

Abstract
As the incidence of hepatocellular carcinoma (HCC) caused by infection with the hepatotropic viruses hepatitis B and hepatitis C decreases, greater attention has become focused on HCC caused by nonalcoholic steatohepatitis (NASH), an advanced form of nonalcoholic fatty liver disease which has shown increasing prevalence in correspondence with the overall increase in metabolic syndrome over the recent decades. Several clinical population studies have shown a positive relationship between NASH and HCC, while also providing initial insights into the underlying mechanisms of HCC development from NASH. Research into the pathological progression of NASH to HCC has advanced by use of several beneficial rodent models. In this review, we summarize the established mouse models for preclinical research of NASH-associated HCC and discuss the underlying hepatic mechanisms of NASH-related tumorigenesis identified to date that could lead to new targets for treatment and prevention.
Keywords: Hepatocellular carcinoma, Nonalcoholic steatohepatitis, Nonalcoholic fatty liver disease
Core tip: This review provides a brief overview of the molecular mechanisms underlying progression to hepatocellular carcinoma from nonalcoholic steatohepatitis that have been identified to date using the array of mouse models currently available and popular in the experimental field.
INTRODUCTION
As Western diet and problems with food satiation have spread across the globe in recent years, there has been a concomitant increase in patients with nonalcoholic fatty liver disease (NAFLD) and its progressive form of nonalcoholic steatohepatitis (NASH). This increase is the result of prevailing metabolic syndrome, including obesity, diabetes and hyperlipidemia[1-4]. The distinctive characteristic of NAFLD is its diversity of conditions, from simple fatty accumulation in the liver to hepatic injury and inflammation with or without fibrosis[2,5-7]. The sequential progression to NASH puts the sufferer at risk for irreversible liver cirrhosis and hepatocellular carcinoma (HCC)[4,7], causing the patient to require more medical attention due to the increased morbidity and mortality[8]. Indeed, HCC is a leading indication for liver transplantation, especially in developed countries[9,10].
Compared with the long history of both clinical and laboratory investigations to elucidate the molecular pathogenesis of HCC derived from chronic hepatotropic virus infections, particularly with hepatitis B virus and hepatitis C virus, and from alcoholic liver disease, the pathologic mechanisms of NASH-associated HCC (NASH-HCC) remain largely uninvestigated and unknown. The public health threat associated with the increasing incidence of NASH-HCC[11], however, highlights the urgent need to gain a more comprehensive and detailed understanding of the mechanisms which mediate NASH-HCC progression. Several experimental mouse models exist for such studies[12-15] and should be continuously applied to preclinical investigations into the pathogenic pathways of NASH-HCC to advance the subsequent development of methods to manage the modern increasing clinical trend.
Here, we summarize the established mouse models for preclinical research of NASH-HCC progression (Table 1) and discuss the revealed mechanisms and the future prospective of NASH-related tumorigenesis in liver which could lead to new targets for treatment or prevention (Figure 1). Of note, we recognize the existence of other available rodent models which can also be used for assessing the mechanisms of NASH-HCC; however, we focused this review on the ones which are most representative of metabolic syndrome-associated steatohepatitis and which generate HCC unfailingly from NASH status within a certain period of time.
Table 1.
Mouse models of nonalcoholic steatohepatitis-associated hepatocellular carcinoma
| List | Backgrounds | Inducer of NASH/HCC | Carcinogenic duration | HCC occurrence (%) | Ref. |
| PTEN null mice | Genetic | Spontaneous | 40 wk | 66 (74-78 wk) | [12,17,18,21,22] |
| MC4R KO mice | Genetic | HFC diet | 1 yr | 100 | [13,29,31] |
| STAM mice | DM/HL | Streptozotocin, HFC diet | 20 wk | 100 | [14,32-36] |
| ALR KO mice | Genetic | Spontaneous | 1 yr | 60 | [15] |
HFC: High fat/calorie; DM: Diabetes; HL: Hyperlipidemia; HCC: Hepatocellular carcinoma; NASH: Nonalcoholic steatohepatitis.
Figure 1.
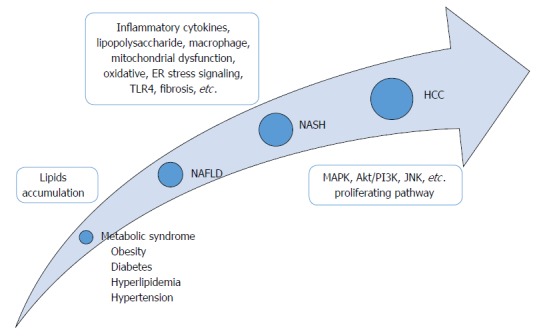
Developmental process of hepatocellular carcinoma via nonalcoholic steatohepatitis. Based on excessive lipids accumulation, several factors such as inflammatory cytokines, oxidative stress or proliferating pathways are involved in the whole process of hepatocellular carcinoma development from nonalcoholic steatohepatitis status via nonalcoholic fatty liver disease. NAFLD: Nonalcoholic fatty liver disease; HCC: Hepatocellular carcinoma; NASH: Nonalcoholic steatohepatitis.
CONFIRMED TUMORIGENIC MECHANISMS OF CURRENT NASH-HCC MOUSE MODELS
The established mouse models for preclinical research of NASH-HCC progression are listed below (Table 1).
PTEN null mice
PTEN, a tumor suppressor gene which antagonizes the PI3K/Akt pathway, is mutated in many human cancers, including HCC, and is essential for maintaining homeostasis and preventing oncogenesis in the liver. Decreased Pten expression leads to increased tumor grade, advanced stage and poor prognosis. Hepatocyte-specific Pten null mice were generated by Horie et al[12], wherein steatohepatitis emerges at 10 wk old and hepatic tumors at 40-44 wk old. The liver tumors become adenomas in 100% of these mice or HCC in 66% at 74-78 wk old, due to the Pten deficiency (Pten knock-out, KO) causing lipid accumulation in hepatocytes. In general, these mice have revealed that Pten function is crucial for preventing tumorigenesis in liver.
Several other research groups have uncovered different mechanisms of NASH-HCC by using the Pten null mouse model. For example, a study of eicosapentaenoic acid (EPA; a typical dietary n-3 polyunsaturated fatty acid contained in fish oil and a reagent for upgrading lipid metabolism[16]) performed by Ishii et al[17] showed the effect of EPA on steatohepatitis and tumor formation in Pten null mice. The data confirmed that the steatotic change, accumulation of inflammatory cells and presence of ballooning hepatocytes were significantly decreased in the EPA group compared with the control group. In addition, liver adenomas developed in 63% of the control group mice, as compared with 0% of the EPA group mice, by 40 wk of age. HCC developed in 75% of the control group and 13% of the EPA group of the Pten KO mice at 76 wk old. In addition, MAPK and Akt, which are both downstream signaling molecules of Ras, were found to be activated in hepatocytes of the Pten KO mice, thereby promoting tumorigenesis[18]. Collectively, these data suggested that EPA alters fatty acid composition in liver and suppresses the development of HCC by inactivating these signaling pathways in Pten null mice.
In another study of the Pten null mice, reduction of glucose-regulated protein 78 (GRP78; a molecular chaperone elevated in several human cancers, including HCC[19,20], and which is critical for endoplasmic reticulum folding, stress signaling and PI3K/Akt activation) promoted liver steatosis and liver injury at 3 mo of age and liver tumors at 6 mo of age[21]. These effects proceeded HCC or cholangiocarcinoma, which developed at 8-9 mo of age and was accompanied by elevation of p-JNK; in contrast, the GRP78 normal Pten null mice never generated tumor lesions in liver, as assessed out to 14 mo of age[21]. Collectively, these data suggested that JNK might contribute to acceleration of tumorigenesis in liver. Accordingly, these data demonstrated GPR78 as a regulator for Pten loss-mediated liver steatosis and tumor progression on the basis of p-JNK elevation.
In a third study of the Pten null nice, Miura et al[22] showed that liver tumors emerged after 36 wk of age, although no liver tumors were found in Pten normal mice until 72 wk of age. Toll-like receptor (TLR) 4 expressed on macrophages was found to contribute to the development of steatohepatitis and HCC in Pten KO mice. In general, gut-derived materials stimulate the immune system, including the TLRs which recognize bacterial components. TLR4, in particular, senses components of Gram-negative bacteria, including the lipopolysaccharide (LPS)[23]. In this way, TLRs affect the development of liver diseases. Moreover, macrophages are known to be a major source of proinflammatory cytokines which facilitate the progression of steatohepatitis[24,25] and Ly6C is a marker for inflammatory macrophages[26]. Hepatic macrophages isolated from the Pten null mice showed an increased expression of Ly6C. In addition, TLR4 signaling was shown to promote hepatic inflammation as well as subsequent liver tumor growth in the Pten null mice. Antibiotic treatment suppressed the tumor growth, in concert with a decreasing LPS level in the portal vein, suggesting that the gut microbiota serves as a source of TLR4 ligand(s) and that the Ly6C-positive macrophages play a role in tumor development in Pten null mice. Collectively, these data indicate that gut-derived LPS-induced inflammation via TLR4 on macrophages and TLR4-mediated inflammation result in HCC.
Melanocortin 4 receptor KO mice
Melanocortin 4 receptor (MC4R), a seven-transmembrane G protein-coupled receptor, is involved in regulation of body weight; hence, MC4R gene mutation is the major monogenic origin of obesity in human[27,28]. Feeding of a high-fat diet to MC4R-deficient (MC4R-KO) mice for 20 wk and 1 year leads to NASH and multiple well-differentiated HCC formations in the liver, respectively[13]. Similar to the findings in Pten null mice, Konuma et al[29] found that highly-purified EPA treatment of MC4R-KO mice effectively inhibited the development of liver fibrosis without affecting body weight.
According to their previous study, hepatic crown-like structures (hCLSs), a unique histological feature, were found to play a pivotal role in the progression from simple steatosis to NASH[30], with EPA markedly suppressing hCLS formation and fibrosis via prevention of hepatocyte injury. Thus, it was concluded that the beneficial effect of EPA involved the hCLSs. In addition, canagliflozin (CANA, a sodium glucose cotransporter 2 inhibitor and antidiabetic drug) was shown to attenuate NASH-HCC in another study[31]. Based on the evidence that CANA induces adipose expansion without promoting macrophage augmentation, inflammation or fibrosis and altered glutathione metabolism to reduce oxidative stress in adipose tissue, the authors concluded that the decreased hepatic fat accumulation upon CANA treatment suppresses hepatic inflammation, fibrosis at 20 wk and subsequent NASH-HCC at 52 wk in Western diet-fed MC4R-KO mice.
STAM mice
The STAM mouse model was generated by neonatal male C57BL/6J mice exposure to low-dose streptozotocin at 2 d after birth followed by feeding with a high-fat diet after 4 wk of age[14]. As a result, NASH developed at 8 wk and HCC at 16-20 wk. This mouse model has specific positive features, such as the average duration of HCC occurrence being within 16-20 wk of age, the number of HCC nodules being over 4 in any single mouse, the basal liver function being relatively preserved and there being no visible metastasis in the entire body[32]. Moreover, this model has the substantial benefit of its HCC development from NASH being identical to the known progression in human patients, but with the whole process being completed within a relatively short period of time.
By using the STAM model, four studies have uncovered several of the mechanisms underlying NASH-HCC. First, Lau et al[33] demonstrated that cancer-associated fibroblasts, which regulate liver tumor-initiating cells, are augmented in parallel with increasing human growth factor (HGF) level during fibrosis and that HGF-induced FRA1 activation is related to fibrosis-dependent HCC development. These data suggest that cancer-associated fibroblast-derived, HGF-mediated FRA1 can be a new therapeutic target for NASH-HCC. Second, Fernandes et al[34] showed that solithromycin, a novel macrolide antibiotic, suppressed NASH, fibrosis and NASH-HCC by modulating the gluconeogenesis pathway, in particular the components of fructose 1, 6-biphosphatase and glucose-6-phosphatase which are regulated by protein kinase C epsilon. Solithromycin improved the hepatic morphological features, such as the hepatocyte ballooning degeneration, and functions, as evidenced by reduction in NAFLD activity score along with decreased inflammation, fibrosis and HCC progression. This mechanism was ultimately suggested as a candidate factor of novel treatment of NASH-HCC.
Third, Conti et al[35] revealed that aberrant expression of hepatic micro (mi)RNAs, such as miR-34a-5p, miR-93-5p, miR-221-3p and miR-222-3p, indicates their mechanistic significance in NASH-HCC tumorigenesis; specifically, 10 over-expressed miRNAs were identified. It is well known that human HCC tumorigenesis is associated with extensive genomic alterations. Therefore, the authors concluded that the altered expression profile of these miRNAs could be a surrogate marker for the initiation and progression of NASH-HCC.
Finally, based on the confirmed finding that NASH-HCC is associated with metabolic alterations in hepatic lipid homeostasis, Pogribny et al[36] indicated that one of the specific features of NASH-HCC is a significant dysregulation of 1-carbon homeostasis, with decreased expression of key 1-carbon metabolism genes, especially of the S-adenosylhomocysteine hydrolase (Ahcy) gene, and increased expression of the S-adenosyl-L-homocysteine (SAH) gene. Their results suggest that the inhibition of Ahcy expression may be a trigger of SAH elevation and subsequent progression of NASH-HCC.
Augmenter of liver regeneration-KO mice
Augmenter of liver regeneration (ALR), a hepatic growth factor, is widely known as a pleiotropic protein. ALR is critical for mitochondrial function, lipid homeostasis and cell survival. Gandhi et al[15] generated a liver-specific ALR-L-KO mouse and reported that depletion of hepatic ALR caused steatosis, mitochondrial degeneration and apoptosis of hepatocytes at 2 wk of age. These effects were followed by consecutive cell death, sustained inflammation at 4 wk, fibrosis/cirrhosis at 8 wk and eventually HCC formation (in 60%) at 1 year. Thus, it was theorized that inhibition of ALR synthesis in hepatocytes could lead to mitochondrial dysfunction and cell death, resulting in consecutive NASH and HCC occurrence.
FUTURE PERSPECTIVES FOR THE STUDY OF NASH-HCC BY ANIMAL MODELS
The “two-hit” hypothesis of the underlying mechanism of NASH-HCC involves the excessive accumulation of lipids in liver as the first step, thereby promoting sensitization to LPS, oxidative stress and inflammatory cytokines, representing the second hit[37-39] (Figure 1). Recently, Tilg and Moschen[40] proposed a “multiple-hit” hypothesis, in which various factors derived from gut and adipose tissue might take place in parallel during the progression from NAFLD to NASH. However, the definitive mechanisms in the progression from simple fatty liver to NASH and HCC are still under investigation, due to the inherent complexity of the functional combination of several factors. For some time, it was believed that the lack of appropriate animal models which were able to sufficiently reflect the actual process of human NASH-HCC progression was the main obstacle to such research[41]. In recent years, however, the situation has changed according to the development and availability of several rodent models. Each model harbors different specific characteristics, including genetic background, obesity status, diet induction, etc. Thus, researchers can now evaluate the mechanisms of NASH-HCC related to a specific factor/parameter by using these animal models.
According to the overall analyses of hepatocarcinogenesis in each of the mouse models discussed above, it is the STAM mice that generate HCC unfailingly and most rapidly. The considerable demerit of this mouse model, however, is the obscurity of the original gene of tumorigenesis for HCC due to lack of genetic manipulation and the inclusion of diabetes and hyperlipidemia in the background. Genetic manipulation in mouse models, such as of the PTEN-KO or ALR-KO, is a useful means by which to clarify the role of a specific gene in the molecular foundation of NASH-HCC progression; although, the sequential progression to HCC in these models has a relatively long duration and HCC occurrence is uncertain.
It is still questionable whether or not these available mouse models represent the initiating and/or progression processes of bona fide human NASH-HCC. Furthermore, it is noteworthy that among actual NASH patients there are individual differences in degree of fibrosis and timing of tumorigenesis in liver. At the present time, however, it is undoubted that these mouse models are essential for investigating the underlying mechanisms of NASH-HCC. Therefore, the future research targets may move forward towards gaining a more comprehensive NASH-HCC evaluation by using these mouse models.
CONCLUSION
Several mouse models have become available in recent years that support investigation into the underlying mechanisms of NASH-HCC. In response to the growing demand for better management of NASH-HCC, further inquiries are expected by researchers upon selecting an appropriate NASH mouse model according to the specific mechanisms and/or therapeutic targets of interest. After that, we hope to get some breakthrough for new treatment or prevention of NASH-HCC in the near future.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: All of the authors declare no potential conflicts of interest relevant to this article.
Peer-review started: April 4, 2018
First decision: April 27, 2018
Article in press: May 6, 2018
P- Reviewer: Gonzalez-Reimers E, Namisaki T, Peltec A S- Editor: Gong ZM L- Editor: A E- Editor: Huang Y
Contributor Information
Kazuki Takakura, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan. ktakakura@jikei.ac.jp.
Tsunekazu Oikawa, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Yoichi Tomita, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Yusuke Mizuno, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Masanori Nakano, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Chisato Saeki, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Yuichi Torisu, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
Masayuki Saruta, Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo 105-8461, Japan.
References
- 1.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Neuschwander-Tetri BA. Non-alcoholic fatty liver disease. BMC Med. 2017;15:45. doi: 10.1186/s12916-017-0806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 5.Rozman D. From nonalcoholic Fatty liver disease to hepatocellular carcinoma: a systems understanding. Dig Dis Sci. 2014;59:238–241. doi: 10.1007/s10620-013-2998-x. [DOI] [PubMed] [Google Scholar]
- 6.Noureddin M, Mato JM, Lu SC. Nonalcoholic fatty liver disease: update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp Biol Med (Maywood) 2015;240:809–820. doi: 10.1177/1535370215579161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duvnjak M, Lerotić I, Barsić N, Tomasić V, Virović Jukić L, Velagić V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–4550. doi: 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Li H, Garzel B, Yang H, Sueyoshi T, Li Q, Shu Y, Zhang J, Hu B, Heyward S, et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol Pharmacol. 2015;87:674–682. doi: 10.1124/mol.114.097287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148:547–555. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, Charlton M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology. 2017;152:1090–1099.e1. doi: 10.1053/j.gastro.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 12.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, Terai S, Sakaida I, Ogawa Y. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol. 2011;179:2454–2463. doi: 10.1016/j.ajpath.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46:141–152. doi: 10.1007/s00795-013-0016-1. [DOI] [PubMed] [Google Scholar]
- 15.Gandhi CR, Chaillet JR, Nalesnik MA, Kumar S, Dangi A, Demetris AJ, Ferrell R, Wu T, Divanovic S, Stankeiwicz T, et al. Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology. 2015;148:379–391.e4. doi: 10.1053/j.gastro.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carpentier YA, Portois L, Malaisse WJ. n-3 fatty acids and the metabolic syndrome. Am J Clin Nutr. 2006;83:1499S–1504S. doi: 10.1093/ajcn/83.6.1499S. [DOI] [PubMed] [Google Scholar]
- 17.Ishii H, Horie Y, Ohshima S, Anezaki Y, Kinoshita N, Dohmen T, Kataoka E, Sato W, Goto T, Sasaki J, et al. Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J Hepatol. 2009;50:562–571. doi: 10.1016/j.jhep.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 18.Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, Wange RL. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol Cell Biol. 2000;20:6945–6957. doi: 10.1128/mcb.20.18.6945-6957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su R, Li Z, Li H, Song H, Bao C, Wei J, Cheng L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer. 2010;10:20. doi: 10.1186/1471-2407-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luk JM, Lam CT, Siu AF, Lam BY, Ng IO, Hu MY, Che CM, Fan ST. Proteomic profiling of hepatocellular carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78) up-regulation and their associated prognostic values. Proteomics. 2006;6:1049–1057. doi: 10.1002/pmic.200500306. [DOI] [PubMed] [Google Scholar]
- 21.Chen WT, Zhu G, Pfaffenbach K, Kanel G, Stiles B, Lee AS. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene. 2014;33:4997–5005. doi: 10.1038/onc.2013.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miura K, Ishioka M, Minami S, Horie Y, Ohshima S, Goto T, Ohnishi H. Toll-like Receptor 4 on Macrophage Promotes the Development of Steatohepatitis-related Hepatocellular Carcinoma in Mice. J Biol Chem. 2016;291:11504–11517. doi: 10.1074/jbc.M115.709048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 24.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–589. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 27.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 28.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konuma K, Itoh M, Suganami T, Kanai S, Nakagawa N, Sakai T, Kawano H, Hara M, Kojima S, Izumi Y, et al. Eicosapentaenoic acid ameliorates non-alcoholic steatohepatitis in a novel mouse model using melanocortin 4 receptor-deficient mice. PLoS One. 2015;10:e0121528. doi: 10.1371/journal.pone.0121528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, Sakugawa H, Kanai S, Hamaguchi M, Fukaishi T, et al. Hepatic crown-like structure: a unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS One. 2013;8:e82163. doi: 10.1371/journal.pone.0082163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiba K, Tsuchiya K, Komiya C, Miyachi Y, Mori K, Shimazu N, Yamaguchi S, Ogasawara N, Katoh M, Itoh M, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci Rep. 2018;8:2362. doi: 10.1038/s41598-018-19658-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takakura K, Koido S, Fujii M, Hashiguchi T, Shibazaki Y, Yoneyama H, Katagi H, Kajihara M, Misawa T, Homma S, et al. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res. 2014;34:4849–4855. [PubMed] [Google Scholar]
- 33.Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, Chai S, Lin CH, Tsang SY, Ma S, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016;15:1175–1189. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 34.Fernandes P, Oldach D, Hashiguchi T, Shirakata Y, Yoneyama H, Gholam PM. Solithromycin Diminishes Steatohepatitis by Modulating Gluconeogenesis and Inhibits Tumor Growth in a Diabetic Mouse Model of Non-Alcoholic Steatohepatitis. J Immunol Infect Inflam Dis. 2016;14:17–19. [Google Scholar]
- 35.de Conti A, Ortega JF, Tryndyak V, Dreval K, Moreno FS, Rusyn I, Beland FA, Pogribny IP. MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget. 2017;8:88517–88528. doi: 10.18632/oncotarget.19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pogribny IP, Dreval K, Kindrat I, Melnyk S, Jimenez L, de Conti A, Tryndyak V, Pogribna M, Ortega JF, James SJ, et al. Epigenetically mediated inhibition of S-adenosylhomocysteine hydrolase and the associated dysregulation of 1-carbon metabolism in nonalcoholic steatohepatitis and hepatocellular carcinoma. FASEB J. 2018;32:1591–1601. doi: 10.1096/fj.201700866R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 38.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 40.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 41.Varela-Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martínez-Chantar ML. Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int J Biochem Cell Biol. 2009;41:969–976. doi: 10.1016/j.biocel.2008.10.027. [DOI] [PubMed] [Google Scholar]