

Abstract

There is a significant unmet medical need for more efficacious and rapidly acting antidepressants. Toward this end, negative allosteric modulators of the N-methyl-d-aspartate receptor subtype GluN2B have demonstrated encouraging therapeutic potential. We report herein the discovery and preclinical profile of a water-soluble intravenous prodrug BMS-986163 (6) and its active parent molecule BMS-986169 (5), which demonstrated high binding affinity for the GluN2B allosteric site (Ki = 4.0 nM) and selective inhibition of GluN2B receptor function (IC50 = 24 nM) in cells. The conversion of prodrug 6 to parent 5 was rapid in vitro and in vivo across preclinical species. After intravenous administration, compounds 5 and 6 have exhibited robust levels of ex vivo GluN2B target engagement in rodents and antidepressant-like activity in mice. No significant off-target activity was observed for 5, 6, or the major circulating metabolites met-1 and met-2. The prodrug BMS-986163 (6) has demonstrated an acceptable safety and toxicology profile and was selected as a preclinical candidate for further evaluation in major depressive disorder.
Keywords: BMS-986163, BMS-986169, GluN2B, negative allosteric modulator, ketamine, depression, preclinical candidate
Major depressive disorder (MDD) is a debilitating psychiatric condition affecting more than 350 million people worldwide.1 Treatment typically includes a combination of psychotherapy and antidepressant medications. Most of the commonly prescribed antidepressants modulate monoamine neurotransmitters and can require several weeks of repeat dosing to reach full therapeutic effect.2 Unfortunately, 20–30% of MDD patients fail to achieve an adequate response to multiple courses of drug therapy and may be diagnosed with treatment-resistant depression (TRD).3 Options for managing TRD can include switching medications, combination therapy, and augmentation with atypical antipsychotics.4 Each option can take months to adequately explore and may not prove effective. Consequently, there is clear and present need for the discovery of more efficacious and rapidly acting antidepressants.
Ketamine, a clinical anesthetic, has received a great deal of attention as an investigational treatment for TRD.5 Over the past two decades, seven clinical trials have demonstrated that intravenous (i.v.) infusion of ketamine at a subanesthetic dose can impart rapid, robust, and sustained antidepressant effects in treatment resistant patients.6 While these findings have fueled off-label clinical use, (±)-ketamine has not been reviewed or approved by the FDA for use as an antidepressant. Ongoing trials may provide additional support for expanded labeling, but the dissociative side effects experienced after ketamine infusion, along with its abuse potential, are significant hurdles.7 Importantly, a nonracemic form of ketamine, S-ketamine (esketamine), has received breakthrough therapy designations with the FDA for both MDD and TRD indications. Esketamine has recently demonstrated robust and durable antidepressant efficacy as adjunctive therapy for patients with TRD and is currently being evaluated in six ongoing phase 3 trials.8
While the anesthetic properties of ketamine are attributed to noncompetitive N-methyl-d-aspartate (NMDA) receptor binding, the neuropharmacology of its antidepressant activity remains under discussion.9 A substantial body of research suggests the mechanism of action may involve (i) initial NMDA receptor antagonism producing a glutamate surge, (ii) activation of AMPA receptors, and (iii) enhanced synaptogenesis in the prefrontal cortex (PFC), resulting from activation of the mTOR pathway.10 Other preclinical research has attributed the antidepressant activity of ketamine to early and sustained activation of AMPA receptors by one of its metabolites, (2R,6R)-hydroxynorketamine.11
A variety of NMDA receptor subtypes with distinct pharmacology and unique expression profiles coexist in the central nervous system.12 Consequently, the pursuit of subtype-selective modulators having ketamine-like activity and reduced liabilities has been an active area of research.5 With respect to GluN2B-subtype receptors, significant evidence including gene deletion experiments in rodents,13 reduced expression levels in the PFC of MDD patients,14 and pharmacological inhibition in rodent models of depression has made GluN2B a compelling target for antidepressant therapy.15 Toward this end, many GluN2B-selective negative allosteric modulators (NAMs), including compounds 1–4, have been reported in the literature (Figure 1).16 In a proof-of-concept study, Traxoprodil (1) as adjunctive therapy for TRD has demonstrated a 60% response rate, with 78% of responders having maintained a response for at least 1 week.17 Despite these encouraging results, the development of Traxoprodil was eventually terminated due to cardiovascular safety concerns.18 In a pilot trial for MDD, CERC-301 (formally, MK-0657) showed antidepressant effects as determined by the Hamilton Depression Rating Scale, but statistical significance was not achieved using the Montgomery–Aberg Depression Rating Scale.19 CERC-301 has fast-track designation for MDD and is continuing in Phase II studies. In this Letter, we disclose medicinal chemistry efforts that culminated in the discovery of an optimized GluN2B NAM BMS-986169 (5) and its water-soluble prodrug BMS-986163 (6).20
Figure 1.

Negative allosteric modulators of GluN2B 1–5 and water-soluble phosphate prodrug BMS-986163 (6).
The ligand-based pharmacophore model for Ifenprodil-like GluN2B NAMs consists of two nonpolar aromatic rings connected by a linker that typically contains a basic nitrogen and spans 9–11 Å.21 Guided by this model, we designed and prepared a variety of novel GluN2B NAMs with heteroatom containing linkers having nonplanar conformational restrictions that might impart superior selectivity and retain drug-like properties. Incorporation of two independent ring constraints produced the piperidinyl pyrrolidinone lead BMT-108908 (7, Table 1). This compound displayed potent rat GluN2B binding affinity (Ki = 1.4 nM) and robust inhibition of GluN2B receptor function (IC50 = 4.2 nM) in oocytes. BMT-108908 also demonstrated dose-dependent ex vivo GluN2B receptor occupancy (60 and 95%) in mouse brain after i.v. dosing (0.3 and 1 mg/kg, respectively). In the mouse forced swim test (mFST), a model commonly used for the evaluation of antidepressant efficacy, BMT-108908 produced a dose-dependent decrease in immobility with a minimum effective dose (MED) of 0.56 mg/kg (i.v.).22 At the conclusion of the mFST MED experiment, the average plasma concentration of BMT-108908 was 150 nM and brain GluN2B receptor occupancy was 75%. Data from this study and others revealed that GluN2B receptor occupancy levels in excess of 70% correlated with significant immobility in the mFST. Despite efforts to impart target selectivity through conformational restriction, safety characterization of BMT-108908 revealed significant inhibition of hERG channels (IC50 = 620 nM). Given a narrow multiple of 34-fold for plasma free drug concentration at the MED versus the hERG IC50, further development of BMT-108908 was halted. Continued medicinal chemistry efforts focused on ameliorating hERG inhibition by modification of BMT-108908 and related molecules through rational design.
Table 1. Data for BMT-108908.
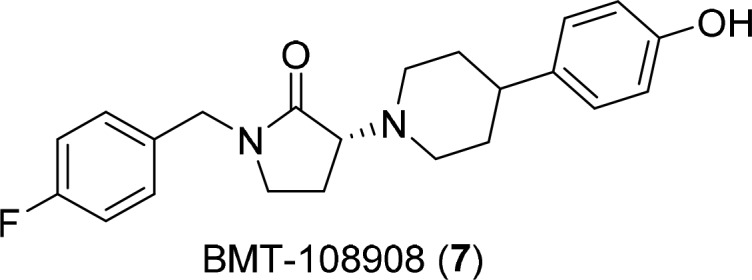
| assay | result | |
|---|---|---|
| rat forebrain GluN2B Ki (nM) | 1.4 ± 0.5 | |
| GluN2B oocyte EP IC50 (nM) | 4.2 | |
| mouse occupancy study dose (mg/kg, i.v.) | 0.3, | 1 |
| ex vivo GluN2B occupancy (%) | 60, | 95 |
| total plasma conc. of 7 (nM) | 70 ± 5, | 340 ± 35 |
| mFST MED (mg/kg, i.v.) | 0.56 | |
| ex vivo GluN2B occupancy (%) | 75 | |
| total plasma conc. of 7 (nM) | 150 ± 70 | |
| plasma free fraction:human, rat, mouse (%) | 10, 11, 12 | |
| pKa (protonated amine), spectrophotometric | 6.8 | |
| hERG PC IC50 (nM) | 620 | |
The classic hERG motif,23 which includes a basic nitrogen flanked by aromatic or hydrophobic groups, overlaps considerably with the GluN2B pharmacophore.21 Indeed, achieving selectivity against hERG had been a clear objective of many past efforts to develop Ifenprodil-like GluN2B NAMs.15 To disrupt interactions of the basic nitrogen in the piperidinyl pyrrolidinone scaffold with hERG residues, we explored both steric and electronic modulation of the piperidine ring. Ultimately, inductive effects of β-fluorine atom substitution to reduce the basicity of the piperidine was most effective in attenuating hERG activity. The exercise was undertaken in context of the 4-methylbenzyl substituted pyrrolidinone analog 8, which demonstrated GluN2B binding (Ki = 1.4 nM), receptor occupancy (92% at 1 mg/kg, i.v.), and hERG inhibition (IC50 = 400 nM) similar to BMT-108908 (Table 2). All new analogs were screened for GluN2B binding affinity and ex vivo receptor occupancy in mice. The occupancy data served as a higher throughput translational surrogate for the mFST. The 3-monofluoro- and 3,3-difluoropiperidinyl diastereomers 5 and 9–12 demonstrated strong GluN2B binding affinity that was within 6-fold of the parent molecule 8. Importantly, compounds 5, and 10−12 all exhibited weak hERG inhibition (IC50’s > 7.8 μM) and high ex vivo brain GluN2B occupancy (87–95%, 3 mg/kg, i.v.). Overall, BMS-986169 (5) with (3S,4S) stereochemistry displayed the best combination of potent target affinity (Ki = 4.0 nM), high occupancy (95% at 3 mg/kg, i.v.), and weak hERG inhibition (IC50 = 28 μM). Incorporation of the (3S,4S)-fluoropiperidine motif into the prior lead BMT-108908 produced compound 13, which had slightly inferior binding affinity and no advantages compared to BMS-986169.
Table 2. Screening Data for Fluorinated Piperidinyl Pyrrolidinones 5 and 8–13.

Single most active diastereomer of all four isomers tested; stereochemistry was undetermined.
In light of the encouraging results with BMS-986169, this compound was subjected to rigorous preclinical characterization.20 In addition to potent rat brain GluN2B binding, compound 5 maintained high affinity for GluN2B receptors in brain preparations from monkey and human (Table 3).20 BMS-986169 also demonstrated potent and selective functional inhibition of GluN2B in comparison to other NMDA receptor subtypes, including GluN2A, GluN2C, GluN2D.20 In vitro screening of BMS-986169 against a broad panel of G-protein coupled receptors (including adrenergic α1), ion channels, nuclear hormone receptors, and enzymes revealed no other significant activity.20 BMS-986169 was negative in an exploratory AMES study. Serum protein binding of BMS-986169 was within a 2-fold multiple across species with average free fractions between 6 and 12%. BMS-986169 did not exhibit potent inhibition of cytochrome P450 enzymes in human liver microsomes (multiple CYPs IC50 > 18 μM), nor did it demonstrate time-dependent CYP inhibition. BMS-986169 was not a substrate for the Caco2 efflux transporter. With respect to physiochemical properties, BMS-986169 has a low polar surface area of 44 Å2 and a LogD = 2.9, which are properties consistent with good brain uptake. The pKa of 4.4 for the protonated piperidine nitrogen was approximately 2 log units lower than the piperidine of BMT-108908 (7, Table 1) and reflects the inductive effects of fluorine substitution. The melting point of crystalline BMS-986169 was 186 °C, and the aqueous solubility at pH 7.4 was 2 μg/mL.
Table 3. Detailed Profile for BMS-986169 (5).
| assay | result |
|---|---|
| GluN2B binding Ki (nM) | |
| cyno cortex | 4.2 ± 0.8 |
| human cortex | 6.3 ± 1.5 |
| GluN2B oocyte EP IC50 (nM) | 24 nM |
| GluN2A/2C/2D oocyte EP at 3 μM (% inhibition) | –2.4, 8.6, 7 |
| AMES result | negative |
| metabolic stability T1/2: human, rat mouse, cyno, dog (min) | 25, 2.7, 3.5, 12, 6.0 |
| plasma free fraction: human, rat, mouse, cyno, dog (%) | 6, 9, 12, 11, 7 |
| CYP 450 HLM inhibition IC50 (μM)a | >18 |
| Caco-2, efflux ratio | 1.1 |
| PSA (Å2) | 44 |
| pKa, spectrophotometric (protonated amine) | 4.4 |
| shake flask log D at pH 7.4b | 2.9 |
| melting point (°C) | 186 |
| aqueous solubility at pH 7.4 (mg/mL) | 0.002 |
| mFST MED (mg/kg, i.v.) | 1 |
| GluN2B occupancy (%) | 73 |
| total plasma conc. of 5 (nM) | 270 ± 130 |
| brain conc. 5 /plasma conc. 5 | 2.8 |
| i.v. pharmacokineticsc | speciesd |
|||
|---|---|---|---|---|
| parameter | mouse | rat | monkey | dog |
| t1/2 (h) | 3.6 | 0.9 | 1.5 | 2.1 |
| CL (mL/min/kg) | 75 | 54 | 15 | 17 |
| Vss (L/kg) | 8.2 | 2.3 | 1.7 | 2.1 |
| AUClast (nM·h) | 530 | 1,300 | 2,700 | 2,500 |
Panel of CYP human liver microsomes: 3A4-BFC and BZR, 1A2, 2B6, 2C8, 2C9, 2C19, 2D6.
Octanol/water partitioning at pH 7.4.
1 mg/kg, 5 min i.v. infusion, vehicle = 10% DMAC, 10% EtOH, 30% HPBCD, 50% water.
CD1 mice, Sprague–Dawley rat, male Cynomolgus monkey, male Beagle dog.
In the mFST, BMS-986169 demonstrated an MED of 1.0 mg/kg (i.v.) at 73% ex vivo GluN2B receptor occupancy and an average terminal plasma concentration of 270 nM (Table 3).20 The brain-to-plasma ratio in mice was favorable at 2.8. In context of plasma free drug concentration at the MED versus hERG IC50, the multiple was 875-fold. In a follow-up cardiovascular safety assessment, BMS-986169 did not produce an increase in QTcB in anesthetized guinea pig after i.v. administration at all doses tested (2, 5, and 10 mg/kg) and with free drug plasma concentrations up to 19 μM. Consistent with ketamine-like synaptic strengthening,24 BMS-986169 demonstrated enhanced ex vivo hippocampal long-term potentiation (LTP) that was sustained for 24 h after dosing in rat.20 In clear differentiation from ketamine, BMS-986169 did not produce dissociative effects in monkeys and only displayed marginal effects on locomotor hyperactivity in mice.20
The metabolic stability of BMS-986169 was poor in rodent liver microsomes (T1/2 = 2.7–3.5 min) and marginally better in microsomes from higher species (T1/2 = 6.0–25 min) (Table 3). Biotransformation studies in microsomes and hepatocytes revealed rapid turnover of BMS-986169 to greater than 15 unique metabolites. Across species, the prominent metabolites were the carboxylic acid met-1 and N–H lactam met-2 (Figure 2). These metabolites resulted from oxidation of the p-tolyl methyl group and N-dealkylation of the lactam, respectively. The structures of both metabolites were confirmed through independent syntheses. The metabolites did not show in vitro activity against GluN2A-D nor a broad panel of other pharmacologically relevant targets.25
Figure 2.

Two major metabolites of BMS-986169 (5).
In terms of i.v. pharmacokinetics, BMS-986169 demonstrated high clearance in rodents and moderate clearance in higher species (Table 3). Volume of distribution was high in mouse but moderate in rat, monkey, and dog. The drug plasma half-life was short across all species. Total drug exposure (AUClast) was at least 2-fold better in monkey and dog than rodents. In a rat PK study at 5 mg/kg (p.o.), BMS-986169 displayed poor oral bioavailability (F = 2.5%). The observed PK parameters of BMS-986169 supported an intravenous route of administration for which >70% GluN2B receptor occupancy would be targeted for a transient period defined by the dose and infusion rate in a clinical setting.
While the aqueous solubility of BMS-986169 was sufficient to support low-dose i.v. preclinical experiments, formulation and dose escalation for toxicology studies and potential clinical investigations was not feasible. To address the issue, we explored solubilizing prodrug modifications of the phenol group present in BMS-986169. It was paramount to this strategy that the prodrug be rapidly converted to parent after i.v. administration to recapitulate the exposure profile observed upon i.v. dosing of BMS-986169. While several phenolic prodrugs of BMS-986169 were prepared, the dihydrogen phosphate BMS-986163 (6) exhibited optimal properties for an intravenous agent. The prodrug 6 was a zwitterionic monohydrate with a sharp melting point of 172 °C. Crystalline material had aqueous solubility of 19.9 mg/mL at pH 7.4. The prodrug 6 was physically and chemically stable in the solid state at 50 °C for up to 4 weeks. Aqueous solutions of 6 were stable for up to 90 days at 25 °C. The prodrug itself demonstrated no relevant in vitro biological activity. Prodrug to parent conversion was rapid in rat, monkey, dog, and human blood at 37 °C. After i.v. administration of prodrug 6 (1.2 mg/kg) in Cynomolgous monkey, prodrug concentrations in the plasma were undetectable 10 min postdose, and the plasma AUCtotal exposure of active parent 5 was approximately 63% of that observed from direct administration of 5 (1.0 mg/kg, i.v.).26 The loss in exposure was not accounted for, but it may be attributed to metabolism of the prodrug prior to phosphate cleavage. Exposure data for the metabolites met-1 and met-2 was collected in the monkey PK study. The carboxylic acid met-1 displayed a plasma AUCtotal that was 36% of parent 5 and AUCtotal for the lactam met-2 was 130% of parent. While in vivo conversion of prodrug to parent was not quantitative, the high solubility of 6 and its rapid conversion to 5 supported additional in vivo studies and dose escalating toxicology experiments. Specific to on-target pharmacology, the prodrug 6 produced robust levels of ex vivo GluN2B receptor occupancy in rat, in vivo functional inhibition of [3H]MK-801 binding in rat, and dose-dependent activity in novelty-suppressed feeding (NSF) in mice after i.v. dosing.20
The safety and tolerability of BMS-986163 (6) was evaluated in two studies. In a 4-day rat experiment, where prodrug 6 was dosed at 10, 30, and 100 mg/kg/day (i.v.), there were no significant clinical-pathological and histopathological observations at any dose. An average total plasma exposure of parent 5 in excess of 48 μM·h was observed at day 4 for the 100 mg/kg/day dose group. In a monkey single-dose toxicokinetic tolerability study at 3.6, 12, and 36 mg/kg (i.v.), all doses were well tolerated with no significant drug related effects. The high dose group exhibited an average Cmax = 19 μM and total plasma AUC24 = 88 μM·h for parent 5.
The chemical syntheses of BMS-986169 (5) and its corresponding prodrug BMS-986163 (6) are outlined in Scheme 1. Suzuki coupling of the commercial boronate ester 14 with 4-benzyloxybromobenzene, followed by hydroboration/oxidation of the resulting product 15 provided the racemic trans-hydroxypiperidine (±)-16. The first eluting enantiomer (S,S)-16, obtained from preparative chiral supercritical fluid chromatography (SFC), was fully deprotected to afford the phenolic piperidine (S,S)-17. The bromopyrrolidinone (±)-21, obtained in two steps from benzylamine 18, was coupled with (S,S)-17 to provide hydroxypiperidine 22 as a mixture of diastereomers. Retentive deoxyfluorination of 22 and separation of the lactam diastereomers provided compound 23 and BMS-986169 (5). The relative and absolute stereochemistry of BMS-986169 was unambiguously established through single crystal X-ray diffraction using anomalous scattering refinement.27 Preparation of the prodrug BMS-986163 (6) was accomplished through reaction with phosphorus oxychloride and hydrolytic workup.28
Scheme 1. Synthesis of GluN2B NAM BMS-986169 (5) and Prodrug BMS-986163 (6).

Reagents: (a) 1-(benzyloxy)-4-bromobenzene, [(PPh3)P]2PdCl2, Na2CO3, DME/H2O (82%); (b) BF3OEt2, NaBH4, NaOH, H2O2, THF/EtOH (87%); (c) SFC: Lux Cellulose-2 (43%); (d) H2, Pd/C, MeOH (85%); (e) HCl dioxane/MeOH (87%); (f) 2,4-dibromobutanoyl chloride, TEA, Et2O (95%); (g) NaH, THF (50%); (h) DIEA, DMF (74%); (i) Deoxofluor, CH2Cl2 (27%); (j) SFC: Chiralpak AD (48%); (k) pyridine, DMAP, POCl3, then H2O (5%).
In summary, rational design efforts afforded a novel class of conformationally restricted, piperidinyl pyrrolidinone GluN2B NAMs. Fluorine substitution of the piperidine core reduced nitrogen basicity and ameliorated hERG inhibition of this chemotype. The optimized lead BMS-986169 (5) demonstrated single digit nanomolar affinity for GluN2B receptors, selective functional inhibition of GluN2B receptors in cells, high brain GluN2B receptor occupancy in rodents, and significant in vivo activity in rodent behavioral models used for the evaluation of antidepressant drugs. The poor aqueous solubility of BMS-986169 was addressed through preparation of the water-soluble phosphate prodrug BMS-986163 (6). The prodrug was stable in solution but was rapidly converted to parent after i.v. administration. The prodrug was safe and well tolerated in rat and monkey toxicology studies. The phosphate prodrug BMS-986163 (6) is an optimized agent for the delivery of GluN2B receptor NAM BMS-986169 (5) and merits further evaluation in MDD and TRD.
Acknowledgments
We thank Mathiazhagan Annadurai and Poornima Sheety for synthetic chemistry efforts.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00080.
Experimental details for synthetic procedures and associated chemical data for compounds 5–23, pharmacological screening data, and biological methods (PDF)
Author Present Address
∥ Editas Medicine, Inc., Cambridge, Massachusetts 02141, United States.
Author Present Address
⊥ Sanofi, Waltham, Massachusetts 02451, United States.
Author Present Address
# Sunovion Pharmaceuticals, Marlborough, Massachusetts 01752, United States.
Author Present Address
∇ Fulcrum Therapeutics, Cambridge, Massachusetts 02139, United States.
Author Present Address
○ King Abdullah International Med Research Center, KSAU-HS, Ministry of National Guard-Health Affairs, Riyadh, Saudi Arabia.
Author Contributions
◆ Senior contributing author. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ferrari A. J.; Charlson F. J.; Norman R. E.; Patten S. B.; Freedman G.; Murray C. J. L.; Vos T.; Whiteford H. A. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013, 10, e1001547. 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racagni G.; Popoli M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin Neurosci 2008, 10, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush A. J.; Trivedi M. H.; Wisniewski S. R.; Nierenberg A. A.; Stewart J. W.; Warden D.; Niederehe G.; Thase M. E.; Lavori P. W.; Lebowitz B. D.; McGrath P. J.; Rosenbaum J. F.; Sackeim H. A.; Kupfer D. J.; Luther J.; Fava M. Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am. J. Psychiatry 2006, 163, 1905–1917. 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Preston T. C.; Shelton R. C. Treatment resistant depression: strategies for primary care. Curr. Psychiatry Rep 2013, 15, 370. 10.1007/s11920-013-0370-7. [DOI] [PubMed] [Google Scholar]
- Lener M. S.; Kadriu B.; Zarate C. A. Jr Ketamine and Beyond: Investigations into the Potential of Glutamatergic Agents to Treat Depression. Drugs 2017, 77, 381–401. 10.1007/s40265-017-0702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport D. J.; Carpenter L. L.; McDonald W. M.; Potash J. B.; Tohen M.; Nemeroff C. B. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am. J. Psychiatry 2015, 172, 950–966. 10.1176/appi.ajp.2015.15040465. [DOI] [PubMed] [Google Scholar]
- Zhang M. W.; Harris K. M.; Ho R. C. Is off-label repeat prescription of ketamine as a rapid antidepressant safe? Controversies, ethical concerns, and legal implications. BMC Med. Ethics 2016, 17, 4. 10.1186/s12910-016-0087-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly E. J.; Fedgchin M.; Manji H.; Drevets W. C.; Singh J. B.; Cooper K.; Lim P.; Shelton R. C.; Thase M. E.; Winokur A.; Van Nueten L. Efficacy and Safety of Intranasal Esketamine Adjunctive to Oral Antidepressant Therapy in Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2018, 75, 139–148. 10.1001/jamapsychiatry.2017.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler M. W.; Yourish H. B.; Ionescu D. F.; Haggarty S. J. Classics in Chemical Neuroscience: Ketamine. ACS Chem. Neurosci. 2017, 8, 1122–1134. 10.1021/acschemneuro.7b00074. [DOI] [PubMed] [Google Scholar]
- Abdallah C. G.; Adams T. G.; Kelmendi B.; Esterlis I.; Sanacora G.; Krystal J. H. Ketamine’s Mechanism of Action: A Path to Rapid-Acting Antidepressants. Depression Anxiety 2016, 33, 689–697. 10.1002/da.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P.; Moaddel R.; Morris P. J.; Georgiou P.; Fischell J.; Elmer G. I.; Alkondon M.; Yuan P.; Pribut H. J.; Singh N. S.; Dossou K. S. S.; Fang Y.; Huang X.-P.; Mayo C. L.; Wainer I. W.; Albuquerque E. X.; Thompson S. M.; Thomas C. J.; Zarate C. A. Jr.; Gould T. D. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature (London, U. K.) 2016, 533, 481–486. 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P.; Bellone C.; Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Miller O. H.; Yang L.; Wang C.-C.; Hargroder E. A.; Zhang Y.; Delpire E.; Hall B. J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 2014, 3, e03581. 10.7554/eLife.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyissa A. M.; Chandran A.; Stockmeier C. A.; Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 70–75. 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppa K. B.; King D.; Olson R. E. NMDA antagonists of GluN2B subtype and modulators of GluN2A, GluN2C, and GluN2D subtypes-recent results and developments. Annu. Rep. Med. Chem. 2012, 47, 89–103. 10.1016/B978-0-12-396492-2.00007-2. [DOI] [Google Scholar]
- Mony L.; Kew J. N. C.; Gunthorpe M. J.; Paoletti P. Allosteric modulators of NR2B-Containing NMDA receptors: Molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–1317. 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn S. H.; Baker B.; Kolluri S.; Menniti F. S.; Krams M.; Landen J. W. An Innovative Design to Establish Proof of Concept of the Antidepressant Effects of the NR2B Subunit Selective N-Methyl-D-Aspartate Antagonist, CP-101,606, in Patients With Treatment-Refractory Major Depressive Disorder. J. Clin. Psychopharmacol. 2008, 28, 631–637. 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R.; Henter I. D.; Zarate C. A. Jr. New targets for rapid antidepressant action. Prog. Neurobiol. (Oxford, U. K.) 2017, 152, 21–37. 10.1016/j.pneurobio.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L.; Diaz Granados N.; Jolkovsky L.; Brutsche N.; Luckenbaugh D. A.; Herring W. J.; Potter W. Z.; Zarate C. A. Jr A Randomized, Placebo-Controlled, Crossover Pilot Trial of the Oral Selective NR2B Antagonist MK-0657 in Patients With Treatment-Resistant Major Depressive Disorder. J. Clin. Psychopharmacol. 2012, 32, 551–557. 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow L. J.; Gulia J.; Weed M. R.; Srikumar B. N.; Li Y.-W.; Graef J. D.; Naidu P. S.; Sanmathi C.; Aher J.; Bastia T.; Paschapur M.; Kalidindi N.; Kumar K. V.; Molski T.; Pieschl R.; Fernandes A.; Brown J. M.; Sivarao D. V.; Newberry K.; Bookbinder M.; Polino J.; Keavy D.; Newton A.; Shields E.; Simmermacher J.; Kempson J.; Li J.; Zhang H.; Mathur A.; Kallem R. R.; Sinha M.; Ramarao M.; Vikramadithyan R. K.; Thangathirupathy S.; Warrier J.; Islam I.; Bronson J. J.; Olson R. E.; Macor J. E.; Albright C. F.; King D.; Thompson L. A.; Marcin L. R.; Sinz M. Preclinical Characterization of (R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one (BMS-986169), a Novel, Intravenous, Glutamate N-Methyl-d-Aspartate 2B Receptor Negative Allosteric Modulator with Potential in Major Depressive Disorder. J. Pharmacol. Exp. Ther. 2017, 363, 377–393. 10.1124/jpet.117.242784. [DOI] [PubMed] [Google Scholar]
- Burger P. B.; Yuan H.; Karakas E.; Geballe M.; Furukawa H.; Liotta D. C.; Snyder J. P.; Traynelis S. F. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol. Pharmacol. 2012, 82, 344–359. 10.1124/mol.112.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Table S1 in the Supporting Information for BMT-108908 full dose response data in mFST.
- Matyus P.; Borosy A. P.; Varro A.; Papp J. G.; Barlocco D.; Cignarella G. Development of pharmacophores for inhibitors of the rapid component of the cardiac delayed rectifier potassium current. Int. J. Quantum Chem. 1998, 69, 21–30. . [DOI] [Google Scholar]
- Graef J. D.; Newberry K.; Newton A.; Pieschl R.; Shields E.; Luan F.-n.; Simmermacher J.; Luchetti D.; Schaeffer E.; Li Y.-W.; Kiss L.; Bristow L. J. Effect of acute NR2B antagonist treatment on long-term potentiation in the rat hippocampus. Brain Res. 2015, 1609, 31–39. 10.1016/j.brainres.2015.03.019. [DOI] [PubMed] [Google Scholar]
- See Table S2 in the Supporting Information for assay targets and corresponding profiling data.
- See Table S4 in the Supporting Information for the monkey PK profile of parent 5 after i.v. administration of prodrug 6.
- Full crystallographic data have been deposited to the Cambridge Crystallographic Data Center (CCDC reference number 1831022). Copies of the data can be obtained free of charge via the Internet at www.ccdc.cam.ac.uk/.
- For an optimized synthesis of BMS-986163 and mechanistic rationale for the retentive deoxyfluorination step see:; Kempson J.; Zhang H.; Wong M. K. Y.; Li J.; Li P.; Wu D.-R.; Rampulla R.; Galella M. A.; Dabros M.; Traeger S.; Vetrichelvan M.; Gupta A.; Nayagam A. P.; Islam I.; Thangathirupathy S.; Warrier J.; Macor J. E.; Thompson L. A.; Marcin L. R.; Mathur A.. The Evolution of a Practical Synthesis to a Potent NR2B Inhibitor and its Prodrug. Submission pending. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.