, CH═), 6.32 (d, J = 10.5 Hz, 1H, ═CH), 6.61 (d, J = 1.8 Hz, 2H, Ar-H), 6.65 (d, J = 1.5 Hz, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 38.2, 55.3, 55.6, 90.4, 107.2, 110.2, 115.7, 118.7, 119.1, 127.7, 130.6, 136.1, 146.5, 158.2, 160.9; HRMS (ESI) m/z calcd for C19H23NO4S [M + H] 362.1348, found 362.1357.
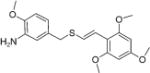
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfide (18)
The title compound was obtained by the reduction of 12 following the procedure as described in compound 17. Yield, 94%; white solid, m.p.: 106–108 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.73 (s, 9H, 3× OCH3), 3.76 (s, 3H, OCH3), 3.80 (s, 2H, CH2), 6.03 (s, 2H, Ar-H), 6.65–6.76 (m, 3H, Ar-H and ═CH), 7.00 (d, J = 15.9 Hz, 1H, CH═), 7.19 (s, 1H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 22.7, 29.7, 31.9, 37.2, 55.3, 55.6, 55.7, 90.8, 107.9, 110.2, 115.6, 118.6, 119.0, 125.5, 130.5, 136.2, 146.5, 158.6, 159.5; HRMS (ESI) m/z calcd for C19H23NO4S [M + H] 362.1348, found 362.1345.
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfoxide (19)
The title compound was obtained by the reduction of 13 following the procedure as described in compound 17. Yield, 95%; white solid, m.p.: 108–109 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.69 (s, 3H, OCH3), 3.74 (s, 6H, 2× OCH3), 3.76 (s, 3H, OCH3), 3.83 (d, 2H, CH2), 6.04 (s, 2H, Ar-H), 6.13 (d, J = 10.5 Hz, 1H, CH═), 6.57–6.68 (m, 3H, Ar-H), 6.79 (d, J = 10.8 Hz, 1H, ═CH); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.4, 55.5, 55.6, 58.4, 90.4, 105.2, 110.3, 116.7, 120.5, 123.3, 128.3, 134.8, 136.2, 147.3, 158.4, 162.1; HRMS (ESI) m/z calcd for C19H23NO5S [M + H] 378.1297, found 378.1280.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfoxide (20)
The title compound was obtained by the reduction of 14 following the procedure as described in compound 17. Yield, 94%; white solid, m.p.: 131–133 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.84 (br s, 12H, 4× OCH3), 3.99 (d, J = 12.6 Hz, 2H, CH2S), 6.11 (s, 2H, Ar-H), 6.67–6.76 (m, 3H, Ar-H), 7.15 (d, J = 15.6 Hz, 1H, CH═), 7.50 (d, J = 15.6 Hz, 1H, ═CH); 13C NMR (75 MHz, CDCl3): δ (ppm): 55.4, 55.5, 55.7, 61.4, 90.4, 105.3, 110.4, 116.7, 120.4, 123.0, 123.3, 127.9, 130.2, 136.3, 147.3, 160.5, 162.2; HRMS (ESI) m/z calcd for C19H23NO5S [M + H] 378.1297, found 378.1292.
(Z)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfone (21)
The title compound was obtained by the reduction of 15 following the procedure as described in compound 17. Yield, 90%; white solid; m.p.: 142–144 °C; 1H NMR (300 MHz, CDCl3) δ (ppm); 3.84 (s, 9H, 3× OCH3), 3.87 (s, 3H, OCH3), 4.14 (s, 2H, CH2), 6.13 (s, 2H, Ar-H), 6.20 (d, J = 11.7 Hz, 1H, ═CH), 6.78–6.83 (m, 3H, Ar-H), 7.02 (d, J = 11.7 Hz, 1H, CH═); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.3, 55.5, 55.6, 55.7, 60.3, 90.3, 103.0, 110.4, 117.2, 120.7, 121.2, 134.2, 136.4, 147.7, 158.8, 162.6; HRMS (ESI) m/z calcd for C19H23NO6S [M + H] 394.1246, found 394.1240.
(E)-2′,4′,6′-Trimethoxystyryl-4-methoxy-3-aminobenzyl sulfone (22)
The title compound was obtained by the reduction of 16 following the procedure as described in compound 17. Yield, 95%; white solid, m.p.: 184–186 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 6H, 2× OCH3), 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 4.13 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 6.67–6.76 (m, 3H, Ar-H), 7.06 (d, J = 15.6 Hz, 1H, ═CH), 7.85 (d, J = 15.9 Hz, 1H, CH═); 13C NMR (75 MHz, CDCl3) δ (ppm): 55.5, 55.5, 55.7, 61.7, 90.3, 103.8, 110.2, 117.2, 121.0, 121.1, 122.8, 135.1, 136.2, 147.6, 161.4, 163.7; HRMS (ESI) m/z calcd for C19H23NO6S [M + H] 394.1246, found 394.1252.
(Z)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (23)
Sodium acetate (0.25 g, 3.1 mmol) was dissolved in methanol (15 mL). Methyl 2-bro-moacetate (0.67 g, 4 mmol) was added to the above solution and refluxed for 10 min. To the cooled reaction mixture compound (Z)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzyl-sulfone (21, 0.43 g, 1 mmol) was added and then refluxed for 4 h. The reaction mixture was concentrated under vacuum and poured into ice water. The formed precipitate was filtered, washed with water and dried under vacuum. The crude product on purification by silica gel flash column chromatography (ethyl acetate/hexane) afforded pure 23. Yield, 74%; white solid, m.p.: 157–158 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.73 (s, 2H, CH2), 3.80 (s, 3H, OCH3), 3.83 (s, 9H, 3× OCH3), 3.86 (s, 3H, OCH3), 3.95 (d, J = 5.4 Hz, 2H, NH-CH2), 4.82 (br s, 1H, NH), 6.15 (s, 2H, Ar-H), 6.34 (d, J = 10.5 Hz, 1H, ═CH), 6.41 (d, J = 10.5 Hz, 1H, CH═), 6.48 (s, 1H, Ar-H), 6.66–6.72 (m, 2H, Ar-H); 13C NMR (75 MHz, CDCl3) δ (ppm): 38.6, 45.5, 52.2, 55.4, 55.6, 90.4, 107.2, 109.3, 110.7, 118.0, 118.7, 127.7, 130.7, 137.0, 146.3, 158.2, 160.9, 171.6; HRMS (ESI) m/z calcd for C22H27NO8S [M + H] 466.1457, found 466.1453.
(E)-Methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (24)
The title compound was obtained by the alkylation of (E)-2′,4′,6′-trimethoxystyryl-4-methoxy-3-aminobenzylsulfone 22 with methyl 2-bromoacetate following the procedure as described in compound 23. Yield, 70%; white solid, m.p.: 150–152 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 3.69 (s, 3H, OCH3), 3.75 (s, 6H, 2× OCH3), 3.78 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.81 (d, J = 5.4 Hz, 2H, NH-CH2), 4.09 (s, 2H, CH2), 4.74 (t, J = 5.4 Hz, 1H, NH), 6.01 (s, 2H, Ar-H), 6.41 (d, J = 1.8 Hz, 1H, Ar-H), 6.61–6.68 (m, 2H, Ar-H), 6.97 (d, J = 15.6 Hz, 1H, ═CH), 7.73 (d, J = 15.6 Hz, 1H, CH═); 13C NMR (75 MHz, CDCl3) δ (ppm): 45.3, 52.2, 55.4, 55.5, 55.7, 62.1, 90.4, 104.0, 109.4, 112.3, 120.3, 121.3, 122.9, 135.3, 137.0, 147.4, 161.5, 163.7, 171.4; HRMS (ESI) m/z calcd for C22H27NO8S [M + H] 466.1457, found 466.1456.
Sodium (Z)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (25)
The title compound was obtained by the direct hydrolysis of (Z)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate 23. To a solution of sodium hydroxide (0.036 g, 0.9 mmol) in ethanol (10 mL) was added (Z)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate 23 (0.35 g, 0.75 mmol) and the resulting mixture was stirred at 60 °C for 30 min. After completion of the reaction monitored by TLC (chloroform/methanol, 9 : 1 on silica gel plate), the resulting mixture was cooled to room temperature and stirred further for 2 h. The precipitated white solid was filtered and dried. This compound was taken into 20 mL of anhydrous ethyl acetate and stirred for 2 h at room temperature and the solid was filtered, washed with anhydrous ethyl acetate and dried to get the title compound 25. Yield, 85%; white solid, m.p. 157–158 °C; 1H NMR (300 MHz, D2O) δ (ppm): 3.59 (s, 2H, NH-CH2), 3.66 (s, 6H, 2× OCH3), 3.74 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 4.24 (s, 2H, CH2), 6.16 (s, 2H, Ar-H), 6.38 (d, J = 11.7 Hz, 1H, ═CH), 6.44 (s, 1H, Ar-H), 6.63 (d, J = 7.8 Hz, 1H, Ar-H), 6.81 (d, J = 8.4 Hz, 1H, Ar-H), 7.03 (d, J = 11.4 Hz, 1H, CH═); 13C NMR (75 MHz, DMSO-d6,) δ (ppm): 47.8, 55.2, 55.3, 55.5, 59.2, 90.4, 103.5, 108.9, 111.7, 117.5, 120.8, 128.1, 133.4, 138.0, 146.2, 158.2, 161.9, 171.12; HRMS (ESI) m/z calcd for C21H24NNaO8S [M + H] 474.1120, found 474.1126.
Sodium (E)-2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)-methyl)phenylamino)acetate (26)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-((2′,4′,6′-trimethoxystyrylsulfonyl)methyl)phenylamino)acetate (24) following the procedure as described in compound 25. Yield, 80%; white solid; m.p.: 174–178 °C: 1H NMR (300 MHz, DMSO-d6) δ (ppm): 3.11 (d, J = 3.9 Hz, 2H, NH-CH2), 3.78 (s, 3H, OCH3), 3.84 (s, 6H, 2× OCH3), 3.85 (s, 3H, OCH3), 4.26 (s, 2H, CH2), 5.18 (t, J = 3.9 Hz, NH), 6.28 (s, 2H, Ar-H), 6.32 (d, J = 1.8 Hz, 1H, Ar-H), 6.47 (dd, J = 8.1, 2.1 Hz, 1H, Ar-H), 6.72 (d, J = 8.1 Hz, 1H, Ar-H), 7.10 (d, J = 15.9 Hz, 1H, ═CH), 7.56 (d, J = 15.9 Hz, 1H, CH═); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 47.6, 55.2, 55.6, 56.0, 60.6, 90.9, 102.6, 108.8, 111.8, 117.5, 121.5, 123.6, 132.5, 137.8, 146.1, 160.8, 163.5, 171.8; HRMS (ESI) m/z calcd for C21H24NNaO8S [M + H] 474.1120, found 474.1116.
Biological assay methods
Cytotoxicity assay
We have tested growth inhibition assay in K562 and Du145 tumor cell lines using a dose response end point assay system. The cells were purchased from ATCC and maintained at 37 °C under 5% CO2 in either DMEM or RPMI supplemented with 10% fetal bovine serum and 1 unit per mL penicillin–streptomycin solution. The tumor cells were plated into 6 well dishes at a cell density of 1.0 × 105 cells per mL per well and compounds were added 24 h later at various concentrations. DMSO was used as a negative control. Cell counts were determined from duplicate wells after 96 h of treatment. The total number of viable cells was determined by trypan blue exclusion.
Supplementary Material
Acknowledgments
The authors are thankful to Onconova Therapeutics Inc., Newtown, PA and Mount Sinai School of Medicine, New York for the financial assistance and interest in this project. We are also thankful to Dr Ramana Tantravahi for editorial assistance.
Footnotes
Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c3ob27220f
Conflict of interest
Dr E. P. Reddy is a stockholder, board member, grant recipient and paid consultant of Onconova Therapeutics Inc. Dr M. V. R. Reddy is a stock holder and paid consultant of Onconova Inc. Dr S. Cosenza is a paid consultant of Onconova Therapeutics Inc.
Notes and references
- 1.For more information, see: http://www. onconova.com, http://www. mdsclinicaltrial.com and http://www.clinicaltrials.gov
- 2.Gumireddy K, Reddy MVR, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. Cancer Cell. 2005;7:275. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Prasad A, Park IW, Allen H, Zhang X, Reddy MVR, Boominathan R, Reddy EP, Groopman JE. Onco-gene. 2007;26:5635. [Google Scholar]
- 4.Chapman CM, Sun X, Roschewski M, Aue G, Farooqui M, Stennett L, Gibellini F, Arthur D, Pérez-Galán P, Wiestner A. Clin Cancer Res. 2012;18:1979. doi: 10.1158/1078-0432.CCR-11-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olnes MJ, Shenoy A, Weinstein B, Pfannes L, Loeliger K, Tucker Z, Tian X, Kwak M, Wilhelm F, Yong ASM, Maric I, Maniar M, Scheinberg P, Groopman J, Young NS, Sloand EM. Leuk Res. 2012;36:982. doi: 10.1016/j.leukres.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seetharama M, Fanb AC, Trana M, Xub L, Renschlerb JP, Felsherb DW, Sridhara K, Wilhelm F, Greenberga PL. Leuk Res. 2012;36:98. doi: 10.1016/j.leukres.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma WW, Messersmith WA, Dy1 GK, Weekes CD, Whitworth A, Ren C, Maniar M, Wilhelm F, Eckhardt SG, Adjei1 AA, Jimeno A. Clin Cancer Res. 2012;18:2048. doi: 10.1158/1078-0432.CCR-11-2813. [DOI] [PubMed] [Google Scholar]
- 8.Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, Hidalgo M, Baker SD, Donehower RC. J Clin Oncol. 2008;26:5504. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy MVR, Venkatapuram P, Mallireddigari MR, Pallela VR, Cosenza SC, Robell KA, Akula B, Hoffman BS, Reddy EP. J Med Chem. 2011;54:6254. doi: 10.1021/jm200570p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Becker HD, Soerensen H, Sandros K. J Org Chem. 1986;51:3223. [Google Scholar]; (b) Kropp PJ. Org Photochem. 1979;4:1. [Google Scholar]; (c) Nakagawa CS, Sigal P. J Chem Phys. 1970;52:3277. [Google Scholar]; (d) Sistla A, Shenoy N. Drug Dev Ind Pharm. 2005;31:1001. doi: 10.1080/03639040500306260. [DOI] [PubMed] [Google Scholar]; (e) Dong J, Abulwerdi F, Baldridge A, Kowalik J, Solntsev KM, Tolbert LM. J Am Chem Soc. 2008;130:14096. doi: 10.1021/ja803416h. [DOI] [PubMed] [Google Scholar]; (f) Furukawa N, Fukumura M Mitsuo, Nishio T, Oae S. Phosphorus, Sulfur Relat Elem. 1978;5:191. [Google Scholar]
- 11.(a) Whitesides GM, Goe GL, Cope AC. J Am Chem Soc. 1969;91:2608. [Google Scholar]; (b) Zhang L, Borysenko CW, Albright TA, Bittner ER, Lee TR. J Org Chem. 2001;66:5275. doi: 10.1021/jo0014818. [DOI] [PubMed] [Google Scholar]
- 12.Trost BM, Lavoie AC. J Am Chem Soc. 1983;105:5075. [Google Scholar]
- 13.Miller RD, Hassig R. Tetrahedron Lett. 1985;26:2395. [Google Scholar]
- 14.(a) Yamaguchi H, Minoura Y. J Appl Polym Sci. 2003;15:1869. [Google Scholar]; (b) Holly ED. J Polym Sci, Part A: Polym Chem. 2003;36:329. [Google Scholar]
- 15.(a) Sader HS, Johnson DM, Jones RN. Antimicrob Agents Chemother. 2004;48:53. doi: 10.1128/AAC.48.1.53-62.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Magnus P, Quagliato D. J Org Chem. 1985;50:1621. [Google Scholar]; (c) Ceruti M, Balliano G, Rocco F, Milla P, Arpicco S, Cattel L, Viola F. Lipids. 2001;36:629. doi: 10.1007/s11745-001-0767-8. [DOI] [PubMed] [Google Scholar]; (d) Johannesson P, Lindeberg G, Johansson A, Nikiforovich GV, Gogoll A, Synnergren B, Le Greves M, Nyberg F, Karlen A, Hallberg A. J Med Chem. 2002;45:1767. doi: 10.1021/jm011063a. [DOI] [PubMed] [Google Scholar]; (e) Marcantoni E, Massaccesi M, Petrini M, Bartoli G, Bellucci MC, Bosco M, Sambri L. J Org Chem. 2000;65:4553. doi: 10.1021/jo000116d. [DOI] [PubMed] [Google Scholar]; (f) Lam HW, Cooke PA, Pattenden G, Bandaranayake WM, Wickramasinghe WA. J Chem Soc, Perkin Trans. 1999;1:847. [Google Scholar]; (g) Mugesh G, du Mont WW, Sies H. Chem Rev. 2001;101:2125. doi: 10.1021/cr000426w. [DOI] [PubMed] [Google Scholar]
- 16.(a) Comasseto JV, Petragnani N. J Organomet Chem. 1978;152:295. [Google Scholar]; (b) Comasseto JV. J Organomet Chem. 1983;253:131. [Google Scholar]; (c) Mizuno H, Domon K, Masuya K, Tanino K, Kuwajima I. J Org Chem. 1999;64:2648. doi: 10.1021/jo981478c. [DOI] [PubMed] [Google Scholar]; (d) Comasseto JV, Ling LW, Petragnani N, Stefani HA. Synthesis. 1997:373. [Google Scholar]; (e) Takeda T, Fujiwara T. Chem Lett. 1982;11:1113. [Google Scholar]
- 17.(a) Benati L, Capella L, Montevecchi PC, Spagnolo P. J Org Chem. 1994;59:2818. [Google Scholar]; (b) Benati L, Capella L, Montevecchi PC, Spagnolo P. J Chem Soc, Perkin Trans. 1995;1:1035. [Google Scholar]
- 18.(a) Yamashita F, Kuniyasu H, Terao J, Kambe N. Org Lett. 2008;10:101. doi: 10.1021/ol7025069. [DOI] [PubMed] [Google Scholar]; (b) Sabarre A, Love J. Org Lett. 2008;10:3941. doi: 10.1021/ol8012843. [DOI] [PubMed] [Google Scholar]; (c) Cao C, Fraser LR, Love JA. J Am Chem Soc. 2005;127:17614. doi: 10.1021/ja055096h. [DOI] [PubMed] [Google Scholar]; (d) Shoai S, Bichler P, Kang B, Buckley H, Love JA. Organometallics. 2007;26:5778. [Google Scholar]; (e) Ogawa A, Ikeda T, Kimura K, Hirao T. J Am Chem Soc. 1999;121:5108. [Google Scholar]; (f) Kondoh A, Yorimitsu H, Oshima K. Org Lett. 2007;9:1383. doi: 10.1021/ol0702876. [DOI] [PubMed] [Google Scholar]; (g) Kuniyasu H, Ogawa A, Sato KI, Ryu I, Kambe N, Sonoda N. J Am Chem Soc. 1992;114:5903. [Google Scholar]; (h) Cai M, Wang Y, Hao W. Green Chem. 2007;9:1180. [Google Scholar]; (i) Guo SR, Yuan YQ. Synth Commun. 2008;38:2722. [Google Scholar]; (j) Ogawa A. J Organomet Chem. 2000;611:463. [Google Scholar]; (k) Kondo T, Mitsudo T. Chem Rev. 2000;100:3205. doi: 10.1021/cr9902749. [DOI] [PubMed] [Google Scholar]; (l) Beller M, Seayad J, Tillack A, Jiao H. Angew Chem, Int Ed. 2004;43:3368. doi: 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]; (m) Alonso F, Beletskaya IP, Yus M. Chem Rev. 2004;104:3079. doi: 10.1021/cr0201068. [DOI] [PubMed] [Google Scholar]; (n) Ananikov VP, Orlov NV, Beletskaya IP. Organometallics. 2006;25:1970. [Google Scholar]; (o) Ding Q, He X, Wu J. J Comb Chem. 2009;11:587. doi: 10.1021/cc900027c. [DOI] [PubMed] [Google Scholar]; (p) Ding Q, Wu J. J Comb Chem. 2008;10:541. doi: 10.1021/cc800043r. [DOI] [PubMed] [Google Scholar]; (q) Weiss CJ, Marks TJ. J Am Chem Soc. 2010;132:10533. doi: 10.1021/ja103979b. [DOI] [PubMed] [Google Scholar]
- 19.(a) Aucagne V, Tatibouet A, Rollin P. Tetrahedron. 2004;60:1817. [Google Scholar]; (b) Stephan E, Olaru A, Jaouen G. Tetrahedron Lett. 1999;40:8571. [Google Scholar]; (c) Silveira CC, Begnini ML, Boeck P, Braga AL. Synthesis. 1997:221. [Google Scholar]; (d) Ishida M, Iwata T, Yokoi M, Kaga K, Kato S. Synthesis. 1985:632. [Google Scholar]; (e) Mikolajczyk M, Grzejszczak S, Midura W, Zatorski A. Synthesis. 1975:278. [Google Scholar]; (f) Kumamoto T, Hosoi K, Mukaiyam T. Bull Chem Soc Jpn. 1968;41:2742. [Google Scholar]
- 20.(a) Cristau HJ, Chabaud B, Labaudiniere R, Christol H. J Org Chem. 1986;51:875. [Google Scholar]; (b) Ogawa T, Hayami K, Suzuki H. Chem Lett. 1989:769. [Google Scholar]
- 21.Di Giuseppe A, Castarlenas R, Pérez-Torrente TJ, Crucianelli M, Polo V, Sancho R, Lahoz FJ, Oro LA. J Am Chem Soc. 2012;134:8171. doi: 10.1021/ja300396h. [DOI] [PubMed] [Google Scholar]
- 22.(a) Ranu BC, Chattopadhyay K, Banerjee S. J Org Chem. 2006;71:423. doi: 10.1021/jo052087i. [DOI] [PubMed] [Google Scholar]; (b) Boubia B, Mioskowski C, Manna S, Falck JR. Tetrahedron Lett. 1989;30:6023. [Google Scholar]; (c) Wang L, Wang M, Huang F. Synlett. 2005:2007. [Google Scholar]; (d) Ichinose Y, Wakamatsu K, Nozaki K, Birbaum JL, Oshima K, Utimoto K. Chem Lett. 1987:1647. [Google Scholar]; (e) Bates CG, Saejueng P, Doherty MQ, Venkataraman D. Org Lett. 2004;6:5005. doi: 10.1021/ol0477935. [DOI] [PubMed] [Google Scholar]; (f) Kabir MS, Linn MLV, Monte A, Cook JM. Org Lett. 2008;10:3363. doi: 10.1021/ol801149n. [DOI] [PubMed] [Google Scholar]
- 23.(a) Murahashi SI, Yamamura M, Yanagisawa KI, Mita N, Kondo K. J Org Chem. 1979;44:2408. [Google Scholar]; (b) Wang Z, Mo H, Bao W. Synlett. 2007:91. [Google Scholar]; (c) Yang Y, Rioux RM. Chem Commun. 2011;47:6557. doi: 10.1039/c1cc11605c. [DOI] [PubMed] [Google Scholar]; (d) Trostyanskaya IG, Beletskaya IP. Synlett. 2012:535. [Google Scholar]; (e) Gerber R, Frech CM. Chem–Eur J. 2012;18:8901. doi: 10.1002/chem.201200388. [DOI] [PubMed] [Google Scholar]
- 24.(a) Burling S, Field LD, Messerle BA, Vuong KQ, Turner P. Dalton Trans. 2003:4181. doi: 10.1039/b821188d. [DOI] [PubMed] [Google Scholar]; (b) Field LD, Messerle BA, Vuong KQ, Turner P. Dalton Trans. 2009:3599. doi: 10.1039/b821188d. [DOI] [PubMed] [Google Scholar]
- 25.(a) Truce WE, Simms JA, Boudakian MM. J Am Chem Soc. 1956;78:695. [Google Scholar]; (b) Truce WE, Simms JA. J Am Chem Soc. 1956;78:2756. [Google Scholar]
- 26.Kondoh A, Takami K, Yorimitsu H, Oshima K. J Org Chem. 2005;70:6468. doi: 10.1021/jo050931z. [DOI] [PubMed] [Google Scholar]
- 27.Griesbaum K. Angew Chem, Int Ed. 1970;9:273. [Google Scholar]
- 28.Majumdar KC, Debnath P. Tetrahedron. 2008;64:9799. [Google Scholar]
- 29.Ichinose Y, Wakamatsu K, Nozaki K, Birbaum JL, Oshima K, Utimoto K. Chem Lett. 1987:1647. [Google Scholar]
- 30.Benati L, Montevecchi PC, Spagnolob P. J Chem Soc, Perkin Trans. 1991:1. 2103. [Google Scholar]
- 31.(a) Reddy MVR, Reddy S. Synthesis. 1983:322. [Google Scholar]; (b) Reddy MVR, Reddy S, Reddy PVR, Reddy DB, Reddy NS. Phosphoros, Sulfur Silicon Relat Elem. 1989;44:123. [Google Scholar]; (c) Reddy MVR, Mallireddigari MR, Pallela VR, Venkatapurm P, Boominathan R, Bell SC, Reddy EP. Bioorg Med Chem. 2005;13:1715. doi: 10.1016/j.bmc.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Bhadra S, Ranu BC. Can J Chem. 2009;87:1605. [Google Scholar]
- 33.Ranjit R, Duan Z, Zhang P, Liu X. Org Lett. 2010;12:4134. doi: 10.1021/ol101729k. [DOI] [PubMed] [Google Scholar]
- 34.(a) Blanksby SJ, Ellison GB. Acc Chem Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]; (b) Massi A, Nanni D. Org Biomol Chem. 2012;10:3791. doi: 10.1039/c2ob25217a. [DOI] [PubMed] [Google Scholar]; (c) Melandri D, Montevecchi PC, Navacchia ML. Tetrahedron. 1999;55:12227. [Google Scholar]
- 35.(a) Fairbanks BD, Sims Evan A, Anseth KS, Bowman CN. Macromolecules. 2010;43:4113. [Google Scholar]; (b) Ito O, Omori R, Matsuda M. J Am Chem Soc. 1982;104:3934. [Google Scholar]
- 36.Chen Z, Zhang Y, An Y, Song XL, Wang YH, Zhu LL, Guo L. Eur J Org Chem. 2009:5146. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.