

Abstract
The rapid evolution and widespread use of next generation sequencing (NGS) in clinical laboratories has allowed an incredible progress in the genetic diagnostics of several inherited disorders. However, the new technologies have brought new challenges. In this review we consider the important issue of NGS data analysis, as well as the interpretation of unknown genetic variants and the management of the incidental findings. Moreover, we focus the attention on the new professional figure of bioinformatics and the new role of medical geneticists in clinical management of patients. Furthermore, we consider some of the main clinical applications of NGS, taking into consideration that there will be a growing progress in this field in the forthcoming future.
Key words: next generation sequencing, genetics, inherited disorders, causative mutations, sequence depth, coverage, incidental findings, variants interpretation, diagnostics, genetic medicine
INTRODUCTION
The next-generation sequencing (NGS) has been introduced in genomic laboratories about 10 years ago. Its impact on technological revolution has important implications in human biology and medicine [1]. After improvements in accuracy, robustness and handling, it became a widely used and an alternative approach to the direct Sanger sequencing [2,3].
The progress of NGS is leading to the increase of discovery of number of genes associated to human inherited disorders and to the elucidation of molecular basis of complex disease [4]. Moreover, since on NGS platforms it is possible to perform a parallel sequencing of different target regions, NGS is widely used in diagnostics. Recently, the use of NGS in clinical laboratories has became increasingly widespread, used in diagnostics of infectious diseases, immune disorders, human hereditary disorders and in non-invasive prenatal diagnosis, and, more recently, in the therapeutic decision making for somatic cancers [5–12].
A great advantage of NGS approach is based on its ability to deliver clinical diagnosis in a short time [3].
Currently, there are several NGS platforms available for routine diagnostic applications. These sequencers allow performing an high-throughput analysis within few days, considerably decreasing costs [13]. These new technologies are different from Sanger sequencing because they are based on a massively parallel analysis and high throughput. Today two different NGS technologies are mainly used in clinical laboratories: Ion Torrent and Illumina systems [14]. The Ion Torrent Personal Genome Machine (PGM) was launched in 2011, while the widely used Illumina benchtops for diagnostic purpose are MiSeq, marketed in 2011, MiniSeq, launched in 2016, or iSeq100, debuting in the end of 2017. The Ion Torrent exploited the emulsion PCR using native dNTP chemistry that releases hydrogen ions during base incorporation by DNA polymerase and a modified silicon chip detecting the pH modification [15], while Illumina technology is based on the existing Solexa sequencing by synthesis chemistry with the use of very small flow-cells, reduced imaging time and fast sequencing process [14].
NGS APPROACH IN CLINICAL LABORATORIES
The increase in number of causative genes associated with human inherited disorders is directly associated with the implementation of NGS.
Until now Sanger sequencing has been the gold standard in clinical laboratories for single-gene tests and it serves as the standard methods by which NGS data should be compared and validated [16]. However, Sanger sequencing achieves the diagnostic goal when there is a clear phenotypic indication of a classical Mendelian disorder and the single-gene test approach is preferred. It eliminates the problem of incidental findings, that we will discuss later, but it may push the patients into a “diagnostic odyssey”, where they could be evaluated by multiple providers, sometimes for years, without a genetic diagnosis [13].
Today there is a different scenario, in which genomic technologies can be very useful to detect genetic variations in patients with a high accuracy and an important reduction of costs, thanks to the first-generation sequencing approach. In particular, next-generation sequencing will increasingly be used for clinically heterogeneous inherited disorders, resulting in an increase in number of reported disease-causing genes [6]. Indeed, in the majority of human inherited diseases not merely one gene but a number of genes may interact leading to overlapping pathological phenotypes [2]. NGS approach is tempting when there is a genetic contribution in heterogeneous and complex diseases, such as in cardiomyopathies, in cardiac arrhythmias, in connective tissue disorders, in mental retardation or autism, where a large number of genes are involved in a large phenotypic spectrum [10,11,17]. In these cases, NGS approaches allows to test a large number of genes simultaneously in a cost-effective manner [13]. An important issue is to decide which kind of NGS testing strategy is best suited for each clinical case. Two options are currently available: targeted gene panels or whole-exome sequencing (WES) [13].
Targeted sequencing of selected genes offers a good coverage (mean 300X, depending on platforms and number of analyzed samples) for the entire analyzed panel and specific regions refractory to NGS can be sequenced by Sanger sequencing, in order to cover the gap and to validate the NGS data [18,19]. So far, targeted resequencing has been adopted to develop tests for genetic disorders, such as non-syndromic deafness [20,21], common and heterogeneous diseases, such as hypertension and diabetes [22], or in traditional cytogenetic and Mendelian disorder diagnosis [23,24]. The main limitation of targeted sequencing is the rigidity of testing only a selected number of genes. Since the genetic field is rapidly evolving, new genes may be associated with a clinical phenotype and as such redesigning and revalidation of the panel is needed [13,16]. On the contrary a clear advantage of the use of targeted panel is the reduction of number of incidental findings and/or the number of variants of unknown significance, that will be discuss later in this review.
On the other hand, the benefit of WES is testing a greater number of genes, even if, in practice, complete coverage of all coding exons is infeasible. The WES application may be useful, for example, in negative cases in targeted sequencing or in a rare disease, especially in exploiting trios approach. Indeed, it allowed the identification of genes responsible for the dominant Freeman-Sheldon syndrome, the recessive Miller Syndrome and the dominant Schinzel-Giedion Syndrome [25]. However it is important to keep in mind that about 10% of targeted bases sequenced in WES do not get the 20 read depth [26], required for clinical confidence and interpretation, and approximately only 85% of genes associated to human diseases into the principle database (OMIM) receive the adequate coverage [27]. Poor coverage in WES can due to several factors: probes that are not tiled for particular genes probably not included during assay development or because repetitive sequences prevented inclusion or poorly performing probes owing to GC-richness and low mapping quality [6].
However it is important to consider that both of these approaches can significantly reduce costs and turn-around time for a genetic test [13].
THE MAIN ISSUE OF NGS: THE INTERPRETATION OF GENETIC DATA FOR A CLINICAL UTILITY
In the NGS process one limiting step is without doubt the complexity of genetic variation interpretation in whole exome, due to the presence of thousands of rare single nucleotide variations without pathogenic effect. Moreover, in the majority of human diseases the pathological phenotype may be caused by a pathogenic rare mutation with a strong effect or it may be caused by a co-presence of multiple genetic variations [28][29].
Reliable interpretation of the multiple and de novo variants identified through NGS will require additional experience and validation before it reaches the clinical stage on a large scale, particularly for diagnosis of complex traits [30]. In the recent past, genetic data did not drive diagnosis but had a primarily confirmatory role. Today the major challenge is to convert pathogenic genetic data into a primary diagnostic tool that can shape clinical decisions and patients management [31].
Actually, the interpretation of genetic variants is based on criteria published by the American college of medical genetics and genomics (ACMG). The ACMG recommends that the variants be allocated to one of the categories reported below [32]:
disease causing (class V): the sequence variation is previously reported and recognized as causative of the disorder;
likely disease causing (class IV): the sequence variation is not previously reported as expected to cause the disorder, frequently in a known disease gene;
variant of unknown clinical significance (VUS; class III): the sequence variation is unknown or expected to be causative of disease and is found to be connected with a clinical presentation;
likely not disease causing (class II): the sequence variation is not previously reported and it is probably not causative of the pathology;
not disease causing (class I): the sequence variation is already reported and documented as neutral variant.
Moreover, most of these classes of variants are subject to supplementary interpretation focusing on literature reported, population frequencies, clinical findings, mutation databases and possibly case-specific research data [31]. The principal human variant databases are useful to annotate both common and pathogenic variants, such as dbSNP, gnomAD or ExAC database (Exome Aggregation Consortium) [33], and to classify variants previously associated with human disorders, such as Human Gene Mutation Database (HGMD) [34] and ClinVar.
The variants of unknown significance (VUS) represent a problem for the interpretative process. Indeed it is known that hundreds of loss of function variants with unknown clinical significance are present in each individual’s genome and today their prioritization remains a primary challenge [35].
In some cases, the interpretation of VUS can be useful in commencing the segregation analysis in large families including affected members or the identification of the occurrence of de novo variation in the affected patient. Unfortunately, in many cases the interpretation of VUS remains unresolved and its identification cannot be used for the clinical management of patients and families [29,36].
Until now few clear guidelines are published for the VUS interpretation [36]. Today, in order to try to assign a pathological score to VUS, it is important to consider, for example, its allelic frequency in a control population (1000 Genomes or exome sequencing project consortium [ExAC]), the amino acidic conservation, the predicted effect on protein function and the results of published functional assay [37,38].
Up to now in silico prediction algorithms, such as Polyphen, Sift, Mutation Taster or UMD predictor, have been developed and they are widely used for the missense variants interpretation [37]. However, they present some intrinsic caveat and limitations, affecting their specificity and sensitivity, that can lead to possible false-positive and false-negative interpretations [39]. Another existing problem involves the allelic frequency, that is mainly estimated from the 1000 Genome project and ExAC, that represents only a fraction of the worldwide population, so the declared allelic frequency available is not stratified according to the real population groups [29].
Since the problem of the management of VUSs is not yet resolved, it would be fundamental to collect and share VUSs and available clinical data, allowing a progressive and definitive classification of these variants, as deleterious (class V) or neutral ones (class I) [29,30].
Another important challenge of the use of NGS approach in clinical diagnostic is the management of the amount of data generated [40]. Indeed generation, analysis and also storage of NGS data require sophisticated bioinformatics infrastructure [41].
A skilled bioinformatics staff is needed to manage and analyze NGS data, and so both computing infrastructure and manpower impact on costs of NGS applications in clinical diagnostics. Bioinformaticians are to be mandatory in the organization chart of clinical laboratories in the NGS era, where they have to closely collaborate with clinicians and laboratory staff to optimize the panel testing and the NGS data analyses [42].
Bioinformatics has been recently defined as the discipline that develops and applies advanced computational tools to manage and analyze the NGS data. Bioinformatics pipeline developed for NGS are aimed to convert the raw sequencing signals to data, data to information, and information to knowledge [43].
This process can be developed in three different steps - primary, secondary, and tertiary analyses [44]:
The primary analysis is the process of raw data produced by NGS instruments,
the secondary analysis is the alignment to a reference sequence and the calling variants and, finally,
the tertiary analysis is the confirmation or validation of detected variants, providing evidence to facilitate interpretation [41].
All clinical bioinformatics systems require these three steps that should be properly validated and documented. In particular, it requires determination of variant calling sensitivity, specificity, accuracy and precision for all variants reported in the clinical assay [44]. The quality criteria of the performed sequencing test have to be described on the report for clinicians and patients. In particular, it is needed to declare the sensitivity and specificity of the techniques used considering both technical and bioinformatics parameters. It is important to report which target region was not sequenced, the number of reads obtained, the quality of the sequence, the limitations of the chosen sequencing method and of the settings of used bioinformatics pipeline [16,45].
ETHICAL CONSIDERATIONS AND MANAGEMENT OF INCIDENTAL FINDINGS
The development and the widespread use of NGS in clinical laboratories are paired with debate on the ethics for reporting incidental findings [46,47]. In 2013 the ACMG has highlighted the question of the incidental findings (IF), defining them as “genetic variations identified by genomic sequencing but not related to the disease being investigated” [48].
According to the European Society of Human Genetics (ESHG) guidelines, the targeted diagnostic testing should be performed minimizing the likelihood of detecting incidental findings, focusing only on genes clinically actionable [49]. It means that genetic testing should aim to analyze the causative genes associated to the primary clinical questions, even if a broader panel of genes or the whole exome sequencing has been performed [49]. It is the role of responsible clinicians requesting the test to disclose an incidental finding to a patient, not the role of the clinical laboratory.
The impact of the IF determines how the genetic finding should be disclosed or not to a patient, also to avoid unwarranted psychological stress. In particular, if it can bring minor consequences or if a clinical intervention is possible, then the variant should be reported.
On the contrary, if the variant is associated to a late onset disorder or has major consequences, counselling and consent will determine if and when the variant can and should be reported to the patient [36]. This implies that genetic tests should be ordered by medical professionals who are capable of performing appropriate counselling [50]. For that reason, the counselling and the informed consent are critical steps.
There is a difference between recording and reporting a variant, as well as between who receives this information, clinicians or patients, and when. When a variant is reported to a clinician, it does not mean that it will be revealed to patient. Indeed, the clinician should evaluate the impossible clinical implication of this information, based on the clinical history of patient. For example, the impact of an IF in a case without a known family history for a specific disorder is different from the case in which the patient is already aware of a preexisting familial condition.
Another interesting example is the acute neonatal care, in which immediate reporting of all IFs to patients’ families may not be appropriate and the genetic information may be reconsider later in baby’s life. Similarly, the report of IFs may be postponed in cases where parents or patients are given a diagnosis linked to poor prognosis or in case of post-mortem genetic testing.
Additional contexts in which the reporting of incidental findings may have an influence on the patients management are carrier testing, prenatal diagnosis, pharmacogenetics testing and additional non-diagnostic testing such as medical research (dependent on the study design), forensic testing, parental and genealogical testing. In conclusion, the issue of IFs requires an appropriate pre and post counselling to correctly inform the patient [16].
The widespread implementation of NGS approach in diagnosis of human pathologies raises the problem of management of IFs and VUSs and it is needed to have clear guidelines for the handling of NGS data in the diagnostics approach (Figure 1).
Figure 1.
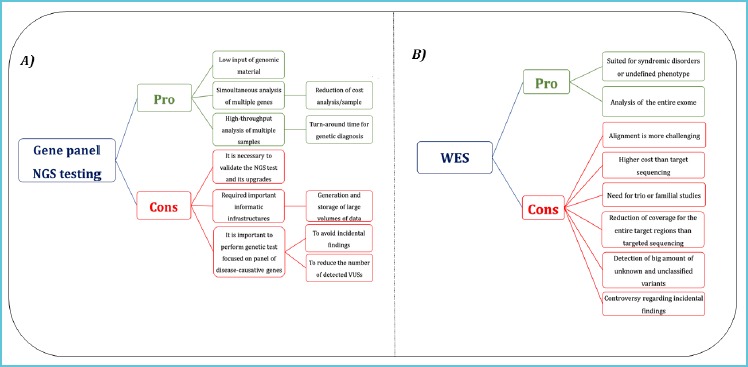
Advantages and challenges of the use of gene panel NGS testing and WES
So far the application of WES in clinical diagnostics presents more open challenges (B) than targeted sequencing (A).
CONCLUSIONS
Until now Sanger sequencing has been the gold standard in molecular diagnostics and it has been used in clinical testing method for Mendelian disorders, in which most of causative variants are identified in the principal causative genes. Since the rapid and incremental improvements in instrumentations, methodologies and throughput and the significant reduction of costs, the NGS technologies are being integrated into patient care and clinical management. NGS allows sequencing of all genes relevant to a given phenotype starting from a small amount of total DNA. In that way, the limitation factors are no longer the size of the gene or its causative contribution but the actual knowledge of the genetic basis of patient’s disease [6].
In the past, clinicians considered genetic tests with a marginal diagnostic value, only if a definitive diagnosis was not yielded or if it had implications on future family planning. Often the positive genetic test results did not influence clinical management of the patient.
However today, with the potentiality of NGS, the parallel sequencing of large multi-genes panel, that may describe a broader range of phenotypes, the clinicians are changing their point of view on the role of the genetics in patients care. Indeed, nowadays the genetic testing may be useful for the evaluation of a clinical case and, if the result were to be positive, it may save time and money in identifying the etiology.
Today physicians often begin their clinical evaluations with the genetic tests. For example, the evaluation of patients with left ventricular hypertrophy begins with genetic testing, given that the genetic diagnosis is achieved in about 80% of hypertrophic cardiomyopathy cases [51].
The results of most targeted genetic tests may be available for clinicians in 2-8 weeks, which is an impressive improvement compared to the time taken for direct Sanger sequencing and the odyssey lived by some patients before to understand the cause of their rare disorder [6].
This strategy of approaching the clinical evaluation has also economically beneficial in patients without diagnosis [52].
The euphoria of the widespread use of the NGS applications to the clinical diagnosis is combined with the awareness of emerged challenges, such as the validation of large number of genetic variations detected, that can be IF or VUSs, the use of standardization processes in clinical diagnostics, the management of terabytes of data and variants interpretation.
In the NGS approach, the analysis of data requires the development of a standard pipeline to process sequencing data. The flow chart analysis includes mapping, variant calling and annotation. Today there are various public database, such as dnSNP [53], the 1000 Genome Project [54], ExAC, as well as several internal control databases.
Targeted panel sequencing or clinical exome sequencing identifies several variations in each person, but as far there are no clear guidelines to filter variants and to delineate their possible pathological meanings. For this reason, the pathogenic validation may be the limiting step. Because of these considerations, it is important to apply the NGS approach in clinical diagnostics for that disorders of which the main causative genes have been identified. Indeed, in this case the genetic tests can successfully reveal a useful result.
Moreover, another consideration involves the fundamental change of the figure of medical geneticist in the NGS era. Indeed, the NGS applications into diagnostic field can lead to useful results for patient’s care with genetic disorders. As such, the geneticists will become a pivotal part of the collaborative team of clinicians and their role will be fundamental for the clinical interpretation of NGS data to guide patient care [25].
Consequently, clinical medical geneticists have to complement their skills with expertise in the clinical interpretation of NGS data.
Moreover we have to keep in mind that the medical geneticist has an important and crucial role also in the pre-test counseling, to deliver reliable information to patients [29]. Indeed it is important to clearly explain to the patient and his family the medical implications of the identification of a genetic alteration, regarding the degree of risk for a disease and also the significance of a possible negative results, both in pretest and in the post test counseling [29].
In meanwhile, the NGS approach becomes a cornerstone for the genetic diagnosis, a more efficient and powerful third-generation technologies are expected to further revolutionize genome sequencing [55]. The three commercially available third-generation DNA sequencing technologies are Pacific Biosciences (Pac Bio), Single Molecule Real Time (SMRT) sequencing, the Illumina Tru-seq Synthetic Long-Read technology, and the Oxford Nanopore Technologies sequencing platform.
Third-generation sequencing was made feasible in part by increasing capacity of existing technologies and improvements in chemistry and it allows to sequence a single nucleic acid molecule, eliminating the DNA amplification step, with a longer and easier mapping of sequencing reads with lower costs [55].
Moreover, the use of longer reads than the second-generation allow to overcome the important limitation of NGS in copy number variation analysis (CNV) [56], even if these single-molecule sequencing approaches have to become even more robust for a wider use.
Lastly, few years ago a new technique called Spatial Transcriptomics was developed and gave rise to fourth generation sequencing, also known as single-cell sequencing [55,57]. In this new technology, NGS chemistry is applied to the sequencing of nucleic acid composition directly in fixed cells and tissues providing a throughput analysis, opening great opportunity mainly for the analysis of tumor cells variability in situ [58]. In forthcoming future, it holds exciting prospective for research and new insights regarding genomic diagnostics.
REFERENCES
- 1.Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155: 27–38. doi:10.1016/j.cell.2013.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grumbt B, Eck SH, Hinrichsen T, Hirv K. Diagnostic applications of next generation sequencing in immunogenetics and molecular oncology. Transfus Med hemotherapy Off Organ der Dtsch Gesellschaft fur Transfusions-medizin und Immunhamatologie. 2013;40: 196–206. doi:10.1159/000351267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vrijenhoek T, Kraaijeveld K, Elferink M, de Ligt J, Kranen-donk E, Santen G, et al. Next-generation sequencing-based genome diagnostics across clinical genetics centers: implementation choices and their effects. Eur J Hum Genet. Nature Publishing Group; 2015;23: 1142–1150. doi:10.1038/ejhg.2014.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coonrod EM, Durtschi JD, Margraf RL, Voelkerding K V. Developing genome and exome sequencing for candidate gene identification in inherited disorders: an integrated technical and bioinformatics approach. Arch Pathol Lab Med. 2013;137: 415–433. doi:10.5858/arpa.2012-0107-RA [DOI] [PubMed] [Google Scholar]
- 5.Peters DG, Yatsenko SA, Surti U, Rajkovic A. Recent advances of genomic testing in perinatal medicine. Semin Perinatol. 2015;39: 44–54. doi:10.1053/j.semperi.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet. 2013;14: 295–300. doi:10.1038/nrg3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorburn F, Bennett S, Modha S, Murdoch D, Gunson R, Murcia PR. The use of next generation sequencing in the diagnosis and typing of respiratory infections. J Clin Virol. 2015;69: 96–100. doi:10.1016/j.jcv.2015.06.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LePichon J-B, Saunders CJ, Soden SE. The Future of Next-Generation Sequencing in Neurology. JAMA Neurol. 2015; doi:10.1001/jamaneurol.2015.1076 [DOI] [PubMed] [Google Scholar]
- 9.Gorokhova S, Biancalana V, Lévy N, Laporte J, Bartoli M, Krahn M. Clinical massively parallel sequencing for the diagnosis of myopathies. Rev Neurol (Paris). 171: 558–571. doi:10.1016/j.neurol.2015.02.019 [DOI] [PubMed] [Google Scholar]
- 10.Harripaul R, Noor A, Ayub M, Vincent JB. The Use of Next-Generation Sequencing for Research and Diagnostics for Intellectual Disability. Cold Spring Harb Perspect Med. Cold Spring Harbor Laboratory Press; 2017;7: a026864. doi:10.1101/cshperspect.a026864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Resta C, Pietrelli A, Sala S, Della Bella P, De Bellis G, Ferrari M, et al. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum Mol Genet. 2015;24: 5828–5835. doi:10.1093/hmg/ddv302 [DOI] [PubMed] [Google Scholar]
- 12.Carrera P, Di Resta C, Volonteri C, Castiglioni E, Bonfiglio S, Lazarevic D, et al. Exome sequencing and pathway analysis for identification of genetic variability relevant for bronchopulmonary dysplasia (BPD) in preterm new-borns: A pilot study. Clin Chim Acta. 2015;451: 39–45. doi:10.1016/j.cca.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 13.Williams ES, Hegde M. Implementing genomic medicine in pathology. Adv Anat Pathol. 2013;20: 238–244. doi:10.1097/PAP.0b013e3182977199 [DOI] [PubMed] [Google Scholar]
- 14.Quail M, Smith ME, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genomics. BioMed Central; 2012;13: 341. doi:10.1186/1471-2164-13-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merriman B, Rothberg JM. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis. 2012;33: 3397–3417. doi:10.1002/elps.201200424 [DOI] [PubMed] [Google Scholar]
- 16.Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15: 733–747. doi:10.1038/gim.2013.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin X, Tang W, Ahmad S, Lu J, Colby CC, Zhu J, et al. Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hear Res. 2012;288: 67–76. doi:10.1016/j.heares.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aziz N, Zhao Q, Bry L, Driscoll DK, Funke B, Gibson JS, et al. College of American Pathologists’ Laboratory Standards for Next-Generation Sequencing Clinical Tests. Arch Pathol Lab Med. 2015;139: 481–493. doi:10.5858/arpa.2014-0250-CP [DOI] [PubMed] [Google Scholar]
- 19.Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91: 97–108. doi:10.1016/j.ajhg.2012.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vona B, Nanda I, Hofrichter MAH, Shehata-Dieler W, Haaf T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol Cell Probes. Academic Press; 2015;29: 260–270. doi:10.1016/J.MCP.2015.03.008 [DOI] [PubMed] [Google Scholar]
- 21.Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M, et al. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014;22: 768–775. doi:10.1038/ejhg.2013.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szopa M, Ludwig-Gałęzowska A, Radkowski P, Skupień J, Zapała B, Płatek T, et al. Genetic testing for monogenic diabetes using targeted next-generation sequencing in patients with maturity-onset diabetes of the young. Pol Arch Med Wewn. 2015;125: 845–851. Available: http://www.ncbi.nlm.nih.gov/pubmed/26552609 [DOI] [PubMed] [Google Scholar]
- 23.Cook JR, Kelley TW. The impact of molecular cytogenetics and next generation sequencing in hematopathology: accomplishments and challenges. Pathology. Elsevier; 2014;46: S22. doi:10.1097/01.PAT.0000454125.58964.86 [Google Scholar]
- 24.Ma ESK, Wan TSK, Au CH, Ho DN, Ma SY, Ng MHL, et al. Next-generation sequencing and molecular cytogenetic characterization of ETV6-LYN fusion due to chromosomes 1, 8 and 12 rearrangement in acute myeloid leukemia. Cancer Genet. 2017;218–219: 15–19. doi:10.1016/j.cancergen.2017.09.001 [DOI] [PubMed] [Google Scholar]
- 25.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14: 681–691. doi:10.1038/nrg3555 [DOI] [PubMed] [Google Scholar]
- 26.Yao R, Zhang C, Yu T, Li N, Hu X, Wang X, et al. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol Cytogenet. BioMed Central; 2017;10: 30. doi:10.1186/s13039-017-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh RR, Luthra R, Routbort MJ, Patel KP, Medeiros LJ. Implementation of next generation sequencing in clinical molecular diagnostic laboratories: advantages, challenges and potential. Expert Rev Precis Med Drug Dev. Taylor & Francis; 2016;1: 109–120. doi:10.1080/23808993.2015.1120401 [Google Scholar]
- 28.Sommariva E, Pappone C, Martinelli Boneschi F, Di Resta C, Rosaria Carbone M, Salvi E, et al. Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. Eur J Hum Genet. 2013;21: 911–917. doi:10.1038/ejhg.2012.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frebourg T. The challenge for the next generation of medical geneticists. Hum Mutat. 2014;35: 909–911. doi:10.1002/humu.22592 [DOI] [PubMed] [Google Scholar]
- 30.Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. Nature Publishing Group; 2016;24: 2–5. doi:10.1038/ejhg.2015.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet. 2013;14: 415–426. doi:10.1038/nrg3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17: 405–424. doi:10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song W, Gardner SA, Hovhannisyan H, Natalizio A, Weymouth KS, Chen W, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: insights of relevance to variant classification. Genet Med. 2016;18: 850–854. doi:10.1038/gim.2015.180 [DOI] [PubMed] [Google Scholar]
- 34.Stenson PD, Mort M, Ball E V, Howells K, Phillips AD, Thomas NS, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1: 13. doi:10.1186/gm13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12: 628–640. doi:10.1038/nrg3046 [DOI] [PubMed] [Google Scholar]
- 36.Hegde M, Bale S, Bayrak-Toydemir P, Gibson J, Bone Jeng LJ, Joseph L, et al. Reporting Incidental Findings in Genomic Scale Clinical Sequencing—A Clinical Laboratory Perspective. J Mol Diagnostics. 2015;17: 107–117. doi:10.1016/j.jmoldx.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Resta C, Manzoni M, Zoni Berisso M, Siciliano G, Benedetti S, Ferrari M. Evaluation of damaging effects of splicing mutations: Validation of an in vitro method for diagnostic laboratories. Clin Chim Acta. 2014;436C: 276282. doi:10.1016/j.cca.2014.05.026 [DOI] [PubMed] [Google Scholar]
- 38.Thompson BA, Spurdle AB, Plazzer J-P, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46: 107–115. doi:10.1038/ng.2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7: 248–249. doi:10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein LD. The case for cloud computing in genome informatics. Genome Biol. 2010;11: 207. doi:10.1186/gb-2010-11-5-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014;15: 256–278. doi:10.1093/bib/bbs086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Koning TJ, Jongbloed JDH, Sikkema-Raddatz B, Sinke RJ. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: the opportunities and challenges. Expert Rev Mol Diagn. 2015;15:61–70. doi:10.1586/14737159.2015.976555 [DOI] [PubMed] [Google Scholar]
- 43.Oliver GR, Hart SN, Klee EW. Bioinformatics for clinical next generation sequencing. Clin Chem. 2015;61: 124–135. doi:10.1373/clinchem.2014.224360 [DOI] [PubMed] [Google Scholar]
- 44.Moorthie S, Hall A, Wright CF. Informatics and clinical genome sequencing: opening the black box. Genet Med. 2013;15: 165–171. doi:10.1038/gim.2012.116 [DOI] [PubMed] [Google Scholar]
- 45.Allyse M, Michie M. Not-so-incidental findings: the ACMG recommendations on the reporting of incidental findings in clinical whole genome and whole exome sequencing. Trends Biotechnol. 2013;31: 439–441. doi:10.1016/j.tibtech.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolf SM, Annas GJ, Elias S. Point-counterpoint. Patient autonomy and incidental findings in clinical genomics. Science. 2013;340: 1049–1050. doi:10.1126/science.1239119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGuire AL, Joffe S, Koenig BA, Biesecker BB, McCullough LB, Blumenthal-Barby JS, et al. Point-counterpoint. Ethics and genomic incidental findings. Science. 2013;340: 1047–1048. doi:10.1126/science.1240156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15: 565–574. doi:10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claustres M, Kožich V, Dequeker E, Fowler B, HehirKwa JY, Miller K, et al. Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular genetic). Eur J Hum Genet. 2014;22:160–170. doi:10.1038/ejhg.2013.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hehir-Kwa JY, Claustres M, Hastings RJ, van Ravenswaaij-Arts C, Christenhusz G, Genuardi M, et al. Towards a European consensus for reporting incidental findings during clinical NGS testing. Eur J Hum Genet. Macmillan Publishers Limited; 2015; doi:10.1038/ejhg.2015.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15: 158–170. doi:10.1016/j.jmoldx.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 52.Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16: 176–182. doi:10.1038/gim.2013.99 [DOI] [PubMed] [Google Scholar]
- 53.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29: 308–311. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=29783&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467: 1061–1073. doi:10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. American Association for the Advancement of Science; 2016;353: 78–82. doi:10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- 56.Jia W, Qiu K, He M, Song P, Zhou Q, Zhou F, et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013;14: R12. doi:10.1186/gb-2013-14-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ke R, Mignardi M, Hauling T, Nilsson M. Fourth Generation of Next-Generation Sequencing Technologies: Promise and Consequences. Hum Mutat. Wiley-Black-well; 2016;37: 1363–1367. doi:10.1002/humu.23051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mignardi M, Nilsson M. Fourth-generation sequencing in the cell and the clinic. Genome Med. BioMed Central; 2014;6: 31. doi:10.1186/GM548 [DOI] [PMC free article] [PubMed] [Google Scholar]