

Abstract
Innate lymphoid cells (ILCs) are defined as a distinct arm of innate immunity. According to their profile of secreted cytokines and lineage-specific transcriptional factors, ILCs can be categorized into the following three groups: group 1 ILCs (including natural killer (NK) cells and ILC1s) are dependent on T-bet and can produce interferon-γ; group 2 ILCs (ILC2s) are dependent on GATA3 and can produce type 2 cytokines, including interleukin (IL)-5 and IL-13; and, group 3 ILCs (including lymphoid tissue-like cells and ILC3s) are dependent on RORγt and can produce IL-22 and IL-17. Collaborative with adaptive immunity, ILCs are highly reactive innate effectors that promptly orchestrate immunity, inflammation and tissue repair. Dysregulation of ILCs might result in inflammatory disorders. Evidence regarding the function of intrahepatic ILCs is emerging from longitudinal studies of inflammatory liver diseases wherein they exert both physiological and pathological functions, including immune homeostasis, defenses and surveillance. Their overall effect on the liver depends on the balance of their proinflammatory and antiinflammatory populations, specific microenvironment and stages of immune responses. Here, we review the current data about ILCs in chronic liver disease progression, to reveal their roles in different stages as well as to discuss their therapeutic potency as intervention targets.
Keywords: Innate lymphoid cells, Chronic liver disease, Hepatitis, Liver fibrosis, Liver cancer
Core tip: Innate lymphoid cells (ILCs), mirroring both the phenotypes and functions of T cells, have been defined as a distinct arm of innate immunity. There has been a marked increase in the studies investigating the dysregulation of ILCs in chronic liver pathologies. This manuscript presents a comprehensive overview of the state of ILCs, including the fundamental concepts as well as summarizing their ambiguous roles in the progression of the chronic liver hepatitis, fibrosis and carcinoma. It also provides an insight into the current research gaps and indicates the therapeutic potency and development direction of future research of ILCs.
INTRODUCTION
Liver diseases usually evolve from inflammation to fibrosis, with cirrhosis manifested in the advanced stage, and serving as a well-determined major risk factor of liver cancer. Liver disease remains a major health problem, affecting millions of people worldwide. Ongoing chronic inflammation in the liver induced by infections, hepatotoxic drugs, autoimmunity, alcohol abuse or toxins will result in liver fibrosis, which is the consequence of an irreversible, progressive condition occurring in most types of chronic liver diseases and characterized by excessive deposition of extracellular matrix (ECM) proteins, mainly composed of collagen[1]. The situation where ECM formation is prompted by activated hepatic stellate cells (HSCs) outweighs the collagen degradation by matrix metalloproteases (MMPs) and will lead to structural distortion of the normal liver tissue and functional impairment; furthermore, it is associated with an increased risk of cirrhosis, portal hypertension and subsequent liver failure and liver cancer[2-4]. Tremendous efforts have been made to design strategies which could prevent liver disease progression.
Innate lymphoid cells (ILCs) are a recently identified group of mononuclear hematopoietic cells which encompass not only cytotoxic natural killer (NK) cells but also noncytotoxic ILCs, and are involved in immunity and tissue remodeling. Though characterized with lymphoid morphology, ILCs lack the rearranged antigen receptors and are defined as cell lineage marker-negative (Lin-) cells[5]. ILCs mirror both the phenotypes and functions of T cells, for which noncytotoxic ILCs have been proposed as the innate counterparts of CD4+ T helper (Th) cells, whereas NK cells are considered to be the innate equivalents of CD8+ cytotoxic T (Tc) cells[6].
Group1 ILCs comprise both Eomes-dependent NK cells and T-bet-dependent ILC1s[7]. Upon stimulation by interleukin (IL)-12, IL-15 and IL-18 derived from both myeloid cells and nonhematopoietic cells, the ILC1s can produce Th1 cell-associated cytokines, such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α, which play critical roles in clearing intracellular pathogens[8,9]. Distinguished from ILC1s, NK cells depend on Eomes to develop and exert their cytotoxic functions by secreting granzymes and perforin[10,11]. Group 2 ILCs (ILC2s), being dependent on GATA3 and ROR-α and respondent to epithelium-derived cytokines IL-25, IL-33 and thymic stroma lymphopoietin (TSLP), can produce Th2 effector cytokines (IL-4, IL-5, IL-9, IL-13 and amphiregulin), thus playing a critical role in antihelminth immunity and allergic inflammation as well as tissue repair[12-15]. Finally, group 3 ILCs are dependent on RORγt and mainly respondent to myeloid cell-derived IL-1β and IL-23. These ILCs include lymphoid tissue-like (LTi) cells and ILC3s, which can produce IL-22, IL-17, granulocyte macrophage colony-stimulating factor and TNF, thus showing great significance in antibacterial immunity[16-18] (Figure 1).
Figure 1.
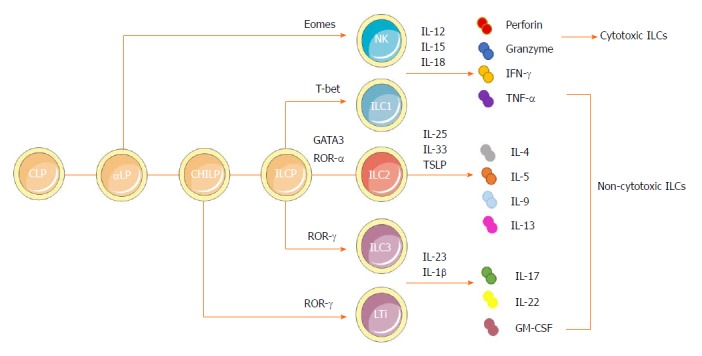
Developmental pathways and classification of innate lymphoid cells. ILCs are derived from a CLP. With the same phenotype as CLP as well as expressing α4β7 integrin, αLP gives rises to cytotoxic NK cells and differentiates into a CHILP, which gives rise to all noncytotoxic ILCs. The transcription factor PLZF further divides the progeny of CHILPs into a PLZF+ ILCPs that are restricted to ILCs except LTi cells and PLZF-independent LTi cells. Group 1 ILCs comprise both Eomes-dependent NK cells and T-bet-dependent ILC1s. They could produce IFN-γ and TNF-α in response to the stimulation by IL-12, IL-15 and IL-18. NK cells can also secrete granzymes and perforin to exert cytotoxic functions. Dependent on GATA3 and ROR-α as well as respondent to cytokines IL-25, IL-33 and TSLP, group 2 ILCs could produce Th2 effector cytokines (IL-4, IL-5, IL-9 and IL-13 and amphiregulin). Group 3 ILCs encompass both RORγ-dependent LTi cells and ILC3s. They can produce IL-22, IL-17 and GM-CSF, mainly in response to IL-1β and IL-23. αLP: α-lymphoid progenitor; CHILP: Common helper-like innate lymphoid progenitor; CLP: Common lymphoid progenitor; GM-CSF: Granulocyte macrophage colony-stimulating factor; IL: Interleukin; ILC: Innate lymphoid cell; ILCP: Innate lymphoid cell precursor; INF: Interferon; LTi: Lymphoid tissue-like; NK: Natural killer; PLZF: Promyeloid leukemia zinc finger; Th: T helper; TNF: Tumor necrosis factor; TSLP: Thymic stroma lymphopoietin.
Dysregulation of ILCs can cause severe inflammation and injury in gut[19], lung[20], skin[21] and liver[22]. During the past 5 years, a growing number of studies have investigated the roles of ILCs in inflammatory, fibrotic and cancerous liver diseases[23-27]. Herein, we summarize the present knowledge of ILCs to reveal their complicated and versatile effects and the underlying mechanism in chronic liver diseases, in order to provide perspectives of new therapeutic strategies.
LIVER INFLAMMATION
Group 1 ILCs
There are two distinct NK populations in murine liver, CD49a-CD49b+ and CD49a+CD49b- cells. The CD49a-CD49b+ subset represents conventional (c)NK cells, which circulate in the blood. The CD49a+CD49b- subset has ever been considered as tissue-resident (tr)NK cells or ILC1s in previous studies, which are beside dendritic cells (DCs) localizing in the sinusoids of the portal area[28,29]. Both cNK and trNK cells express natural cytotoxicity receptors and require IL-15 signaling for their development. Compared to cNK cells, trNK cells have relatively lower expression of CD11b, Ly49 receptors, CD43 and KLRG1, but higher expression of CXCR3 and CXCR6, which is a chemokine receptor for CXCL16 responsible for the enrichment of natural killer T (NKT) cells in the liver and can provide intravascular immune surveillance[30,31]. In parallel, human livers contain CD56bright and CD56dim (accounting for 90%) NK cells, respectively representing the equivalents of murine cNK and trNK cells. With respect to function, CD56bright NK cells are prominent cytokine producers, whereas CD56dim NK cells are efficient killers[32].
Liver is an immune-tolerant organ predisposing for chronic infections of certain clinically significant pathogens. As an organ with predominant innate immunity, the liver is enriched with NK cells, which account for 25%-50% of total intrahepatic lymphocytes and are responsible for killing transformed cells and viruses. The cytotoxicity of NK cells is regulated by both cytokines and surface receptors[33,34]. IFN-γ is one of the most prominent cytokines derived from NK cells to exert antiviral, antifibrotic and antitumorigenic effects. Furthermore, it has been demonstrated that cNK rather than trNK cell-derived IFN-γ promotes Th1 polarization and secondary CD8+ cytotoxic lymphocyte (CTL) responses, the major effectors for clearance of hepatic B virus (HBV) in transgenic HBV mouse models[35]. Meanwhile, the interactions of NK cells with hepatocytes via the NKG2A inhibitory receptor could prime DCs to induce CD4+CD25+ regulatory T cells (Tregs), which will in turn up-regulate the expression of NKG2A on NK cells via IL-10 production, thus impairing the antiviral ability of NK cells[36,37].
In the pathogenesis of chronic HBV infection (CHB), ILC1s have potential proinflammatory effects that mirror Th1 cells in adaptive immunity exactly. First, in patients with CHB, liver injury has been significantly associated with enhanced ILC1s’ response, as reflected by markedly elevated levels of T-bet, IFN-γ and IL-12 signaling. Besides, decreased ILC1-produced IFN-γ has been found to have a connection with the telbivudine-induced alleviation of liver injury in CHB patients[23]. These results could be explained by the study of Krueger et al[38], in which it was demonstrated that CD49a+ ILC1s could inhibit expression of CXCL9, which was further required for robust accumulation of IFN-γ+CD49b+ NK cells during the early phase of adenovirus infection. In this way, ILC1s played a role in maintaining the liver as a tolerogenic site as a result of increased expression of NKG2A receptors compared with NK cells, which would further suppress the activation of liver CD103+ DCs, thus interrupting the priming of antigen-specific, antiviral CD8+ T cells and the clearance of virus. The mechanism was found to be the same in hepatitis C virus infection for which NKG2A-/- patients showed resistance[39,40]. To conclude, ILC1s help to maintain the tolerance of liver in normal conditions, and blockage of NKG2A signaling to generate potent anti-viral CD8+ T cell responses required for the elimination of persistent liver pathogens may prove to be a novel therapeutic strategy (Figure 2A).
Figure 2.

Protective and pathogenic roles of innate lymphoid cells in hepatic inflammation. A: cNK cells could produce IFN-γ to enhance the priming of CD8+ T cells to clear HBV. The interactions of NK cells with hepatocytes via NKG2A inhibitory receptor could prime DCs to induce CD4+CD25+ Tregs, which would in turn up-regulate the expression of NKG2A on NK cells via IL-10 production, thus impairing the antiviral ability of NK cells. Because of increased expression of NKG2A on ILC1s in hepatic Ad as well as hepatitis C virus infection, ILC1s play a role in maintaining the liver as a tolerogenic site by inhibiting CXCL9 expression, which is required for the accumulation of cNK cells. This would further impair the activation of liver CD103+ DCs, thus interrupting the proliferation of virus-specific CD8+ T cells and the clearance of virus; B: In ConA-induced immune hepatitis, hepatic ILC2s could amplify inflammation through the expression of IL-5 to recruit eosinophils in response to IL-33 released upon liver tissue damage. The inflammatory activity of endogenous ILC2s in immune-mediated hepatitis might be regulated by IL-33-elicited ST2+ Tregs. Besides, in Ad-induced viral hepatitis, a strong expression of ILC2s was induced by IL-33 to exert a protective role through down-regulation of the hepatotoxic cytokine TNF-α in T cells and macrophages. Both the proinflammatory and protective roles of ILC2s in hepatitis are part of IL-33 action; C: In immune hepatitis, ILC3-derived IL-22 has a protective role in ConA- and carbon tetrachloride-induced hepatitis, while IL-17 plays a pathological role in ConA-induced hepatitis. Besides, Notch-mediated IL-22 is an important mediator of the inflammatory response in HBV infection, being responsible for the recruitment of antigen-nonspecific inflammatory cells into the liver and subsequent liver injury. In Ad-induced acute hepatitis, the IL-17A/F signaling is critical for adaptive T response and is responsible for affected lymphocyte infiltration and hepatic inflammation. Ad: Adenovirus; cNK: Conventional natural killer; ConA: Concanavalin A; DC: Dendritic cell; HBV: Hepatitis B virus; IL: Interleukin; ILC: Innate lymphoid cell; NK: Natural killer; Tregs: T regulatory cells.
Group 2 ILCs
IL-33 belongs to the IL-1 superfamily, which is alarmins secreted by epithelial cells upon cellular stress and tissue damage. Upon binding to its specific heterodimeric receptor which comprises the ST2 and IL-1 receptor accessory protein, IL-33 is able to induce strong expression of Th2-like cytokines, thus balancing the Th1 immune response[41,42]. Epithelial cells, hepatocytes and HSCs have been reported as the main sources of IL-33 in the liver[43,44]. The expression of IL-33 shows significant connection with chronic liver diseases, such as persistent viral infection[45-47], liver fibrosis[43] and liver failure[48].
It has been demonstrated that through interaction with ST2, IL-33 induces production of the inflammatory cytokine IFN-γ, as well as Fas-FasL interaction between hepatocytes and NKT cells in concanavalin A (ConA)-induced hepatitis, which is a well-established murine model of T-cell mediated hepatitis, resembling the pathology of immune-mediated hepatitis in humans[49,50]. Neumann et al[24] further demonstrated that IL-33-elicited ILC2s were also involved in the pathogenic process of murine immune-mediated hepatitis. In response to the release of IL-33 upon liver tissue damage induced by CD4+ T cells, hepatic ILC2s were found to be poised to produce type 2 cytokines, including IL-13 and IL-5. Recruitment of eosinophils induced by IL-5 could be one mechanism by which ILC2 amplifies inflammation during immune hepatitis, and IL-13 was indicated to have a prominent role in chronic diseases[51]. Exogenous IL-33-elictied hepatic ILC2s appear to aggravate immune-mediated hepatitis, while the inflammatory activities of endogenous ILC2s might be regulated by IL-33-elicited ST2+ Tregs, which showed strong expansion in immune-mediated hepatitis as well. These findings are consistent with those of the previous study that revealed IL-33/ST2 axis to exert a protective role in ConA-induced hepatitis by preventing Th1 and Th17 cell-mediated hepatic immune responses, promoting IL-4 production of CD4+ liver-infiltrating T cells, elevating the total number of CD4+Foxp3+ Tregs together with affecting the expression of apoptotic or antiapoptotic proteins[52]. These results suggest that the proinflammatory role of ILC2s in immune-mediated hepatitis is part of the action mechanism of IL-33. Multiple modules of the immune response should be taken into consideration when investigating its overall protective or pathogenic effect on the liver.
What’s more, the IL33/ST2 axis has also been shown to play a crucial role in driving antiviral CD8+ and CD4+ T cell responses[53,54]. On one hand, Liang et al[55] demonstrated that, as a newly discovered damage-associated molecular pattern molecule, IL-33 can promote innate IFN-γ production by γδT cells and NK cells. It could also modulate DC responses to enhance the plurifunctionality of antiviral T cells in lymphocytic choriomeningitis virus-induced hepatitis in mice, while it was also further demonstrated that ILC2s were not involved in this process[56]. On the other hand, IL-33 was able to directly engage multiple arms of immune mechanisms to mediate potent hepatoprotective effects in adenovirus-induced hepatitis, wherein strong CTL, CD4+ Th and B lymphocyte responses share common characteristics with a number of hepatotropic viruses, including hepatitis A virus, HBV, cytomegalovirus, herpes simplex and Epstein-Barr virus. It significantly inhibited the expression of TNF-α in T cells and macrophages and induced a strong expression of IL-5- or IL-13-expressing-Lin- nuocytes to further down-modulate the hepatotoxic cytokine TNF-α[57]. An increased number of Lin-13+ or Lin-5+ cells were found in the livers of Lin- cells adoptively-transferred mice. Though the serum level of alanine aminotransferase and hepatic TNF-α presented a downward trend, there was no statistical significance compared with control groups[58]. These results also suggest that the potential protective role of ILC2s in viral hepatitis might only be one facet of the complex mechanisms of IL-33, but this still remains to be further elucidated (Figure 2B).
Group 3 ILCs
Dependent on RORγt and IL-7, ILC3s induce the production of IL-17 and IL-22 upon stimulation by IL-23 and IL-1β. IL-22 is a member of the IL-10 cytokine family and has a crucial role in inflammation, immune surveillance and tissue homeostasis. In the inflammatory context, IL-22 has both proinflammatory and protective properties[59,60]. The proinflammatory nature of IL-22 has been shown in mouse models with diseases such as psoriasis and rheumatoid arthritis[61,62]. In contrast, the protective role has been shown in inflammatory bowel disease[63], hepatitis[64] and pathogenic bacterial infection[65,66]. Hepatocytes are important targets of IL-22, for it can induce the expression of acute-phase proteins, several antiapoptotic proteins and mitogenic proteins, to protect cells against liver tissue damage[67-69]. IL-22 can also act on liver stem or progenitor cells, which are important in chronic and severe liver injury[70].
In ConA-induced acute immune hepatitis, the expression of IL-23 combined with activated Notch signaling resulted in an aryl hydrocarbon receptor (AHR)-dependent production of IL-22, as well as in an RORγt-dependent production of IL-17. IL-22 was shown to play a protective role, while IL-17 was shown to be critical for the pathogenesis in liver tissue[71]. The protective role of IL-22 in hepatitis was consistent with findings of a previous study that identified NKT and T cells, rather than ILC3s, as the main sources of IL-22[64]. Later, it was confirmed by Abe et al[72] that, combined with the suppression of IFN-γ from NKT cells induced by AHR, IL-22-producing ILC3s were also involved in the protective process in ConA-induced acute hepatic injury, as high IL-22 mRNA levels were found in CD3-Sca1+Thy1+ cells rather than in CD3+ T cells after stimulation by IL-23. Besides, the same results were obtained in RORγt-/- mice; specifically, there was almost no IL-22 production in the hepatic mononuclear cells of Ahr-/- or Ahr and recombination activation gene (RAG) double-negative mice, thus further suggesting that the major sources of IL-22 were both RORγt- and AHR-dependent ILC3s. In addition, the decreased frequency of IL-22-producing ILC3s (Lin- SCA-1+ Thy1high ILCs) was consistent with the severity of carbon tetrachloride-induced hepatitis of RAG-2-/-*RORγt-/- mice compared with that of RAG-2-/- mice[72]. Taken together, all the results of these studies considering RORγt+ hepatic ILC3s in immune-mediated hepatitis suggest their protective roles against liver injury via IL-22 production. Compared with IL-22-producing Th17 cells, which can play a protective role against liver injury as well, hepatic ILC3s may be able to act in the early innate immune response stage[73]. On the contrary, IL-17, another ILC3s-derived cytokine, has shown a pathological role in ConA-induced hepatitis. The overexpression of IL-17A resulted in massive hepatocyte necrosis, and antiIL-17A blockage significantly ameliorated liver injury[74]. In addition, Lafdil et al[75] showed that liver injury was alleviated in ConA-induced hepatitis among IL-17-deficient mice.
On the other hand, however, IL-22 was identified as a potent mediator of the inflammatory response in HBV infection, following the recognition of HBV by T cells in the liver[67,76]. It was further confirmed that inhibition of Notch signaling in vivo would lead to decreased ILC22 and LTi4 cells, along with down-regulated expressions of IL-22 and related proinflammatory cytokines and chemokines in the liver. As a result, subsequent liver injury was alleviated due to blockage of the recruitment of antigen-nonspecific inflammatory cells into the liver, without affecting HBV antigen production in HBV infection[77]. These results suggest the potential proinflammatory role of Notch-mediated IL-22 and provide a new potential therapeutic approach for the treatment of HBV. What’s more, intrahepatic early IL-17 was found to be important for activating antigen presenting cells in viral infection, but the sources and regulation of IL-17 surges were not well defined at first[78]. It was further shown that ILC3s, including a large proportion of NKP46- ILC3s and a small part of the CD4+ LTi cells, secreted IL-17A and IL-17F shortly after adenovirus infection, in addition to γδT cells. In adenovirus-induced acute hepatitis, the IL-17A/F signaling was found to be critical for adaptive T response and was responsible for affected lymphocyte infiltration and hepatic inflammation, except in viral clearance. The study also revealed the existence of the compensatory IL-17F production for IL-17A deficiency underlying the previous contradictory result that IL-17A deficiency did not appear to thwart T cell activation and liver inflammation[64,79]. Though there have been studies showing Th17-derived IL-17 causes liver damage by IL-23 activation, the role of ILC3-derived IL-17 remains to be further clarified in chronic infection models, such as for lymphocytic choriomeningitis virus and HBV infections[80]. To conclude, the downstream effector cytokines of ILC3s may exert both proinflammatory and protective roles according to the specific microenvironment, and more studies are required to clarify their explicit role in liver inflammation (Figure 2C).
LIVER FIBROSIS
Group 1 ILCs
NK cells can directly decrease the proliferation and activation as well as induce cell cycle arrest of HSCs through IFN-γ[81,82]. They can also induce apoptosis of activated HSCs through the TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand pathways[83,84]. These interactions between NK cells and HSCs are regulated by NK cell receptors and cytokines. The activation of HSCs in response to hepatocyte damage leads to increased NK cell stimulation and decreased NK cell inhibition. Firstly, increased amounts of retinoic acid derived from activated HSCs was found to be associated with elevated expression of RAE-1, a ligand for the NKG2D-activating receptor. Together with MICA, RAE-1 could promote the killing of activated HSCs by NK cells[85,86]. The NKp46 and NKp30 activating receptors have also been shown to be involved in the amelioration of liver fibrosis by inducing HSC killing by NK cells, in both humans and mice[87,88]. Secondly, engagement of Ly49 inhibitory NK cell receptors was found to be reduced by the mechanism of siRNA-mediated silencing, as a result of down-regulated major histocompatibility complex (MHC) class I in activated HSCs[89,90]. Besides, elevated surface expression of TRAIL in NK cells via IFN-α, simultaneous with increased expression of TRAIL receptors, in activated HSCs could also enhance HSC killing by NK cells[91,92]. Instead, tumor growth factor (TGF)-β down-regulates the surface expression of NKG2D and 2B4 to suppress the antifibrotic role of NK cells[93]. Whether trNK cells exert a protective role in liver fibrosis remains unclear. As a member of the IFN-γ-producing group 1 ILCs, these cells may contribute to the activation of NK cells by their production of IFN-γ, which is crucial for the antifibrotic roles of NK cells[81].
Group 2 ILCs
In the study by Marvie et al[43], the over-expression of IL-33 was shown to be closely associated with hepatic fibrosis, in both human and mouse cases. Besides, type 2 cytokines including IL-4 and IL-13 have been considered as representatives of the most potent fibrogenic factors[94]. As the major sources of Th2-type cytokines, ILC2s, which also require IL-33 for activation, were proposed as potent profibrogenic factors in hepatic fibrosis[95]. It has been demonstrated that, in response to ST2-dependent signaling owing to chronic hepatocellular stress and tissue damage, IL-33 release leads to accumulation and activation of IL-13-producing liver resident ILC2s. The downstream effector cytokine IL-13 can further trigger the activation and transdifferentiation of HSCs in an IL-4Ra- and STAT6-dependent manner to induce potent fibrogenic responses, suggesting a pathogenic capacity of ILC2s in the context of the tissue damage response[22]. In parallel, the study of human liver fibrosis by Forkel et al[96] has identified primary hepatocytes, HSCs and Kupffer cells as cellular sources of IL-33 and TSLP, which could further potentially cause the accumulation of ILC2s in fibrotic livers following TLR3-activation, as a model for hepatitis C infection. There was also a direct correlation found between the increase in frequency of intrahepatic ILC2s and the severity of liver fibrosis. The induction of IL-13 by intrahepatic ILC2s in response to IL-33 and TSLP was also confirmed, suggesting the possibility of a similar mechanism in humans and mice[96]. These results provide an avenue for investigation into the application of serum IL-33 as a possible noninvasive diagnostic biomarker for uncovering early inflammatory and fibrogenic events. Furthermore, targeting ILC2s may represent a novel therapeutic strategy for liver fibrosis treatment.
Group 3 ILCs
As one of the ILC3s’ downstream effector cytokines, IL-22 has been shown to promote the survival and proliferation of epithelial cells (e.g., hepatocytes), suggesting its possibility of involvement in liver fibrosis[64,68]. Upon binding to IL-22R1 and IL-10R2 on HSCs, IL-22 can induce senescence of the HSCs following the activation of a STAT3/SOCS3/p53 signaling axis, which represents an important strategy to ameliorate liver fibrosis[97]. It could also inhibit HSC apoptosis, without affecting HSC proliferation. By means of enhancement of in vivo clearance of senescent HSCs, most probably by NK cells, simultaneous to reduction of released tissue inhibitor of metalloprotease 2 to promote MMP activities and down-regulate the deposition of collagen, the senescence of activated HSCs played an important role in limiting liver fibrosis[86,98]. The expression of α-smooth muscle actin was also down-regulated in response to IL-22, but this effect was not associated with senescent HSCs[97]. Besides, elevated systemic IL-22 level - independent of age, liver-related complications, C-reactive protein, creatinine and model for end-stage liver disease score - could be predictive for reduced survival prognosis in patients with liver cirrhosis. Thus, it is possible that systemic IL-22 level could be applied as a negative indicator for evaluating the prognosis of advanced liver cirrhosis[99]. Though no direct evidence has linked ILC3s with liver fibrosis, ILC3s may exert a protective role since they are the source of IL-22. Nonetheless, considering IL-22 can be produced by Th17 cells as well, it is important to use specific gene knock-out mice to determine which type of cell plays the pivotal role.
In experimental liver fibrosis, upon stimulation of IL-17A, both HSCs and Kupffer cells could produce TGF-β, TNF-α and IL-6 following the activation of STAT3 and nuclear factor-κB. Accordingly, mouse models with IL-17A and IL-17RA deficiency have displayed reduced liver fibrosis, suggesting a profibrotic role[100]. In addition, IL-17A can also exert an antifibrotic effect in normal fibroblast cultures directly, by down-regulating the expressions of collagen and connective tissue growth factor, which was shown to be impaired in the isolated primary fibroblasts from patients with scleroderma[101]. Considering the complicated and versatile effects in fibrosis, it might be necessary to determine the exact roles of such cytokines in different stages of liver fibrosis (Figure 3).
Figure 3.

Contributions of innate lymphoid cells in liver fibrosis. NK cells can decrease the proliferation and activation as well as induce cell cycle arrest of HSCs through IFN-γ. They can also induce the apoptosis of activated HSCs through the TRAIL and Fas ligand pathways. The expression of RAE-1, which is the ligand for the NKG2D activating receptor, is increased on activated HSCs, thus promoting killing by NK cells. IFN-α could increase the surface expression of TRAIL on NK cells to enhance HSCs killing by NK cells, while TGF-β down-regulates the surface expression of NKG2D and 2B4 to suppress the antifibrotic role of NK cells. In both human and mouse cases, IL-33 released from hepatocytes, HSCs and Kupffer cells in response to chronic hepatocellular stress leads to the accumulation and activation of IL-13-producing liver resident ILC2s via ST2-dependent signaling. IL-13 further triggers the activation and transdifferentiation of HSCs into myofibroblasts, to induce potent fibrogenic responses. ILC3s play more complicated roles in fibrosis. On one hand, ILC3s exert an antifibrotic effect by inducing fibroblasts senescence through IL-22 signaling and by down-regulating the expressions of collagen and CTGF through IL-17 signaling. On the other hand, IL-17 can promote inflammation and induce activation of fibroblasts, indicating a profibrotic role for the ILC3s. CTGF: Connective tissue growth factor; HSC: Hepatic stellate cell; IL: Interleukin; IFN: Interferon; NK: Natural killer; TGF: Tumor growth factor; TRAIL: TNF-related apoptosis-inducing ligand.
LIVER CANCER
Group 1 ILCs
Considering the potent tumor surveillance properties of NK cells, a group of NK cell-associated genes in hepatocellular carcinoma (HCC) tissues was positively associated with prolonged survival[102]. Evidence of dysfunction of NK cells in HCC has been observed, as well, suggesting a strong connection between NK cells and HCC progression[103].
Although there currently is no direct evidence revealing connections between ILC1s and liver tumor immunity, the effects of their secreted cytokines have been extensively investigated, among which IFN-γ was shown to have prominent antiproliferative, antiangiogenic and proapoptotic effects against cancer cells[104-106]. This cytokine can promote the up-regulation of MHC molecules to induce the priming as well as antigen processing and presentation of professional antigen presenting cells, and has been shown to increase the immunogenicity of tumor cells, thereby enhancing antitumor responses[107,108]. In addition to promoting the polarization of CD4+ T cells into Th1 cells, it can also boost the responses of macrophages, NK cells and CTLs against tumor tissues[109,110]. TNF-α, another cytokine secreted by ILC1s, can also play an antitumor role by interfering with angiogenesis, cellular growth and migration. Further, it can induce the recruitment of macrophages and DCs, as well as the generation of CTLs, leading to a strong antitumor immune response[111,112]. Combined with the previously reported research findings, a recent study which showed the NK1.1+CD49a+CD103+ ILC1-like cells could lyse tumor cells, dependent on the activation of granzyme B and TRAIL, in an oncogene-induced cancer model also supports the protective function of ILC1s in antitumor immunity[113]. However, its protective function can be hampered in cancer patients, as ILC1s of acute myeloid leukemia patients were found to be dramatically impaired in their production of IFN-γ and TNF-α compared to those of healthy control subjects[114].
It was also demonstrated that both IFN-γ and TNF-α could play ambiguous roles in cancer immunity. The protumor function of IFN-γ involves increased proliferative and antiapoptotic signals, as well as escape of the tumor cells from recognition and cytolysis by CTLs and NK cells[115]. TNF-α is also involved in tumor formation, growth and spread considering its versatile impacts on the expression of angiogenic and growth factors, cytokines, adhesion receptors and proteases[111,116,117]. Recently, Gao et al[118] demonstrated that CD49a-CD49b+Eomes+ NK cells could convert into intermediate CD49a+CD49b+Eomes+ type 1 innate lymphoid cells (intILC1s) and CD49a+CD49b-Eomes- ILC1s in tumor microenvironment in a TGF-β signaling-dependent manner. Strikingly, distinguishable from the potent tumor surveillance properties of NK cells, intILC1s and ILC1s were incapable of controlling local tumor growth and metastasis, uncovering an unknown mechanism by which tumors can escape surveillance by the innate immune system. This study also provided a new insight into the phenotypic and functional plasticity of tumor group 1 ILCs, while the precious roles and interactions of ILC1s in tumor microenvironment - especially in the liver - still needs to be further elucidated[118,119].
Group 2 ILCs
Considering the potential profibrogenic properties of ILC2s elicited by IL-33 in hepatic fibrosis under the circumstances of tissue damage, it is possible that these cells are involved in the progression from liver fibrosis to cancer as well. The precise role of ILC2s in carcinogenesis remains unclear; however, it can be supported by the evidence that has emerged from studies addressing factors that trigger their activation and proliferation, as well as their downstream effector molecules.
When it comes to the liver, IL-33 was shown to be involved in the initiation of cancer, based on a previous study wherein increased expressions of Th2 cytokines and hepatic IL1RL1 mRNA encoding ST2 were detected in a subgroup of patients at the time of diagnosis of biliary atresia[120]. ILC2s were further identified as important mediators of the IL-33-depedent proliferative response for their production of high levels of IL-13, which in turn promoted cholangiocyte proliferation and epithelial hyperplasia in mice by involving the activation of IL-4R and the downstream target Stat6. Administration of IL-33 with constitutive activation of AKT and Yes-associated protein in biliary epithelium would lead to the development of cholangiocarcinoma, which resembles both the morphological and biochemical features of human disease in a mouse model of experimental carcinogenesis[121,122]. Thus, the activation of the IL-33/ILC2s/IL-13 circuit may promote epithelial repair, and the disruption of IL-33 or other elements of the paracrine circuit may constitute potential new therapeutic targets against cholangiocarcinoma. Furthermore, the level of IL-33 was found to be increased in patients with HCC as well[123].
IL-33 can also increase the intratumor accumulation of myeloid-derived suppressor cells (MDSCs), which require arginase and nitric oxide synthase II from IL-13 for their activation[124,125]. Together with the proangiogenesis process, MDSCs can also produce TGF-β to support tumor progression[126]. Besides, IL-13 can induce the polarization of macrophages to the M2 phenotype, and the production of growth and angiogenic factors to promote tumor initiation, progression and metastasis[127,128]. It has also been demonstrated that amphiregulin, another cytokine secreted by ILC2s, could enhance the activities of Tregs in vivo, which would further inhibit antitumor immune responses induced by DC vaccination[129,130].
IL-33 also plays a role in antitumor immune responses via effector functions of both CD8+ T cells and ILC2s, dependent on its dose[131,132]. The latter was confirmed in a study by Kim et al[133], which demonstrated that ILC2s were involved in IL-33-mediated antitumor responses. A massive amount of CXCR2 ligands released from ILC2s interacted with CXCR2 expressed by tumor cells by means of a dysfunctional angiogenesis/hypoxia/reactive oxygen species axis triggered by IL-33, subsequently leading to the apoptosis of active tumor cells[133]. Taking these results into consideration, ILC2s could exert both immune suppression and antitumor functions according to different tumor microenvironments, while its precious role, especially in the context of human livers, remains to be further confirmed.
Group 3 ILCs
The protective role of ILC3s has been directly revealed for its contribution to the formation of protective tumor-associated tertiary lymphoid structures in non-small cell lung cancer (NSCLC)[134]. The ILC3s are able to up-regulate adhesion molecules in the tumor microenvironment to enhance leukocyte invasion, and have been characterized as important mediators of the efficacy of a combination therapy of chemotherapy and tumor antigen-targeted monoclonal antibodies[135]. Though there is no direct evidence linking ILC3s and liver cancer, the connection can be inferred according to their role in colorectal cancer, as well as the ambiguous roles of their downstream effector cytokines, including IL-22 and IL-17.
It has been demonstrated that ILC3-derived IL-22 is crucial for promoting bacterial inflammation-induced colorectal cancer in Rag-/- mice through the activation of epithelial cells via STAT3 signaling[136]. Furthermore, deficiency of soluble IL-22 binding protein (IL-22BP) secreted by immature DCs was found to be associated with increased colitis-associated colon cancer due to the aberrant proliferation induced by IL-22. However, IL-22 was demonstrated as important for colonic epithelial cell repair in the early stage of colitis, using the same model; in particular, IL22-/- mice were shown to have enhanced cancer development[137]. These results suggest that IL-22 is crucial for regulating intestinal tissue repair during the peak of damage, while prolonged IL-22 in the recovery phase would be expected to favor tumorigenesis.
Paralleling the dual effects of IL-22 on tumorigenesis of colitis-associated colon cancer, IL-22 has also been reported to induce tissue regeneration or tumorigenesis and metastasis in the liver. Firstly, characterized with the protective role of hepatocyte proliferation and tissue regeneration during hepatitis and after hepatectomy, the functions of IL-22 may be exploited in liver cancer, as suggested by the significant up-regulation of IL-22 detected in human HCC tumor-infiltrated leukocytes[70,138,139]. Besides, there is a positive correction between IL-22 expression and the oncogenesis and staging of tumors, according to the finding that both IL-23 and IL-22BP are highly expressed in tumor tissue[68]. Secondly, though the induction of MMP enables IL-22 to protect against tumor formation in chronic liver fibrotic diseases, by the same mechanism it can increase the metastatic capacity of established tumor cells by digesting ECM, invading surrounding tissue and escaping the primary site. This has been shown in the A549 lung carcinoma cell line and pancreatic cancer, while whether the same mechanism also exists in hepatic tumor tissue remains unknown[97,140,141].
The same balance also exists in the antiviral activity and associated oncogenesis. IL-22 disturbs the establishment of chronic inflammation to prevent liver cancer. As was shown in acute infection of HBV, IL-22 acts as the mediator of an acute phase reaction to clear the virus via the recruitment of T cells[142]. However, it plays a contrary role in the progressive diseases, as IL-22 level was elevated and high serum IL-22 level indicated a poor prognosis both in patients with HBV and hepatitis C virus-associated HCC, suggesting that expression of IL-22 during progressive disease may reflect increased aggressiveness of HCC instead of predisposal to cirrhosis[76,143].
IL-22 can also influence the outcome of tumorigenesis by the mechanisms of pro- and antiinflammatory functions, angiogenesis, epithelial-mesenchymal transition, dysplasia and metabolic functions that remain less clear in the liver[142]. All these results suggest whether the effect of IL-22 is tumorigenic or antitumorigenic seems to depend on the stage of their responses and the specific tumor microenvironment.
Thy1+IL-23R+ ILC3s are important for IL-23-induced initiation of gut tumorigenesis, as substantial inhibition of tumorigenesis in RAG-/-*IL17-/- double knock-out mice provided evidence for an important contribution of IL-17 expression in ILC3s, which consistently occurred before the recruitment of overt inflammatory infiltrates[144]. When it comes to liver, the connection between IL-17 and angiogenesis has emerged in the context of HCC[145]. Besides, it has been shown to have protumor activity in proliferation, immune-resistance, tumorigenesis and metastasis as well[146]. On the contrary, IL-17 also plays a vital role in antitumor activity via the stimulation of tumor-specific CTLs, which were associated with establishment of a tumor-protective immunity in hematopoietic cancer[147]. These results also lead to the suggestion that there is a balance between protective CTLs’ formation during the acute phase of hepatitis and angiogenic activity during the chronic phase, which would determine the outcome of tumors.
Overall, ample evidence has pointed towards ILC3s having an important role in tumor progression. The elements consisting of specific tumor microenvironment as well as the timing of responses count when considering their ambiguous roles, while their explicit functions in humans, especially in human liver cancer, are incompletely understood and remain to be fully elucidated (Figure 4).
Figure 4.
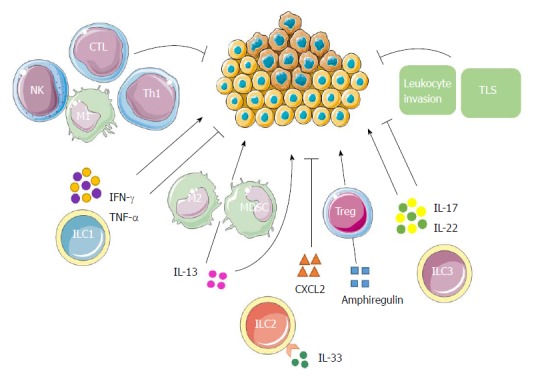
Possible innate lymphoid cell interactions in liver tumor immunity. ILC1s have both antitumor and protumor effects according to the properties of cytokines secreted. IFN-γ and TNF-α have antiproliferative, antiangiogenic and proapoptotic effects against cancer cells. In addition to promoting the polarization of CD4+ T cells into Th1 cells, they can also boost the responses of macrophages, NK cells and CTLs, leading to a strong antitumor response. On the contrary, their ambiguous roles enable them to enhance tumor formation, growth and spread. ILC2s may contribute to tumor progression either directly through the tumorigenic effects of IL-13, or indirectly by stimulating M2 macrophages and MDSCs through IL-13. The production of amphiregulin suggests that ILC2s may further inhibit antitumor immune responses either directly or via stimulation of Tregs. IL-33 could also induce the secretion of a massive amount of CXCR2 ligands from ILC2s as well as create a tumor microenvironment wherein tumor cells express CXCR2, leading to apoptosis of active tumor cells. ILC3s might promote antitumor responses by enhancing leukocyte invasion, promoting TLSs’ induction and through the antitumor effects of IL-17 and IL-22. Conversely, ILC3s may promote tumor formation and progression by IL-17 and IL-22 as well according to the phase of responses and specific tumor microenvironment. CTL: Cytotoxic T lymphocyte; IL: Interleukin; ILC: Innate lymphoid cell; IFN: Interferon; MDSC: Myeloid-derived suppressor cell; NK: Natural killer; Th: T helper; TLS: Tertiary lymphoid structure; TNF: Tumor necrosis factor; Tregs: T regulatory cells.
CONCLUSION AND FUTURE PERSPECTIVES
The recent research studies on the roles of ILCs in development of chronic liver diseases have made great progress, especially for hepatitis and liver fibrosis. Different or even the same ILC subsets have shown complex functions at a certain stage of chronic liver diseases. Also, the same ILC subsets have exhibited both pathological and protective functions during the dynamic development of chronic liver diseases. When considering their effects on liver, both the downstream effector cytokines and the molecules involved in the upstream signaling must be taken into consideration simultaneously, and more research is required to further elucidate the underlying mechanisms and signaling pathways. Apparently, there is a balance between the protective and pathological properties of ILCs, according to the specific liver tissue microenvironment at different stages of liver diseases, whereby effector cytokines, surrounding interaction cells and functional state of cell receptors vary remarkably. What’s more, the basis of the functions of ILCs and their downstream effector cytokines in hepatitis and liver fibrosis can represent the foundation of future research interests for investigating their roles in tumorigenesis.
Different methods have been applied to detect ILCs and measure their activities. Intrahepatic as well as peripheral blood mononuclear cells are isolated for further in vitro staining with fluorescence-labeled antibodies according to the cluster of differentiation on the surface of different ILC subsets and intracellular contents. Flow cytometry is further applied to detect the frequency and cellularity of ILCs and analyse the expression of their transcription factors and effector cytokines induced by PMA/ionomycin once they have been sorted in vitro. By observation of the differences of these factors between patients with chronic liver diseases and healthy control groups, their changes before and after the inducing factors and their consistency with liver injury, the researchers could validate the activities and functions of ILCs in the liver[23,38]. Considering limited accessibility of primary intrahepatic ILCs, the expansion of cell lines of primary intrahepatic ILCs is also an alternative to assess the function of this small cell population and to seek their secretion profile through the stimulation of PMA/ionomycin[96]. Besides, there have been studies exploring the roles of ILCs in initiation of liver injury including both hepatitis and liver fibrosis by the mechanism of in vivo depletion of ILCs using specific antibodies[22,73] or targeted transcription factor gene-deficient mice[72]. The protective or pathological roles of ILCs are determined by comparison of the severity of liver injury before and after the depletion of ILCs as indicated by histological analysis of liver tissue and expression of liver injury serum biomarkers as well as inflammatory cytokines in RAG1-/- mice which are reconstituted with CD4+ T cells. In vivo experiments to distribute the signaling pathway of ILCs through the blockade of upstream cytokines and surface receptors of ILCs via targeted gene-knock mice are also important methods, in which the expansion of ILCs and their expression of transcription factors and downstream effector cytokines are further detected by flow cytometry and quantitative real-time PCR analysis[24,52]. Additionally, the activities of ILCs could also be monitored by transfer experiments, in which purified ILCs sorted by MACS/FACS are adoptively transferred into recipient mice before the challenge of stimulus including ConA and carbon tetrachloride to further investigate the function of ILCs in the liver[22,24,58].
There still remains a lot to be fully elucidated. Firstly, the functions of some ILC subsets at a particular stage of chronic liver diseases have only been inferred by their downstream effector cytokines, while lacking direct and potent evidence. Secondly, given their distribution characteristics, evidence with regard to the functions of ILCs in tumorigenesis is emerging from studies that have mostly investigated chronic inflammation and the procarcinogenic role of secreted cytokines in skin, gut and lung, and less so for the liver. Thirdly, as the innate counterpart of CD4+ Th cells, the same effector cytokines can be produced by both adaptive lymphoid cells and ILCs. It is important to identify the sources of effector cytokines, while the results from the most recent in vivo studies were obtained from RAG-/- mice or antibodies that are specific to ILCs’ genes leading to broad immune deficiencies. Thus, it is necessary to apply ILCs’ specific gene-knockout or transgenic models to reveal the precise and direct actions of each in the liver.
New strategies targeting ILCs have been designed for diagnosis and treatment, to prevent or stop the progression of chronic liver diseases. The inhibition of NKG2A receptors on ILC1s to further promote robust CD8+ T cell responses has been considered a potential therapeutic strategy against persistent liver pathogens in patients with hepatitis. Besides, for liver fibrosis treatment, serum IL-33 may be a possible noninvasive diagnostic biomarker for uncovering early inflammatory and fibrogenic events. Targeting ILC2s may represent a novel strategy as well. Further in-depth studies to elucidate the distinct and explicit effects of each of the ILC subsets at different stages of chronic liver diseases are required in order to promote the exploration and realization of their therapeutic potency.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Supported by National Nature Science Foundation of China, No. 81670541; and National Science and Technology Major Project, No. 2013ZX10002004 and No. 2017ZX10203202.
Conflict-of-interest statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Peer-review started: March 30, 2018
First decision: April 19, 2018
Article in press: May 6, 2018
P- Reviewer: Carvalho-Filho RJ, Osna NA S- Editor: Gong ZM L- Editor: A E- Editor: Huang Y
Contributor Information
Yue Shen, Department of Gastroenterology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jing Li, Department of Gastroenterology, Zhongshan Hospital, Fudan University, Shanghai 200032, China; Department of Gastroenterology, Tongji Hospital, Tongji University, Shanghai 200000, China.
Si-Qi Wang, Department of Gastroenterology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Wei Jiang, Department of Gastroenterology, Zhongshan Hospital, Fudan University, Shanghai 200032, China. jiang.wei@zs-hospital.sh.cn.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–S50. doi: 10.1053/j.gastro.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kew MC. The role of cirrhosis in the etiology of hepatocellular carcinoma. J Gastrointest Cancer. 2014;45:12–21. doi: 10.1007/s12029-013-9556-9. [DOI] [PubMed] [Google Scholar]
- 5.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 6.Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157:340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity. 2013;38:769–781. doi: 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14:221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 10.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508:397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211:563–577. doi: 10.1084/jem.20131560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, et al. Transcription factor RORα is critical for nuocyte development. Nat Immunol. 2012;13:229–236. doi: 10.1038/ni.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Licona-Limón P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. 2013;14:536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 16.Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, Cua D, Di Santo JP, Eberl G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 17.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 19.Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36:451–463. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 21.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5:170ra16. doi: 10.1126/scitranslmed.3005374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, Voehringer D, McKenzie AN, Neurath MF, Pflanz S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Tang T, Wei X, Yang S, Tian Z. Type 1 innate lymphoid cells contribute to the pathogenesis of chronic hepatitis B. Innate Immun. 2015;21:665–673. doi: 10.1177/1753425915586074. [DOI] [PubMed] [Google Scholar]
- 24.Neumann K, Karimi K, Meiners J, Voetlause R, Steinmann S, Dammermann W, Lüth S, Asghari F, Wegscheid C, Horst AK, et al. A Proinflammatory Role of Type 2 Innate Lymphoid Cells in Murine Immune-Mediated Hepatitis. J Immunol. 2017;198:128–137. doi: 10.4049/jimmunol.1600418. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Tang J, Tian Z, van Velkinburgh JC, Song J, Wu Y, Ni B. Innate Lymphoid Cells: A Promising New Regulator in Fibrotic Diseases. Int Rev Immunol. 2016;35:399–414. doi: 10.3109/08830185.2015.1068304. [DOI] [PubMed] [Google Scholar]
- 26.van Beek JJP, Martens AWJ, Bakdash G, de Vries IJM. Innate Lymphoid Cells in Tumor Immunity. Biomedicines. 2016;4:pii: E7. doi: 10.3390/biomedicines4010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vallentin B, Barlogis V, Piperoglou C, Cypowyj S, Zucchini N, Chéné M, Navarro F, Farnarier C, Vivier E, Vély F. Innate Lymphoid Cells in Cancer. Cancer Immunol Res. 2015;3:1109–1114. doi: 10.1158/2326-6066.CIR-15-0222. [DOI] [PubMed] [Google Scholar]
- 28.Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, Sun R, Yokoyama WM, Tian Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. 2013;123:1444–1456. doi: 10.1172/JCI66381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3:e01659. doi: 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. 2016;13:347–353. doi: 10.1038/cmi.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Björkström NK, Kekäläinen E, Mjösberg J. Tissue-specific effector functions of innate lymphoid cells. Immunology. 2013;139:416–427. doi: 10.1111/imm.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watzl C. How to trigger a killer: modulation of natural killer cell reactivity on many levels. Adv Immunol. 2014;124:137–170. doi: 10.1016/B978-0-12-800147-9.00005-4. [DOI] [PubMed] [Google Scholar]
- 34.Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. 2015;12:292–302. doi: 10.1038/cmi.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng M, Sun R, Wei H, Tian Z. NK Cells Help Induce Anti-Hepatitis B Virus CD8+ T Cell Immunity in Mice. J Immunol. 2016;196:4122–4131. doi: 10.4049/jimmunol.1500846. [DOI] [PubMed] [Google Scholar]
- 36.Jinushi M, Takehara T, Tatsumi T, Yamaguchi S, Sakamori R, Hiramatsu N, Kanto T, Ohkawa K, Hayashi N. Natural killer cell and hepatic cell interaction via NKG2A leads to dendritic cell-mediated induction of CD4 CD25 T cells with PD-1-dependent regulatory activities. Immunology. 2007;120:73–82. doi: 10.1111/j.1365-2567.2006.02479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li F, Wei H, Wei H, Gao Y, Xu L, Yin W, Sun R, Tian Z. Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology. 2013;144:392–401. doi: 10.1053/j.gastro.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 38.Krueger PD, Narayanan S, Surette FA, Brown MG, Sung SJ, Hahn YS. Murine liver-resident group 1 innate lymphoid cells regulate optimal priming of anti-viral CD8+ T cells. J Leukoc Biol. 2017;101:329–338. doi: 10.1189/jlb.3A0516-225R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut. 2006;55:869–877. doi: 10.1136/gut.2005.076463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thoens C, Berger C, Trippler M, Siemann H, Lutterbeck M, Broering R, Schlaak J, Heinemann FM, Heinold A, Nattermann J, et al. KIR2DL3+NKG2A- natural killer cells are associated with protection from productive hepatitis C virus infection in people who inject drugs. J Hepatol. 2014;61:475–481. doi: 10.1016/j.jhep.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 42.Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–37. doi: 10.1016/j.coi.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Marvie P, Lisbonne M, L’helgoualc’h A, Rauch M, Turlin B, Preisser L, Bourd-Boittin K, Théret N, Gascan H, Piquet-Pellorce C, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–1739. doi: 10.1111/j.1582-4934.2009.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arshad MI, Piquet-Pellorce C, L’Helgoualc’h A, Rauch M, Patrat-Delon S, Ezan F, Lucas-Clerc C, Nabti S, Lehuen A, Cubero FJ, et al. TRAIL but not FasL and TNFα, regulates IL-33 expression in murine hepatocytes during acute hepatitis. Hepatology. 2012;56:2353–2362. doi: 10.1002/hep.25893. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Cai Y, Ji H, Feng J, Ayana DA, Niu J, Jiang Y. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis B. J Interferon Cytokine Res. 2012;32:248–253. doi: 10.1089/jir.2011.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Zhao P, Guo H, Sun X, Jiang Z, Xu L, Feng J, Niu J, Jiang Y. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis C. Mediators Inflamm. 2012;2012:819636. doi: 10.1155/2012/819636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB. Interleukin-33: a novel mediator with a role in distinct disease pathologies. J Intern Med. 2011;269:29–35. doi: 10.1111/j.1365-2796.2010.02316.x. [DOI] [PubMed] [Google Scholar]
- 48.Roth GA, Zimmermann M, Lubsczyk BA, Pilz J, Faybik P, Hetz H, Hacker S, Mangold A, Bacher A, Krenn CG, et al. Up-regulation of interleukin 33 and soluble ST2 serum levels in liver failure. J Surg Res. 2010;163:e79–e83. doi: 10.1016/j.jss.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Duan L, Xiong A, Zhang H, Zheng F, Tan Z, Gong F, Fang M. Blockade of IL-33 ameliorates Con A-induced hepatic injury by reducing NKT cell activation and IFN-γ production in mice. J Mol Med (Berl) 2012;90:1505–1515. doi: 10.1007/s00109-012-0938-4. [DOI] [PubMed] [Google Scholar]
- 50.Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gieseck RL 3rd, Ramalingam TR, Hart KM, Vannella KM, Cantu DA, Lu WY, Ferreira-González S, Forbes SJ, Vallier L, Wynn TA. Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis. Immunity. 2016;45:145–158. doi: 10.1016/j.immuni.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, Pejnovic N, Arsenijevic N, Lukic ML. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J Hepatol. 2012;56:26–33. doi: 10.1016/j.jhep.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 53.Bonilla WV, Fröhlich A, Senn K, Kallert S, Fernandez M, Johnson S, Kreutzfeldt M, Hegazy AN, Schrick C, Fallon PG, et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science. 2012;335:984–989. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
- 54.Baumann C, Bonilla WV, Fröhlich A, Helmstetter C, Peine M, Hegazy AN, Pinschewer DD, Löhning M. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc Natl Acad Sci USA. 2015;112:4056–4061. doi: 10.1073/pnas.1418549112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang Y, Jie Z, Hou L, Yi P, Wang W, Kwota Z, Salvato M, de Waal Malefyt R, Soong L, Sun J. IL-33 promotes innate IFN-γ production and modulates dendritic cell response in LCMV-induced hepatitis in mice. Eur J Immunol. 2015;45:3052–3063. doi: 10.1002/eji.201545696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu ZX, Govindarajan S, Okamoto S, Dennert G. Fas- and tumor necrosis factor receptor 1-dependent but not perforin-dependent pathways cause injury in livers infected with an adenovirus construct in mice. Hepatology. 2000;31:665–673. doi: 10.1002/hep.510310317. [DOI] [PubMed] [Google Scholar]
- 58.Liang Y, Jie Z, Hou L, Aguilar-Valenzuela R, Vu D, Soong L, Sun J. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J Immunol. 2013;190:5666–5675. doi: 10.4049/jimmunol.1300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Connor W Jr, Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol. 2010;11:471–476. doi: 10.1038/ni.1882. [DOI] [PubMed] [Google Scholar]
- 60.Geboes L, Dumoutier L, Kelchtermans H, Schurgers E, Mitera T, Renauld JC, Matthys P. Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum. 2009;60:390–395. doi: 10.1002/art.24220. [DOI] [PubMed] [Google Scholar]
- 61.Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–3702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 62.Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 63.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz SM, Köhler G, Schütze N, Knauer J, Straubinger RK, Chackerian AA, Witte E, Wolk K, Sabat R, Iwakura Y, et al. Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J Immunol. 2008;181:7891–7901. doi: 10.4049/jimmunol.181.11.7891. [DOI] [PubMed] [Google Scholar]
- 66.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Cobleigh MA, Lian JQ, Huang CX, Booth CJ, Bai XF, Robek MD. A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology. 2011;141:1897–1906. doi: 10.1053/j.gastro.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 69.Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, Ki SH, Yoo SH, Dooley S, Wang FS, et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. 2011;54:252–261. doi: 10.1002/hep.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, Gershwin ME, Gao B. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012;143:188–198.e7. doi: 10.1053/j.gastro.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu M, Morishima N, Mizoguchi I, Chiba Y, Fujita K, Kuroda M, Iwakura Y, Cua DJ, Yasutomo K, Mizuguchi J, et al. Regulation of the development of acute hepatitis by IL-23 through IL-22 and IL-17 production. Eur J Immunol. 2011;41:2828–2839. doi: 10.1002/eji.201141291. [DOI] [PubMed] [Google Scholar]
- 72.Abe H, Kimura A, Tsuruta S, Fukaya T, Sakaguchi R, Morita R, Sekiya T, Shichita T, Chayama K, Fujii-Kuriyama Y, et al. Aryl hydrocarbon receptor plays protective roles in ConA-induced hepatic injury by both suppressing IFN-γ expression and inducing IL-22. Int Immunol. 2014;26:129–137. doi: 10.1093/intimm/dxt049. [DOI] [PubMed] [Google Scholar]
- 73.Matsumoto A, Kanai T, Mikami Y, Chu PS, Nakamoto N, Ebinuma H, Saito H, Sato T, Yagita H, Hibi T. IL-22-producing RORγt-dependent innate lymphoid cells play a novel protective role in murine acute hepatitis. PLoS One. 2013;8:e62853. doi: 10.1371/journal.pone.0062853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan S, Wang L, Liu N, Wang Y, Chu Y. Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol Cell Biol. 2012;90:421–428. doi: 10.1038/icb.2011.59. [DOI] [PubMed] [Google Scholar]
- 75.Lafdil F, Wang H, Park O, Zhang W, Moritoki Y, Yin S, Fu XY, Gershwin ME, Lian ZX, Gao B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology. 2009;137:2125–2135.e1-2. doi: 10.1053/j.gastro.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, Jin L, Zhou C, Fu J, Gao B, et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology. 2014;59:1331–1342. doi: 10.1002/hep.26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei X, Wang JP, Hao CQ, Yang XF, Wang LX, Huang CX, Bai XF, Lian JQ, Zhang Y. Notch Signaling Contributes to Liver Inflammation by Regulation of Interleukin-22-Producing Cells in Hepatitis B Virus Infection. Front Cell Infect Microbiol. 2016;6:132. doi: 10.3389/fcimb.2016.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hou L, Jie Z, Desai M, Liang Y, Soong L, Wang T, Sun J. Early IL-17 production by intrahepatic T cells is important for adaptive immune responses in viral hepatitis. J Immunol. 2013;190:621–629. doi: 10.4049/jimmunol.1201970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jie Z, Liang Y, Hou L, Dong C, Iwakura Y, Soong L, Cong Y, Sun J. Intrahepatic innate lymphoid cells secrete IL-17A and IL-17F that are crucial for T cell priming in viral infection. J Immunol. 2014;192:3289–3300. doi: 10.4049/jimmunol.1303281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Q, Zhou J, Zhang B, Tian Z, Tang J, Zheng Y, Huang Z, Tian Y, Jia Z, Tang Y, et al. Hepatitis B virus induces IL-23 production in antigen presenting cells and causes liver damage via the IL-23/IL-17 axis. PLoS Pathog. 2013;9:e1003410. doi: 10.1371/journal.ppat.1003410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 82.Gao B, Radaeva S, Jeong WI. Activation of natural killer cells inhibits liver fibrosis: a novel strategy to treat liver fibrosis. Expert Rev Gastroenterol Hepatol. 2007;1:173–180. doi: 10.1586/17474124.1.1.173. [DOI] [PubMed] [Google Scholar]
- 83.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 84.Glässner A, Eisenhardt M, Krämer B, Körner C, Coenen M, Sauerbruch T, Spengler U, Nattermann J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. 2012;92:967–977. doi: 10.1038/labinvest.2012.54. [DOI] [PubMed] [Google Scholar]
- 85.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–G816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 86.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-Tair L, Safadi R, Mandelboim O. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut. 2012;61:885–893. doi: 10.1136/gutjnl-2011-301400. [DOI] [PubMed] [Google Scholar]
- 88.Mantovani S, Mele D, Oliviero B, Barbarini G, Varchetta S, Mondelli MU. NKp30 isoforms in patients with chronic hepatitis C virus infection. Immunology. 2015;146:234–242. doi: 10.1111/imm.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 90.Muhanna N, Abu Tair L, Doron S, Amer J, Azzeh M, Mahamid M, Friedman S, Safadi R. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2011;60:90–98. doi: 10.1136/gut.2010.211136. [DOI] [PubMed] [Google Scholar]
- 91.Stegmann KA, Björkström NK, Veber H, Ciesek S, Riese P, Wiegand J, Hadem J, Suneetha PV, Jaroszewicz J, Wang C, et al. Interferon-alpha-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138:1885–1897. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- 92.Ahlenstiel G, Edlich B, Hogdal LJ, Rotman Y, Noureddin M, Feld JJ, Holz LE, Titerence RH, Liang TJ, Rehermann B. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology. 2011;141:1231–1239, 1239.e1-1239.e2. doi: 10.1053/j.gastro.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, Wei H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012;8:e1002594. doi: 10.1371/journal.ppat.1002594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA. 2014;111:367–372. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hammerich L, Tacke F. Interleukins in chronic liver disease: lessons learned from experimental mouse models. Clin Exp Gastroenterol. 2014;7:297–306. doi: 10.2147/CEG.S43737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forkel M, Berglin L, Kekäläinen E, Carlsson A, Svedin E, Michaëlsson J, Nagasawa M, Erjefält JS, Mori M, Flodström-Tullberg M, et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur J Immunol. 2017;47:1280–1294. doi: 10.1002/eji.201646890. [DOI] [PubMed] [Google Scholar]
- 97.Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, Gao B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–1159. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schnabl B, Purbeck CA, Choi YH, Hagedorn CH, Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology. 2003;37:653–664. doi: 10.1053/jhep.2003.50097. [DOI] [PubMed] [Google Scholar]
- 99.Kronenberger B, Rudloff I, Bachmann M, Brunner F, Kapper L, Filmann N, Waidmann O, Herrmann E, Pfeilschifter J, Zeuzem S, et al. Interleukin-22 predicts severity and death in advanced liver cirrhosis: a prospective cohort study. BMC Med. 2012;10:102. doi: 10.1186/1741-7015-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, Wang X, Ryffel B, Sun B. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. 2013;191:1835–1844. doi: 10.4049/jimmunol.1203013. [DOI] [PubMed] [Google Scholar]
- 101.Nakashima T, Jinnin M, Yamane K, Honda N, Kajihara I, Makino T, Masuguchi S, Fukushima S, Okamoto Y, Hasegawa M, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. 2012;188:3573–3583. doi: 10.4049/jimmunol.1100591. [DOI] [PubMed] [Google Scholar]
- 102.Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, Weber A, Slankamenac K, Poon RT, Yang H, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut. 2012;61:427–438. doi: 10.1136/gutjnl-2011-300509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. 2015;36:1191–1199. doi: 10.1038/aps.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levy EM, Roberti MP, Mordoh J. Natural killer cells in human cancer: from biological functions to clinical applications. J Biomed Biotechnol. 2011;2011:676198. doi: 10.1155/2011/676198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hobeika AC, Etienne W, Torres BA, Johnson HM, Subramaniam PS. IFN-gamma induction of p21(WAF1) is required for cell cycle inhibition and suppression of apoptosis. J Interferon Cytokine Res. 1999;19:1351–1361. doi: 10.1089/107999099312812. [DOI] [PubMed] [Google Scholar]
- 106.Beatty G, Paterson Y. IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J Immunol. 2001;166:2276–2282. doi: 10.4049/jimmunol.166.4.2276. [DOI] [PubMed] [Google Scholar]
- 107.Seliger B, Ruiz-Cabello F, Garrido F. IFN inducibility of major histocompatibility antigens in tumors. Adv Cancer Res. 2008;101:249–276. doi: 10.1016/S0065-230X(08)00407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, Ugel S, Cingarlini S, Bronte V, Zanovello P, et al. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010;28:3548–3557. doi: 10.1016/j.vaccine.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 109.Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–1059. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reiter Z. Interferon--a major regulator of natural killer cell-mediated cytotoxicity. J Interferon Res. 1993;13:247–257. doi: 10.1089/jir.1993.13.247. [DOI] [PubMed] [Google Scholar]
- 111.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 112.Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, Mak TW, Ohashi PS. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest. 2007;117:3833–3845. doi: 10.1172/JCI32567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell. 2016;164:365–377. doi: 10.1016/j.cell.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Trabanelli S, Curti A, Lecciso M, Salomé B, Riether C, Ochsenbein A, Romero P, Jandus C. CD127+ innate lymphoid cells are dysregulated in treatment naïve acute myeloid leukemia patients at diagnosis. Haematologica. 2015;100:e257–e260. doi: 10.3324/haematol.2014.119602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. 2011;17:6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leibovich SJ, Polverini PJ, Shepard HM, Wiseman DM, Shively V, Nuseir N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature. 1987;329:630–632. doi: 10.1038/329630a0. [DOI] [PubMed] [Google Scholar]
- 117.Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, Zhang G, Bu XZ, Cai SH, Du J. Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS One. 2013;8:e56664. doi: 10.1371/journal.pone.0056664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18:1004–1015. doi: 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 119.Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, Colonna M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol. 2017;18:995–1003. doi: 10.1038/ni.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li J, Bessho K, Shivakumar P, Mourya R, Mohanty SK, Dos Santos JL, Miura IK, Porta G, Bezerra JA. Th2 signals induce epithelial injury in mice and are compatible with the biliary atresia phenotype. J Clin Invest. 2011;121:4244–4256. doi: 10.1172/JCI57728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, Bessho K, Wang YH, Glaser SS, Shivakumar P, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–3251. doi: 10.1172/JCI73742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, Borad MJ, Bezerra JA, Chen X, Gores GJ. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61:1627–1642. doi: 10.1002/hep.27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang P, Liu XK, Chu Z, Ye JC, Li KL, Zhuang WL, Yang DJ, Jiang YF. Detection of interleukin-33 in serum and carcinoma tissue from patients with hepatocellular carcinoma and its clinical implications. J Int Med Res. 2012;40:1654–1661. doi: 10.1177/030006051204000504. [DOI] [PubMed] [Google Scholar]
- 124.Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int. 2011;24:1027–1039. doi: 10.1111/j.1432-2277.2011.01306.x. [DOI] [PubMed] [Google Scholar]
- 125.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dhakal M, Hardaway JC, Guloglu FB, Miller MM, Hoeman CM, Zaghouani AA, Wan X, Rowland LM, Cascio JA, Sherman MP, et al. IL-13Rα1 is a surface marker for M2 macrophages influencing their differentiation and function. Eur J Immunol. 2014;44:842–855. doi: 10.1002/eji.201343755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. 2015;112:10762–10767. doi: 10.1073/pnas.1509070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, van Bergen en Henegouwen PM, Roovers RC, Coffer PJ, Sijts AJ. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38:275–284. doi: 10.1016/j.immuni.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, Lu J, Qin W, Qi Y, Xie F, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. 2015;194:438–445. doi: 10.4049/jimmunol.1401344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, Yan J, Morrow MP, Weiner DB. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res. 2014;74:1789–1800. doi: 10.1158/0008-5472.CAN-13-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim J, Kim W, Moon UJ, Kim HJ, Choi HJ, Sin JI, Park NH, Cho HR, Kwon B. Intratumorally Establishing Type 2 Innate Lymphoid Cells Blocks Tumor Growth. J Immunol. 2016;196:2410–2423. doi: 10.4049/jimmunol.1501730. [DOI] [PubMed] [Google Scholar]
- 134.Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. 2015;6:8280. doi: 10.1038/ncomms9280. [DOI] [PubMed] [Google Scholar]
- 135.Moskalenko M, Pan M, Fu Y, de Moll EH, Hashimoto D, Mortha A, Leboeuf M, Jayaraman P, Bernardo S, Sikora AG, et al. Requirement for innate immunity and CD90+ NK1.1- lymphocytes to treat established melanoma with chemo-immunotherapy. Cancer Immunol Res. 2015;3:296–304. doi: 10.1158/2326-6066.CIR-14-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W Jr, Murphy AJ, Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham C, Flavell RA. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–909. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 139.Ren X, Hu B, Colletti LM. IL-22 is involved in liver regeneration after hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G74–G80. doi: 10.1152/ajpgi.00075.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ye ZJ, Zhou Q, Yin W, Yuan ML, Yang WB, Xiang F, Zhang JC, Xin JB, Xiong XZ, Shi HZ. Interleukin 22-producing CD4+ T cells in malignant pleural effusion. Cancer Lett. 2012;326:23–32. doi: 10.1016/j.canlet.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 141.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 142.Lim C, Savan R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. 2014;25:257–271. doi: 10.1016/j.cytogfr.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 143.Waidmann O, Kronenberger B, Scheiermann P, Köberle V, Mühl H, Piiper A. Interleukin-22 serum levels are a negative prognostic indicator in patients with hepatocellular carcinoma. Hepatology. 2014;59:1207. doi: 10.1002/hep.26528. [DOI] [PubMed] [Google Scholar]
- 144.Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, Vives F, Moon C, Penaflor E, Turner S, et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. 2014;7:842–856. doi: 10.1038/mi.2013.101. [DOI] [PubMed] [Google Scholar]
- 145.Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 146.Song Y, Yang JM. Role of interleukin (IL)-17 and T-helper (Th)17 cells in cancer. Biochem Biophys Res Commun. 2017;493:1–8. doi: 10.1016/j.bbrc.2017.08.109. [DOI] [PubMed] [Google Scholar]
- 147.Satoh T, Tajima M, Wakita D, Kitamura H, Nishimura T. The development of IL-17/IFN-γ-double producing CTLs from Tc17 cells is driven by epigenetic suppression of Socs3 gene promoter. Eur J Immunol. 2012;42:2329–2342. doi: 10.1002/eji.201142240. [DOI] [PubMed] [Google Scholar]