

Abstract
We present the results of the first independent assessment of protein assemblies in CASP. A total of 1624 oligomeric models were submitted by 108 predictor groups for the 30 oligomeric targets in the CASP12 edition. We evaluated the accuracy of oligomeric predictions by comparison to their reference structures at the interface patch and residue contact levels. We find that interface patches are more reliably predicted than the specific residue contacts. Whereas none of the 15 hard oligomeric targets have successful predictions for the residue contacts at the interface, six have models with resemblance in the interface patch. Successful predictions of interface patch and contacts exist for all targets suitable for homology modeling, with at least one group improving over the best available template for each target. However, the participation in protein assembly prediction is low and uneven. Three human groups are closely ranked at the top by overall performance, but a server outperforms all other predictors for targets suitable for homology modeling. The state of the art of protein assembly prediction methods is in development and has apparent room for improvement, especially for assemblies without templates.
Keywords: CASP, protein assembly, protein interfaces, protein symmetry, structure prediction
1 | INTRODUCTION
Most proteins carry out their functions in the cell as part of an oligomeric complex.1 A survey on the Protein Data Bank (PDB)2 reveals that >50% of deposited structures are oligomeric, at the time of this article. Therefore, in order to understand the mechanism of activity of proteins, a detailed knowledge of the molecular partners and modes of association is needed.
A biological assembly is the complete functional unit of a protein in the cell, defined by the protein composition and mode of association. The composition (stoichiometry) of protein assemblies can be obtained by common experimental techniques, like size exclusion chromatography (SEC)3 or analytical ultracentrifugation (AUC).4 However, describing the mode of association of proteins in a biological assembly is a more difficult task, since it requires structural information of the macromolecular complex at a reasonable resolution, obtained by the use of techniques like X-ray crystallography or electron microscopy (EM). For crystallographic techniques, a careful analysis of the protein–protein interfaces in the crystal lattice is needed to distinguish biologically relevant interfaces from crystal contacts, known as the interface classification problem.5 The error rate of annotations in the PDB has been estimated to be at least 6.6% at the protein–protein interface level6 and 14.7% at the biological assembly level.7 Accurate biological assembly assignment is of particular importance when generating the reference quaternary structures to which prediction models are compared.
Protein assembly is also key for the interpretation of structural models and the modeling process itself. In recent years, the protein prediction community has placed huge efforts to incorporate assembly modeling in prediction methods. Homology modeling techniques have included quaternary structure templates in their pipelines8 and the analysis of co-evolving residues has allowed the prediction of interface contacts without a template.9 These approaches are added to the already established methods of the docking community and bring the CASP and CAPRI experiments closer together.
The goal of the CASP experiment is to provide an objective and fair comparison of the state of the art in protein prediction techniques, to give an account of the method capabilities and limitations and to stimulate method development. Groups that participate in the protein assembly category are given the protein sequences and the stoichiometry (composition), and are asked to submit predictions of the three-dimensional structures of the macromolecular assembly, in the form of multi-chain models. Each predictor can submit up to five different models for each target assembly, but they are requested to designate what they believe to be their best model as the number one. For compatibility with servers that can only predict structures of a single protein sequence, heteromeric assemblies are presented as separate targets. Predictors are informed of the targets that participate together in an assembly and they are instructed to submit separate models for each of the targets in the same coordinate reference frame.
Previous CASP assessors have tried to evaluate the performance of assembly prediction. A first attempt was explored in CASP9,10 as part of the template-based assessment category. The participation and performance in assembly prediction were underwhelming, with only six groups submitting oligomeric models for more than half of the oligomeric targets, and only a single group showing better overall performance than the naïve predictor. A second endeavor to evaluate protein assembly predictions was in collaboration with CAPRI in CASP11.11 Although the participation of CASP groups stayed low, with only five groups submitting oligomeric models for more than half of the 17 CASP oligomeric targets included in the CAPRI assessment, four of them submitted accurate models by CAPRI standards. In addition, this time CAPRI predictors improved over the sequence and structural templates for all except one target.
For the first time, there is an independent assessment category fully dedicated to protein assembly. The assessment of protein assembly in CASP12 consisted in (1) generating the target assemblies to use as the reference, (2) evaluating the model accuracy by comparison to the references, and (3) ranking the groups. We distinguish protein assembly modeling from the classical protein–protein docking, where two protein subunits, named ligand and receptor, are in contact through a single interface. In our assessment, we want to acknowledge accurate predictions of the full protein assembly, as the functional biological unit, which involves both predictions of individual protein–protein interfaces and overall assembly topology. We present the results of our assessment of the CASP12 predictions for the target protein assemblies, analyzing the state of the art of the techniques, and highlighting areas with potential for future development.
2 | METHODS
2.1 | Generation of target assemblies
Information about the oligomeric state of the target structures was collected in collaboration with experimentalists. Biological assemblies of the oligomeric targets were determined from their experimental structures. Structures obtained by electron microscopy (EM) (T0918 and T0930) or nuclear magnetic resonance (NMR) (T0865 and T0929) were considered to be in the correct oligomeric state. For protein structures obtained using crystallographic techniques we carefully analyzed the crystal lattice using computational tools, additional experimental evidence, such as SEC data, and visual inspection.
The Evolutionary Protein–Protein Interface Classifier (EPPIC)12 was used as a starting point to analyze the protein–protein interfaces in the crystal and generate the most probable biological assemblies. Low confidence predictions were then cross-checked using PISA13 and by manual inspection. If available, the biological assemblies of homologs were also used in the final decision. Supporting information file S1 contains the information used, author annotations, and final assignments for the CASP12 oligomeric targets. If multiple copies of the biological assembly were present in the asymmetric unit, we selected as a reference the assembly with the strongest biological evidence as determined by EPPIC and PISA.
Six targets (T0859, T0863, T0864, T0886, T0887, and T0896) were initially released as monomeric and later, when the experimental structure was made available to CASP, re-released as new multimeric-prediction only targets (T0929, T0930, T0932, T0933, T0931, and T0934, respectively) with the corrected oligomeric state information. Only submissions for the re-released targets were used for assessment. Additionally, three oligomeric targets (T0865, T0871, and T0902) were released as monomeric, but we decided not to rerelease them due to reasons described in Supporting information file S1.
2.2 | Target prediction difficulty
We have classified the oligomeric targets in a discrete prediction difficulty scale, in order to be able to distinguish template-based from template-free assembly predictions (Figure 1), with the following three levels:
Easy: Targets with at least one template with detectable sequence similarity and the same protein assembly. Easy oligomeric targets were considered suitable for homology modeling.
Medium: Targets with at least one template with detectable sequence similarity that shares a partial subset of chains in the same association mode. Medium oligomeric targets can benefit from homology modeling, but may require additional docking or free modeling to derive the complete biological assembly.
Hard: Targets for which no oligomeric templates exist. Hard targets form completely novel assemblies, so they are not suitable for homology modeling.
FIGURE 1.
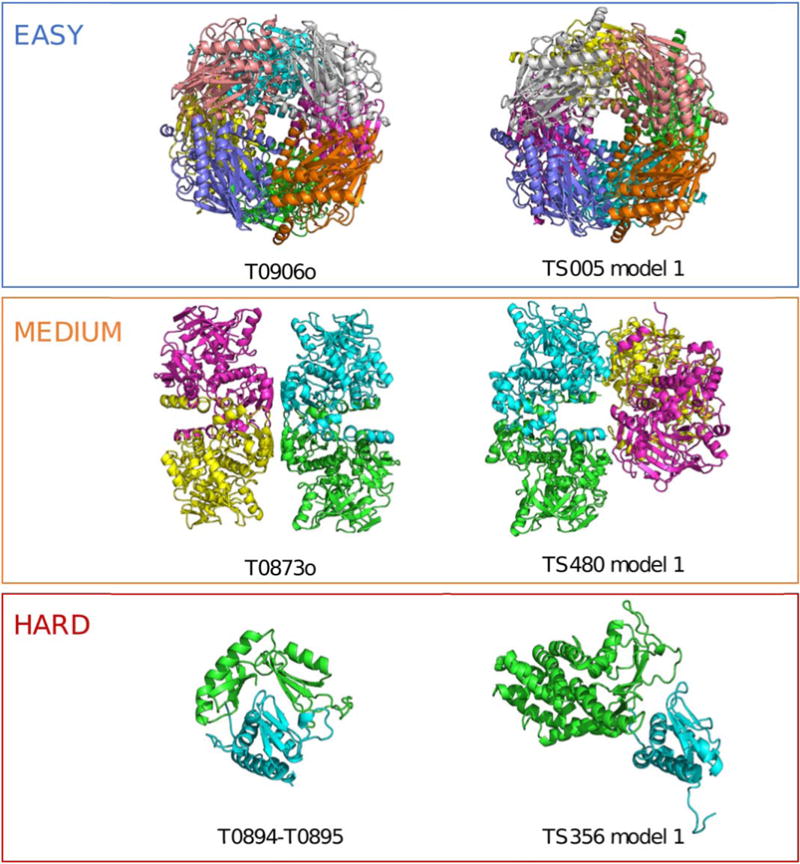
One target for each prediction difficulty with an example prediction. (Top) Fructose biphosphatase had templates with 44% sequence identity that shared the same octameric assembly. Predictions for this target were highly successful. (Middle) A dimeric template was available for TtnD, but modeling the complete dihedral tetramer required the prediction of a novel secondary interface. Successful models of the monomer or dimer were submitted, but none successfully predicted the full tetrameric assembly. (Bottom) The contact-dependent inhibitor toxin/immunity protein complex lacked templates for the toxin (green). Some models with partial resemblance to the target assembly were submitted, but none as the first model
There were three oligomeric targets with available structural templates of their assembly, but not detectable using regular sequence similarity methods. The two fiber heads, T0880o and T0888o, were classified as medium difficulty, since their homotrimeric assembly was well known. On the other hand, the viral capsid coat protein, T0929o, was significantly harder to predict, as the poor predictions for its tertiary structure show, and it was classified as hard.
2.3 | Assembly scoring
2.3.1 | Contacts and clashes
A contact is defined as a residue of one protein chain with at least one heavy atom within a distance threshold of 5 Å to a heavy atom of a residue from another protein chain. If the interatomic distance is below 3 Å, it is regarded as a clash.
2.3.2 | Interface contact
An interface contact set, C, is defined as all pairs of residues from two protein chains that are in contact. For a particular pair of chains, this can be defined for both the prediction model (CM) and the experimental structure of the target (CT).
The contact precision, P, is the fraction of contacts in the model CM that are also present in the target CT.
| (1) |
The recall R is the fraction of contacts in the target CT correctly reproduced by the model CM.
| (2) |
The Interface Contact Similarity (ICS) score is the combination of precision and recall of contact predictions using the F1-measure.
| (3) |
The ICS score can have a value between 0 and 1, where 1 is the best possible score. For reference, the ICS score is equal to the F1 score reported by the Prediction Center divided by 100.14
2.3.3 | Interface patch
An interface patch is defined as the set of residues I of one protein in contact to its partners. This is the equivalent of taking all the residues in the interface contact set disregarding their pairing.
The Interface Patch Similarity (IPS) score measures the similarity between the interface patches of the model IM and the target IT using a Jaccard coefficient of the interface residues.
| (4) |
It is a less stringent metric than the ICS since rotations and translations of the partner subunits on the interface plane are less heavily penalized. The IPS score ranges between 0 and 1, where 1 is the best possible score. For reference, the IPS score is equal to one minus the Jaccard distance score reported by the Prediction Center.
2.3.4 | Symmetry
The quaternary structure symmetry detector algorithm from the Bio-Java library15 (version 5) was used to determine the point group and symmetry axes of the target and model assemblies. In addition, the symmetry RMSD and TM-score16 were computed as the average alpha-carbon RMSD and TM-score of each protein subunit after rotating around the symmetry axes of the assembly.
2.3.5 | Interface RMSD
To assess higher resolution details of the interfaces we calculated an interface RMSD. The two protein partners of an interface from the model and the target are superimposed based only on the residues in contact at the target interface. The RMSD over alpha-carbon atoms of the residues used in the superposition is then obtained. A similar score, the I-rms, is used as a ranking criterion in CAPRI, described more extensively by Lensink et al.11
2.4 | Quaternary structure alignment
In order to detect partial assembly predictions, we used the scalable quaternary structure alignment algorithm developed by A. Lafita17 and available as part of BioJava (version 5). The output of the alignment is a maximal subset of protein subunits of the model that are equivalent to the target assembly and an associated alpha-carbon RMSD of their superposition.
2.5 | Baseline performance
In order to quantify the accuracy improvement of predictions from the best available templates we designed a naïve predictor. The procedure starts by using HHSearch18 to identify sequence homologs from the PDB. For each target, the biological assembly with the highest sequence similarity to the target, if any, was selected as the template. Afterwards, templates and prediction models were compared with the target structures using the QS-score metric.19 The sequence templates and QS-scores for each target assembly suitable for homology modeling are provided in Supporting information Table S1.
QS-score considers the assembly interface as a whole (without decomposing it to binary interactions) and is suitable for comparing homo- or hetero-oligomers with identical or different stoichiometries, alternative relative orientations of chains, and distinct amino acid sequences (that is, homologous complexes). This flexibility allows using a single metric for the comparison. The method identifies the mapping between chains in two input oligomers, performs an alignment of the sequences of the equivalent chains, and computes a weighted fraction of shared Cb contacts in the overall interface. Oligomers that do not have the same stoichiometry or the same interacting contacts will be penalized.
A QS-score close to 1 indicates that the compared interfaces are very similar, that is, the complexes have equal stoichiometry and a majority of the interfacial contacts are identical. A QS-score close to 0 indicates a radically diverse quaternary structure, different stoichiometries and/or alternative binding conformations.
2.6 | Ranking
The final ranking was computed based on both the interface contact and patch similarity scores. First, raw scores were normalized to a Z-score for each target after removing outliers. Next, negative scores were flattened to zero to avoid penalizing prediction attempts. Finally, a weighted sum over all targets was computed, with equal weight on each score. Total score was used rather than average score to reward groups with a higher coverage of targets.
| (5) |
where MT represents the first model of group G for target T.
For heteromeric assemblies, the models for each target participating in the assembly were merged. As a quality criteria and to filter out models which were not intended as assembly predictions, oligomeric models with >20 clashes or fewer contacts than clashes were not considered for ranking purposes. Only the first model was considered for ranking, to reward good model selection and to avoid penalizing groups that submitted fewer models. Seven targets (T0866o, T0912o, T0913o, T0930o, T0932o, T0933o, and T0934o) where no groups successfully predicted interface contacts (<4% ICS) were not included in the ranking to avoid instabilities in their Z-scores. All models were scored and shown in the summary tables available at the Prediction Center website (http://predictioncenter.org/casp12).
3 | RESULTS AND DISCUSSION
3.1 | Oligomeric targets
A total of 30 oligomeric targets were suitable for assembly prediction and assessment in CASP12. The diversity of the oligomeric targets was remarkable (Table 1). Eight heteromeric assemblies, one of them (T0861o-T0862o-T070o) involving three targets interacting in a hexamer, were released for prediction. Among the remaining 22 homomeric targets, there are four dihedral symmetry assemblies (T0873o, T0889o, T0906o, and T0913o) and five viral fiber heads (T0860o, T0867o, T0880o, T0881o, and T0888o) with the same trimeric arrangement, but different amino-acid sequence. The two membrane dimers (T0930o and T0945o) are also of special interest due to the modeling challenges of membrane interfaces.20 The CASP12 target distribution of symmetry, composition, and macromolecular size closely resembles that of the PDB.
TABLE 1.
Oligomeric targets in the CASP12 edition. The symmetry (Symm.), stoichiometry (Stoich.) and prediction difficulty of each target, together with the number of groups that submitted predictions and models for each target
| Target | Stoich | Symm | Difficulty | Groups | Models |
|---|---|---|---|---|---|
| T0860o | A3 | C3 | Easy | 15 | 71 |
| T0861o-T0862o-T0870o | A2B2C2 | C2 | Medium | 38 | 72 |
| T0866o | A6 | C6 | Hard | 4 | 16 |
| T0867o | A3 | C3 | Easy | 17 | 75 |
| T0868-T0869 | AB | C1 | Hard | 60 | 143 |
| T0873o | A4 | D2 | Medium | 7 | 24 |
| T0875o | A2 | C2 | Hard | 12 | 46 |
| T0880o | A3 | C3 | Medium | 8 | 32 |
| T0881o | A3 | C3 | Easy | 11 | 48 |
| T0884-T0885 | AB | C1 | Hard | 27 | 60 |
| T0888o | A3 | C3 | Medium | 11 | 43 |
| T0889o | A4 | D2 | Easy | 2 | 10 |
| T0893o | A2 | C2 | Easy | 11 | 49 |
| T0894-T0895 | AB | C1 | Hard | 58 | 120 |
| T0897-T0898 | AB | C1 | Hard | 44 | 115 |
| T0903o-T0904o | A2B2 | C2 | Medium | 37 | 72 |
| T0906o | A8 | D4 | Easy | 8 | 35 |
| T0909o | A3 | C3 | Easy | 8 | 31 |
| T0912o | A2 | C2 | Hard | 2 | 7 |
| T0913o | A6 | D3 | Hard | 7 | 27 |
| T0914-T0915 | AB | C1 | Hard | 60 | 130 |
| T0917o | A2 | C2 | Easy | 12 | 56 |
| T0921-T0922 | AB | C1 | Easy | 36 | 108 |
| T0929o | A2 | C2 | Hard | 9 | 32 |
| T0930o | A2 | C2 | Hard | 8 | 32 |
| T0931o | A2 | C2 | Medium | 12 | 50 |
| T0932o | A2 | C2 | Hard | 9 | 30 |
| T0933o | A6 | C6 | Hard | 8 | 30 |
| T0934o | A2 | C2 | Hard | 9 | 37 |
| T0945o | A2 | C2 | Hard | 6 | 23 |
| Total | 108 | 1624 |
3.2 | Participation
The number of predictions was uneven among the targets, as it can be observed in Table 1. There is no target with participation from all the groups, and for almost half of the targets less than 10 distinct groups submitted predictions. Overall, we observe a higher participation for target assemblies suitable for homology modeling (easy and medium difficulty). T0868-T0869 (hard difficulty) is the heteromeric target with the highest participation with 60 predictors and >140 models, while T0867o (easy difficulty) is the homomeric target with the highest participation with >70 valid models from 17 different groups.
The larger participation for heteromeric assemblies can be explained by the model submission format, which required separate models for each target in the assembly in the same reference coordinate frame. This means that groups that did not voluntarily participate in assembly prediction, but their monomeric target models happened to be in compatible reference frames, might still be included in the assessment. The number of such groups can be large, considering that there were only 22 distinct groups that participate in assembly prediction for homomeric targets. We believe that in the future, assembly predictions should be submitted as complete assembly models including all the targets in the complex.
The low participation for targets T0889o and T0912o is because they were released with unknown assembly information. Therefore, only groups capable of predicting the oligomeric state from the sequence alone were expected to submit assembly models. In addition, the oligomeric state annotation of some targets was changed during the prediction season. This is due to the unavailability of some experimental structures when the targets are first released and makes assembly predictions more challenging and assessment possibly unfair. However, due to the intrinsic organization of the CASP experiment, it is difficult to solve this problem. We propose to provide only information about the interacting targets for hetero-oligomers, and refrain from releasing the oligomeric state information.
In total, 108 predictor groups submitted a combined 1624 models for the target protein assemblies. However, we only consider as actively participating assembly predictors those that submitted models for at least three distinct oligomeric targets. Therefore, a total of 68 groups and 1510 assembly models are considered for the assessment and ranking (Table 2). The participation among groups is generally low and uneven. No group submitted predictions for all the oligomeric targets and only 10 groups submitted models for more than 10 targets. There is also underrepresentation of servers among the top participating groups, only three out of 10, which we attribute to the challenges of systematically parsing the assembly information and adapting the automated modeling pipelines for multimeric model submission.
TABLE 2.
Participation of CASP groups in the assembly prediction category. Number of distinct oligomeric targets (Tr) and total valid models (Md) submitted per group, classified into the prediction difficulty categories. Table truncated to groups with 7 or more total target predictions
| Group | Type | ID | Easy Tr | Easy Md | Medium Tr | Medium Md | Hard Tr | Hard Md | Total Tr | Total Md |
|---|---|---|---|---|---|---|---|---|---|---|
| TSlab-assembly | Human | 356 | 8 | 40 | 6 | 30 | 13 | 62 | 27 | 132 |
| Bates_BMM | Human | 1 | 8 | 40 | 6 | 27 | 13 | 64 | 27 | 131 |
| Seok-assembly | Server | 495 | 8 | 39 | 6 | 26 | 11 | 55 | 25 | 120 |
| Seok | Human | 23 | 7 | 35 | 5 | 25 | 11 | 55 | 23 | 115 |
| FONT | Human | 480 | 7 | 18 | 5 | 10 | 10 | 20 | 22 | 48 |
| chuo-u | Human | 188 | 6 | 28 | 4 | 10 | 10 | 30 | 20 | 68 |
| BAKER-ROSETTASERVER | Server | 5 | 9 | 40 | 3 | 10 | 7 | 22 | 19 | 72 |
| YASARA | Server | 446 | 5 | 20 | 3 | 11 | 9 | 26 | 17 | 57 |
| McGuffin | Human | 17 | 1 | 1 | 4 | 4 | 10 | 11 | 15 | 16 |
| KIAS-Gdansk | Human | 60 | 6 | 30 | 3 | 13 | 5 | 23 | 14 | 66 |
| BAKER | Human | 247 | 1 | 5 | 3 | 15 | 5 | 11 | 9 | 31 |
| StructPre | Human | 367 | 1 | 1 | 3 | 9 | 5 | 10 | 9 | 20 |
| UNRES | Human | 162 | 2 | 10 | 1 | 5 | 5 | 25 | 8 | 40 |
| MULTICOM-CLUSTER | Server | 287 | 1 | 1 | 2 | 2 | 5 | 14 | 8 | 17 |
| GOAL_COMPLEX | Server | 430 | 5 | 25 | 1 | 5 | 1 | 5 | 7 | 35 |
| FLOUDAS_SERVER | Server | 357 | 1 | 5 | 1 | 4 | 5 | 23 | 7 | 32 |
| Seok-naive_assembly | Server | 284 | 6 | 24 | 0 | 0 | 1 | 5 | 7 | 269 |
| MULTICOM-NOVEL | Server | 345 | 1 | 5 | 2 | 2 | 4 | 14 | 7 | 21 |
| tsspred2 | Server | 464 | 1 | 2 | 2 | 3 | 4 | 15 | 7 | 20 |
| Total | 9 | 483 | 6 | 293 | 15 | 490 | 30 | 1624 |
3.3 | Target-based performance
For most targets, there are only a few good predictions among all models submitted. In general, the distance between the top scoring models and the median for each target is large (Figure 2). Extreme cases are predictions for target assemblies T0893o and T0921-T0922.
FIGURE 2.
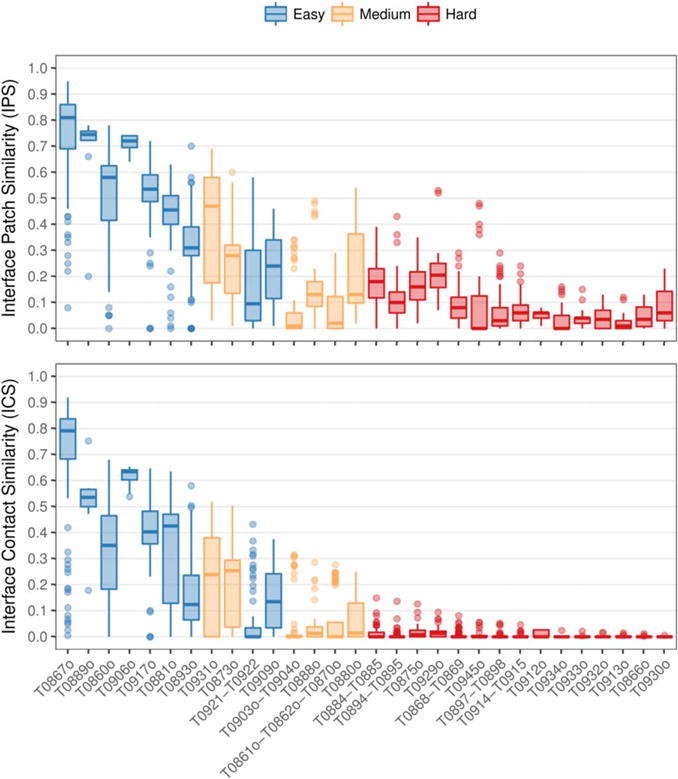
Distribution of IPS (top) and ICS (bottom) scores of all submitted models for each target assembly as a boxplot. The higher the IPS and ICS scores, the more accurate the model. Targets colored by prediction difficulty and sorted decreasingly by maximum ICS score
We also find that prediction of protein contacts at the interface without a template is extremely challenging and that the prediction of interface patches is more successful. Considering patches, there are predictions with an IPS above 0.3 for the majority of targets, including six out of the 15 difficult ones. On the contrary, considering interface contacts, there are models with an ICS higher than 0.25 for all easy and medium targets, while the hard targets are always below 0.2. This trend can be further observed in Figure 3. Both scores are highly correlated for high accuracy models (top right), but for lower accuracy models (lower left) values of the IPS can be about 0.5 while the ICS is lower than 0.1.
FIGURE 3.
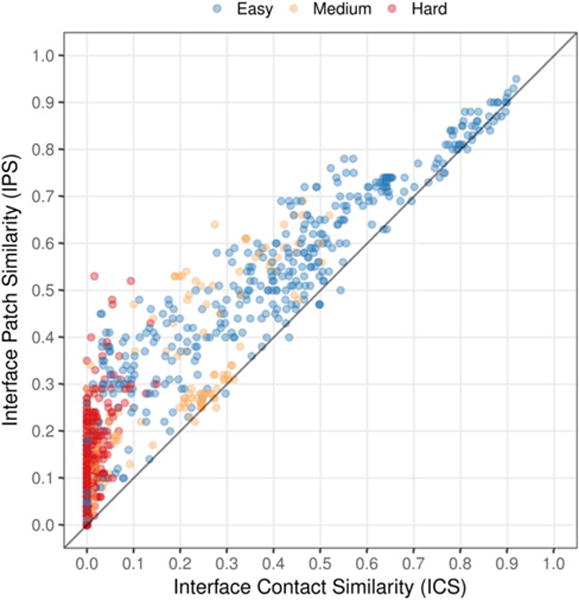
Correlation between the IPS and ICS scores for all the oligomeric models submitted to CASP12. Models are colored by target difficulty. The correlation drops for low accuracy models due to the more challenging interface contact prediction
An explanation can be that methods require the prediction of interaction patches in the protein subunits before predicting the assembly. For instance, a prediction for the dimeric hard target T0945o docked the two monomers at the correct interface, but differing at an angle of about 60° from the correct orientation (Figure 4). As a result, the prediction is successful for the interface patch, but unsuccessful for the interface contacts.
FIGURE 4.
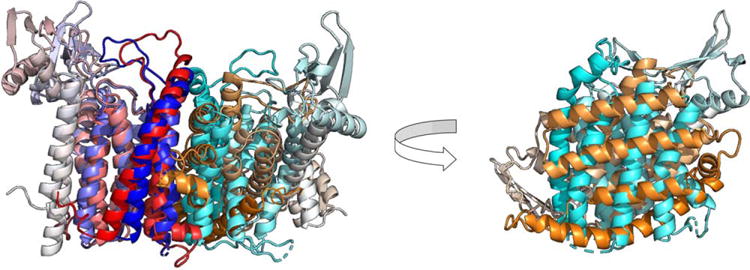
Example of a model with good interface patch prediction (high IPS score) and bad interface contact prediction (low ICS score). Superposition of target dimer T0945o (blue/cyan) and its best-scoring IPS model (red/orange) based on a single chain (left). The non-superposed chains, as seen from the interface (right). The model reproduces more accurately the interface patch of the chains (IPS = 0.48) than the residue contacts at the interface (ICS = 0.05), due to a rotation at the interface plane
3.4 | Group ranking
The overall predictor ranking is given in Figure 5. The FONT, Seok, and TSlab-assembly human groups showed the best overall performance among all target prediction difficulties. The top-ranked server, BAKER-ROSETTASERVER, primarily submitted high accuracy models for easy targets. However, it is ranked in the overall fourth position due to its lower performance in medium and hard targets.
FIGURE 5.
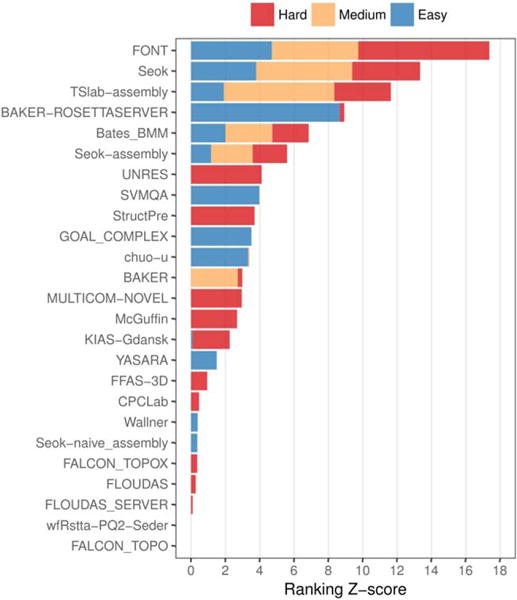
Ranking of CASP groups in the assembly prediction category. Groups are sorted decreasingly by sum of Z-scores, so that the best overall performing groups are at the top. Among the actively participating groups, only predictors that submitted oligomeric first models for a minimum of three distinct targets are shown. For each prediction difficulty, a minimum of three oligomeric first models is required to obtain the Z-score
Similarly to BAKER-ROSETTASERVER, most of the other groups seem to perform differently for easy and hard targets. Groups like SVMQA, GOAL_COMPLEX, and chuo-u perform well only for easy targets, while groups like UNRES and StructPre perform well only for hard targets. This opens the possibility to separate the ranking into template-based and template-free prediction methods, analogously to the tertiary structure assessment. We did not explore this ranking alternative due to the low and uneven participation across targets and because five out of the six top-ranking groups do perform equally across prediction difficulties. However, we think this is a possibility for future rankings.
3.5 | Baseline performance comparison
In CASP12, assembly prediction methods improve consistently the best available template. The median of the group performance distributions for each target is above the naïve predictor baseline in 12 out of the 15 oligomeric targets suitable for homology modeling (Figure 6). Furthermore, for every target there is always a group that improves over the baseline, with large differences for many targets. We observe no significant differences between human and server predictors.
FIGURE 6.
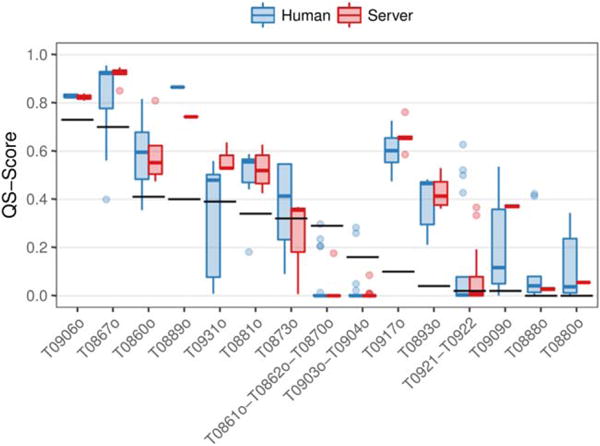
Performance comparison analysis of CASP12 oligomeric predictions against a naïve predictor. Distribution of the QS-score of the best model from each group as a boxplot. The QS-score of the best available template for each target is shown as horizontal black lines. Only targets suitable for homology modeling are shown, sorted decreasingly by the baseline QS-score. Predictions colored by the group type: human or server
3.6 | Symmetry modeling
Protein assemblies typically have high levels of symmetry.21 Independently of the assessment for the correct assembly, we analyze how prediction methods consider symmetry in their models (Supporting information Figure S4). We identify (i) groups that constrain the models to be almost perfectly symmetric (for example, StructPre), (ii) groups that never produce asymmetric models, but allow differences (flexibility) in the tertiary structure of the subunits (for example, YASARA), and (iii) groups that may consider symmetry in their modeling, but allow asymmetric assembly models (for example, chuo-u).
The experimental structures of CASP12 target assemblies are symmetric with overall low RMSD deviations among the subunits (Supporting information Table S2). The higher symmetry RMSD of target assembly T0903o-T0904o can be explained by its complex crystal lattice (four copies of the tetramer in the asymmetric unit), which creates distinct crystal environments for each of the protein subunits in the assembly. We believe that assembly models should be always symmetric, except if strong evidence of the contrary exists. Nearly all predictor groups that submitted models for the symmetric target assemblies seem to be already following this rule (Supporting information Figure S4).
3.7 | Assembly prediction highlights
The prediction of viral fiber trimers was very successful. High accuracy predictions (above 0.6 ICS and 0.7 IPS in Figure 2) were submitted for viral fiber targets with available sequence templates: T0860o, T0867o, and T0881o. Indeed, the target with the most accurate models among all CASP12 target assemblies is the viral fiber T0867o. One of the models from BAKER-ROSETTASERVER achieves nearly perfect accuracy in interface patch and contact similarity to the target experimental structure (Supporting information Figure S1). Good predictions (above 0.25 ICS and below 0.6 IPS in Figure 2) were also submitted for targets without sequence templates, T0880o and T0888o. In addition, in Figure 6 we observe that the majority of the models for the five viral fibers are above the baseline performance.
The positive outliers in interface patch dissimilarity for the hard targets T0929o and T0945o stand out from other predictions. Models from TSlab-assembly accurately reproduced the tertiary structure of the T0929 monomers and their dimeric assembly, although a threading error led to poor monomeric and interface contact scores (Supporting information Figure S2). The top-scoring IPS model for T0945o from Bates_BMM was described earlier and shown in Figure 4. Knowledge that the target is a membrane protein could have been used to model the correct docking orientation, since transmembrane helices of membrane dimers are usually parallel to each other and perpendicular to the membrane plane.
Assembly prediction for the oligomeric target T0866o was a negative result, in contrast to the outstanding predictions for its tertiary structure. This target is a cyclic homo-hexamer without structural templates, but a large amount of sequence homologs, which could facilitate the use of sequence evolution information, for instance the coevolution of residues to predict interface contacts. High accuracy monomeric predictions were submitted for its tertiary structure, but the assembly prediction was totally unsuccessful. Although oligomeric models reproduce correctly the cyclic symmetry of the target assembly, they all failed in the prediction of both the subunit fold (tertiary structure) and mode of association (Supporting information Figure S3). Therefore, assembly prediction methods, contrary to tertiary structure prediction methods, could not benefit from large sequence evolution information.
4 | CONCLUSIONS
The inclusion of quaternary structure prediction as a full assessment category in CASP12 is both an acknowledgment of the essential role that quaternary structure plays in protein function, as well as the fact that numerous prediction methods are now able to successfully predict quaternary structure. We introduced a set of scores and analysis tools to compare the accuracy of protein assembly models to their experimentally determined structures. In addition, we defined a difficulty classification scheme for targets to distinguish homology modeling from template-free assembly predictions. The two scores we used for model accuracy evaluation captured all the spectrum of prediction accuracy, thanks to their different levels of detail. We observed that, although good predictions for the interface patches exist across the prediction difficulty levels, the prediction of residue contacts at the interface without a template remains a challenge. For template-based predictions, at least one group improves over the best available template for all target assemblies, proving their usefulness in protein assembly studies.
The low and uneven participation among groups for oligomeric targets made assessment and ranking of the methods more difficult. We think there were two major challenges for participating groups: (1) the assembly information and (2) the submission of assembly models. For the first challenge, we propose either assembly prediction without providing the oligomeric state information or a two-stage experiment with a short stage to predict the oligomeric state of the target followed by a longer prediction stage after revealing the assembly information. For the second issue, we suggest to allow the submission of complete assembly models for heteromeric assemblies. We also suggest that groups declare explicitly at the beginning of the experiment their intentions to participate in assembly prediction, in order to avoid results-based inferences of the assembly predictor groups. Many predictors only perform well in either targets suitable for homology modeling or template-free targets, preventing them from ranking higher. That is why we propose to introduce separate rankings for template-based and template-free predictions, which will also highlight the methodological differences and difficulty of the problems. Another suggestion, in line with current trends in tertiary structure assessment, is to include a self-accuracy estimate for multimeric predictions.
Although the prediction of protein assemblies by CASP groups in this edition has been overall good, we believe that there is apparent room for improvement. For instance, we did not observe better prediction accuracy for targets with large sequence evolution information. We believe that using co-evolution of residues to predict interface contacts can improve the quality of the models, especially for assemblies without structural templates. The prediction of novel assemblies is an open challenge for the community, as the results for this edition show. We hope that CASP continues its effort to encourage development of protein assembly prediction methods and that our work can set a precedent for future assessments. We look forward to seeing how the state of the art of assembly prediction methods improves in future editions of the experiment.
Supplementary Material
Acknowledgments
We thank experimental groups and predictors that participated in this CASP edition. We are grateful to CASP organizers, for their invitation to participate in CASP12 and their unfailing support. S.B. was supported by the Intramural Research Program of the National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health USA. This work was partially supported by the US National Institute of General Medical Sciences (NIGMS/NIH), grant R01GM100482 to A.K. M.B. was supported by the “Fellowship for Excellence” international PhD program of the Biozentrum Basel. J.D. was supported by the RCSB PDB, funded by a grant (DBI-1338415) from the National Science Foundation, the National Institutes of Health, and the US Department of Energy. Financial support to G.C. from the Swiss National Science Foundation and the Research Committee of the Paul Scherrer Institute is gratefully acknowledged.
Funding information
Intramural Research Program of the National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, USA; US National Institute of General Medical Sciences (NIGMS/NIH), Grant/Award Number: R01GM100482; Fellowship for Excellence international PhD program of the Biozentrum Basel; RCSB PDB; National Science Foundation, the National Institutes of Health, and the US Department of Energy, Grant/Award Number: DBI-1338415; Research Committee of the Paul Scherrer Institute; Swiss National Science Foundation
Footnotes
AUTHOR CONTRIBUTIONS
G.C. initiated the research, planned the assessment protocol and assigned target assemblies. Tragically, he sickened and passed away on May 2, 2017. A.L. assigned target assemblies, developed software for the alignment, and scoring of assemblies and analyzed results. S. B. and J.D. analyzed results. A.K. and B.M. developed software for assessing model accuracy, ran the automatic evaluation pipeline and created the web interface for interactive analysis of the results. M.B. developed the QS-score used in the baseline performance analysis. All authors except G.C. contributed to the manuscript.
ORCID
Aleix Lafita, http://orcid.org/0000-0003-1549-3162
Spencer Bliven, http://orcid.org/0000-0002-1200-1698
Andriy Kryshtafovych, http://orcid.org/0000-0001-5066-7178
Martino Bertoni, http://orcid.org/0000-0003-0720-1027
Jose M. Duarte, http://orcid.org/0000-0002-9544-5621
Torsten Schwede, http://orcid.org/0000-0003-2715-335X
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Neelan JM, Sunde M, Matthews JM. The power of two: Protein dimerization in biology. Trends Biochem Sci. 2004;29(11):618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fekete S, Beck A, Veuthey J-L, et al. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J Pharmaceut Biomed Anal. 2014;101:161–173. doi: 10.1016/j.jpba.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Schuck P. Analytical ultracentrifugation as a tool for studying protein interactions. Biophys Rev. 2013;5(2):159–171. doi: 10.1007/s12551-013-0106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capitani G, Duarte JM, Baskaran K, et al. Understanding the fabric of protein crystals: Computational classification of biological interfaces and crystal contacts. Bioinformatics. 2016;32(4):481–489. doi: 10.1093/bioinformatics/btv622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baskaran K, Duarte JM, Biyani N, et al. A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct Biol. 2014;14(1):1–11. doi: 10.1186/s12900-014-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy ED. PiQSi: Protein quaternary structure investigation. Structure. 2007;15(11):1364–1367. doi: 10.1016/j.str.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(W1):W252. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. eLife. 2014;3(3) doi: 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariani V, Kiefer F, Schmidt T, et al. Assessment of template based protein structure predictions in CASP9. Proteins. 2011;79(Suppl 10):37–58. doi: 10.1002/prot.23177. [DOI] [PubMed] [Google Scholar]
- 11.Lensink MF, et al. Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins. 2016;84(Suppl. 1):323–348. doi: 10.1002/prot.25007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duarte JM, Srebniak A, Schärer MA, et al. Protein interface classification by evolutionary analysis. BMC Bioinformatics. 2012;13(1):334. doi: 10.1186/1471-2105-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Kryshtafovych A, Monastyrskyy B, Fidelis K. CASP11 statistics and the prediction center evaluation system. Proteins: Structure, Function and Bioinformatics. 2016;84:15–19. doi: 10.1002/prot.25005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prlić A, et al. BioJava: An open-source framework for bioinformatics in 2012. Bioinformatics. 2012;28(20):2693–2695. doi: 10.1093/bioinformatics/bts494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 17.Lafita A. Assessment of protein assembly prediction in CASP12 & Conformational dynamics of integrin α-I domains. ETH Zürich. 2017;99 [Google Scholar]
- 18.Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 19.Bertoni M, et al. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. 2017;7:10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duarte JM, Biyani N, Baskaran K, et al. An analysis of oligomerization interfaces in transmembrane proteins. BMC Struct Biol. 2013;13(1):21. doi: 10.1186/1472-6807-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy ED, Teichmann S. Structural, evolutionary, and assembly principles of protein oligomerization. Prog Mol Biol Transl Sci. 2013;117:25–51. doi: 10.1016/B978-0-12-386931-9.00002-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.