

Abstract
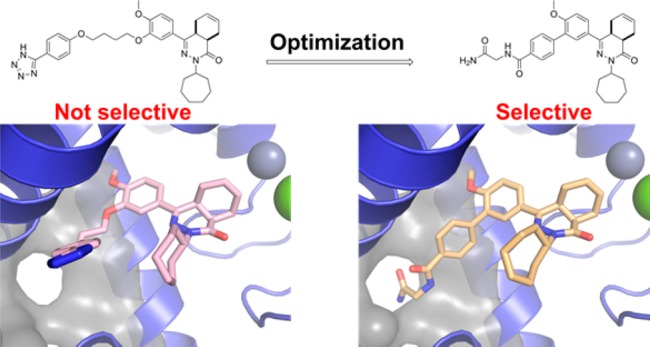
Several trypanosomatid cyclic nucleotide phosphodiesterases (PDEs) possess a unique, parasite-specific cavity near the ligand-binding region that is referred to as the P-pocket. One of these enzymes, Trypanosoma brucei PDE B1 (TbrPDEB1), is considered a drug target for the treatment of African sleeping sickness. Here, we elucidate the molecular determinants of inhibitor binding and reveal that the P-pocket is amenable to directed design. By iterative cycles of design, synthesis, and pharmacological evaluation and by elucidating the structures of inhibitor-bound TbrPDEB1, hPDE4B, and hPDE4D complexes, we have developed 4a,5,8,8a-tetrahydrophthalazinones as the first selective TbrPDEB1 inhibitor series. Two of these, 8 (NPD-008) and 9 (NPD-039), were potent (Ki = 100 nM) TbrPDEB1 inhibitors with antitrypanosomal effects (IC50 = 5.5 and 6.7 μM, respectively). Treatment of parasites with 8 caused an increase in intracellular cyclic adenosine monophosphate (cAMP) levels and severe disruption of T. brucei cellular organization, chemically validating trypanosomal PDEs as therapeutic targets in trypanosomiasis.
Introduction
The family of trypanosomatids is responsible for three major neglected tropical diseases (NTDs) caused by protozoan parasites, namely, Chagas disease (Trypanosoma cruzi), leishmaniasis (Leishmania spp.), and human African trypanosomiasis, also called African sleeping sickness (Trypanosoma brucei rhodesiense and T. brucei gambiense).1 Although millions of people, mostly in underdeveloped countries, are at risk, current options for drug treatment of these infectious diseases remain limited.2
Recently, the inhibition of 3′,5′-cyclic nucleotide phosphodiesterases (PDEs) has emerged as a new approach to target these kinetoplastid protozoans.3−6 All three parasites, T. cruzi, Leishmania spp., and T. brucei spp., have the same set of four different class I PDE families in their genomes.7 Using an inducible siRNA approach, Seebeck and co-workers demonstrated that simultaneous inhibition of T. brucei PDE B1 (TbrPDEB1) and TbrPDEB2 blocks parasite proliferation and eliminates parasitemia from infected mice,8 thereby establishing the parasite PDEs as promising drug targets.
Subsequent efforts to identify TbrPDEB1 inhibitors have resulted in the repurposing of ligands that were originally developed as inhibitors for human PDE4 (hPDE4) as TbrPDEB1 inhibitors (Figure 1).9−12 However, until now, all compounds reported as TbrPDEB1 inhibitors remain more active on hPDE4. The most potent TbrPDEB1 inhibitor reported to date, NPD-001 (1), is 10-fold more potent on hPDE4 (hPDE4B1 IC50 = 0.6 nM) than on TbrPDEB1 (IC50 = 4 nM).11 In a series of analogs of 1 a close correlation between TbrPDEB1 inhibitory potency and antiparasitic activity has been found.11 Although these TbrPDEB1 inhibitors raise cAMP levels in T. brucei and show strong antiparasitic effects in vitro,10−12 their potent inhibition of hPDE4 is undesirable.13−16 Considerable screening and synthetic efforts have been reported,9−12 but no TbrPDEB1-selective (over hPDEs) inhibitors have been identified to date. Despite the high similarity of hPDE4 and TbrPDEB1 substrate binding pockets,17 tantalizing evidence for discriminating selectivity is evident from the observation that established hPDE4 inhibitors such as rolipram and etazolate do not inhibit either the enzyme TbrPDEB1 or T. brucei cellular PDE activity and do not kill the parasite.9,10
Figure 1.

Representative hPDE4 inhibitors and reported TbrPDEB1 inhibitors.9,10,12
In an effort to gain structural understanding to aid in the design of selective TbrPDEB1 inhibitors, we previously reported the crystal structure of the apo-TbrPDEB1 catalytic domain as part of a structure-based virtual screening effort.17 The high-resolution structure of the TbrPDEB1 catalytic domain revealed an open cavity formed between helix 14 (H14), helix 15 (H15), and the M-loop (Figure 2). This cavity is also present in the apo structure of related parasite PDEs, including Leishmania major PDE B1 (LmPDEB1)18 and T. cruzi PDE C (TcrPDEC),19 but is not present in any of the 11 human PDEs.20,21 Being parasite-specific, this cavity has been named the P-pocket. As the most prominent structural difference between parasite and human PDE enzymes, the P-pocket has been considered a promising feature for selective TbrPDEB1 inhibitor design.17 Molecular docking studies suggested that the phenyltetrazole-containing tail group of inhibitors 1 and 2 can interact with residues deep inside the P-pocket.11,12 However, the preferential inhibition of hPDE4 over TbrPDEB1 by these compounds is not consistent with this hypothesis, and therefore the usefulness of targeting the P-pocket to achieve selectivity remains to be demonstrated.
Figure 2.
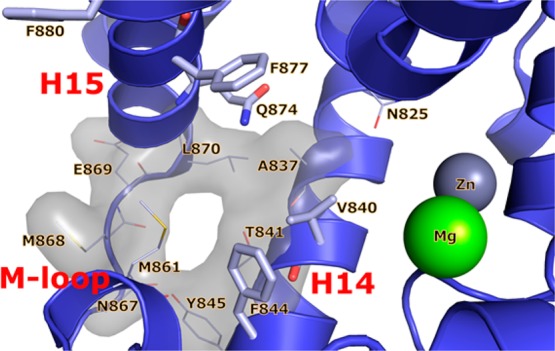
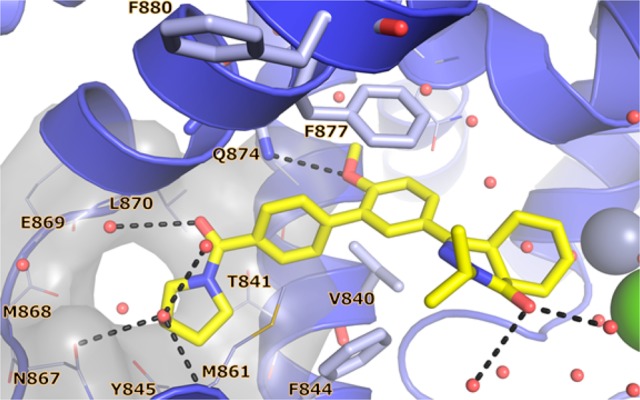
Crystal structure of apo-TbrPDEB1 (PDB code 4I15) active site highlighting the parasite P-pocket. Helix 14 (H14), helix 15 (H15), and the M-loop are labeled in red, the main binding site features are labeled in black, water molecules have been omitted for clarity. The conserved Gln874Q.50, the hydrophobic clamp residues Val840HC.32 and Phe877HC.52, and the distal aromatic residues Phe844S.35 and Phe880HC2.54 are shown as sticks. P-pocket residues Ala837Q1.30, Thr841Q2.33, Tyr845Q2.36, Asn867Q2.43, Met868Q2.44, Glu869Q2.45, and Leu870Q2.46 are shown as lines. All binding site residues have been named according to the PDEStrIAn nomenclature convention.21
Here, we present detailed structural insights in TbrPDEB1 ligand binding by reporting the first inhibitor-bound TbrPDEB1 crystal structures, including cocrystal structures with 1 and 2. These structures clearly explain the lack of selectivity of the previously reported inhibitors and guide the design and synthesis of novel P-pocket targeting inhibitors with, for the first time, selectivity over hPDE4B. We show that these novel compounds inhibit cAMP degradation in T. brucei with consequent trypanocidal activity.
Results and Discussion
The most potent TbrPDEB1 inhibitor reported to date, NPD-001 (1), is a nanomolar inhibitor of TbrPDEB1 and a sub-nanomolar inhibitor of hPDE4 subtypes (IC50 < 1 nM) but is over 100-fold less active against all other human PDEs.10,11 The structurally related pyrazolone analogs, such as NPD-340 (2), are also potent TbrPDEB1 inhibitors, but these too are more potent at hPDE4.12 Our earlier molecular docking studies suggested that the central dialkoxyphenyl moiety of 1 and 2 forms a hydrogen bond to the PDE family wide conserved Gln874Q.50 residue while occupying the hydrophobic clamp, formed by Val840HC.32 and Phe877HC.52 in TbrPDEB1.11,12 In addition to these hallmark interactions of PDE inhibitor binding, the docking studies also suggested that 1 and 2 bind in the P-pocket of TbrPDEB1 with their tetrazole-containing flexible tail group. The TbrPDEB1 activity of these compounds is promising, but their even higher potency against hPDE4 seemed inconsistent with successful targeting of the P-pocket as a means of obtaining selectivity.
In order to gain a better understanding of the lack of selectivity of these compounds and to guide our ongoing medicinal chemistry efforts, we obtained the crystal structure of 1 in complex with the catalytic domain of TbrPDEB1 at a resolution of 1.73 Å (Figure 3a). The overall fold of the protein was highly similar (rms deviation of Cα = 0.38 Å over 630 atoms) to that observed for the apo structure of TbrPDEB1 (PDB code 4I15). Two protein molecules pack in the crystal asymmetric unit of TbrPDEB1 where the binding site of one of the molecules lies close to a symmetry-related molecule. We refer to the interactions of TbrPDEB1 with ligands at the chain that is free from such crystal packing effects, unless noted otherwise. Consistent with biochemical data, 1 was found to bind in the substrate-binding pocket. The dialkoxyphenyl moiety of 1 was situated in the hydrophobic clamp formed by Val840HC.32 and Phe877HC.52, and its two ether functionalities were within hydrogen bonding distance of the conserved Gln874Q.50 residue. The (4aS,8aR)-enantiomer of 1 was observed in the crystal structure. In contrast to the docking pose suggested by the earlier computational studies,11,12 the tail group of 1 is oriented away from the P-pocket, with the tetrazole moiety instead forming a stacking interaction with Phe880HC2.54 in H15.
Figure 3.

Crystal structures of nonselective inhibitors with TbrPDEB1, hPDE4B, and hPDE4D. (a) TbrPDEB1–1, including |Fo – Fc|αcalc electron density map contoured at 2.5σ. (b) TbrPDEB1–2. (c) hPDE4B–1. (d) hPDE4D–1. In all panels: key binding site amino acid residues are shown as sticks, minor amino acid residues are shown as lines. The P-pocket is shown as a gray surface with P-pocket residues represented as lines, and key polar interactions are depicted with black dashed lines. Water molecules are displayed as red spheres, zinc cations as metallic blue spheres, and magnesium cations as green spheres.
The crystal structure of the TbrPDEB1 catalytic domain with 2 was determined at 2.25 Å resolution (Figure 3b). Compounds 1 and 2 adopted an identical binding mode with conservation of the key interactions of the catechol and phenyltetrazole tail with TbrPDEB1.
We also determined the crystal structure of 1 in complex with hPDE4B (Figure 3c) and hPDE4D (Figure 3d) at 2.4 and 2.25 Å resolution, respectively. As expected, the hPDE4B structure displayed a similar overall fold to the TbrPDEB1 structure bound to 1 (rms deviation of Cα = 1.26 Å over 247 atoms). In line with its high potency as a hPDE4 inhibitor, 1 was observed in the cAMP-binding site and interacted with hPDE4B through a bifurcated hydrogen bond to the invariant Gln615Q.50. As in the TbrPDEB1 structure, there are key interactions with the hydrophobic clamp, formed by Ile582HC.32 and Phe618HC.52, in addition to a well-positioned stacking interaction of the tetrazole moiety with Tyr621HC2.54. In the catalytic domain of hPDE4D the interaction pattern exhibited by 1 was identical to that observed in the hPDE4B structure. In all structures, only the single (4aS,8aR)-enantiomer of 1 was observed. The similar binding mode observed in these structures unequivocally establish that the tetrazole tail present in 1 and 2 does not target the P-pocket in TbrPDEB1, which potentially explains the lack of selectivity of these compounds for TbrPDEB1 over hPDE4B.
Inspection of the binding site characteristics of TbrPDEB1 and the binding modes observed for 1 and 2 highlighted that the hydrophobic clamp and the extended hydrophobic region (HC2) (Figure S1) provide a favorable environment for the flexible linker of these inhibitors to fold away from the P-pocket and interact with the aromatic residue found in H15, being Phe880HC2.54 in TbrPDEB1 and Tyr621HC2.54 in hPDE4B. We hypothesized that modification of the flexible alkyl linker could prevent such a hydrophobic collapse and utilized the Phosphodiesterase Structure and Ligand Interaction Annotated (PDEStrIAn)21 tool for the design of such ligands. More specifically, it was observed that some cocrystallized human PDE inhibitors (for example, PDB codes 3IAD and 4AEL) have a central aromatic moiety bound in the hydrophobic clamp while having additional (hetero)aryl groups along the vector that would point toward the P-pocket in TbrPDEB1. It was hypothesized that replacing the flexible linker at the 3-position of the dialkoxyphenyl ring of 1 with a phenyl ring would provide a biphenyl system with the desired rigidity and vector to target the P-pocket.
Accordingly, we produced an initial series of biphenyl analogs 3–6 and screened them as inhibitors of TbrPDEB1 and hPDE4B1 (Table 1). Introduction of the biphenyl linker indeed led to some selectivity for TbrPDEB1 over hPDE4B1 in carboxylic acids 3 and 4. The TbrPDEB1 selectivity was higher for the para-substituted 3 over its meta-substituted analog (4). The potency at TbrPDEB1 increased 10-fold upon introduction of a carboxamide group at the para-position in compound 5. In accordance with the overlapping pharmacological profiles of hPDE4 subtypes, 5 showed the same potency against all tested hPDE4s but was selective over the other human PDE subtypes (Table S5).
Table 1. Introduction of the Biphenyl Linkera.


pKi values are averages (n ≥ 2, with SD less than ±0.3 for all).
Compounds are racemic mixtures of cis-isomers.
Selectivity is calculated as hPDE4B1 (Ki)/TbrPDEB1 (Ki).
These findings led to further design and synthesis of a series of para-substituted carboxamide analogs, using 5 as a template to further improve TbrPDEB1 potency and selectivity over hPDE4B (Table 2). Extending the carboxamide of 5 with a methoxyethyl substituent (7) was found to increase the selectivity for TbrPDEB1 7-fold, while the potency was comparable to that of its parent compound. Gratifyingly, glycinamide 8, and its isopropyl-substituted counterpart 9, were even more potent TbrPDEB1 inhibitors (Ki = 100 nM and 99 nM, respectively). The cycloheptyl-substituted compound 8 displayed 10-fold selectivity over hPDE4B1 and was also selective over the other hPDE4 subtypes (Table S5). The potency of 8 was fully explained by the biological activity of a single enantiomer; (8a) was found to be 100-fold less potent against TbrPDEB1 than its optical isomer 8b. Both enantiomers of 8 were selective TbrPDEB1 inhibitors, with 8b showing 16-fold selectivity. Furthermore, isopropyl analog 9 showed a 19-fold preference for TbrPDEB1 over hPDE4B1. These glycinamide compounds are the most selective TbrPDEB1 inhibitors reported to date. The introduction of N-methyl, N,N-dimethyl, or N-isopropyl substituents on different positions of the P-pocket tail (compounds 10–13) led to decreases in potency as well as selectivity. Alkylation was least tolerated at the terminal nitrogen (12, 13), indicating that changes in hydrophobicity or hydrogen bonding properties are unfavorable. Increasing the chain length of glycinamide 8 with an additional carbon, to give 14, resulted in a substantial decrease in potency at TbrPDEB1 but not at hPDE4B1, decreasing selectivity for TbrPDEB1 to only 2-fold. Inversion of the carboxamide of 14 affected the hPDE4B1 potency and restored the TbrPDEB1 selectivity back to 5-fold in compound 15. Finally, further extension of the glycinamide of 8 with a hydroxyethyl (16) or methoxyethyl (17) substituent was tolerated and resulted in ∼8 fold selective TbrPDEB1 inhibitors. These findings highlight that selectivity for TbrPDEB1 over hPDE4B1 is very sensitive to small changes in the inhibitor tail group.
Table 2. Exploration of SAR in the P-Pocketa.


pKi values are averages (n ≥ 2, with SD less than ±0.3 for all).
Compounds are racemic mixtures of cis-isomers unless otherwise noted.
Selectivity is calculated as hPDE4B1 (Ki)/TbrPDEB1 (Ki).
In order to validate the P-pocket targeting approach to selectivity, we obtained the crystal structures of 8 (Figure 4a) and 9 (Figure 4b) with TbrPDEB1 at a resolution of 1.8 and 2.0 Å, respectively. The overall fold of these structures was highly similar to the apo-TbrPDEB1 (PDB code 4I15) and the TbrPDEB1–1 crystal structures (rms deviations of Cα < 0.3 Å). Both ligands bonded to the invariant Gln874Q.50 and occupied the hydrophobic clamp with the methoxyphenyl ring of their biphenyl linker. The (4aR,8aS)-enantiomer was observed in the crystal structures of both inhibitors. In the case of 8, we therefore propose the 100-fold more potent enantiomer 8b to be (4aR,8aS)-configured. Compounds 8 and 9 successfully targeted the P-pocket. The glycinamide tail of 8 formed a direct hydrogen bond to Tyr845Q2.36 and three water-mediated hydrogen bonds to the following residues: (1) Thr841Q2.33 and Met861S.40; (2) Leu870Q2.46 and Gln874Q.50; and (3) Gly873Q2.49. Alignment of the crystal structure of 8 with the nonselective inhibitor 1 clearly highlights the distinctive binding mode of the tail groups (Figure 5). However, the 4a,5,8,8a-tetrahydrophthalazinone and methoxy-substituted phenyl rings of 1 and 8 closely aligned in the active site of TbrPDEB1 and therefore will be competitive inhibitors of cAMP breakdown. The tail group of 9 was observed in a slightly different conformation, with direct hydrogen bonds formed to Tyr845Q.2.36 and Glu869Q2.45 and two water-mediated hydrogen bonds to (1) Thr841Q2.33 and Met861S.40 and (2) Leu870Q2.46 and Gln874Q.50. Average temperature factors (B factors) indicated greater flexibility in the P-pocket region of TbrPDEB1 compared to the rest of the protein. The observed electron density for the glycinamide tail of these inhibitors was somewhat weaker than that for the rest of the ligand, indicative of greater ligand flexibility in the P-pocket region. Both findings have been supported by observations from molecular dynamics (MD) simulations (data not shown).
Figure 4.

TbrPDEB1 crystal structures of selective inhibitors. (a) TbrPDEB1–8, including |Fo – Fc|αcalc electron density map contoured at 2.5σ; (b) TbrPDEB1–9.
Figure 5.
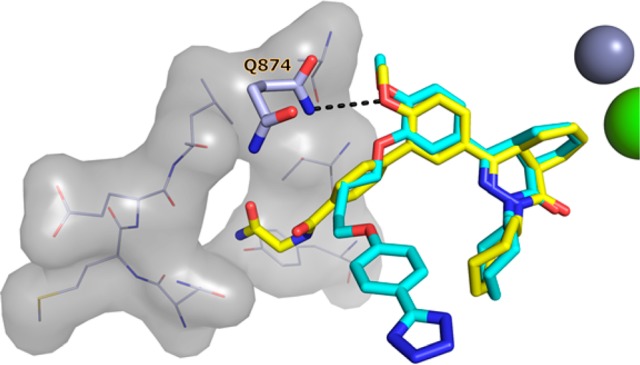
Alignment of the TbrPDEB1 crystal structures of 1 (cyan) and 8 (yellow). Coordinates of Gln874Q.50, P-pocket residues, and metal cations were derived from TbrPDEB1–8 and are shown for clarity.
Attempts to elongate the glycinamide tail of compound 8 with a 2-hydroxyethyl substituent to further probe the P-pocket led to compound 16. In the 2.1 Å crystal structure of 16 with TbrPDEB1, the elongated tail was found to penetrate the P-pocket (Figure 6). The overall binding mode for the single enantiomer (4aR,8aS)-16 was similar to that observed for glycinamide analogs 8 and 9. This modification was not accompanied by an increase in TbrPDEB1 activity.
Figure 6.
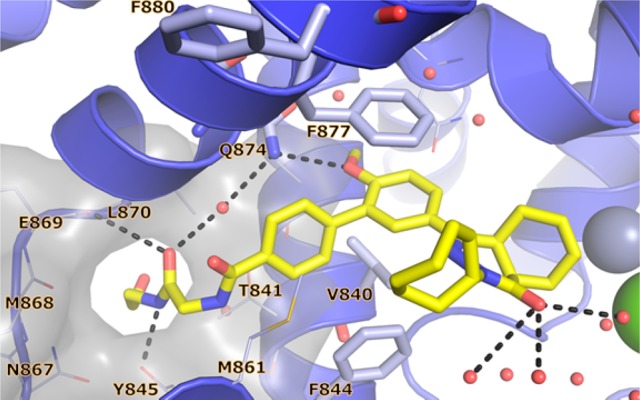
X-ray crystal structure of TbrPDEB1–16.
The main residues lining the P-pocket in TbrPDEB1 are Ala837Q1.30, Thr841Q2.33, Tyr845Q2.36, Asn867Q2.43, Met868Q2.44, Glu869Q2.45, and Leu870Q2.46, with residues from the adjacent M-loop potentially playing a minor role. The character of these residues and the flexibility of the P-pocket prompted us to direct chemistry efforts toward a series of aliphatic heterocyclic tail substituents in order to introduce rigidity into the tail region to further interrogate the P-pocket (Table 3).
Table 3. SAR of Aliphatic Heterocycles in the P-Pocketa.


pKi values are averages (n ≥ 2, with SD less than ±0.3 for all).
Compounds are racemic mixtures of cis-isomers.
Selectivity is calculated as hPDE4B1 (Ki)/TbrPDEB1 (Ki).
Compound 18 and its N-methylated counterpart 19 were designed as constrained analogs of 8. However, cyclization of the glycinamide led to a loss of selectivity for TbrPDEB1 over hPDE4B1 and reduced potency. As seen for the noncyclized analogs, N-methylation (19) led to a further reduction in potency. The pyrrolidin-3-one analog (20) was comparable to 18 and 19 in TbrPDEB1 potency but was ∼3-fold selective over hPDE4B1, indicating that a nitrogen in this ring is not necessary for selectivity. Going from a pyrrolidin-3-one to a (R)-pyrrolidin-3-ol substituent (21) increased the TbrPDEB1 selectivity to 9-fold and led to a more potent TbrPDEB1 inhibitor. Interestingly, its isopropyl-substituted counterpart (22) was even less potent at hPDE4B1 and displayed a 15-fold selectivity for TbrPDEB1. Finally, we explored two (R)-pyrrolidine-2-carboxamide analogs, 23 and 24, and both were more than 10-fold selective for TbrPDEB1, but overall less potent.
In order to investigate how these heterocycles interact with the P-pocket in TbrPDEB1, we determined the crystal structure of 22 (Figure 7) bound to TbrPDEB1 at a resolution of 1.8 Å. Only the (4aS,8aR)-enantiomer was observed in the crystal structure. The (R)-3-hydroxypyrrolidine tail group targeted the P-pocket through direct hydrogen bonds to the backbone carbonyl of Met861S.40 and to the side chain of Asn867Q2.43. A water molecule was also observed making a polar interaction with the hydroxyl substituent. Furthermore, the carbonyl oxygen of the tail was within hydrogen bonding distance of a water molecule coordinated by Glu869Q2.45, Lys872Q2.48, and Gly873Q2.49. It should be noted that in this crystal structure, 22 was only present at the chain with a symmetry-related molecule close to the ligand binding site. However, the pose of 22 was in line with the overall binding modes observed for the other TbrPDEB1 inhibitors in this series.
Figure 7.
X-ray crystal structure of TbrPDEB1–22.
We have previously shown that inhibition of TbrPDEB1 and TbrPDEB2 by 1 in T. b. brucei leads to a rapid and dose-dependent increase in intracellular cAMP levels.10,11,22 Incubation of bloodstream form trypanosomes with 8 increased intracellular cAMP levels in these parasites in a dose-dependent fashion (Figure 8). Compound 8 showed a small but significant effect at 100 nM (P < 0.05) and a strong increase of cAMP level at 10 μM (P < 0.001), a concentration that was equally effective as 0.3 μM of 1, used as positive control. The effect on the intracellular cAMP levels of the antitrypanosomal compound pentamidine, used as a negative control, was not statistically different from untreated parasites.
Figure 8.

Intracellular cAMP levels in T. brucei after treatment with 8 (NPD-008), with 0.3 μM of compound 1 (NPD-001) used as a positive control, and pentamidine as a negative control.
Having identified and characterized 8 as a selective TbrPDEB1 inhibitor, we initiated further phenotypic profiling. It has been reported that inhibition of cAMP metabolism causes severe cellular defects in T. brucei, particularly the ability of the cells to complete cell division, as cytokinesis and abscission are impaired, while nuclear and kinetoplast division remain apparently unaffected.6,8,10 This leads to misshapen and nonviable cells with multiple nuclei and kinetoplasts. Incubation with 8 at a concentration of 10 μM had the same effect: after incubation of healthy trypanosomes (Figure 9a) for 6 h (roughly equivalent to one doubling time under standard culture conditions) with 10 μM compound 8, cells could be seen to have undergone at least one round of nuclear and kinetoplast division but without completing cell division into daughter cells (Figure 9b), indicating a defect in cytokinesis. At 12 h, this had resulted in rounded cells with multiple flagella, some of them detached, and at least 4 nuclei (Figure 9c). At 24 h, only a few live cells could be detected, all of which were severely misshapen (Figure 9d). These results were consistent with the observed antitrypanosomal activity of 8 (IC50 = 5.5 ± 3.3 μM). The isopropyl analog 9 displayed similar anti-T. brucei effects (IC50 = 6.7 ± 1.6 μM) in the same cytokinesis assay. While exhibiting a potency in the sub-micromolar range against TbrPDEB1, the observed lower antitrypanosomal activity might be due to reduced cell penetration properties. At these concentrations, off-target effects can also not be ruled out. However, the cytotoxic effects of 8 and 9 against human MRC-5 cells were comparable for both compounds (CC50 = 36 and 35 μM, respectively) and clearly lower than the antiparasitic effects. Yet, because of the relatively narrow selectivity, these compounds do not meet the published criteria of drug candidates for the treatment of human African trypanosomiasis,23 and their trypanocidal activity, in particular, requires further phenotypic optimization.
Figure 9.
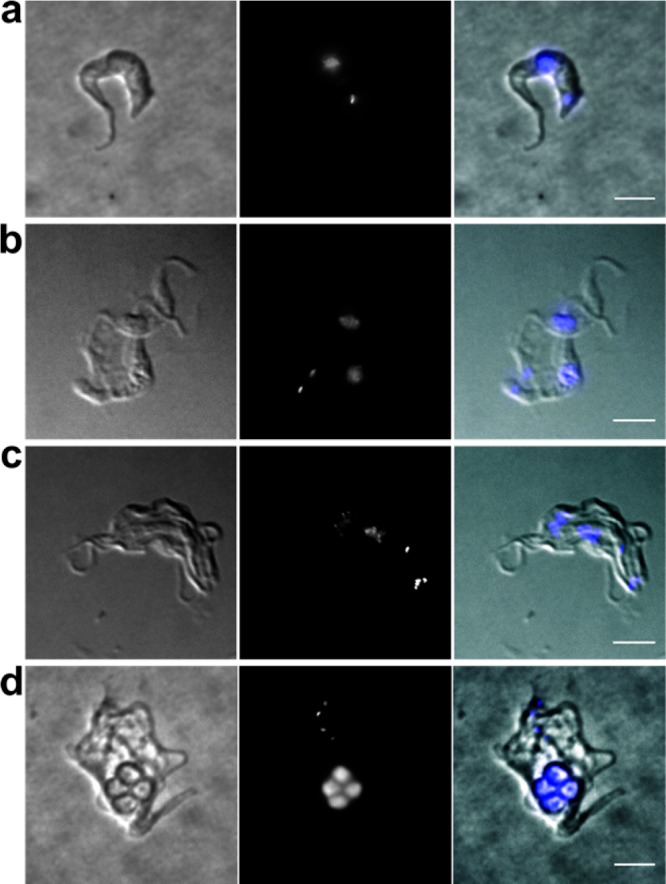
Fluorescence microscopy of wild-type T. brucei incubated with 8 (10 μM) for the following periods: (a) 0 h (control); (b) 6 h; (c) 12 h; (d) 24 h. Left panels are brightfield images, middle panels are fluorescence images after DAPI staining, and the right-hand panels are the merged images. Scale bars, 5 μm.
Chemistry
The chemical synthesis of compounds 3–6 and intermediate 28 proceeded as shown in Scheme 1. The key 4a,5,8,8a-tetrahydrophthalazin-1(2H)-one building block (25)24−26 was N-alkylated with the appropriate alkyl halides, and a Suzuki cross-coupling reaction afforded the biaryl systems.
Scheme 1.

Reagents and conditions: (a) alkyl halide, NaH, DMF, RT, 4 h, 65–85%; (b) arylboronic acid, Pd(dppf)Cl2·CH2Cl2, Na2CO3, DME, H2O, 100 °C, 16 h, 48–81%.
Compounds 7–10, 14–18, 21–24, and 29 were prepared using carbodiimide-mediated amide coupling methods using 4 or 28 as starting material (Scheme 2). Amide coupling was performed without the addition of DIPEA in the synthesis of 15, 21, 23, and 24 or without Et3N in the case of analog 22.
Scheme 2.

Reagents and conditions: (a) amine, EDC·HCl, HOAt, DIPEA, CH2Cl2, RT, 3 h, 38–88% (compounds 7, 10, 14, 15, 17, 18, 21, 23, 24, and 29), or amine, EDC·HCl, HOBt hydrate, Et3N, CH2Cl2, RT, 18 h, 54–65% (compounds 8, 9, 16, and 22), or separation of the enantiomers of 4 by preparative chiral HPLC, then 4a or 4b, 2-aminoacetamide·HCl, EDC·HCl, HOAt, DIPEA, CH2Cl2, RT, 24 h, 50–55% (8a, 8b).
To enable access to the enantiomers of 8, racemic 4 was separated into its enantiomers 4a and 4b using preparative chiral HPLC. Each of the enantiomers of 4 was reacted with 2-aminoacetamide, as depicted in Scheme 2. Enantiomer 8a was obtained with 99.6% ee, while its optical isomer 8b was obtained with 96.6% ee. The X-ray crystal structures indicate that TbrPDEB1 exhibits chiral discrimination, allowing the tentative assignment of the absolute configuration of (4aR,8aS) to 8b, as this enantiomer possesses a 100-fold higher potency than 8a.
The synthesis of 11–13 is outlined in Scheme 3. Methyl ester 29 was treated with aqueous base to give intermediate 30, and subsequent carbodiimide-mediated amide coupling yielded 11–13. Compound 13 was prepared without the addition of DIPEA.
Scheme 3.

Reagents and conditions: (a) NaOH, EtOH, RT, 2 h, 89%; (b) amine, EDC·HCl, HOAt, DIPEA, CH2Cl2, RT, 36 h, 29–54%.
Scheme 4 depicts the synthesis of 19 and 20. Analog 19 was prepared by the N-alkylation of 18 using iodomethane. Finally, compound 20 was attained by the oxidation of 21 using Dess–Martin periodinane.
Scheme 4.

Reagents and conditions: (a) iodomethane, NaH, DMF, RT, 1.5 h, 54%; (b) Dess–Martin periodinane, CH2Cl2, RT, 4 h, 58%.
Conclusion
In summary, we have identified the first series of selective TbrPDEB1 inhibitors reported to date and explain their specificity for TbrPDEB1 over hPDE4 isoforms on the basis of the cocrystal structures obtained. We have also explained why previously reported inhibitors failed to show any selectivity. Our novel trypanocidal TbrPDEB1 inhibitors feature a tail group containing a rigid biphenyl system with polar substituents for targeting the P-pocket. Structural data currently available on the PDEs of L. major and T. cruzi indicate the presence of similar P-pockets in those enzymes,18,19 while human PDEs do not have such a pocket.21 Therefore, this work identifies important possibilities for the development of parasite-selective PDE inhibitors for a variety of neglected tropical diseases.
Experimental Section
Chemistry
All reagents and solvents were obtained from commercial suppliers and were used as received. All reactions were magnetically stirred and carried out under an inert atmosphere. Reaction progress was monitored using thin-layer chromatography (TLC) and LC-MS analysis. Silica gel column chromatography was carried out manually or with automatic purification systems using the indicated eluent. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance 500 (500 MHz for 1H and 126 MHz for 13C) or Bruker Avance 600 (600 MHz for 1H and 151 MHz for 13C) instruments equipped with a Bruker CryoPlatform, or a Bruker DMX300 (300 MHz). Chemical shifts (δ in ppm) and coupling constants (J in Hz) are reported with residual solvent as internal standard (δ 1H NMR, CDCl3 7.26, DMSO-d6 2.50; δ 13C NMR, CDCl3 77.16, DMSO-d6 39.52). Various compounds exhibited rotamers leading to more complicated 1H NMR spectra and less accurate integrations. LC-MS analysis was performed on a Shimadzu LC-20AD liquid chromatograph pump system, equipped with an Xbridge (C18) 5 μm column (50 mm, 4.6 mm), connected to a Shimadzu SPD-M20A diode array detector, and MS detection using a Shimadzu LC-MS-2010EV mass spectrometer. The LC-MS conditions were as follows: solvent B (acetonitrile with 0.1% formic acid) and solvent A (water with 0.1% formic acid), flow rate of 1.0 mL/min, start 5% B, linear gradient to 90% B in 4.5 min, then 1.5 min at 90% B, then linear gradient to 5% B in 0.5 min, then 1.5 min at 5% B; total run time of 8 min. Analytical chiral HPLC was performed with a Chiralpak AD-H column (250 mm × 4.6 mm, 5 μm) with the following conditions: flow, 1 mL/min; column temperature, 35 °C; detection, 270 nm; eluent, heptane/isopropanol 9:1; runtime, 30 min. Exact mass measurement (HRMS) was performed on a Bruker micrOTOF-Q instrument with electrospray ionization (ESI) in positive ion mode and a capillary potential of 4500 V. Systematic names for molecules were generated with ChemBioDraw Ultra 14.0.0.117 (PerkinElmer, Inc.). The reported yields refer to isolated pure products; yields were not optimized. The purity, reported as the peak area % at 254 nm, of all final compounds was ≥95% based on LC-MS.
Synthetic Procedures
Building block 25,24−26 NPD-001 (1),11 and NPD-340 (2)12 were prepared as described elsewhere. All other compounds were prepared as described below.
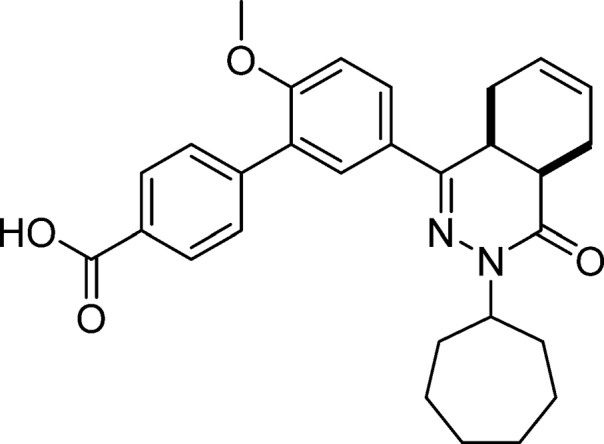
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-3-carboxylic Acid (3, NPD-740)
Prepared from 27 (0.5 g, 1.2 mmol) and 3-boronobenzoic acid (0.25 g, 1.5 mmol) as described for 28. The title compound was obtained in 65% yield. 1H NMR (500 MHz, DMSO-d6) δ 13.09 (s, 1H), 8.05 (s, 1H), 7.92 (d, J = 7.7 Hz, 1H), 7.88 (dd, J = 8.7, 2.3 Hz, 1H), 7.80–7.71 (m, 2H), 7.56 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 8.7 Hz, 1H), 5.73–5.56 (m, 2H), 4.74–4.62 (m, 1H), 3.83 (s, 3H), 3.53–3.44 (m, 1H), 2.79 (t, J = 6.1 Hz, 1H), 2.76–2.67 (m, 1H), 2.21–2.05 (m, 2H), 1.96–1.63 (m, 7H), 1.62–1.36 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 167.8, 166.0, 157.7, 154.0, 138.4, 134.1, 131.3, 130.5, 129.5, 128.9, 128.5, 128.3, 128.0, 127.6, 126.4, 124.5, 112.5, 56.3, 55.3, 34.2, 33.3, 33.0, 30.3, 28.7, 28.7, 24.9, 24.8, 23.0, 22.4. LC-MS (ESI): tR = 5.44 min, area >98%, m/z 473 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C29H33N2O4 473.2435, found 473.2414.
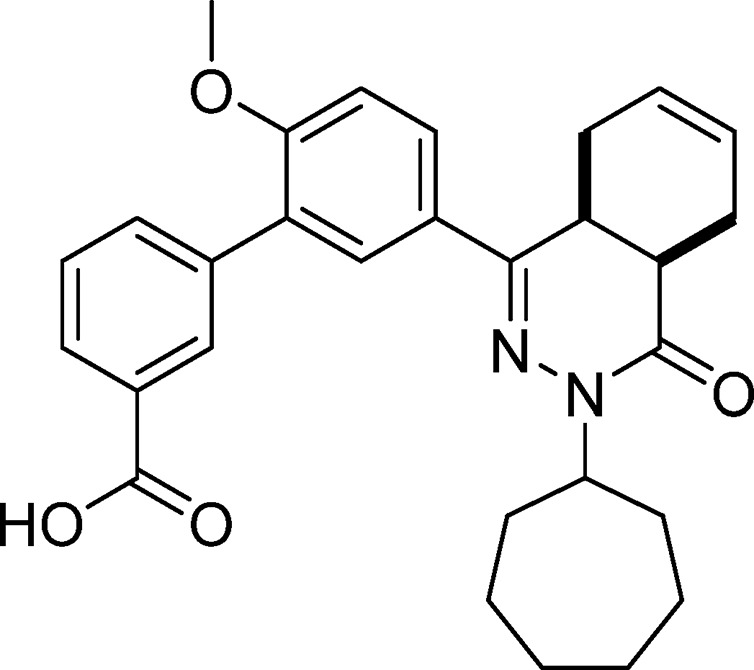
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxylic Acid (4, NPD-733)
Prepared from 27 (1.00 g, 2.32 mmol) and 4-boronobenzoic acid (0.66 g, 4.0 mmol) as described for 28. The title compound was obtained in 66% yield. 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 8.5 Hz, 2H), 7.84 (dd, J = 8.7, 2.4 Hz, 1H), 7.77 (d, J = 2.3 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.7 Hz, 1H), 5.85–5.61 (m, 2H), 4.88–4.74 (m, 1H), 3.88 (s, 3H), 3.38–3.27 (m, 1H), 3.09–2.95 (m, 1H), 2.76 (t, J = 5.9 Hz, 1H), 2.27–2.14 (m, 3H), 2.12–1.96 (m, 2H), 1.96–1.85 (m, 1H), 1.85–1.70 (m, 4H), 1.69–1.43 (m, 5H), 1.34–1.18 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 177.1, 166.0, 157.6, 153.5, 143.8, 130.0, 129.7, 129.7, 128.3, 128.1, 128.0, 127.2, 126.1, 123.9, 111.3, 56.3, 55.8, 34.7, 33.2, 33.0, 31.9, 29.0, 28.4, 28.4, 25.1, 25.0, 22.7, 22.4. LC-MS (ESI): tR = 5.41 min, area: 98%, m/z 473 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C29H33N2O4 473.2435, found 473.2432.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (5, NPD-584)
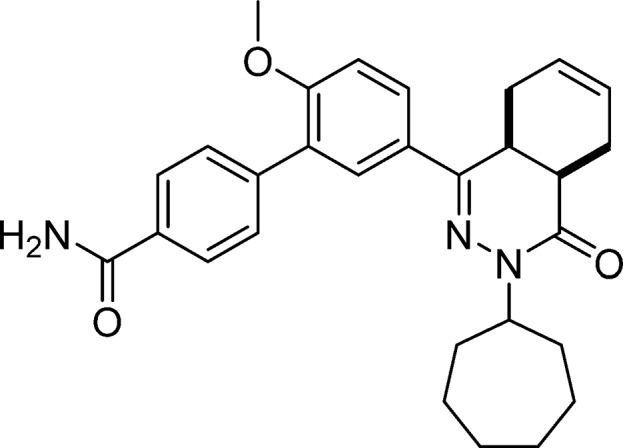
Prepared from 27 (200 mg, 0.464 mmol) and (4-carbamoylphenyl)boronic acid (116 mg, 0.703 mmol) as described for 28. The title compound was isolated in 48% yield. 1H NMR (500 MHz, CDCl3) δ 7.90 (d, J = 8.3 Hz, 2H), 7.84 (dd, J = 8.7, 2.3 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.06 (d, J = 8.7 Hz, 1H), 6.13 (s, 1H), 5.84–5.66 (m, 2H), 5.62 (s, 1H), 4.87–4.77 (m, 1H), 3.89 (s, 3H), 3.38–3.27 (m, 1H), 3.06–2.97 (m, 1H), 2.76 (t, J = 6.0 Hz, 1H), 2.26–1.70 (m, 8H), 1.69–1.60 (m, 4H), 1.57–1.42 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 168.9, 165.9, 157.6, 153.3, 142.0, 131.9, 129.8, 129.7, 128.2, 128.1, 127.2, 127.1, 126.1, 123.9, 111.3, 56.2, 55.8, 34.7, 33.2, 33.0, 31.1, 28.43, 28.36, 25.04, 24.98, 23.1, 22.4. LC-MS (ESI): tR = 5.34 min, area: 96%, m/z 472 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C29H34N3O3 472.2595, found 472.2593.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carbonitrile (6, NPD-744)
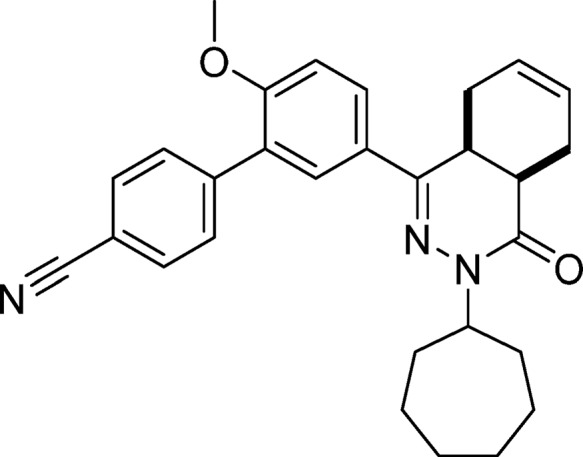
Prepared from 27 (1.0 g, 2.3 mmol) and 4-cyanophenylboronic acid (0.51 g, 3.5 mmol) as described for 28. The title compound was obtained in 81% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.92 (dd, J = 8.7, 2.3 Hz, 1H), 7.89 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 2.3 Hz, 1H), 7.71 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.8 Hz, 1H), 5.73–5.55 (m, 2H), 4.72–4.62 (m, 1H), 3.83 (s, 3H), 3.54–3.44 (m, 1H), 2.80–2.68 (m, 2H), 2.20–2.07 (m, 2H), 1.96–1.61 (m, 7H), 1.61–1.35 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 170.7, 162.4, 158.6, 147.8, 137.2, 135.5, 133.3, 133.1, 132.9, 132.9, 131.1, 129.2, 124.1, 117.4, 115.0, 61.1, 60.2, 39.0, 38.0, 37.8, 35.0, 33.3, 33.3, 29.7, 29.6, 27.7, 27.2. LC-MS (ESI): tR = 5.93 min, area: >98%, m/z 454 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C29H32N3O2 454.2489, found 454.2477.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-N-(2-methoxyethyl)-[1,1′-biphenyl]-4-carboxamide (7, NPD-734)
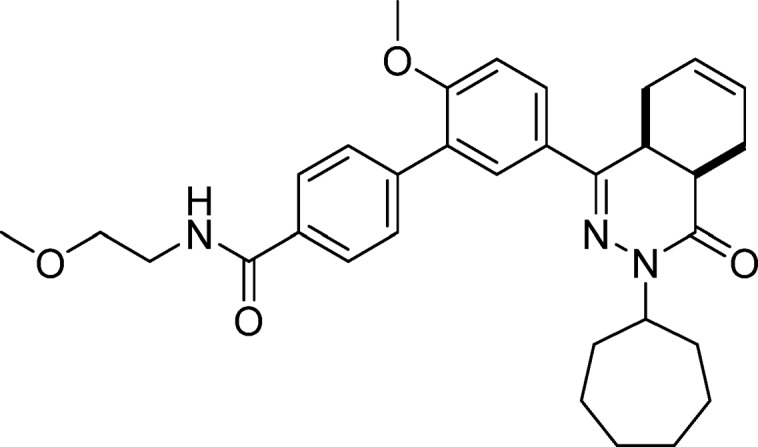
EDC·HCl (49 mg, 0.26 mmol) was added to a solution of 4 (0.10 g, 0.21 mmol), HOAt (29 mg, 0.21 mmol), 2-methoxyethanamine (0.024 g, 0.32 mmol), and DIPEA (0.11 mL, 0.64 mmol) in CH2Cl2 (5 mL). The mixture was stirred at RT for 3 h, diluted with CH2Cl2 (100 mL), and washed with water (2 × 50 mL) and brine (50 mL). The organic phase was dried over Na2SO4, filtered, concentrated, and purified using a reverse phase C18-silica gel column eluting with water/MeCN + 0.1% HCOOH (gradient, 95:5 to 5:95), to afford 7 in 79% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.57 (t, J = 5.1 Hz, 1H), 7.90 (d, J = 8.0 Hz, 2H), 7.87 (dd, J = 8.7, 2.3 Hz, 1H), 7.76 (d, J = 2.3 Hz, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.7 Hz, 1H), 5.74–5.55 (m, 2H), 4.74–4.59 (m, 1H), 3.82 (s, 3H), 3.54–3.40 (m, 5H), 3.28 (s, 3H), 2.83–2.66 (m, 2H), 2.22–2.06 (m, 2H), 1.96–1.62 (m, 7H), 1.62–1.34 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.0, 164.6, 156.3, 152.5, 139.5, 132.0, 128.2, 126.8, 126.5, 126.2, 126.0, 124.9, 123.1, 111.0, 69.5, 57.0, 54.9, 54.0, 38.0, 32.8, 31.9, 31.6, 28.9, 27.2, 27.2, 23.5, 23.5, 21.6, 21.0. LC-MS (ESI): tR = 5.30 min, area: >98%, m/z 530 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C32H40N3O4 530.3013, found 530.3005.
N-(2-Amino-2-oxoethyl)-5′-(cis-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (8, NPD-008)
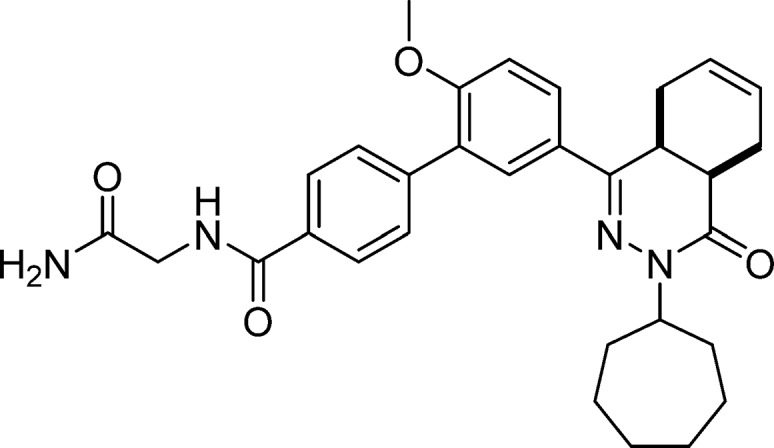
EDC·HCl (183 mg, 0.952 mmol) was added to a solution of 4 (300 mg, 0.635 mmol), HOBt hydrate (97 mg, 0.63 mmol), 2-aminoacetamide·HCl (105 mg, 0.952 mmol), and Et3N (0.265 mL, 1.90 mmol) in CH2Cl2 (6 mL). The reaction mixture was stirred at RT for 18 h. EtOAc (100 mL) was added, and the resulting solution was washed with water (2 × 50 mL) and brine (50 mL). The organic phase was dried over Na2SO4, filtered and concentrated to obtain the crude product as a light brown oil. The crude oil was purified on a silica gel column eluting with CH2Cl2/MeOH (gradient, 100:0 to 95:5), to give 8 as a white solid in 65% yield. 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 2H), 7.81 (dd, J = 8.7, 2.3 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.29–7.24 (m, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.54 (s, 1H), 5.82–5.61 (m, 3H), 4.85–4.74 (m, 1H), 4.23 (d, J = 4.7 Hz, 2H), 3.85 (s, 3H), 3.35–3.27 (m, 1H), 3.04–2.94 (m, 1H), 2.73 (t, J = 6.0 Hz, 1H), 2.24–2.12 (m, 2H), 2.10–1.94 (m, 2H), 1.93–1.83 (m, 1H), 1.82–1.68 (m, 4H), 1.67–1.42 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 171.2, 167.6, 165.9, 157.6, 153.4, 141.9, 132.0, 129.8, 129.7, 128.2, 128.1, 127.1, 127.0, 126.1, 123.9, 111.3, 56.3, 55.8, 43.3, 34.7, 33.2, 33.0, 31.9, 31.1, 28.43, 28.36, 25.1, 25.0, 23.1, 22.4. LC-MS (ESI): tR = 4.69 min, area: >98%, m/z 529 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C31H37N4O4 529.2809, found 529.2811.
N-(2-Amino-2-oxoethyl)-5′-((4aS,8aR)-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (8a, NPD-949) and N-(2-Amino-2-oxoethyl)-5′-((4aR,8aS)-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (8b, NPD-1373)

The enantiomers of racemic 4 were separated into 4a (first eluting isomer) and 4b (second eluting isomer) using preparative chiral chromatography. Analytical chiral HPLC indicated 4a and 4b to have the same retention times as their respective counterparts in racemic mixture 4. The title compounds were synthesized from separate solutions of enantiomerically pure 4 (124 mg, 0.262 mmol) and 2-aminoacetamide HCl (34.8 mg, 0.315 mmol) as described for 7, and were isolated in 50–55% yield. Absolute configuration was tentatively assigned to enantiomer 8b. Compound 8a, Chiralpak AD-H 99.6% ee; compound 8b, Chiralpak AD-H tR = 19.71 min, 96.9% ee.
N-(2-Amino-2-oxoethyl)-5′-(cis-3-isopropyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (9, NPD-039)
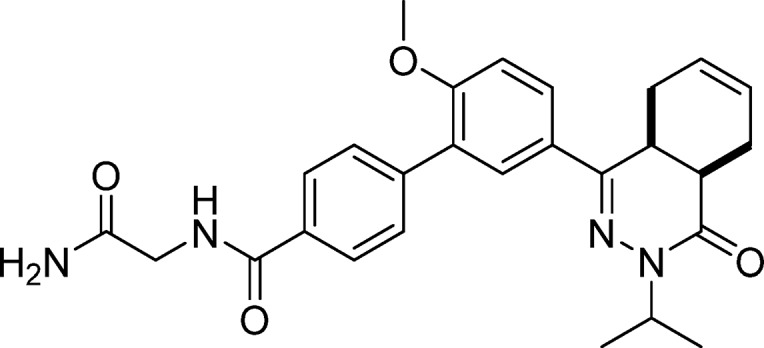
Prepared from 28 (300 mg, 0.717 mmol) as described for 8. The title compound was isolated in 59% yield. 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 8.1 Hz, 2H), 7.84 (dd, J = 8.7, 2.3 Hz, 1H), 7.80 (d, J = 2.3 Hz, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.05 (d, J = 8.7 Hz, 1H), 6.99–6.93 (m, 1H), 5.99 (s, 1H), 5.85–5.64 (m, 2H), 5.48 (s, 1H), 5.06 (hept, J = 6.8 Hz, 1H), 4.22 (d, J = 4.8 Hz, 2H), 3.89 (s, 3H), 3.39–3.30 (m, 1H), 3.08–2.97 (m, 1H), 2.78 (t, J = 6.0 Hz, 1H), 2.24–2.18 (m, 2H), 2.13–2.00 (m, 1H), 1.34 (d, J = 6.5 Hz, 3H), 1.22 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.7, 167.4, 166.4, 157.6, 153.4, 141.9, 132.0, 129.84, 129.76, 128.2, 128.0, 127.0, 126.9, 126.0, 123.9, 111.2, 55.8, 46.7, 43.3, 34.8, 31.1, 23.2, 22.4, 20.6, 20.2. LC-MS (ESI): tR = 4.13 min, area: >95%, m/z 475 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C27H31N4O4 475.2340, found 475.2344.
N-(2-Amino-2-oxoethyl)-5′-(cis-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-N-methyl-[1,1′-biphenyl]-4-carboxamide (10, NPD-935)
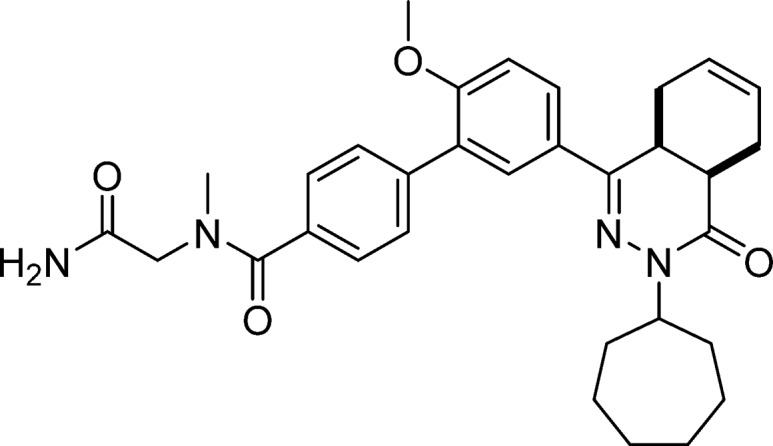
Prepared from 4 (0.20 mg, 0.42 mmol) and 2-(methylamino)acetamide·HCl (63 mg, 0.51 mmol) as described for 7. The title compound was obtained in 38% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.88 (dd, J = 8.8, 2.3 Hz, 1H), 7.79–7.70 (m, 1H), 7.62–7.38 (m, 5H), 7.26–7.08 (m, 2H), 5.73–5.56 (m, 2H), 4.67 (tt, J = 8.9, 4.7 Hz, 1H), 4.04 (s, 1H), 3.89–3.78 (m, 4H), 3.49 (dt, J = 11.4, 5.7 Hz, 1H), 3.01–2.91 (m, 3H), 2.81–2.68 (m, 2H), 2.21–2.07 (m, 2H), 1.96–1.65 (m, 7H), 1.60–1.40 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 171.3, 170.3, 166.0, 157.7, 154.0, 139.2, 135.5, 129.6, 129.5, 128.3, 128.0, 127.5, 127.4, 126.9, 126.3, 124.5, 112.5, 56.3, 55.4, 54.0, 39.0, 34.3, 34.3, 33.3, 33.0, 30.3, 28.6, 28.6, 25.0, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 4.77 min, area: >95%, m/z 543 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C32H39N4O4 543.2966, found 543.2969.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-N-(2-(methylamino)-2-oxoethyl)-[1,1′-biphenyl]-4-carboxamide (11, NPD-936)
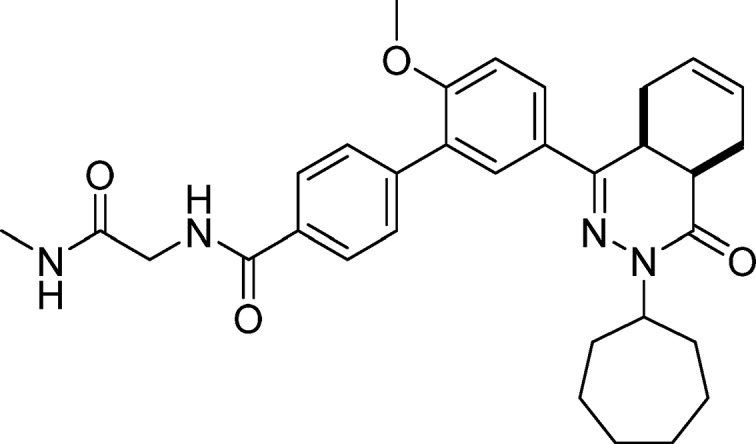
EDC·HCl (43 mg, 0.22 mmol) was added to a solution of 30 (0.10 g, 0.19 mmol), HOAt (26 mg, 0.19 mmol), methyl amine·HCl (28 mg, 0.42 mmol), and DIPEA (0.066 mL, 0.38 mmol) in CH2Cl2 (4 mL). The reaction mixture was stirred at RT for 36 h, CH2Cl2 (100 mL) was added, and the resulting solution was washed with water (2 × 50 mL) and brine (50 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The product was purified on a silica gel column eluting with EtOAc, to give 11 as a white solid in 29% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.86 (t, J = 6.0 Hz, 1H), 8.04–7.97 (m, 2H), 7.96–7.88 (m, 2H), 7.83 (d, J = 2.4 Hz, 1H), 7.70–7.63 (m, 2H), 7.28 (d, J = 8.8 Hz, 1H), 5.80–5.63 (m, 2H), 4.73 (td, J = 8.8, 4.5 Hz, 1H), 3.91 (d, J = 5.9 Hz, 2H), 3.89 (s, 3H), 3.55 (dt, J = 11.6, 5.7 Hz, 1H), 2.88–2.75 (m, 2H), 2.67 (d, J = 4.6 Hz, 3H), 2.27–2.13 (m, 2H), 2.02–1.72 (m, 7H), 1.67–1.45 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 169.8, 166.6, 166.0, 157.7, 153.9, 141.1, 133.1, 129.6, 129.6, 128.2, 128.0, 127.7, 127.6, 126.3, 124.5, 112.5, 56.3, 55.4, 43.2, 34.2, 33.3, 33.1, 30.3, 28.6, 28.6, 26.0, 24.9, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 4.86 min, area: >98%, m/z 543 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C32H39N4O4 543.2966, found 543.2963.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-N-(2-(dimethylamino)-2-oxoethyl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (12, NPD-939)
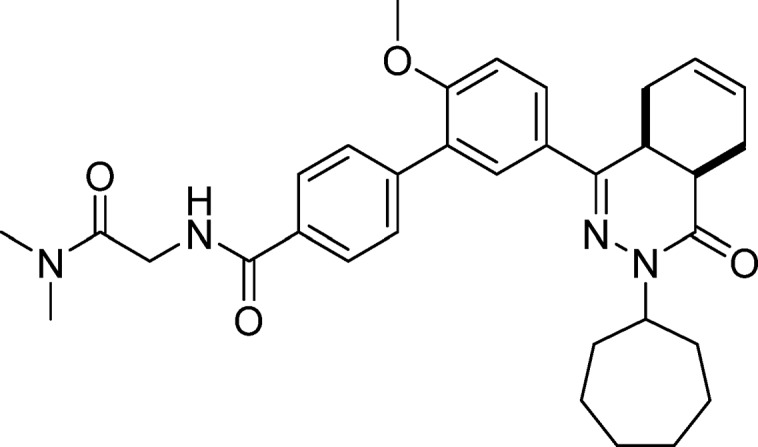
Prepared from 30 (25 mg, 0.048 mmol) and dimethylamine·HCl (8.5 mg, 0.11 mmol) as described for 11. The title compound was obtained in 54% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.64 (t, J = 5.7 Hz, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.95 (dd, J = 8.7, 2.3 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.71–7.63 (m, 2H), 7.29 (d, J = 8.8 Hz, 1H), 5.80–5.64 (m, 2H), 4.74 (tt, J = 8.6, 4.8 Hz, 1H), 4.19 (d, J = 5.6 Hz, 2H), 3.89 (s, 3H), 3.56 (dt, J = 11.5, 5.7 Hz, 1H), 3.09 (s, 3H), 2.92 (s, 3H), 2.88–2.76 (m, 2H), 2.27–2.12 (m, 2H), 2.03–1.73 (m, 7H), 1.67–1.45 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 168.7, 166.5, 166.0, 157.7, 154.0, 141.1, 133.2, 129.7, 129.6, 128.3, 128.0, 127.6, 127.5, 126.3, 124.5, 112.5, 56.3, 55.4, 41.4, 36.2, 35.6, 34.2, 33.3, 33.1, 30.3, 28.6, 28.6, 25.0, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 5.01 min, area: >95%, m/z 557 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H41N4O4 557.3122, found 557.3114.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-N-(2-(isopropylamino)-2-oxoethyl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (13, NPD-942)

Prepared from 30 (0.10 g, 0.19 mmol) and isopropyl amine (0.034 mL, 0.42 mmol) as described for 11, but excluding DIPEA. The title compound was obtained in 38% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.69 (t, J = 5.9 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.88 (dd, J = 8.7, 2.3 Hz, 1H), 7.82–7.73 (m, 2H), 7.65–7.54 (m, 2H), 7.22 (d, J = 8.8 Hz, 1H), 5.77–5.56 (m, 2H), 4.74–4.62 (m, 1H), 3.91–3.83 (m, 3H), 3.83 (s, 3H), 3.49 (dt, J = 11.6, 5.8 Hz, 1H), 2.81–2.69 (m, 2H), 2.20–2.08 (m, 2H), 1.95–1.65 (m, 7H), 1.61–1.36 (m, 6H), 1.07 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 168.2, 166.5, 166.0, 157.7, 154.0, 141.1, 133.2, 129.6, 129.6, 128.2, 128.0, 127.6, 127.6, 126.3, 124.5, 112.5, 56.3, 55.4, 43.0, 40.9, 33.3, 33.1, 30.3, 28.6, 28.6, 24.9, 24.9, 23.0, 22.9, 22.4. LC-MS (ESI): tR = 5.17 min, area: >98%, m/z 571 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C34H43N4O4 571.3279, found 571.3262.
N-(3-Amino-3-oxopropyl)-5′-(cis-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (14, NPD-800)

Prepared from 4 (80 mg, 0.17 mmol) and 3-aminopropanamide·HCl (42 mg, 0.34 mmol) as described for 7. The title compound was obtained in 64% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.55 (t, J = 5.6 Hz, 1H), 7.87 (dd, J = 8.7, 2.5 Hz, 3H), 7.76 (d, J = 2.3 Hz, 1H), 7.62–7.54 (m, 2H), 7.43–7.35 (m, 1H), 7.22 (d, J = 8.8 Hz, 1H), 6.86 (s, 1H), 5.73–5.58 (m, 2H), 4.73–4.62 (m, 1H), 3.82 (s, 3H), 3.54–3.41 (m, 3H), 2.81–2.68 (m, 2H), 2.36 (t, J = 7.2 Hz, 2H), 2.22–2.08 (m, 2H), 1.97–1.63 (m, 8H), 1.63–1.39 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 173.0, 166.4, 166.0, 157.7, 154.0, 140.9, 133.6, 129.6, 128.2, 128.0, 127.6, 127.4, 126.3, 124.5, 112.5, 56.3, 55.4, 36.5, 35.5, 34.2, 33.3, 33.1, 30.3, 28.7, 28.6, 28.6, 25.0, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 4.72 min, area: >95%, m/z 543 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C32H39N4O4 543.2966, found 543.2966.
N-(2-Acetamidoethyl)-5′-(cis-3-cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (15, NPD-801)

Prepared from 4 (80 mg, 0.17 mmol) and N-(2-aminoethyl)acetamide (35 mg, 0.34 mmol) as described for 7, but excluding DIPEA. The title compound was obtained in 78% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.55 (t, J = 5.6 Hz, 1H), 8.00 (t, J = 5.8 Hz, 1H), 7.91–7.84 (m, 3H), 7.76 (d, J = 2.3 Hz, 1H), 7.62–7.55 (m, 2H), 7.22 (d, J = 8.9 Hz, 1H), 5.74–5.56 (m, 2H), 4.67 (tt, J = 8.5, 4.9 Hz, 1H), 3.82 (s, 3H), 3.49 (dt, J = 11.4, 5.7 Hz, 1H), 3.31 (q, J = 6.1 Hz, 2H), 3.21 (q, J = 6.3 Hz, 2H), 2.81–2.68 (m, 2H), 2.21–2.07 (m, 2H), 1.93–1.66 (m, 11H), 1.60–1.40 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 170.0, 166.5, 166.0, 157.7, 154.0, 141.0, 133.6, 129.6, 129.6, 128.2, 128.0, 127.6, 127.4, 126.3, 124.5, 112.5, 56.3, 55.4, 39.7, 38.7, 34.2, 33.3, 33.1, 30.3, 28.6, 28.6, 25.0, 24.9, 23.1, 23.0, 22.4. LC-MS (ESI): tR = 4.84 min, area: >95%, m/z 557 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H41N4O4 557.3122, found 557.3128.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-N-(2-((2-hydroxyethyl)amino)-2-oxoethyl)-2′-methoxy-[1,1′-biphenyl]-4-carboxamide (16, NPD-937)

Prepared from 4 (200 mg, 0.423 mmol) and 2-amino-N-(2-hydroxyethyl)acetamide·HCl (65 mg, 0.42 mmol) as described for 8. The title compound was obtained in 54% yield. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.1 Hz, 2H), 7.75 (dd, J = 8.7, 2.3 Hz, 1H), 7.69 (d, J = 2.2 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.14 (t, J = 5.2 Hz, 1H), 6.97 (d, J = 8.7 Hz, 1H), 6.77 (t, J = 4.7 Hz, 1H), 5.75–5.56 (m, 2H), 4.79–4.68 (m, 1H), 4.14 (d, J = 4.9 Hz, 2H), 3.79 (s, 3H), 3.70 (t, J = 4.9 Hz, 2H), 3.43 (q, J = 5.2 Hz, 2H), 3.28–3.21 (m, 1H), 2.99–2.88 (m, 1H), 2.67 (t, J = 6.0 Hz, 1H), 2.19–2.05 (m, 2H), 2.04–1.73 (m, 8H), 1.66–1.36 (m, 7H). 13C NMR (126 MHz, CDCl3) δ 169.7, 167.8, 165.9, 157.6, 153.3, 142.1, 131.9, 129.9, 129.7, 128.2, 128.1, 127.1, 127.0, 126.1, 123.9, 111.3, 61.9, 56.2, 55.8, 43.8, 42.5, 34.7, 33.2, 33.0, 31.1, 28.43, 28.37, 25.1, 25.0, 23.1, 22.4. LC-MS (ESI): tR = 4.63 min, area: >98%, m/z 573 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H41N4O5 573.3071, found 573.3057.
5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-N-(2-((2-methoxyethyl)amino)-2-oxoethyl)-[1,1′-biphenyl]-4-carboxamide (17, NPD-878)

Prepared from 4 (0.15 g, 0.32 mmol) and 2-amino-N-(2-methoxyethyl)acetamide (50 mg, 0.38 mmol) as described for 7. The title compound was obtained in 32% yield. 1H NMR (500 MHz, DMSO-d6) δ 8.77 (t, J = 6.0 Hz, 1H), 8.00 (t, J = 5.7 Hz, 1H), 7.94 (d, J = 8.4 Hz, 2H), 7.88 (dd, J = 8.7, 2.3 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.65–7.55 (m, 2H), 7.22 (d, J = 8.8 Hz, 1H), 5.74–5.57 (m, 2H), 4.73–4.62 (m, 1H), 3.88 (d, J = 5.9 Hz, 2H), 3.83 (s, 3H), 3.49 (dt, J = 11.5, 5.7 Hz, 1H), 3.40–3.31 (m, 2H), 3.25 (d, J = 5.0 Hz, 5H), 2.82–2.68 (m, 2H), 2.22–2.07 (m, 2H), 1.97–1.64 (m, 7H), 1.61–1.37 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 166.2, 165.6, 157.3, 153.5, 140.7, 132.7, 129.2, 129.2, 127.8, 127.6, 127.2, 125.9, 124.1, 112.0, 70.6, 58.0, 55.9, 55.0, 42.6, 38.4, 33.8, 32.9, 32.6, 29.9, 28.2, 28.2, 24.5, 24.5, 22.6, 22.0. LC-MS (ESI): tR = 4.92 min, area: >98%, m/z 587 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C34H43N4O5 587.3228, found 587.3220.
cis-2-Cycloheptyl-4-(6-methoxy-4′-(4-oxoimidazolidine-1-carbonyl)-[1,1′-biphenyl]-3-yl)-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (18, NPD-060)
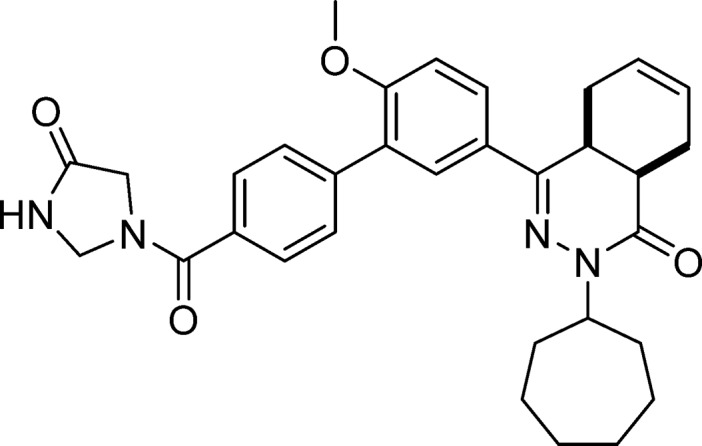
Prepared from 4 (0.20 g, 0.42 mmol) and imidazolidin-4-one·HCl (70 mg, 0.57 mmol) as described for 7. The title compound was obtained in 88% yield. Note: less accurate integrations due to rotamers/diastereomers. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 0.6H), 8.61 (s, 0.4H), 7.90 (dd, J = 8.7, 2.4 Hz, 1H), 7.77 (d, J = 2.3 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.61 (m, 3H), 7.25 (d, J = 8.8 Hz, 1H), 5.73–5.60 (m, 2H), 4.90 (m, 2H), 4.69 (tt, J = 9.0, 4.8 Hz, 1H), 4.08 (s, 0.6H), 3.98 (s, 0.4H), 3.85 (s, 3H), 3.50 (m, 1H), 2.82–2.70 (m, 2H), 2.20–2.10 (m, 2H), 1.97–1.89 (m, 1H), 1.88–1.68 (m, 6H), 1.62–1.41 (m, 6H). LC-MS (ESI): tR = 4.81 min, area: >95%, m/z 541 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C32H37N4O4 541.2809, found 541.2792.
cis-2-Cycloheptyl-4-(6-methoxy-4′-(3-methyl-4-oxoimidazolidine-1-carbonyl)-[1,1′-biphenyl]-3-yl)-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (19, NPD-062)
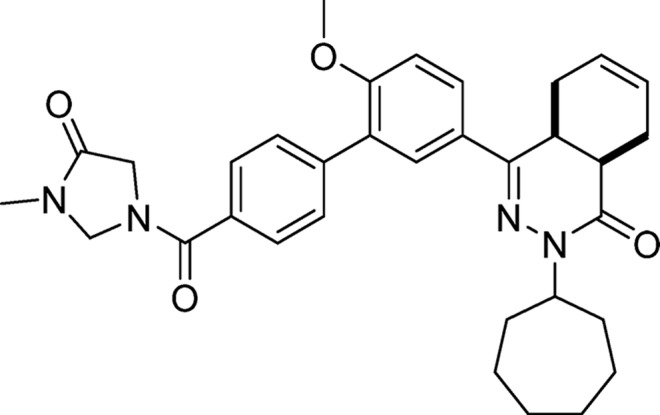
To an ice-cooled solution of 18 (0.10 g, 0.19 mmol) in DMF (4 mL), sodium hydride (60% dispersion in mineral oil, 9.3 mg, 0.21 mmol) was added. The temperature of the mixture was allowed to increase to RT; after 30 min iodomethane (27 mg, 0.19 mmol, 12 μL) was added. This mixture was stirred for 1.5 h, diluted with water (25 mL) and extracted with CH2Cl2 (3 × 45 mL). The combined organic phases were washed with brine (15 mL), dried over Na2SO4, filtered, concentrated, and purified on a silica gel column eluting with CH2Cl2/MeOH (gradient, 100:0 to 96:4). The product was freeze-dried to obtain 19 in 54% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.89 (dd, J = 8.7, 2.2 Hz, 1H), 7.76 (t, J = 2.8 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.65–7.57 (m, 3H), 7.24 (d, J = 8.7 Hz, 1H), 5.73–5.57 (m, 2H), 4.99–4.90 (m, 2H), 4.68 (tt, J = 8.8, 4.8 Hz, 1H), 4.17 (s, 1H), 4.07 (s, 1H), 3.84 (s, 3H), 3.49 (dt, J = 11.6, 5.8 Hz, 1H), 2.86 (s, 2H), 2.81–2.69 (m, 3H), 2.21–2.07 (m, 2H), 1.97–1.65 (m, 7H), 1.62–1.38 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 168.1, 167.4, 166.0, 157.7, 153.9, 140.4, 133.8, 129.8, 129.7, 129.3, 128.3, 128.0, 127.7, 127.6, 127.4, 126.3, 124.4, 112.5, 64.4, 56.3, 55.4, 50.5, 34.2, 33.2, 33.0, 30.3, 28.6, 28.5, 27.4, 24.9, 24.8, 23.0, 22.4. LC-MS (ESI): tR = 3.85 min, area: >95%, m/z 555 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H39N4O4 555.2966, found 555.2957.
cis-2-Cycloheptyl-4-(6-methoxy-4′-(3-oxopyrrolidine-1-carbonyl)-[1,1′-biphenyl]-3-yl)-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (20, NPD-887)
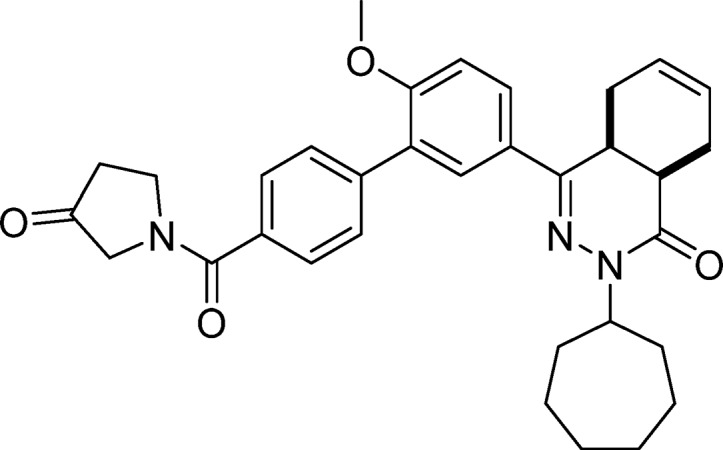
To a solution of 21 (0.58 g, 1.1 mmol) in CH2Cl2 (3 mL), Dess–Martin periodinane (0.11 g, 2.7 mmol) was added, and the reaction mixture was stirred at RT for 4 h. Water (0.019 mL, 1.1 mmol) was added, and the mixture was stirred for an additional 30 min. A mixture of aqueous 10% sodium metabisulfite and saturated aqueous sodium bicarbonate (1:1 ratio, 2 mL total volume) was added, the resulting mixture was extracted with CH2Cl2 (20 mL) and washed with sodium bicarbonate (2 × 10 mL). The organic phase was concentrated, and the product was purified on a silica gel column eluting with EtOAc/heptane (gradient, 6:4 to 9:1), to afford 20 as a white solid in 58% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.95 (dd, J = 8.7, 2.4 Hz, 1H), 7.82 (d, J = 2.3 Hz, 1H), 7.71–7.59 (m, 4H), 7.30 (d, J = 8.8 Hz, 1H), 5.81–5.60 (m, 2H), 4.74 (tt, J = 8.9, 4.8 Hz, 1H), 4.09–3.94 (m, 4H), 3.90 (s, 3H), 3.56 (dt, J = 11.5, 5.8 Hz, 1H), 2.87–2.75 (m, 2H), 2.67 (t, J = 7.8 Hz, 2H), 2.26–2.15 (m, 2H), 2.02–1.73 (m, 7H), 1.66–1.47 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 166.0, 157.7, 154.0, 139.9, 129.7, 129.5, 128.3, 128.0, 127.5, 126.3, 124.5, 112.5, 56.3, 55.6, 55.4, 52.7, 46.4, 42.8, 37.9, 36.1, 34.2, 33.3, 33.1, 30.3, 28.6, 28.6, 25.0, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 5.19 min, area: >95%, m/z 540 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H38N3O4 540.2857, found 540.2880.
cis-2-Cycloheptyl-4-(4′-((R)-3-hydroxy-pyrrolidine-1-carbonyl)-6-methoxy-[1,1′-biphenyl]-3-yl)-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (21, NPD-746)
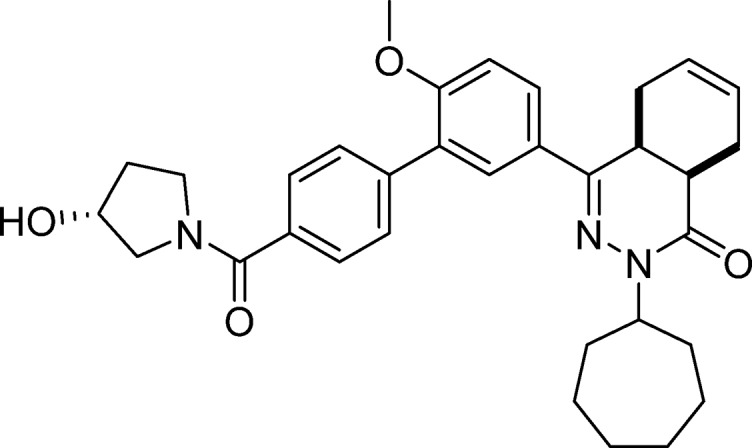
Prepared from 4 (0.83 g, 1.8 mmol) and (R)-pyrrolidin-3-ol (0.18 g, 2.1 mmol) as described for 7, but excluding DIPEA. The title compound was obtained in 75% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.89 (dd, J = 8.7, 2.3 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.62–7.55 (m, 4H), 7.24 (d, J = 8.8 Hz, 1H), 5.75–5.57 (m, 2H), 5.05 (d, J = 3.5 Hz, 0H), 4.96 (d, J = 3.1 Hz, 1H), 4.74–4.63 (m, 1H), 4.35 (s, 1H), 4.26 (s, 1H), 3.84 (s, 3H), 3.68–3.45 (m, 4H), 3.42–3.38 (m, 1H), 3.30–3.25 (m, 1H), 2.83–2.70 (m, 2H), 2.21–2.09 (m, 2H), 1.98–1.65 (m, 9H), 1.63–1.39 (m, 6H). 13C NMR (126 MHz, DMSO) δ 168.7, 166.0, 157.7, 154.0, 139.6, 136.1, 129.6, 129.5, 128.3, 128.0, 127.5, 127.4, 126.4, 124.5, 112.5, 69.8, 68.5, 57.6, 56.3, 55.4, 54.9, 47.4, 44.6, 34.9, 34.3, 33.3, 33.1, 32.6, 30.3, 28.6, 28.6, 25.0, 24.9, 23.0, 22.4. LC-MS (ESI): tR = 4.91 min, area: >98%, m/z 542 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H40N3O4 542.3013, found 542.3013.
cis-4-(4′-((R)-3-Hydroxypyrrolidine-1-carbonyl)-6-methoxy-[1,1′-biphenyl]-3-yl)-2-isopropyl-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (22, NPD-038)
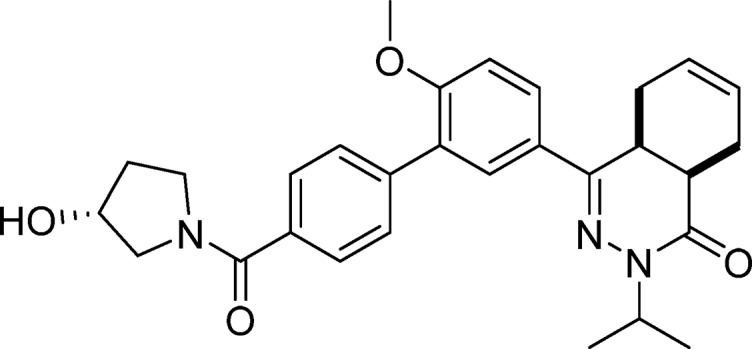
Prepared from 28 (300 mg, 0.717 mmol) and (R)-pyrrolidin-3-ol (62 mg, 0.72 mmol) analogous to the method described for 8 but excluding Et3N. The title compound was isolated in 64% yield. 1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 8.7, 2.3 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.67–7.53 (m, 4H), 7.02 (d, J = 8.7 Hz, 1H), 5.82–5.64 (m, 2H), 5.04 (hept, J = 6.6 Hz, 1H), 4.69–4.41 (m, 1H), 3.86 (s, 3H), 3.88–3.47 (m, 5H), 3.39–3.26 (m, 1H), 3.06–2.93 (m, 1H), 2.75 (t, J = 6.0 Hz, 1H), 2.29–1.96 (m, 5H), 1.32 (d, J = 6.6 Hz, 3H), 1.20 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.9, 166.4, 157.7, 153.5, 139.9, 135.3, 130.1, 129.4, 128.3, 128.0, 127.1, 126.8, 126.0, 124.0, 111.2, 71.0, 69.8, 57.4, 55.8, 55.0, 47.4, 46.7, 34.8, 31.1, 23.2, 22.4, 20.6, 20.2. LC-MS (ESI): tR = 4.31 min, area: >98%, m/z 488 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C29H34N3O4 488.2544, found 488.2534.
(R)-1-(5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carbonyl)pyrrolidine-2-carboxamide (23, NPD-802)
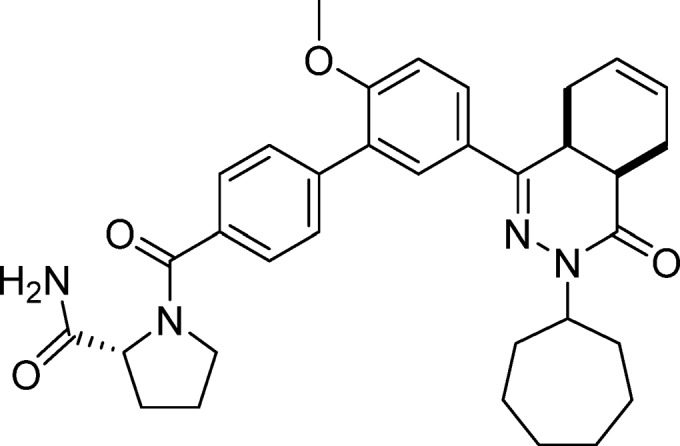
Prepared from 4 (80 mg, 0.17 mmol) and (R)-pyrrolidine-2-carboxamide (38 mg, 0.34 mmol) as described for 7, but excluding DIPEA. The title compound was obtained in 78% yield. Note: less accurate integrations due to rotamers/diastereomers. 1H NMR (500 MHz, DMSO-d6) δ 7.88 (dd, J = 8.8, 2.3 Hz, 1H), 7.76 (d, J = 2.3 Hz, 0.7H), 7.72 (d, J = 2.3 Hz, 0.3H), 7.64 (d, J = 8.1 Hz, 1.7H), 7.57 (d, J = 8.1 Hz, 1.7H), 7.53 (d, J = 7.9 Hz, 0.6H), 7.46–7.39 (m, 1.6H), 7.23 (d, J = 8.7 Hz, 1H), 7.05 (d, J = 3.2 Hz, 0.3H), 6.98 (s, 0.8H), 5.74–5.56 (m, 2H), 4.75–4.63 (m, 1H), 4.38 (dd, J = 8.2, 5.6 Hz, 0.8H), 4.25 (d, J = 8.0 Hz, 0.3H), 3.83 (s, 3H), 3.70–3.54 (m, 1.3H), 3.54–3.44 (m, 1.8H), 2.83–2.68 (m, 2H), 2.27–2.07 (m, 3H), 1.94–1.66 (m, 10H), 1.62–1.37 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 173.7, 168.3, 165.6, 157.3, 153.6, 139.3, 135.4, 129.2, 129.0, 127.9, 127.6, 127.2, 127.1, 126.5, 125.9, 124.1, 112.1, 60.2, 55.9, 55.0, 49.9, 33.8, 32.9, 32.6, 29.9, 29.7, 28.2, 28.2, 25.1, 24.6, 24.5, 22.6, 22.0. LC-MS (ESI): tR = 4.89 min, area: >96%, m/z 569 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C34H41N4O4 569.3122, found 569.3143.
(R)-1-(5′-(cis-3-Isopropyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carbonyl)pyrrolidine-2-carboxamide (24, NPD-885)
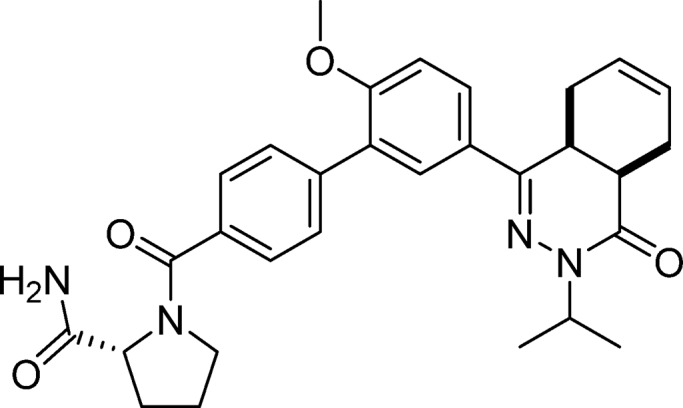
Prepared from 28 (0.10 g, 0.21 mmol) and (R)-pyrrolidine-2-carboxamide (43 mg, 0.38 mmol) as described for 7, but excluding DIPEA. The title compound was obtained in 56% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.83 (dd, J = 8.8, 2.4 Hz, 1H), 7.72 (d, J = 2.3 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 13.1 Hz, 1H), 7.15 (dd, J = 8.9, 2.6 Hz, 1H), 6.92 (s, 1H), 5.67–5.51 (m, 2H), 4.81 (hept, J = 6.7 Hz, 1H), 4.32 (dd, J = 8.3, 5.6 Hz, 1H), 3.77 (d, J = 3.1 Hz, 3H), 3.61–3.49 (m, 1H), 3.43 (dt, J = 11.1, 5.7 Hz, 2H), 2.76–2.63 (m, 2H), 2.18–2.03 (m, 3H), 1.89–1.65 (m, 4H), 1.17 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 174.2, 168.7, 166.5, 157.8, 154.0, 139.7, 135.8, 129.7, 129.4, 128.2, 128.0, 127.7, 127.6, 126.9, 126.3, 124.5, 112.4, 60.6, 56.3, 50.4, 46.2, 34.3, 30.4, 30.2, 25.5, 23.0, 22.4, 20.9, 20.6. LC-MS (ESI): tR = 4.22 min, area: >95%, m/z 515 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C30H35N4O4 515.2653, found 515.2652.
cis-4-(3-Bromo-4-methoxyphenyl)-2-isopropyl-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (26)
To a solution of 25 (50.0 g, 149 mmol) in DMF (500 mL), NaH (60% dispersion in mineral oil, 14.9 g, 373 mmol) was added at RT. The resulting mixture was stirred for 30 min, and 2-bromopropane (21.0 mL, 224 mmol) was added. After stirring the reaction mixture for 4 h, 0.5 M aqueous HCl (1.5 L) was added, and the resulting suspension was filtered. The residue was dissolved in a mixture of MeOH and CH2Cl2, dried over Na2SO4, filtered, and concentrated under reduced pressure. The light brown solid was triturated with Et2O to give 26 as a white solid in 80% yield. 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 2.1 Hz, 1H), 7.74 (dd, J = 8.7, 2.2 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 5.85–5.62 (m, 2H), 5.05 (hept, J = 6.7 Hz, 1H), 3.95 (s, 3H), 3.32–3.21 (m, 1H), 3.07–2.95 (m, 1H), 2.74 (t, J = 6.0 Hz, 1H), 2.28–2.11 (m, 2H), 2.09–1.95 (m, 1H), 1.33 (d, J = 6.6 Hz, 3H), 1.21 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.4, 156.8, 152.3, 130.8, 129.1, 126.2, 126.0, 123.8, 112.2, 111.5, 56.4, 46.8, 34.7, 31.0, 23.0, 22.3, 20.6, 20.2. LC-MS (ESI) m/z 377/379 [M + H]+.
cis-4-(3-Bromo-4-methoxyphenyl)-2-cycloheptyl-4a,5,8,8a-tetrahydrophthalazin-1(2H)-one (27)
Prepared from building block 25 and bromocycloheptane as described for 26. The title compound was prepared in 65% yield. 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 2.2 Hz, 1H), 7.71 (dd, J = 8.7, 2.2 Hz, 1H), 6.91 (d, J = 8.6 Hz, 1H), 5.83–5.55 (m, 2H), 4.85–4.71 (m, 1H), 3.92 (s, 3H), 3.30–3.16 (m, 1H), 3.06–2.89 (m, 1H), 2.69 (t, J = 6.0 Hz, 1H), 2.27–2.06 (m, 2H), 2.06–1.92 (m, 2H), 1.92–1.81 (m, 1H), 1.81–1.67 (m, 4H), 1.67–1.38 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 165.8, 156.8, 152.2, 130.7, 129.1, 126.2, 126.0, 123.8, 112.1, 111.6, 56.4, 56.3, 34.6, 33.2, 33.0, 31.0, 28.34, 28.27, 25.1, 25.0, 23.0, 22.3. LC-MS (ESI) m/z 431/433 [M + H]+.
5′-(cis-3-Isopropyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carboxylic Acid (28)
To a degassed solution of 26 (1.00 g, 2.65 mmol) in DME (15 mL), 4-boronobenzoic acid (0.66 g, 4.0 mmol), Pd(dppf)Cl2·CH2Cl2 (0.13 g, 0.16 mmol), and 2 M aqueous Na2CO3 (5.2 mL) was added. The mixture was stirred at 100 °C for 16 h, then diluted with saturated aqueous NH4Cl solution (50 mL) and water (250 mL) and extracted with CH2Cl2 (150 mL). The organic phase was washed with water (150 mL) and brine (100 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified on a silica gel column eluting with CH2Cl2/MeOH (gradient, 99:1 to 97:3) to afford 28 in 55% yield. 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 8.2 Hz, 2H), 7.85 (dd, J = 8.7, 2.3 Hz, 1H), 7.80 (d, J = 2.2 Hz, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.05 (d, J = 8.6 Hz, 1H), 5.86–5.63 (m, 2H), 5.06 (hept, J = 6.6 Hz, 1H), 3.89 (s, 3H), 3.40–3.29 (m, 1H), 3.10–2.96 (m, 1H), 2.77 (t, J = 6.0 Hz, 1H), 2.28–2.14 (m, 2H), 2.12–1.99 (m, 1H), 1.33 (d, J = 6.6 Hz, 3H), 1.21 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 171.4, 166.5, 157.7, 153.5, 143.7, 130.1, 129.8, 129.7, 128.3, 128.1, 128.0, 127.2, 126.1, 124.0, 111.3, 55.9, 46.8, 34.8, 31.1, 23.2, 22.4, 20.6, 20.2. LC-MS (ESI) m/z 419 [M + H]+.
Methyl (5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carbonyl)glycinate (29)
Prepared from 4 (1.0 g, 2.1 mmol) and glycine methyl ester·HCl (0.32 g, 2.5 mmol) as described for 7. The title compound was obtained in 58% yield. 1H NMR (300 MHz, DMSO-d6) δ 9.00 (t, J = 8.2 Hz, 1H), 7.94 (d, J = 8.5, 2H), 7.90 (dd, J = 8.8, J = 2.3 Hz, 1H) 7.78 (d, J = 2.3 Hz, 1H), 7.63 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.8 Hz, 1H), 5.71–5.60 (m, 2H), 4.73–4.64 (m, 1H), 4.05 (d, J = 5.8 Hz, 2H), 3.83 (s, 3H), 3.67 (s, 3H), 3.51–3.46 (m, 1H), 2.81–2.72 (m, 2H), 2.20–2.11 (m, 2H), 2.00–1.36 (m, 13H).
(5′-(cis-3-Cycloheptyl-4-oxo-3,4,4a,5,8,8a-hexahydrophthalazin-1-yl)-2′-methoxy-[1,1′-biphenyl]-4-carbonyl)glycine (30)
Aqueous sodium hydroxide (6.2 mL, 25 mmol) was added to a suspension of 29 (0.84 g, 1.5 mmol) in EtOH (25 mL) and stirred at RT for 2 h. The reaction mixture was acidified with HCl (2 M) and extracted with EtOAc (2 × 30 mL). The combined organic phases were washed with water, dried over Na2SO4, filtered, and concentrated to give the title compound as a brown solid in 89% yield. 1H NMR (300 MHz, DMSO-d6) δ 8.87 (t, J = 5.8 Hz, 1H), 7.95 (d, J = 8.5, 2H), 7.91 (dd, J = 8.9, J = 2.2 Hz, 1H) 7.79 (d, J = 2.3 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.26 (d, J = 8.8 Hz, 1H), 5.73–5.61 (m, 2H), 4.74–4.65 (m, 1H), 3.97 (d, J = 5.8 Hz, 2H), 3.85 (s, 3H), 3.55–3.47 (m, 1H), 2.81–2.72 (m, 2H), 2.20–2.11 (m, 2H), 2.00–1.36 (m, 13H). LC-MS (ESI): tR = 4.93 min, m/z 530 [M + H]+
Interference Compounds
All final compounds have been examined for the presence of substructures classified as Pan Assay Interference Compounds (PAINS) using a KNIME workflow.27
Phosphodiesterase Activity Assay
To determine the effect of test compounds on the enzymatic activity of full length TbrPDEB1 (Km = 7.97 ± 2.32 μM) and full length recombinant hPDE4B1 (Km = 2.0 ± 0.7 μM), the standard scintillation proximity assay (SPA) was used, as reported previously.10,12 In this assay, the cAMP substrate concentration was 0.5 μM, and the enzyme concentration was adjusted so that <20% of substrate was consumed. The Ki values are represented as the mean of at least two independent experiments with the associated standard deviation (SD) as indicated.
Gene Constructs for Structural Studies
TbrPDEB1 Catalytic Domain
A gene segment coding for TbrPDEB1 catalytic domain residues 565–918 (Uniprot entry Q8WQX9) was PCR amplified with flanking NdeI and EcoRI restriction sites and cloned into a pET28a(+) expression vector (Novagen) previously digested with the same set of restriction enzymes. An N-terminal, thrombin cleavable 6×His tag was kept in-frame with the gene to facilitate subsequent purification of the expressed protein by metal affinity chromatography. The resultant recombinant vector was named pET28a(+)-TbrPDEB1_CD.
hPDE4B Regulatory Domain (UCR2 + Catalytic Domain)
Coding sequence for hPDE4B UCR2 and catalytic domain residues 241–659 (Uniprot entry Q07343) was synthesized and cloned into a pMA vector (GeneArt, Invitrogen, Life Technologies). This vector was then used as a PCR template for subcloning of the gene segment into a pFastBacHTA insect cell expression vector (Invitrogen, Life Technologies) with a C-terminal 6×His purification tag.
hPDE4D2 Catalytic Domain
A gene segment coding for residues 381–740 of hPDE4D2 (Uniprot entry Q08499) was PCR amplified using a forward primer including an NdeI restriction site and a reverse primer including a XhoI restriction site. The PCR product was cloned into a pET15b E. coli expression vector (Novagen) previously digested with the same set of enzymes. The primer design was such to keep an N-terminal 6×His tag from the vector in frame with the target gene. The resultant recombinant vector was named pET15b-hPDE4D_CD.
Protein Expression and Purification for Structural Studies
TbrPDEB1 Catalytic Domain
Escherichia coli BL21 (DE3) cells were transformed with pET28a(+)-TbrPDEB1_CD and allowed to grow in 2 L of 2×YT medium at 37 °C until the optical density at 600 nm reached 0.6–0.8. At this stage, the culture was cooled down, induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and further grown overnight at 16 °C. Cells were collected by centrifugation, resuspended in a buffer containing 20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 10 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol (BME), and protease inhibitor cocktail tablet (Roche), and lysed by passing through a cell disruptor (20 kpsi/pass). Cleared cell lysate was loaded onto a 5 mL HisTrap HP nickel affinity column (GE Healthcare Biosciences) and bound protein was eluted with a linear gradient of 0–1 M imidazole. Fractions containing the target protein, as assessed by SDS gel electrophoresis, were pooled and desalted to remove imidazole. Removal of N-terminal 6×His tag was performed by overnight incubation at 4 °C of the sample with human thrombin (Abcam) at 5 NIH units/mL of the pooled sample followed by a second nickel affinity purification step to remove any remaining tagged fraction. The protein was then dialyzed against ion exchange buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5% glycerol, 2 mM BME), loaded onto a HiTrap Q HP column (GE Healthcare Biosciences), and eluted with a linear gradient of 0–1 M NaCl. A final size exclusion chromatography step was performed on the collected sample using a Superdex 200 increase 10/300 GL column (GE Healthcare Biosciences) pre-equilibrated with 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5% glycerol, and 2 mM BME after which the protein was concentrated using Amicon Ultra concentrators (Millipore) to 7 mg/mL and stored at −80 °C prior to use. For crystallization trials, NaCl was removed from the buffer while keeping other components intact.
hPDE4B Regulatory Domain (UCR2 + Catalytic Domain)
hPDE4B regulatory domain expression and purification was performed according to the method described.28 Recombinant baculovirus generation, insect cell (Sf21) culture, and infection were carried out following manufacturer’s instructions (Bac-to-Bac expression system; Invitrogen, Life technologies). Infected cells were grown for 48 h at 28 °C and harvested by centrifugation at 500g for 15 min. Lysis was performed by resuspending the cells in a hypotonic buffer of 10 mM HEPES, pH 7.5, 50 mM NaCl, and 1 mM tris(2-carboxyethyl) phosphine (TCEP) followed by centrifugation to remove cell debris. The obtained cleared lysate was loaded onto a 5 mL HisTrap HP nickel affinity column (GE Healthcare Biosciences) pre-equilibrated with 100 mM HEPES, pH 7.5, 150 mM NaCl, 50 mM arginine, 10 mM imidazole, and 1 mM TCEP and eluted with a linear gradient of 0–1 M imidazole. Eluted protein was then dialyzed against ion-exchange buffer (100 mM HEPES, 50 mM NaCl, 1 mM dithiothreitol (DTT)) and loaded onto a HiTrap Q HP ion exchange column (GE Healthcare Biosciences). A linear gradient elution of 20–250 mM NaCl was performed followed by dialysis of the collected sample into size exclusion buffer (10 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM DTT). Protein was then passed through a Superdex 200 increase 10/300 GL size exclusion column (GE Healthcare Biosciences). The collected sample was concentrated to 10 mg/mL and stored at −80 °C prior to use in crystallization.
hPDE4D2 Catalytic Domain
BL21 (DE3) Codon Plus cells were transformed with pET15b-hPDE4D_CD and allowed to grow in 1 L of 2×YT medium at 37 °C until the optical density at 600 nm reached 0.6–0.8. At this stage, culture temperature was lowered, and expression was induced by addition of 0.5 mM IPTG followed by further overnight growth at 22 °C. The same cell disruption and protein purification procedures were followed as performed in the case of TbrPDEB1 catalytic domain protein with the following changes in the buffers used: cell resuspension and nickel affinity chromatography buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 5 mM BME, 20 mM imidazole), ion exchange chromatography buffer (50 mM Tris-HCl, pH 8, 50 mM NaCl, 5 mM DTT), and size exclusion chromatography buffer (50 mM Bis-tris, pH 6.8, 100 mM NaCl, 5 mM DTT). Purified protein was concentrated to 9 mg/mL and stored at −80 °C prior to use in crystallization.
Protein Crystallization, Ligand Soaking and Data Collection
All crystallization trials were performed by vapor diffusion hanging drop technique, typically with 500 μL reservoir volume and 2 μL droplets with a protein to crystallization solution ratio of 1:1. Crystals of the TbrPDEB1 catalytic domain were grown in 20% PEG 3350, 400 mM sodium formate, 300 mM guanidine, and 100 mM MES, pH 6.5, at 4 °C. Soaking of these crystals with various compounds was performed maintaining a final compound concentration of 5–15 mM and for varying duration of overnight to 48 h. Soaked crystals were then briefly dipped in growth solution supplemented with 20% (v/v) glycerol or ethylene glycol and were mounted using CryoLoop (Hampton Research) or LithoLoops (Molecular Dimensions) and vitrified in liquid nitrogen for data collection. In the case of hPDE4D2, thick plate-like crystals appeared within 5–6 days in a condition containing 24% PEG 3350, 30% ethylene glycol, and 100 mM HEPES, pH 7.5, at 19 °C. For hPDE4B, a condition containing 20% PEG 400, 50 mM calcium acetate, and 100 mM sodium acetate, pH 4.6, resulted in well-diffracting crystals. Compound soaking and crystal harvesting were performed in the same way as for TbrPDEB1 crystals except that in case of hPDE4D2 no cryopreservative was added prior to vitrification. X-ray diffraction data sets were collected at Diamond Light Source (DLS; Didcot, Oxfordshire, UK) beamlines I03 and I04 at 100 K. The data sets were processed by xia229 or autoPROC,30 which incorporates XDS31 and AIMLESS,32 or were integrated using iMOSFLM33 and reduced using POINTLESS, SCALA, and TRUNCATE,34 all of which are part of CCP4.35
X-ray Crystal Structure Determination, Refinement, and Analysis
The crystal structure of TbrPDEB1 bound to NPD-008 (8) was solved by molecular replacement (MR) using PHASER,36 taking the apo structure (PDB code 4I15) as search model. Reflections for calculating Rfree were selected randomly, and the same set was used in all other ligand-bound TbrPDEB1 data sets except for NPD-038 (22) where the crystal was nonisomorphous. All isomorphous crystal forms were solved by Fourier synthesis using the partially refined, ligand-free 8 model, whereas MR was applied for the 22 data set. hPDE4B and hPDE4D2 structures with NPD-001 (1) were solved by MR using their respective apo models (PDB codes 3G45 and 3SL3, respectively). Ligand descriptions were generated by ACEDRG available within the CCP4 package35 or with the grade Web Server (http://grade.globalphasing.org/). Adjustment of the models and ligand fitting were performed with COOT37 and refinement with REFMAC5.38 The final structures had good geometry and could be refined to low R-factors (Tables S1 and S3). All refined models were validated with MOLPROBITY.39 Data collection and refinement statistics are given in Supplementary Tables S1–S4. Root-mean-square (rms) deviation values were calculated from a sequence alignment, structural superposition, and refinement cycle on Cα carbons with the align function as implemented in PyMOL 1.7.4.4 (The PyMOL Molecular Graphics System, Schrödinger, LLC). All binding site residues have been named according to the PDEStrIAn nomenclature system (http://pdestrian.vu-compmedchem.nl/).21 Structural figures were prepared with PyMOL 1.7.4.4. For clarity, selected residues from the helix capping the substrate binding pocket (i.e., D784, M785, A786, K787, H788, G789, S790, A791, L792, and E793 in TbrPDEB1; D518, M519, S520, K521, H522, M523, S524, and L525 in hPDE4B; D272, M273, S274, K275, H276, M277, N278, and L279 in hPDE4D) have been omitted in the rendering of the figures. Coordinates of the structures have been deposited to the RCSB Protein Data Bank with following accession codes: 5G57 (TbrPDEB1–NPD-001); 5LAQ (hPDE4B–NPD-001); 5LBO (hPDE4D–NPD-001); 5L9H (TbrPDEB1–NPD-340); 5G2B (TbrPDEB1–NPD-008); 5L8C (TbrPDEB1–NPD-039); 5G5V (TbrPDEB1–NPD-038); 5L8Y (TbrPDEB1–NPD-937).
Molecular Dynamics
Molecular dynamics simulations of the TbrPDEB1–NPD-008 (8) crystal structure (PDB code 5G2B) were performed with GROMACS 5.1,40,41 using the Amber ff99SB-ILDN force field42 and a TIP3P water model,43 running with MPI parallelization. Ligand input files, based on 8 extracted from chain A, were prepared with Antechamber as implemented in AmberTools14 (AMBER 14, University of California, San Francisco, 2014) by the calculation of AM1-BCC partial charges, and generation of a GAFF topology. Subsequent preparation of the parameters and ligand topology was performed with LEaP. ACPYPE44 was used to convert the ligand files to GROMACS input format. The protein–ligand complex of 8 and chain A from TbrPDEB1, together with the Mg2+ and Zn2+ ions and the crystal water network directly coordinating the binding site cations consisting of six crystal waters, was centered at least 1.0 nm from the edges of a cubic box, that was fully solvated and charge neutralized by the addition of Na+ ions. The system was energy minimized with a steepest descent algorithm for 5000 steps with an initial step size of 0.01 nm. Bonds were constrained with the P-LINCS algorithm,45 an integration time step of 2 fs was used, and the smooth particle mesh Ewald (PME) method was used for the calculation of long-range electrostatics. The Verlet cutoff scheme was used for neighbor searching; cutoff distances of 1.0 nm were set for the short-range neighbor list as well as for Coulomb and van der Waals interactions. Initial equilibration was performed in three 250 ps steps by gradually increasing the temperature from 100 to 298 K while decreasing the position restraint force constant from 1000 kJ mol–1 nm–1 to 500 kJ mol–1 nm–1 and finally to 50 kJ mol–1 nm–1. In the canonical (NVT) ensemble, a velocity rescaling thermostat was used. The Parrinello–Rahman barostat and a position restraint force constant of 25 kJ mol–1 nm–1 was used in the isothermal–isobaric (NPT) ensemble equilibration for 250 ps at 298 K and 1.0 bar. A final unrestrained equilibration step was executed for 500 ps at 298 K and 1.0 bar, followed by a production run of 100 ns. The first 10 ns of the simulation was omitted from analysis. The stability of the system and the results of the simulations were validated and analyzed with gmx rms, energy, gyrate, rmsf, distance, and hbond tools as implemented in GROMACS 5.1.
Parasite Culturing for cAMP Measurements and Microscopy
Bloodstream forms of T. brucei Lister 427 were cultured in Hirumi-9 (HMI-9) medium (Invitrogen), supplemented with 10% heat inactivated fetal bovine serum (FBS; Gibco) in vented culture flasks (Corning), at 37 °C, in a 5% CO2 atmosphere, as described previously.46
Intracellular cAMP Measurements
Intracellular cAMP was measured as described previously,10 with minor changes. Briefly, log-phase bloodstream form trypanosomes were inoculated into HMI-9/FBS media and incubated at 37 °C overnight. The cells were counted and suspended in HMI-9/FBS at 2 × 106 cells/mL, which was divided into 8 subcultures of 6 mL each. To each culture flask, a small volume of HMI-9 medium containing either NPD-001 (1) (positive control; final concentration 0.3 μM), pentamidine (a known trypanocide not acting on cAMP signaling; final concentration 0.025 μM), NPD-008 (8) (at 0.1, 0.33, 1, 3.3, and 10 μM), or no drug (negative control) was added. The cultures were incubated under standard conditions (37 °C, 5% CO2) for 5 h, after which cell densities were determined in each culture using a hemocytometer; 5 × 106 cells were transferred into new tubes and collected by centrifugation at 1500g for 10 min at 4 °C. Cell pellets were resuspended in 100 μL of 0.1 M HCl and left on ice for 20 min to complete cell lysis. The samples were centrifuged in a microfuge at 12 000g for 10 min at 4 °C, and the supernatants (cell extracts) were stored at −80 °C for the determination of intracellular cAMP. Each cAMP determination was performed in duplicate, data are represented as the mean of four independent cAMP determinations with the standard error of the mean (SEM) and were analyzed with Student’s t-test, differences were considered significant at P < 0.05, with P values as indicated.
Microscopy and DAPI Staining
For the monitoring of the cell cycle (division of nucleus, kinetoplast, and cells) and cellular morphology, trypanosomes were stained with 4′,6-diamidino-2-phenylindole (DAPI) and observed by fluorescence microscopy as described.47 Briefly, a culture of T. brucei bloodstream forms was inoculated at 2 × 105 cell/mL in the presence of 10 μM NPD-008 (8). Samples for microscopy were taken at 0, 6, 12, and 24 h and centrifuged at 2600 rpm for 10 min at 4 °C in a Heraeus Biofuge centrifuge. Supernatant was decanted, and the pellet was washed with phosphate-buffered saline (PBS), pH 7.4, resuspended in 20 μL of PBS and spread on a microscope slide. The slides were air-dried and then fixed in 4% formaldehyde/PBS for 15 min. Slides were rinsed 3 times with PBS after which the samples were mounted in VectaShield (Vector Laboratories Inc., USA) mounting medium with DAPI. Samples were imaged on an Axioskop II microscope (Zeiss, Inc.) and a DeltaVision Core (AppliedPrecision).
Phenotypic Cellular Assays
For the cellular assays, the following reference drugs were used as positive controls: suramin (Sigma-Aldrich, Germany) for T. brucei (pIC50 = 7.4 ± 0.2, n = 5), and tamoxifen (Sigma-Aldrich, Germany) for MRC-5 cells (pIC50 = 5.0 ± 0.1, n = 5). All compounds were tested at five concentrations (64, 16, 4, 1, and 0.25 μM) to establish a full dose-titration and determination of the IC50 and CC50; data are represented as the mean of triplicate experiments ± SD. The final concentration of DMSO did not exceed 0.5% in the assays.
Antitrypanosomal Cellular Assay with T. brucei
Squib-427 strain (suramin-sensitive) was cultured at 37 °C and 5% CO2 in HMI-9 medium, supplemented with 10% fetal calf serum (FCS). About 1.5 × 104 trypomastigotes were added to each well, and parasite growth was assessed after 72 h at 37 °C by adding resazurin. The color reaction was read at 540 nm after 4 h, and absorbance values were expressed as a percentage of the blank controls.
MRC-5 Cytotoxicity Cellular Assay
MRC-5 SV2 cells, originally from a human diploid lung cell line, were cultivated in MEM, supplemented with l-glutamine (20 mM), 16.5 mM sodium hydrogen carbonate, and 5% FCS. For the assay, 104 MRC-5 cells/well were seeded onto the test plates containing the prediluted sample and incubated at 37 °C and 5% CO2 for 72 h. Cell viability was assessed fluorimetrically 4 h after the addition of resazurin. Fluorescence was measured (excitation 550 nm, emission 590 nm), and the results were expressed as percentage reduction in cell viability compared to control.
Statistical Analysis
Details of the applied statistical analyses are provided with each experiment. No statistical methods were used to predetermine the size of samples. The experiments were not randomized, and the investigators were not blinded to allocation during experiments or outcome assessment.
Acknowledgments
We thank W. Uitslag, R. Biharie, M. Mulders, V. Moorman, L. Capoferri, and L. Lemgruber for technical and analytical support and N. N. Mohamed for assistance with manuscript preparation. We also thank the Diamond Light Source for beam time and support on I03 and I04 beamlines. A.R.B. is supported by a grant from The Netherlands Organization for Scientific Research (NWO ECHO). This work was supported by TI Pharma Grant T4-302 and the European Commission seventh Framework Programme FP7-HEALTH-2013-INNOVATION-1 under project reference 602666 “Parasite-specific cyclic nucleotide phosphodiesterase inhibitors to target Neglected Parasitic Diseases” (PDE4NPD).
Glossary
Abbreviations Used
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- hPDE
human phosphodiesterase
- hPDE4
human phosphodiesterase 4
- hPDE4B
human phosphodiesterase 4B
- hPDE4D
human phosphodiesterase 4D
- LmPDEB1
Leishmania major phosphodiesterase B1
- MD
molecular dynamics
- NTD
neglected tropical disease
- PDE
3′,5′-cyclic nucleotide phosphodiesterase
- PDEStrIAn
Phosphodiesterase Structure and Ligand Interaction Annotated tool
- siRNA
small interfering RNA
- TbrPDEB1
Trypanosoma brucei phosphodiesterase B1
- TbrPDEB2
Trypanosoma brucei phosphodiesterase B2
- TcrPDEC
Trypanosoma cruzi phosphodiesterase C
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOAt
1-hydroxy-7-azabenzotriazole
- DIPEA
N,N-diisopropylethylamine
- HOBt
hydroxybenzotriazole
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01670.
Schematic PDE topology and binding site annotation according to PDEStrIAn; stereoviews and difference electron density maps for all X-ray crystal structures; crystal structure data collection and refinement statistics; PDE panel pIC50 data for 5 and 8; chiral HPLC data for 4, 4a and 4b (PDF)
Molecular formula strings (CSV)
Accession Codes
The coordinates of the crystal structures have been deposited to the RCSB Protein Data Bank under the following accession codes: 5G57 (TbrPDEB1–NPD-001); 5LAQ (hPDE4B–NPD-001); 5LBO (hPDE4D–NPD-001); 5L9H (TbrPDEB1–NPD-340); 5G2B (TbrPDEB1–NPD-008); 5L8C (TbrPDEB1–NPD-039); 5G5V (TbrPDEB1–NPD-038); 5L8Y (TbrPDEB1–NPD-937). Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
○ A.R.B. and A.K.S. contributed equally to this work. A.R.B., E.E., K.M.O., J.J.N.V., T.v.d.B., E.d.H., H.C., M.S., G.J.S., and I.J.P.d.E. were involved in compound design, synthesis, and analysis. A.K.S., E.B., and D.G.B. were involved in protein production, crystallization, data collection, and refinement for structural studies. A.R.B., A.K.S., C.J., E.B., and D.G.B. were involved in crystal structure analysis. H.T. was involved in the PDE activity assays. A.R.B. and W.J.M. were involved in the MD studies. D.N.A.T., T.D.K., J.C.M., and H.P.d.K. were involved in the intracellular cAMP assays and microscopy experiments. A.M. and L.M. were involved in the phenotypic cellular assays. E.E., K.M.O., M.W., M.S., C.d.G., L.M., H.P.d.K., G.J.S., I.J.P.d.E., D.G.B., and R.L. supervised the experiments and conceived the project. A.R.B., A.K.S., D.S.B., I.J.P.d.E., D.G.B., and R.L. integrated all data and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Research priorities for Chagas disease, human African trypanosomiasis and leishmaniasis. WHO Technical Report Series 2012, 975, 1–100. [PubMed] [Google Scholar]
- Bilbe G. Overcoming neglect of kinetoplastid diseases. Science 2015, 348, 974–976. 10.1126/science.aaa3683. [DOI] [PubMed] [Google Scholar]
- Shakur Y.; de Koning H. P.; Ke H.; Kambayashi J.; Seebeck T.. Therapeutic potential of phosphodiesterase inhibitors in parasitic diseases. In Handbook of Experimental Pharmacology; Francis S. H., Conti M., Houslay M. D., Eds.; Springer: Berlin, 2011; Vol. 204, pp 487–510. [DOI] [PubMed] [Google Scholar]
- Seebeck T.; Sterk G. J.; Ke H. Phosphodiesterase inhibitors as a new generation of antiprotozoan drugs: exploiting the benefit of enzymes that are highly conserved between host and parasite. Future Med. Chem. 2011, 3, 1289–1306. 10.4155/fmc.11.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoraghi R.; Kunz S.; Gong K.; Seebeck T. Characterization of TbPDE2A, a novel cyclic nucleotide-specific phosphodiesterase from the protozoan parasite Trypanosoma brucei. J. Biol. Chem. 2001, 276, 11559–11566. 10.1074/jbc.M005419200. [DOI] [PubMed] [Google Scholar]
- Zoraghi R.; Seebeck T. The cAMP-specific phosphodiesterase TbPDE2C is an essential enzyme in bloodstream form Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 4343–4348. 10.1073/pnas.062716599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S.; Beavo J. A.; D’Angelo M. A.; Flawia M. M.; Francis S. H.; Johner A.; Laxman S.; Oberholzer M.; Rascon A.; Shakur Y.; Wentzinger L.; Zoraghi R.; Seebeck T. Cyclic nucleotide specific phosphodiesterases of the kinetoplastida: a unified nomenclature. Mol. Biochem. Parasitol. 2006, 145, 133–135. 10.1016/j.molbiopara.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Oberholzer M.; Marti G.; Baresic M.; Kunz S.; Hemphill A.; Seebeck T. The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2: flagellar enzymes that are essential for parasite virulence. FASEB J. 2007, 21, 720–731. 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- Bland N. D.; Wang C.; Tallman C.; Gustafson A. E.; Wang Z.; Ashton T. D.; Ochiana S. O.; McAllister G.; Cotter K.; Fang A. P.; Gechijian L.; Garceau N.; Gangurde R.; Ortenberg R.; Ondrechen M. J.; Campbell R. K.; Pollastri M. P. Pharmacological validation of Trypanosoma brucei phosphodiesterases B1 and B2 as druggable targets for African sleeping sickness. J. Med. Chem. 2011, 54, 8188–8194. 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning H. P.; Gould M. K.; Sterk G. J.; Tenor H.; Kunz S.; Luginbuehl E.; Seebeck T. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J. Infect. Dis. 2012, 206, 229–237. 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerman J.; van den Bergh T.; Orrling K. M.; Jansen C.; Cos P.; Maes L.; Chatelain E.; Ioset J. R.; Edink E. E.; Tenor H.; Seebeck T.; de Esch I. J. P.; Leurs R.; Sterk G. J. Synthesis and evaluation of analogs of the phenylpyridazinone NPD-001 as potent trypanosomal TbrPDEB1 phosphodiesterase inhibitors and in vitro trypanocidals. Bioorg. Med. Chem. 2016, 24, 1573–1581. 10.1016/j.bmc.2016.02.032. [DOI] [PubMed] [Google Scholar]
- Orrling K. M.; Jansen C.; Vu X. L.; Balmer V.; Bregy P.; Shanmugham A.; England P.; Bailey D.; Cos P.; Maes L.; Adams E.; van den Bogaart E.; Chatelain E.; Ioset J. R.; van de Stolpe A.; Zorg S.; Veerman J.; Seebeck T.; Sterk G. J.; de Esch I. J. P.; Leurs R. Catechol pyrazolinones as trypanocidals: fragment-based design, synthesis, and pharmacological evaluation of nanomolar inhibitors of trypanosomal phosphodiesterase B1. J. Med. Chem. 2012, 55, 8745–8756. 10.1021/jm301059b. [DOI] [PubMed] [Google Scholar]
- Houslay M. D.; Schafer P.; Zhang K. Y. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discovery Today 2005, 10, 1503–1519. 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Manallack D. T.; Hughes R. A.; Thompson P. E. The next generation of phosphodiesterase inhibitors: structural clues to ligand and substrate selectivity of phosphodiesterases. J. Med. Chem. 2005, 48, 3449–3462. 10.1021/jm040217u. [DOI] [PubMed] [Google Scholar]
- Tenor H.; Hatzelmann A.; Beume R.; Lahu G.; Zech K.; Bethke T. D.. Pharmacology, clinical efficacy, and tolerability of phosphodiesterase-4 inhibitors: impact of human pharmacokinetics. In Handbook of Experimental Pharmacology; Francis S. H., Conti M., Houslay M. D., Eds.; Springer: Berlin, 2011; Vol. 204, pp 85–119. [DOI] [PubMed] [Google Scholar]
- Maurice D. H.; Ke H.; Ahmad F.; Wang Y.; Chung J.; Manganiello V. C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discovery 2014, 13, 290–314. 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C.; Wang H.; Kooistra A. J.; de Graaf C.; Orrling K. M.; Tenor H.; Seebeck T.; Bailey D.; de Esch I. J. P.; Ke H.; Leurs R. Discovery of novel Trypanosoma brucei phosphodiesterase B1 inhibitors by virtual screening against the unliganded TbrPDEB1 crystal structure. J. Med. Chem. 2013, 56, 2087–2096. 10.1021/jm3017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Yan Z.; Geng J.; Kunz S.; Seebeck T.; Ke H. Crystal structure of the Leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol. Microbiol. 2007, 66, 1029–1038. 10.1111/j.1365-2958.2007.05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Kunz S.; Chen G.; Seebeck T.; Wan Y.; Robinson H.; Martinelli S.; Ke H. Biological and structural characterization of Trypanosoma cruzi phosphodiesterase C and Implications for design of parasite selective inhibitors. J. Biol. Chem. 2012, 287, 11788–11797. 10.1074/jbc.M111.326777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard B. L.; Thompson P. E.; Manallack D. T. Active site similarity between human and Plasmodium falciparum phosphodiesterases: considerations for antimalarial drug design. J. Comput.-Aided Mol. Des. 2011, 25, 753–762. 10.1007/s10822-011-9458-5. [DOI] [PubMed] [Google Scholar]
- Jansen C.; Kooistra A. J.; Kanev G. K.; Leurs R.; de Esch I. J. P.; de Graaf C. PDEStrIAn: a phosphodiesterase structure and ligand interaction annotated database as a tool for structure-based drug design. J. Med. Chem. 2016, 59, 7029–7065. 10.1021/acs.jmedchem.5b01813. [DOI] [PubMed] [Google Scholar]
- Gould M. K.; Bachmaier S.; Ali J. A.; Alsford S.; Tagoe D. N.; Munday J. C.; Schnaufer A. C.; Horn D.; Boshart M.; de Koning H. P. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob. Agents Chemother. 2013, 57, 4882–4893. 10.1128/AAC.00508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaka S.; Hudson A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discovery 2006, 5, 941–955. 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- Hatzelmann A.; Marx D.; Steinhilber W.; Sterk G. J.. (Altana Pharma AG). Novel Phthalazinones. International Patent WO 2002085906, 2002.
- Hatzelmann A.; Boss H.; Häfner D.; Beume R.; Kley H.-P.; Van der laan I. J.; Timmerman H.; Sterk G. J.; Van der Mey M.. (Byk Gulden Lomberg Chemische Fabrik GmbH). Phthalazinone PDE III/IV Inhibitors. WO 199947505, 1999.
- van der Mey M.; Hatzelmann A.; Van Klink G. P.; Van der Laan I. J.; Sterk G. J.; Thibaut U.; Ulrich W. R.; Timmerman H. Novel selective PDE4 inhibitors. 2. Synthesis and structure-activity relationships of 4-aryl-substituted cis-tetra- and cis-hexahydrophthalazinones. J. Med. Chem. 2001, 44, 2523–2535. 10.1021/jm010838c. [DOI] [PubMed] [Google Scholar]
- Saubern S.; Guha R.; Baell J. B. KNIME workflow to assess PAINS filters in SMARTS format. Comparison of RDKit and Indigo cheminformatics libraries. Mol. Inf. 2011, 30, 847–850. 10.1002/minf.201100076. [DOI] [PubMed] [Google Scholar]
- Burgin A. B.; Magnusson O. T.; Singh J.; Witte P.; Staker B. L.; Bjornsson J. M.; Thorsteinsdottir M.; Hrafnsdottir S.; Hagen T.; Kiselyov A. S.; Stewart L. J.; Gurney M. E. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- Winter G.; Lobley C. M.; Prince S. M. Decision making in xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1260–1273. 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct., Funct., Genet. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Sousa da Silva A. W.; Vranken W. F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Alkhaldi A. A.; Creek D. J.; Ibrahim H.; Kim D. H.; Quashie N. B.; Burgess K. E.; Changtam C.; Barrett M. P.; Suksamrarn A.; de Koning H. P. Potent trypanocidal curcumin analogs bearing a monoenone linker motif act on Trypanosoma brucei by forming an adduct with trypanothione. Mol. Pharmacol. 2015, 87, 451–464. 10.1124/mol.114.096016. [DOI] [PubMed] [Google Scholar]
- Alkhaldi A. A.; Martinek J.; Panicucci B.; Dardonville C.; Zikova A.; de Koning H. P. Trypanocidal action of bisphosphonium salts through a mitochondrial target in bloodstream form Trypanosoma brucei. Int. J. Parasitol.: Drugs Drug Resist. 2016, 6, 23–34. 10.1016/j.ijpddr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.