

Abstract
G protein-coupled receptor-35 (GPR35) has emerged as a potential target in the treatment of pain and inflammatory and metabolic diseases. We have discovered a series of potent GPR35 agonists based on a coumarin scaffold and found that the introduction of a 1H-tetrazol-5-yl group significantly increased their potency. We designed and synthesized a new series of N-[2-(1H-tetrazol-5-yl)phenyl]benzamide derivatives through a two-step synthetic approach, and characterized their agonistic activities against GPR35 using a dynamic mass redistribution (DMR) assay. N-(5-bromo-2-(1H-tetrazol-5-yl)phenyl)-4-methoxybenzamide (56) and N-(5-bromo-2-(1H-tetrazol-5-yl)phenyl)-2-fluoro-4-methoxybenzamide (63) displayed the highest agonistic potency agonist GPR35 with an EC50 of 0.059 μM and 0.041 μM, respectively. The physicochemical properties of selected compounds were calculated to evaluate their druglikeness, suggesting that compounds 56 and 63 have good druglike properties. Together, N-[2-(1H-tetrazol-5-yl)phenyl]benzamide derivatives are potentially great candidates for developing potent GPR35 agonists.
Keywords: GPR35, dynamic mass redistribution, N-[2-(1H-tetrazol-5-yl)phenyl]benzamide derivatives, Lipinski’s rule
G protein-coupled receptors (GPCRs) as the most successful druggable receptor family have an important position in drug development.1 However, the physiological functions of many GPCRs, in particular orphan receptors,2 are far from clear. Discovering potent probe molecules for these orphan receptors has great significance in understanding their physiological functions and for drug development. The orphan G protein-coupled receptor 35 (GPR35) was first discovered in 1998, and it later has been implicated in a variety of diseases,3 such as cardiovascular diseases,4 gastric cancer,3,5 artery disease,6 and type 2 diabetes.7
In the past decade, several endogenous ligands have been discovered to activate GPR35; these ligands include kynurenic acid,5 lysophosphatidic acid,8 multiple tyrosine metabolites,9 and the mucosal chemokine CXCL1710 but with variable potency. Several classes of synthetic agonists including the phosphodiesterase 5 inhibitor zaprinast (1),11 the antiasthma drugs doxantrazole (2), and pemirolast (3)12 have also been reported to display GPR35 agonistic activity. Furthermore, zaprinast is the most widely used reference agonist to elucidate the biological functions of GPR35. Many more GPR35 agonists were discovered through high-throughput screening,13,14 and only a few structure–activity relationship (SAR) studies have been reported in the literature so far.15,16
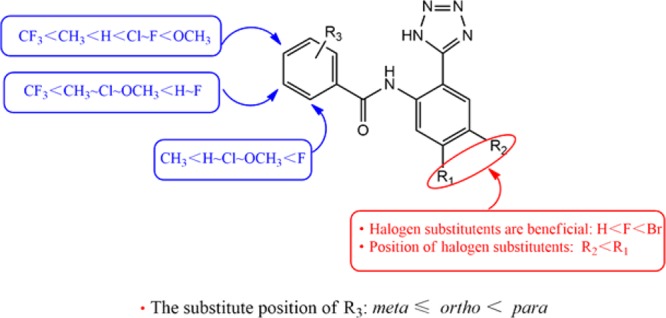
Our previous studies have shown that tetrazole 4 possessed significant agonism at GPR35.17 Several other studies have shown that halogen atom substitution also increased the agonistic activity in certain positions.14−16 The introduction of lipophilic residues and hydrogen bond accepting groups at specific positions in the molecule could probably enhance the activity of compounds.15,16 Inspired by these findings, a variety of N-[2-(1H-tetrazol-5-yl)phenyl]amido derivatives with different halogen atoms at the 4- or 5-positions were synthesized. The compounds were prepared starting from 6a–6c and 8.
The 2-(1H-tetrazol-5-yl)aniline derivatives 7a and 7b were obtained from 6a and 6b via the [3 + 2] cycloaddition of nitriles and sodium azide catalyzed by aluminum chloride at 90 °C with excellent yields (84% and 91%, respectively) (Scheme 1).
Scheme 1. Synthesis of Compounds 7a and 7b.

Reagents and conditions: (a) NaN3, AlCl3, THF, 90 °C, 5 h, yield 81–86%.
The amines 6a–6c and 8 were reacted with various acid chlorides in pyridine as the base and solvent at room temperature, yielding a series of N-(2-cyanophenyl)amido derivatives (19–41) (Scheme 2, Scheme 3). The N-[2-(1H-tetrazol-5-yl)phenyl]benzamide derivatives (42–64) were synthesized from N-(2-cyanophenyl)benzamide derivatives (19–41) via a [3 + 2] cycloaddition, also with good yields (69–91%) (Scheme 2, Scheme 3).
Scheme 2. Synthesis of Compound 14 and its Derivatives.

Reagents and conditions: (a) R3COCl, pyridine, overnight, yield 68–87%. (b) NaN3, AlCl3, THF, 90 °C, 5 h, yield 69–91%
Scheme 3. Synthesis of Compounds 41 and 64.

Reagents and conditions: (a) 4-Methoxybenzoyl chloride, pyridine, overnight. (b) NaN3, AlCl3, THF, 90 °C, 5 h.
All N-[2-(1H-tetrazol-5-yl)phenyl]amido compounds obtained were tested using a dynamic mass redistribution (DMR) assay18 using HT-29, a cell line endogenously expressing GPR35.19 The reference agonist zaprinast was also used to perform a DMR desensitization assay.19 Results showed that all N-[2-(1H-tetrazol-5-yl)phenyl]amido compounds obtained not only gave rise to a dose-dependent DMR in HT-29 but also desensitized the cells to subsequent stimulation with 1 μM zaprinast. Notably, the potency to trigger DMR for all compounds was found to be almost equal to that to desensitize the zaprinast response (Table 1, Table 2), suggesting that these compounds are GPR35 agonists.
Table 1. Potency of 2-Substituted 2-(1H-Tetrazol-5-yl)aniline Derivatives in DMR Assays.

| compd | R1 | R2 | R3 | EC50a (μM) | desensitization IC50b (μM) | antagonist IC50c (μM) |
|---|---|---|---|---|---|---|
| 7a | 29.99 ± 2.80 | 35.56 ± 2.54 | 0.67 ± 0.19 | |||
| 7b | 44.06 ± 4.06 | 75.34 ± 9.31 | 0.67 ± 0.25 | |||
| 14 | Br | H | ethyl | 0.62 ± 0.06 | 0.70 ± 0.07 | 0.47 ± 0.11 |
| 15 | Br | H | isopropyl | 0.38 ± 0.02 | 0.28 ± 0.02 | 0.58 ± 0.06 |
| 16 | Br | H | cyclohexyl | 0.44 ± 0.03 | 0.20 ± 0.01 | 0.43 ± 0.08 |
| 17 | Br | H | furyl | 1.06 ± 0.13 | 0.44 ± 0.03 | 0.29 ± 0.08 |
| 18 | Br | H | thienyl | 0.51 ± 0.02 | 0.26 ± 0.03 | 0.50 ± 0.05 |
EC50 to trigger DMR.
IC50 to desensitize upon cells repeated stimulation with 1 μM zaprinast.
IC50 of known GPR35 antagonist 5 to block the agonism. The data respresent mean ± sd from two independent measurements, each with four replicates (n = 8).
Table 2. Potency of N-(2-(1H-Tetrazol-5-yl)phenyl)benzamide Derivatives in the DMR Assay.

| R3 |
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | ortho | meta | para | EC50a (μM) | desensitization IC50b (μM) | antagonist IC50c (μM) |
| 37 | inactive | |||||||
| 42 | Br | H | H | H | H | 0.87 ± 0.09 | 0.30 ± 0.03 | 0.47 ± 0.06 |
| 43 | Br | H | CH3 | H | H | 2.05 ± 0.11 | 0.81 ± 0.05 | 0.19 ± 0.02 |
| 44 | Br | H | H | CH3 | H | 0.86 ± 0.04 | 0.36 ± 0.02 | 0.12 ± 0.03 |
| 45 | Br | H | H | H | CH3 | 1.33 ± 0.13 | 0.51 ± 0.03 | 0.13 ± 0.04 |
| 46 | Br | H | Cl | H | H | 0.87 ± 0.10 | 0.41 ± 0.03 | 0.77 ± 0.08 |
| 47 | Br | H | H | Cl | H | 2.18 ± 0.37 | 0.48 ± 0.03 | 0.42 ± 0.11 |
| 48 | Br | H | H | H | Cl | 0.52 ± 0.03 | 0.21 ± 0.01 | 0.52 ± 0.05 |
| 49 | Br | H | Cl | H | Cl | 0.31 ± 0.03 | 0.11 ± 0.01 | 1.11 ± 0.42 |
| 50 | Br | H | F | H | H | 0.35 ± 0.02 | 0.17 ± 0.01 | 0.52 ± 0.07 |
| 51 | Br | H | H | F | H | 0.61 ± 0.07 | 0.21 ± 0.02 | 0.57 ± 0.07 |
| 52 | Br | H | H | H | F | 0.45 ± 0.02 | 0.16 ± 0.01 | 0.18 ± 0.06 |
| 53 | Br | H | F | H | F | 0.13 ± 0.01 | 0.06 ± 0.01 | 0.22 ± 0.03 |
| 54 | Br | H | OCH3 | H | H | 0.86 ± 0.06 | 0.40 ± 0.03 | 0.28 ± 0.05 |
| 55 | Br | H | H | OCH3 | H | 1.94 ± 0.08 | 0.81 ± 0.07 | 0.56 ± 0.08 |
| 56 | Br | H | H | H | OCH3 | 0.059 ± 0.007 | 0.026 ± 0.003 | 0.20 ± 0.09 |
| 57 | Br | H | H | OCH3 | OCH3 | 0.83 ± 0.07 | 0.26 ± 0.02 | 1.00 ± 0.16 |
| 58 | H | Br | H | H | OCH3 | 0.30 ± 0.03 | 0.11 ± 0.01 | 1.38 ± 0.15 |
| 59 | F | H | H | H | OCH3 | 0.20 ± 0.01 | 0.082 ± 0.005 | 0.34 ± 0.08 |
| 60 | H | H | H | H | OCH3 | 0.65 ± 0.05 | 0.28 ± 0.02 | 1.00 ± 0.22 |
| 61 | Br | H | H | CF3 | H | 4.60 ± 0.46 | 1.28 ± 0.08 | 0.84 ± 0.06 |
| 62 | Br | H | H | H | CF3 | 3.65 ± 0.19 | 1.36 ± 0.16 | 0.25 ± 0.05 |
| 63 | Br | H | F | H | OCH3 | 0.041 ± 0.005 | 0.014 ± 0.002 | 1.69 ± 0.16 |
| 64 | OCH3 | 17.58 ± 3.30 | 8.36 ± 0.99 | 0.63 ± 0.19 | ||||
EC50 to trigger DMR.
IC50 to desensitize cells upon cells repeated stimulation with 1 μM zaprinast.
IC50 of known GPR35 antagonist 5 to block the agonist-induced DMR. The data represents mean ± sd from two independent measurements, each with four replicates (n = 8).
The DMR antagonist assay further showed that the known GPR35 antagonist ML-145 (5, Figure 1) dose-dependently and completely blocked the DMR arising from all N-[2-(1H-tetrazol-5-yl)phenyl]amido derivatives, each at its respective EC80 to EC100 concentration.20 This suggests that the DMR assays of these N-[2-(1H-tetrazol-5-yl)phenyl]amido derivatives were specific to the activation of GPR35.
Figure 1.

Selected GPR35 agonists with potencies at GPR35 (1–4) and antagonist (5).
Inspired by our previous findings, a tetrazolyl group was first introduced into the simplest compounds 6a and 6b, yielding compounds 7a and 7b. Compared to the inactive compounds 6a and 6b, 7a and 7b both showed moderate potency with an EC50 of 29.99 μM and 44.06 μM, respectively. This result suggests that the introduction of the tetrazolyl to the molecule significantly improves the agonistic activity.
Next, an amide linker was introduced into compound 7b, given that lipophilic residues and hydrogen bond-accepting groups may also play an important role in activating GPR35.15 The propanamide (14, EC50 0.62 μM) showed more than 50-fold increase in potency compared to 7b. The introduction of the bulkier alkyl substituents further improved potency. For instance, isopropyl (15, EC50 0.38 μM) and cyclohexyl (16, EC50 0.44 μM) substituted compounds displayed more than 100-fold increased potency compared to 7b. Compounds with an aromatic group in this position also had relatively higher potency, such as furyl (17, EC50 1.06 μM), thienyl (18, EC50 0.52 μM) and phenyl (42, EC50 0.87 μM).
Since the benzoic acid derivatives are easy to obtain, we chose the phenyl-substituted derivative 42 (N-(5-bromo-2-(1H-tetrazol-5-yl)phenyl)benzamide) for further modification. N-(5-Bromo-2-(1H-tetrazol-5-yl)phenyl)benzamide derivatives with different substituents in the o-, m-, and p-position were investigated (mono- or disubstitutions).
For the monosubstitutions, the same substituent in different positions of the benzene ring could significantly change the activity. The following rank order of potency among compounds with the same substituent in different positions (o-, m-, and p-position) was observed: m-methyl (44, EC50 0.86 μM) ≈ p-methyl (45, EC50 1.33 μM) > o-methyl (43, EC50 2.05 μM) (Figure S1a); p-chloro (48, EC50 0.52 μM) > o-chloro (46, EC50 0.87 μM) > m-chloro (47, EC50 2.18 μM) (Figure S1b); p-fluoro (52, EC50 0.45 μM) ≈ o-fluoro (50, EC50 0.35 μM) > m-fluoro (51, EC50 0.61 μM) (Figure S1c); p-methoxyl (56, EC50 0.059 μM) > o-methoxyl (54, EC50 0.86 μM) > m-methoxyl (55, EC50 1.94 μM) (Figure S 1d); p-trifluoromethyl (54, EC50 3.65 μM) > m-trifluoromethyl (55, EC50 4.60 μM). These results suggest that substituents in the para-position led to a considerable increase in potency except for the methyl-derivatives with better tolerance in the meta-position. For N-(5-bromo-2-(1H-tetrazol-5-yl)phenyl)benzamide derivatives with varying substituents in the para-position, the rank order of potency was as follows: methoxyl (56, EC50 0.059 μM) > fluoro (52, EC50 0.45 μM) > chloro (48, EC50 0.52 μM) > methyl (45, EC50 1.33 μM) > trifluoromethyl (54, EC50 3.65 μM), suggesting that methoxyl in the para-position was favorable for the agonistic activity.
For the disubstitution of the benzamide ring, the rank order of potency of o,p-dichloro (49), o,p-difluoro (53), and m,p-dimethoxy (57) was as follows: o,p-difluoro (53, EC50 0.13 μM) > o,p-dichloro (49, EC50 0.31 μM) > m,p-dimethoxy (57, EC50 0.83 μM), suggesting that the o,p-positions led to a visible increase in activity compared to the o- or p-monosubstituted compounds. However, the dimethoxy in m,p-positions (57) led to a large reduction in activity compared to compound 56, suggesting that bulky m,p-substituted compounds are not well tolerated but o,p-disubstitutions are.
Compounds 50 and 56 showed the highest agonistic potency among the derivatives. Therefore, we devised the synthesis of compound 63, which was substituted with a fluoro in the o-position and a methoxy in the p-position. As expected, compound 63 displayed further improved potency (EC50 0.041 μM, Figure 2).
Figure 2.
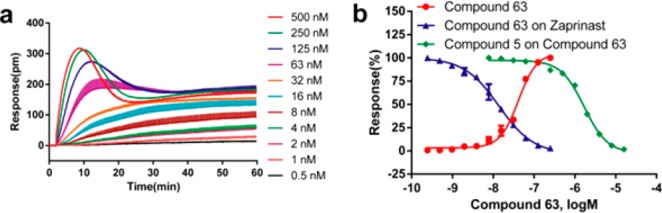
(a) Real time kinetic responses of 63 at different doses in HT-29 cells. (b) DMR amplitudes of compound 63 as a function of doses compared with the dose-dependent desensitization of zaprinast DMR by 63, and the dose-dependent inhibition of the DMR of 100 nM compound 63 by compound 5. The data represents mean ± sd from two independent measurement, each with four replicates (n = 8).
There is evidence that the halogen atom bromine is very important for the retention of compound activity agonist GPR35.14−17,21 In order to study the effect of the change of the bromine atom on the activity of the compound, we chose compound 56 for further study. Results showed that transforming the substitution position of the bromine atom (58, EC50 0.30 μM), replacing the bromine with a fluorine (59, EC50 0.20 μM), or removing the bromine (60, EC50 0.65 μM) all decreased potency.
Compound 37, a synthetic precursor of 60 was also tested and found to be devoid of activity, demonstrating the necessity of the tetrazole. The activity was completely lost when the tetrazolyl was replaced by cyano (Figure S1e). In addition, changing the position of the tetrazoyl also significantly reduced the activity of the compound (64, EC50 17.58 μM).
We further examined the activity of compounds 53 and 56 through the ERK phosphorylation. Compound 53 and compound 56 increased ERK phosphorylation (Figure. 3a). As control, the known GPR35 agonist zaprinast also triggered ERK phosphorylation (Figure 3b). Moreover, the GPR35 antagonist ML-145 attenuated ERK phosphorylation induced by these compounds (Figure. 3b). These results suggested that compounds 53 and 56 induced the phosphorylation of ERK via the activation of GPR35.
Figure 3.

(a) Western blot of ERK1/2 and p-ERK1/2 after treatment with compounds 53 and 56 for 15 min at concentrations of 100 nM, 1 μM respectively. (b) Western blot of ERK1/2 and p-ERK1/2 after treatment with the known GPR35 agonist Zaprinast (Zap) at 1 μM and Western blot of ERK1/2 and p-ERK1/2 after treatment with compounds for 10 min at the same concentrations as in (a) in the presence of ML-145 (25 μM) for 5 min.
In order to evaluate the druglikeness of a compound, several physiochemical properties such as the partition coefficient (clogP), the ligand efficiency (LE) and the ligand-lipophilicity efficiency (LLE) were introduced.22−24 These parameters were calculated for the compounds with relatively high potency, including compounds 14–16, 18, 42, 44, 46, 48–54, 56–60, and 63 (Table 3). Lipinski’s Rule of Five considered that the clogP value of a druglike compound should be lower than 5.25,26 The clogP values of all these selected compounds were found to be within this range. In fact, most of these compounds exhibited a clogP value between 2 and 3, which is favorable for orally administered drugs.
Table 3. Physicochemical Properties of Selected Compounds.
| compd | pEC50 | clogPa | LE | LLE |
|---|---|---|---|---|
| 14 | 6.21 | 1.27 | 0.37 | 4.94 |
| 15 | 6.42 | 1.58 | 0.36 | 4.84 |
| 16 | 6.36 | 2.78 | 0.30 | 3.58 |
| 18 | 6.29 | 1.98 | 0.31 | 4.31 |
| 42 | 6.06 | 2.23 | 0.29 | 3.83 |
| 44 | 6.06 | 2.73 | 0.28 | 3.33 |
| 46 | 6.06 | 2.19 | 0.28 | 3.87 |
| 48 | 6.28 | 3.02 | 0.29 | 3.26 |
| 49 | 6.50 | 2.93 | 0.28 | 3.57 |
| 50 | 6.46 | 2.04 | 0.29 | 4.42 |
| 51 | 6.21 | 2.45 | 0.28 | 3.76 |
| 52 | 6.35 | 2.45 | 0.29 | 3.90 |
| 53 | 6.88 | 2.21 | 0.30 | 4.67 |
| 54 | 6.07 | 2.29 | 0.26 | 3.78 |
| 56 | 7.23 | 2.36 | 0.31 | 4.87 |
| 57 | 6.08 | 2.01 | 0.24 | 4.07 |
| 58 | 6.52 | 2.36 | 0.28 | 4.16 |
| 59 | 6.70 | 1.64 | 0.29 | 5.06 |
| 60 | 6.19 | 1.31 | 0.28 | 4.88 |
| 63 | 7.38 | 2.15 | 0.32 | 5.23 |
Calculated by the Chembiodraw Ultra 11.0.
The LE combines physiochemical with pharmacological properties, and it represents the binding force between each atom and receptors and calculates it as follows: LE = pEC50/N (N = non-hydrogen atoms). The LE values of most selected compounds were found to be about 0.3, except for compounds 54 and 57.
LLE (LEE = pEC50 – clogP), another useful parameter, combines the potency and lipophilicity. Among these compounds, 14, 15, 56, 59, 60, and 63 showed a LLE value of 5. A suitable drug candidate should have a LE > 0.3 and LLE > 5. In summary, the new compounds 56 and 63 with potent potency showed a very good clogP and suitable LE and LLE values.
In conclusion, a series of N-[2-(1H-tetrazol-5-yl)phenyl]amido derivatives were synthesized as potent GPR35 agonists through a two-step synthesis method. SAR analysis showed that a bromine substituent in the 5-position and a p-methoxy-benzamide in the 2-position would significantly improve the activity (56, EC50 0.059 μM). The o,p-distribution such as o,p-dichloro (49) and o,p-difluoro (53) could also improve the potency when compared with the o- or p-monosubstituted analogs. Combining these findings, compound 63 was synthesized and found to display the highest potency (EC50 0.041 μM) and good physicochemical properties. Together, this study provides a new series of potent GPR35 agonists, such as 56 and 63, which may become useful leading ligands to further elucidate the physiological roles of GPR35.
Acknowledgments
This work is supported by Project of National Science Foundation of China (81473436). We are also grateful for the support by the State Key Program of National Natural Science of China (Grant No.U1508221) and innovation program (DICP TMSR201601) of science and research from DICP, CAS.
Glossary
Abbreviations
- GPR35
G protein-coupled receptor 35
- GPCR
G protein-coupled receptor
- SARs
structure activity relationships
- DMR
dynamic mass redistribution
- THF
tetrahydrofuran
- LE
ligand efficiency
- LLE
ligand-lipophilicity efficiency
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00510.
Experimental details and characterization data for the reported compounds; NMR spectra; and biological assays (PDF)
Author Contributions
⊥ L.W. and T.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Rask-Andersen M.; Almen M. S.; Schioth H. B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery 2011, 10, 579–590. 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Chung S.; Funakoshi T.; Civelli O. Orphan GPCR research. Br. J. Pharmacol. 2008, 153, S339–S346. 10.1038/sj.bjp.0707606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd B. F.; Nguyen T.; Marchese A.; Cheng R.; Lynch K. R.; Heng H. H. Q.; Kolakowski L. F.; George S. R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- Min K. D.; Asakura M.; Liao Y. L.; Nakamaru K.; Okazaki H.; Takahashi T.; Fujimoto K.; Ito S.; Takahashi A.; Asanuma H.; Yamazaki S.; Minamino T.; Sanada S.; Seguchi O.; Nakano A.; Ando Y.; Otsuka T.; Furukawa H.; Isomura T.; Takashima S.; Mochizuki N.; Kitakaze M. Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem. Biophys. Res. Commun. 2010, 393, 55–60. 10.1016/j.bbrc.2010.01.076. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Simonavicius N.; Wu X. S.; Swaminath G.; Reagan J.; Tian H.; Ling L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- Sun Y. V.; Bielak L. E.; Peyser P. A.; Turner S. T.; Sheedy P. E.; Boerwinkle E.; Kardia S. L. R. Application of machine learning algorithms to predict coronary artery calcification with a sibship-based design. Genet. Epidemiol. 2008, 32, 350–360. 10.1002/gepi.20309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V.; Federico A.; Pollastro C.; Ziviello C.; Cataldi S.; Formisano P.; Ciccodicola A. Computational Analysis of Single Nucleotide Polymorphisms Associated with Altered Drug Responsiveness in Type 2 Diabetes. Int. J. Mol. Sci. 2016, 17, 1008. 10.3390/ijms17071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S.; Ota R.; Shima M.; Yamashita A.; Sugiura T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. 10.1016/j.bbrc.2010.03.169. [DOI] [PubMed] [Google Scholar]
- Deng H. Y.; Hu H. B.; Fang Y. Multiple tyrosine metabolites are GPR35 agonists. Sci. Rep. 2012, 2, 12. 10.1038/srep00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravillas-Montero J. L.; Burkhardt A. M.; Hevezi P. A.; Carnevale C. D.; Smit M. J.; Zlotnik A. Cutting Edge: GPR35/CXCR8 Is the Receptor of the Mucosal Chemokine CXCL17. J. Immunol. 2015, 194, 29–33. 10.4049/jimmunol.1401704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y.; Tonai-Kachi H.; Shinjo K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006, 580, 5003–5008. 10.1016/j.febslet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- MacKenzie A. E.; Caltabiano G.; Kent T. C.; Jenkins L.; McCallum J. E.; Hudson B. D.; Nicklin S. A.; Fawcett L.; Markwick R.; Charlton S. J.; Milligan G. The antiallergic mast cell stabilizers lodoxamide and bufrolin as the first high and equipotent agonists of human and rat GPR35. Mol. Pharmacol. 2014, 85, 91–104. 10.1124/mol.113.089482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. Y.; Hu H. B.; Ling S. Z.; Ferrie A. M.; Fang Y. Discovery of Natural Phenols as G Protein-Coupled Receptor-35 (GPR35) Agonists. ACS Med. Chem. Lett. 2012, 3, 165–169. 10.1021/ml2003058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. Y.; Hu H. B.; He M. Q.; Hu J. Y.; Niu W. J.; Ferrie A. M.; Fang Y. Discovery of 2-(4-Methylfuran-2(5H)-ylidene)malononitrile and Thieno 3,2-b thiophene-2-carboxylic Acid Derivatives as G Protein-Coupled Receptor 35 (GPR35) Agonists. J. Med. Chem. 2011, 54, 7385–7396. 10.1021/jm200999f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke M.; Thimm D.; Schiedel A. C.; Muller C. E. 8-Benzamidochromen-4-one-2-carboxylic Acids: Potent and Selective Agonists for the Orphan G Protein-Coupled Receptor GPR35. J. Med. Chem. 2013, 56, 5182–5197. 10.1021/jm400587g. [DOI] [PubMed] [Google Scholar]
- Thimm D.; Funke M.; Meyer A.; Muller C. E. 6-Bromo-8-(4-[(3)H]methoxybenzamido)-4-oxo-4H-chromene-2-carboxylic Acid: a powerful tool for studying orphan G protein-coupled receptor GPR35. J. Med. Chem. 2013, 56, 7084–99. 10.1021/jm4009373. [DOI] [PubMed] [Google Scholar]
- Wei L.; Wang J. X.; Zhang X. L.; Wang P.; Zhao Y. P.; Li J. Q.; Hou T.; Qu L. L.; Shi L. Y.; Liang X. M.; Fang Y. Discovery of 2H-Chromen-2-one Derivatives as G Protein-Coupled Receptor-35 Agonists. J. Med. Chem. 2017, 60, 362–372. 10.1021/acs.jmedchem.6b01431. [DOI] [PubMed] [Google Scholar]
- Ferrie A. M.; Wu Q.; Fang Y. Resonant waveguide grating imager for live cell sensing. Appl. Phys. Lett. 2010, 97, 3. 10.1063/1.3522894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. Y.; Hu H. B.; Fang Y. Tyrphostin analogs are GPR35 agonists. FEBS Lett. 2011, 585, 1957–1962. 10.1016/j.febslet.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Deng H. Y.; Fang Y. Discovery of nitrophenols as GPR35 agonists. MedChemComm 2012, 3, 1270–1274. 10.1039/c2md20210g. [DOI] [Google Scholar]
- Deng H. Y.; Fang Y. Synthesis and Agonistic Activity at the GPR35 of 5,6-Dihydroxyindole-2-carboxylic Acid Analogues. ACS Med. Chem. Lett. 2012, 3, 550–554. 10.1021/ml300076u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. P.; Price D. A. Role of physicochemical properties and ligand lipophilicity efficiency in addressing drug safety risks. Annu. Rep. Med. Chem. 2010, 45, 380–391. 10.1016/S0065-7743(10)45023-X. [DOI] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Chen K.; Sharp K. A.; Kollman P. A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9997–10002. 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2012, 64, 4–17. 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.