

Abstract
Gap junctions (GJs) are membrane-spanning channels that allow for the movement of small molecules across cell membranes. Connexin43 (Cx43) is the predominant GJ protein in bone. In vitro studies suggest that gap junctional intercellular communication (GJIC) sensitizes bone cells to mechanical signals. Additionally, mechanical signals detected by osteocytes are communicated to osteoblasts via GJIC, and osteocytic Cx43 hemichannels release anabolic factors, such as PGE2 and ATP, in response to mechanical load. These findings and others have led to near consensus among researchers in the field that GJIC, hemichannels or connexins facilitate the anabolic response of bone to mechanical load and, in their absence, bone would be less sensitive to load. However, recent in vivo evidence suggests the opposite is true. Studies from our laboratory and others demonstrate that Cx43-deficient mice have an increased anabolic response to mechanical load and are protected against the catabolic effects of mechanical unloading. These developments suggest a paradigm shift in our understanding of connexins, GJIC, and mechanotransduction in bone. That is, inhibiting bone cell Cx43 expression or GJIC has a beneficial effect on bone’s response to its mechanical environment, preserving bone during unloading and enhancing its formation during loading. Here, we review literature in support of this hypothesis and suggest a mechanism by which Cx43, through interaction with WNT/β-catenin signaling, moderates both arms of bone remodeling.
Keywords: BONE CELLS, GAP JUNCTIONS, ANIMAL MODELS, AGING
Introduction
Gap junctions (GJs) are membrane-spanning channels that allow for the passage of small molecules between adjacent cells or between the cell and extracellular environment when unopposed as hemichannels.(1–4) An intercellular GJ is composed of two hemichannels termed connexons, which are in turn composed of six subunits termed connexins.(5) Connexins are typically named according to their molecular mass in kilodaltons. More than 20 mammalian connexins have been identified,(6) and the predominant connexin in skeletal tissue is Connexin43 (Cx43). However, bone cells also express Connexin45 (Cx45)(7) and Connexin46 (Cx46), but Cx46 is trapped in the Golgi and does not contribute to intercellular communication.(8) Although functional connexin redundancy has been identified to a very limited degree, in some cell types(9) the functional significance in bone cells is not known. Much work has focused on the role of Cx43 in the activity of osteoblasts and osteocytes,(10) but recent studies have also identified a role for Cx43 in the function of osteoclasts(11,12) and chondrocytes.(13) In addition to functioning in GJ channels or hemichannels, connexins, including Cx43, can also regulate cell behavior by interacting with intercellular signaling molecules.(14)
Cx43 has an integral role in the maintenance of skeletal homeostasis and the activity of mature bone cells.(15–17) However, Cx43 is also a critical factor during skeletal development, as demonstrated by humans with mutations in the Cx43 gene GJA1.(18) These individuals develop a disease called oculodentodigital dysplasia (ODDD), characterized by dental anomalies, including small teeth with enamel hypoplasia, syndactyly, craniofacial abnormalities, missing phalanges of the toes, and the development of a tubular bone morphology.(19) Several animal models have been developed in an attempt to recapitulate the clinical presentation of ODDD. Flenniken and colleagues developed a mouse model containing a point mutation in murine Gja1 that results in production of a mutant protein that acts in a dominant negative fashion to disrupt assembly of GJs composed of Cx43. This model is able to recapitulate many of the classical features of ODDD but also demonstrates cortical thinning, reduced bone mineral density (BMD), and reduced bone strength.(20) Dobrowolski and colleagues inserted a human Cx43G138R point mutation into Gja1.(21) These transgenic mice had a skeletal phenotype characterized by reduced trabecular bone mass and cranial abnormalities. Finally, Watkins and colleagues utilized the Dermo/Twist2 (DM1) promoter, which is expressed during endochondral ossification, to introduce an ODDD mutation into the chondro-osteogenic lineage.(22) These mice were significantly osteopenic, with cortical thinning, expanded marrow cavities, and reduced femur strength but no trabecular phenotype. Cx43 has also been shown to play a role in the development of limb patterning and growth in mice.(23) Inhibition of Cx43 expression, using antisense nucleotides in chick embryos, resulted in a significant decrease in bone formation, whereas zebrafish studies indicate a role for Cx43 in joint positioning.(24) Taken together, these studies demonstrate that Cx43 has a critical role both during skeletal development as well as in the function of mature bone cells.
One suggested role for GJs in mature bone is a contribution to mechanotransduction. For instance, mechanical signals regulate Cx43 levels and GJIC between bone cells;(25–30) GJIC sensitizes bone cell networks to diverse extracellular signals;(31–34) and mechanically induced signals are communicated among bone cells via gap junctions.(10,35,36) This has led to near consensus among researchers in the field that GJIC, hemichannels, or connexins facilitate the anabolic response of bone to mechanical load.(1–4) Indeed, the paradigm that has emerged is that in the absence of connexins and/or GJIC, bone would lose sensitivity to the anabolic effects of mechanical load.(1–4) However, recent developments challenge this and suggest a new paradigm. Here, we describe this new paradigm, review the data supporting this paradigm, and suggest a mechanism underlying this paradigm. We begin with the role of connexins in bone cell differentiation.
Cx43 and Bone Cell Differentiation
Abundant data from in vitro studies have demonstrated that Cx43 expression and GJIC parallel osteoblastic differentiation, and inhibiting GJIC and Cx43 expression in osteoblastic cells, including MC3T3-E1, UMR-106, ROS 17/2.8, human primary culture osteoblastic cells, and murine calvarial cells, with pharmacological agents or genetic manipulation results in decreased expression of phenotypic characteristics of differentiated osteoblasts including alkaline phosphatase, osteocalcin, bone sialoprotein, and PTH responsiveness.(37–43) Furthermore, mineralized extracellular matrix synthesis was impaired in calvarial osteoblasts isolated from Cx43-deficient mice.(44) Additionally, Schiller and colleagues(45) showed that inhibiting GJIC induces the transdifferentiation of osteoblastic MC3T3-E1 cells and primary culture human osteoblastic cells to an adipocytic phenotype. More recently, Inose and colleagues used a microRNA approach to target Cx43, resulting in decreased alkaline phosphatase activity and Bglap (encodes Osteocalcin) expression, whereas restoration of Cx43 rescued osteoblastic differentiation.(46) Cx43 is also important in cell proliferation(47) and survival.(48) Gramsch and colleagues transfected osteoblastic UMR 106 cells with Cx43. At baseline, these cells have high Cx45 expression and low Cx43 expression. A seven-fold increase in proliferation and increased size of mineralized nodules were observed in cells with increased Cx43 expression.(47) Taken together, these studies strongly suggest that Cx43 and GJIC contribute to osteoblastic proliferation and differentiation in vitro.
In vivo studies suggest that GJs may be involved in cell signaling processes important to limb bud differentiation and skeletogenesis in embryonic mice(49) and contribute to cellular differentiation and intramembranous bone formation in developing chick mandible.(50,51) Furthermore, Cx43 null mice display impaired intramembranous bone formation, and osteoblastic cells from these animals express decreased levels of type 1 collagen, osteopontin, and osteocalcin,(52) suggesting a defect in osteoblastic maturation. Although these data strongly suggest that Cx43 contributes to osteoblastic differentiation, they do not exclude the possibility that other connexins or even pannexins also contribute to osteoblastic differentiation.(53)
In addition to modulating bone formation through osteoblastic maturation, Cx43 can also affect bone remodeling and osteoclast activity. Ransjo and colleagues found that Cx43 mRNA was upregulated during parathyroid hormone (PTH)-stimulated bone resorption in vitro, whereas Schilling and colleagues found increased Cx43 expression in the giant osteoclasts of patients with Paget’s disease.(54–55) Consistent with this, inhibitors of GJIC impair osteoclast formation,(55) degree of multinucleation,(54) and resorptive activity.(11,55,56) Ilvesaro and colleagues used Gap27, a synthetic connexin-mimetic peptide, to decrease GJIC and found a lower number of multinucleated, TRAP-positive osteoclasts, along with inhibition of osteoclastic pit formation.(56) We recently demonstrated that Cx43-deficient osteocytic MLO- Y4 cells have an increased receptor activator of nuclear factor-κB ligand (RANKL)/osteoprotegerin (OPG) ratio relative to wild-type (WT) osteocytes in vitro, a finding consistent with increased osteoclastic bone resorption in vivo.(12) Watkins and colleagues have shown that co-culture of osteoblasts and osteoclast precursors, both of which are Cx43 deficient, increases the number of osteoclasts per well relative to WT osteoblasts cultured with either WT or Cx43-deficient osteoclast precursors.(22) An increase in osteoclast number was also observed in Cx43-deficient osteoblasts cultured with WT osteoclast precursors, suggesting that osteoclastogenesis may be modulated by Cx43 expression in osteoblasts rather than osteoclast precursors.(22) Taken together, these data suggest that decreasing GJIC between osteoclast and themselves inhibits osteoclastogenesis, whereas decreasing Cx43 expression in osteoblast or osteocytes increases osteoclastogenesis. However, the relative contribution of each mechanism is not known. In any case, the emerging data suggest a critical role for Cx43 in both arms of bone remodeling.
In Vitro Signaling Through Cx43 Gap Junctions and Hemichannels
Apoptosis
Bisphosphonates are antiresorptive agents that maintain bone mass by inducing osteoclast apoptosis.(57) Conversely, osteocyte and osteoblast apoptosis is inhibited by bisphosphonates, a process that occurs via increased Cx43 hemichannel opening.(58,59) Interestingly, hemichannels are more important than complete GJ channels in cell survival, specifically in modulating the anti-apoptotic effects of bisphosphonates. Transfection of Cx43, but not other connexins, confers resistance to apoptosis in cells treated with bisphosphonates when cultured at a density low enough to prevent GJIC.(59) Bisphosphonates increase Cx43 interaction with Src leading to phosphorylation of Src and ERK1/2,(59) with hemichannel opening itself responsible for increased Src and ERK expression.(59) Upon ERK1/2 activation, osteoblast and osteocyte apoptosis is inhibited by the activation of an anti-apoptotic signaling cascade that includes activation of the kinase p90RSK, phosphorylation of BAD and C/EBPb, and the subsequent binding of C/EBPb to pro-caspases 1 and 8.(60) In addition to inhibiting osteoblast apoptosis, risedronate can enhance transcriptional activation of the Cx43 promoter and increase Cx43 protein expression, while concurrently stimulating osteoblastic differentiation.(61) Together, these data suggest that risedronate, and possibly other bisphosphonates, may modulate bone formation by promoting osteoblastic differentiation and inhibiting osteocyte apoptosis via Cx43.
Second messengers
Although the involvement of Cx43 and GJs in osteogenesis is clear, the small molecular signals that pass through the channels and how they affect osteoblastic differentiation is not well understood. However, previous studies demonstrated that changes in Cx43 expression alter the osteoblastic response to fibroblast growth factor 2 (FGF2) signaling by modulating the transcriptional activity of Runx2, a process that is dependent on ERK and PKCd.(62–64) Furthermore, inhibition of phospholipase Cg1 (PLCg1) attenuates Cx43-mediated, FGF2-induced signaling through Runx2.(65) Interruption of this pathway inhibits PKCd nuclear accumulation and interaction with Runx2, leading to decreased Runx2 transcriptional activity of osteoblastic genes including Col1a1, Osteocalcin (Bglap), and Osx (encodes Osterix) in MC3T3 cells. These data suggest that transmission of inositol phosphates through Cx43 GJs are important in generating signals to activate PKCd and osteogenesis.
Cytosolic calcium also transfers between cells via GJIC and can affect downstream signaling including calmodulin and calcineurin. For instance, studies from our lab demonstrate that mechanically induced calcium signal activation of calcineurin mediates the effect of mechanical signals on mesenchymal stem cell proliferation.(66) Presumably, calcium signals arriving from osteocytic cells via GJIC would have a similar effect. Finally, recent evidence suggests that microRNA can pass from one cell to another via GJIC.(67) Although this has yet to be demonstrated specifically in bone cells, it offers yet another mechanism by which bone cells might communicate with one another.
In Vitro Studies of Cx43 and Mechanotransduction
The spatial localization of osteoblastic Cx43 in areas of contact between cells(40) and at the tips of the dendritic processes in osteocytes(35) suggests an important role for GJs in the bone cell response to biophysical stimuli, including mechanical loading. In vitro studies reveal that GJIC sensitizes bone cell networks to diverse extracellular signals including load-induced signals.(32,33) Furthermore, mechanical signals detected by osteocytes can be communicated to osteoblastic cells via GJIC.(10,35) Mechanical stimulation increased Cx43 expression(26,29) and phosphorylation(26) as well as Cx43-mediated GJIC.(26,29) Conversely, Thi and colleagues showed that shear stress decreased both Cx43 protein expression at the cell membrane and GJIC in MLO-Y4 osteocytic cells.(30) Cx43 protein localization at dendritic tips of osteocytes was decreased when low-magnitude shear stress (5 dynes/cm2) was applied, relative to control cells. Cx43 expression decreased further as the magnitude of shear stress increased; however, differential effects on Cx43 and Cx45 mRNA expression were observed between low- and high-magnitude shear stresses. Cx43 mRNA was increased under low-magnitude shear stress and no change in Cx45 expression was observed, whereas Cx43 mRNA was decreased under high-magnitude shear stress, with a concomitant increase in Cx45 mRNA expression. These findings suggest a possible compensatory response by Cx45 as a result of Cx43 inhibition and indicate that the composition of GJs can be altered in response to specific mechanical stimuli.(30) However, although Cx45 upregulation may compensate for decreased Cx43 expression, the functional significance of this is unclear. Although the expression of Cx43 may vary under different mechanical stimulation conditions, there remains a clear role for Cx43 in the osteocytic response to these signals, as well as subsequent osteoblastic differentiation.(10) Taken together, these studies suggest that Cx43 and GJIC are critical to the anabolic response of bone to mechanical load.
In addition to the role of GJs in bone cell response to mechanical load, emerging evidence suggests that Cx43 hemichannels may also be a key component in the transduction of osteogenic signals in response to mechanical stimulation.(68,69) Osteocytic MLO-Y4 cells cultured at low density exhibited enhanced hemichannel activity in response to fluid flow, a response attenuated by the inhibition of hemichannel activity or by inhibition of Cx43 expression.(70) Additionally, a recent study has demonstrated that direct mechanical perturbation via magnetic beads of integrin a5β1 leads to the opening of the Cx43 hemichannels.(2) Furthermore, estradiol increases Cx43 expression in osteocytic MLO-Y4 cells, and this increases the effect of fluid flow on MAP kinase activity.(71)
Mechanistically, hemichannel activity increases the release of adenosine triphosphate (ATP), which in turn stimulates the production of the bone anabolic agent prostaglandin E2 (PGE2). Demonstrating the interdependent relationship between Cx43 and PGE2 in response to mechanical stimulation, PGE2 treatment stimulates Cx43 expression,(28) whereas Cx43 hemichannels are necessary for the release of PGE2.(72) Moreover, inhibition of PGE2 dramatically reduces Cx43 expression,(73) whereas Cx43 siRNA decreases ATP release from hemichannels and inhibition of ATP release attenuates PGE2 production.(70) These data suggest a role for Cx43 hemichannels in the osteogenic response to mechanical stimulation via the release of anabolic PGE2, ATP, or both, at least in vitro.
In Vivo Studies on Cx43 and Mechanotransduction
In vivo models
Aside from attempts to recapitulate the ODDD phenotype, numerous studies have focused on defining the physiological role of Cx43 in skeletal homeostasis through the creation of mouse strains with Cx43 deletion in specific cell populations (summarized in Table 1). This approach is necessary because global Cx43 knockout is perinatal lethal attributable to the development of severe cardiac dysfunction.(74) Castro and colleagues developed two mouse strains with an osteoblast/osteocyte-specific Cx43 deficiency (cKO), driven by either the 2.3-kb fragment of the a1-collagen promoter (Col1-Cre) or the osteocalcin OG2 promoter (OCN-Cre).(75) The a1-collagen promoter is expressed earlier in osteoblast differentiation than is the osteocalcin promoter.(76) Thus, using these different promoters, one can control the point in the osteoblastic lineage that Gja1 expression is decreased. Because osteocytes are terminally differentiated osteoblasts, the result of using either of these promoters is an osteoblast and osteocyte-specific knockout. This approach avoids the development of skeletal deformities resulting from Cx43 ablation at earlier stages of maturation. Col1-Cre cKO mice have an osteopenic phenotype, with low BMD, cortical thinning, and expansion of the marrow cavity, much like their ODDD counterparts.(31,77) However, these mice do not possess cranial abnormalities or cardiac defects, highlighting the lack of effect on early development or systemic physiology. These mice also have reduced trabecular bone volume fraction (BV/TV), although the morphology of individual trabecular struts (ie, thickness, separation, number) is not different. Although OCN-Cre cKO mice have a similar osteopenic cortical bone phenotype, there is no difference in trabecular microstructure.(78,79) This suggests that the stage of the osteoblastic lineage at which Cx43 is deleted is critical to the bone compartments affected, with the relatively delayed deletion of Cx43 in OCN-Cre cKO mice resulting in a normal trabecular phenotype. It is possible that this is related to the greater rate of turnover of trabecular bone.(80) Col1-Cre cKO mice also weigh approximately 10% less than their WT littermates.(77) We have demonstrated that OCN-Cre mice weigh the same as WT and have a similar body composition (lean, fat, fluid mass) as assessed by 1H-nuclear magnetic resonance (NMR).(78) A detailed assessment of Col1-Cre body composition may reveal differences in fat or lean mass, or it could be that early osteoblast deletion of Cx43 delays overall animal growth by an unknown mechanism. Watkins and colleagues used the DM1-Cre to ablate Cx43 from the chondro-osteogenic lineage. DM1-Cre cKO mice have low bone mass with delayed mineralization, a cortical osteopenic phenotype with characteristic cortical expansion and thinning, and no trabecular phenotype.(22) Finally, Bivi and colleagues created osteocyte-specific Cx43 knockout mice using the Dmp1 promoter (Dmp1-Cre) and found them to have the aforementioned cortical osteopenic phenotype with no effect on trabecular bone microstructural parameters or change in bone mass.(79)
Table 1.
Summary of Murine Models of Altered Cx43 Expression
| Construct | Cells affected | Cortical phenotype | Trabecular phenotype | Loading response | Unloading response | Reference numbers |
|---|---|---|---|---|---|---|
| Global deletion | All | Cardiac defects, delayed ossification of axial and appendicular skeleton | ? | ? | ? | (74) |
| Global ODDD mutation | All | Osteopenic: cortical thinning, reduced BMD, reduced strength | Osteopenic | ? | ? | (20,21) |
| DM1-Cre ODDD | Chondro-osteogenic lineage | Osteopenic: cortical thinning, reduced BMD, expanded marrow cavity, reduced strength | None | ? | ? | (22) |
| DM1-Cre cKO | Chondro-osteogenic lineage | Osteopenic: cortical thinning and marrow expansion, lower BMD | None | ↑ | ? | (22) |
| Col1-Cre cKO | Early osteoblast/osteocyte | Osteopenic: attenuated response to anabolic PTH | Osteopenic | ↑ | ↓ | (31,75,77) |
| OCN-Cre cKO | Late osteoblast/osteocyte | Osteopenic: increased endosteal BFR, increased osteoclastic bone resorption, increased osteocyte apoptosis | None | ↑ | ↓ | (75,78,79) |
| Dmp1-Cre cKO | Osteocyte | No change in bone volume, increased cortical area, increased osteocyte apoptosis | None | ↑ | ? | (79) |
| Global overexpression | All | Cardiac and neural tube defects; skeletal phenotypes have not been examined | ? | ? | ? | (23) |
Despite cortical expansion with increased cross-sectional moment of inertia, mechanical testing has generally demonstrated reduced mechanical strength of femurs from Cx43- deficient mice;(22) however, studies from our laboratory have demonstrated that although OCN-Cre mice have reduced femur strength at 2 months of age,(12) it is actually increased by 7 months.(78) It may be that the cortical phenotype of cKO mice changes with age, such that a confluence of morphological and material changes in bone results in an overall increase in strength.
The common morphological trait among in vivo models of Cx43 deficiency is cortical expansion and thinning, a phenomenon similar to that observed in human long bones with age.(81) In Cx43-deficient mice, bone formation and mineral apposition rate at the periosteal surface are increased, whereas osteoclast indices and serum bone resorption markers at the endocortical surface are also increased.(12,22,31,77–79) The result is the “tubular” phenotype initially described for patients with ODDD. Thus, understanding the role of Cx43 in skeletal homeostasis requires an understanding of its effect on both bone formation and resorption.
A major regulator of bone resorption is the action of RANKL on its receptor RANK on the surface of osteoclasts.(82) RANKL is a critical factor in the coupling of osteoblasts and osteoclasts, ensuring appropriate turnover of bone and maintenance of systemic mineral balance. Osteoblasts and stromal cells have long been thought of as an important source of RANKL, although recent studies have revealed that osteocyte-secreted RANKL may be the more critical regulator of homeostatic bone resorption.(83) The activity of RANKL is modulated by OPG, an endogenous decoy receptor for RANKL.(82) Thus, the RANKL/OPG ratio is a common means of expressing whether the overall balance of factors favors relatively increased or decreased osteoclast activity.
Cx43 deficiency results in an increased RANKL/OPG ratio. This has been demonstrated in vivo through measurement of whole bone mRNA levels of Tnsfs11 (encodes RANKL) and Tnsfs11b (encodes OPG) from DM1-Cre cKO mice(22) and quantification, via immunohistochemistry, of RANKL+ and OPG+ osteocytes in femur cortical bone from Dmp1-Cre cKO mice. (79) In vitro, the Tnsfs11/Tnsfs11b ratio is increased in Cx43-deficient MLOY-4 osteocyte-like cells(12,79) and in primary calvarial cells isolated from Dmp1-Cre cKO mice.(79) Interestingly, although in vivo studies indicate that the increased RANKL/OPG ratio is attributable to decreased OPG in individual osteocytes, with RANKL remaining unchanged, in vitro studies have shown it to be a result of a simultaneous decrease in OPG and increase in RANKL.(12,79) The reason for the difference is not certain. It is possible that RANKL expression in individual osteocytes is actually increased but cannot be detected by the techniques utilized, or it could be that confounding factors present in the in vivo bone milieu attenuate RANKL expression in the absence of Cx43. Furthermore, areas of increased osteoclast activity on the endocortical surface occur adjacent to sites of increased osteocyte apoptosis in cortical bone.(79) During osteocyte apoptosis, release of RANKL and apoptotic bodies occurs, which recruits osteoclasts and induces resorption.(84) This suggests that local osteocyte apoptosis contributes to the skeletal phenotype of Cx43-deficient mice. Regardless, Cx43 deficiency induces a net increase in the RANKL/OPG ratio, which most likely accounts for the increased endosteal resorption present in these mice.
Cx43 not only plays a role in bone resorption but also bone formation. The mechanism by which this occurs is not known; however, emerging evidence suggests an interaction with the Wnt/β-catenin pathway, one of the primary factors regulating bone formation.(85) Secreted Wnt ligand binds to a receptor complex formed by low-density lipoprotein receptor-related protein 5 or 6 (Lrp5/6) and frizzled. This membrane complex binds and inhibits Axin and GSK-3β, which prevents phosphorylation of β-catenin. Without Wnt signaling, phosphorylated β-catenin would be the target of ubiquitination and proteasomal degradation. Free β-catenin then translocates to the nucleus, where it associates with TCF/Lef transcription factors, resulting in increased expression of osteogenic genes including OPG,(85) cyclin D,(86,87) and at least some WNT genes.(88,89) β-catenin nuclear translocation also suppresses Tnfsf11 expression.(90–93) Kramer and colleagues reported that homozygous deletion of β-catenin results in a severe fragile bone phenotype and premature death.(94) Indeed, the forearms of these mice cannot withstand mechanical loading. However, the same study showed that heterozygous mice that have lost only one β-catenin allele had only a modest bone phenotype. Interestingly, a recent study by Javaheri and colleagues(95) confirmed that heterozygous β-catenin-deficient mice had a modest bone phenotype, but this was largely confined to trabecular bone from female mice. More important, heterozygous β-catenin-deficient mice did not display an anabolic response to load. These results led the authors to suggest that a critical threshold of β-catenin is required for bone formation in response to mechanical loading. We will discuss this interesting concept within the context of the new paradigm we propose.
Sclerostin is an osteocyte-derived inhibitor of Wnt binding that suppresses bone formation and is responsive to mechanical signals; in general it is decreased during mechanical loading(96) and increased during mechanical unloading.(96,97) However, a recent study showed mechanical unloading resulted in increased expression of Sost (encodes sclerostin) in diaphyseal cortical bone but decreased expression in cancellous metaphyseal bone, suggesting site-specific differences in response to unloading.(98) Sclerostin expression is reduced in cortical bone of Cx43- deficient mice,(22,79,99) consistent with the increased osteoblastic bone formation in these mice. This may be the result of the significant Cx43 deficiency and related osteocyte apoptosis, as evidenced by the presence of empty lacunae and TUNEL staining throughout the cortical shell, in Dmp1-Cre and OCN-Cre cKO mice.(79,99) Reduced sclerostin expression and osteocyte apoptosis were correlated with areas of increased bone formation at the periosteal surface. Interestingly, the trabecular compartment is unaffected by osteocyte apoptosis; however, this is consistent with a lack of effect of Cx43 deficiency on trabecular microstructure in these mice.(78,79) The remaining osteocytes, however, tend to have less OPG expression, suggesting that other mechanisms independent of osteocyte apoptosis are also important. Finally, Cx43 has been shown to bind β-catenin in cardiac myocytes,(100) mammary epithelial cells,(101) and seminiferous epithelium.(102) These mechanisms may also explain unexpected findings regarding the response of Cx43- deficient mice to mechanical signals.
Loading in vivo
Wolff’s Law stipulates that bone structure is responsive to the physical force it experiences.(103) Practical examples of this can be found in the increased bone mass of the racquet arm of tennis players(104) or the development of increased bone density with resistance training.(105) As a result of the application of load, physical deformation of the bone matrix results in canalicular fluid flow,(106) direct mechanical strain,(107) electromagnetic effects,(34) vibration,(108) and microfractures.(109) These signals are then transduced to effector cells, which initiate bone formation and resorption at appropriate locations. In vitro evidence suggests that Cx43 is critical for sensitizing the skeletal system to mechanical signals. However, bone is a complicated biomechanical structure with many different and interconnected microenvironments that are difficult to reproduce in vitro. For example, osteocytes are normally embedded within the bone matrix and connected to surface osteoblasts and osteoclasts via canaliculi and dendritic processes. This complicated multicellular niche ultimately means that, although in vitro studies are critical to providing the direction future studies should take, eventually one must examine the role of Cx43 in bone in vivo using the aforementioned animal models.
Initial studies by Grimston and colleagues supported the hypothesis suggested by numerous in vitro studies, that the anabolic response to mechanical load is attenuated in Cx43- deficient mice.(110) The investigators subjected WT and Col1-Cre cKO mice to a three-point bending protocol. These mice experienced less of an increase in bone formation rate (BFR) and BMD in response to loading compared with WT mice. However, there are several important caveats to this investigation. First, the bending protocol used was less physiologic when compared with others, such as compression. In addition, the application of load in this study resulted in the production of copious amounts of woven bone. This suggests an injury response resulting from high strain (4665 με was used in these studies), rather than lamellar bone formation, which occurs during normal bone remodeling. In considering this limitation, a subsequent study from our group utilized a cantilever loading regimen of the tibia in OCN-Cre cKO mice.(12) In general, the response to loading is primarily manifested at the periosteal surface, and an active periosteum at baseline has the potential to confound assessment of the response to loading. Therefore, we initiated the loading protocol in mice at 6 months of age, having conducted dynamic histomorphometry on sentinel mice that indicated minimal baseline periosteal bone formation at this age. Load was applied such that the peak strain at the tibia midshaft was approximately 1300 με. In WT mice, this load was not enough to result in any detectable change in periosteal BFR, mineral apposition rate (MAR), or mineralizing surface (MS/BS). Conversely, in OCN-Cre cKO mice, there was a 217% increase in periosteal BFR and 132% increase in MS/BS. These data were the first to suggest that, rather counterintuitively, Cx43 deficiency actually sensitizes bone to the effects of mechanical signals in vivo.
A subsequent study by Grimston and colleagues subjected relatively young (2-month-old) DM1-Cre cKO mice to an axial tibia loading protocol using physiological loads.(111) The investigators found that although 1200 με did not elicit any change in trabecular or cortical bone microstructural parameters in the tibia of WT mice, there were significant gains in bone mass in cKO mice. This included increased trabecular BV/TV and trabecular thickness, as well as increased cortical bone volume and total area. Similarly, 1900 με was required to achieve an increase in BFR and MS/BS in WT mice, whereas only 1200 με was required for cKO mice. BFR and MS/BS decreased at the endocortical surface in WT mice subjected to loading, and decreased by an even greater degree in cKO mice, indicating that Cx43 deficiency results in a loading response that is greater in magnitude than the response in WT mice. The increased response to loading in the tibia is surprising given the lack of trabecular phenotype in these mice.(22,111) These findings suggest that Cx43 has a critical role in the response of bone to acute loading. Compressive loading of the ulnae from Dmp1-Cre cKO mice confirmed these findings, with significantly greater bone formation response compared with WT.(112) However, the investigators did not see any effect of loading on histomorphometric parameters at the endocortical surface.
Taken together, emerging data suggest that Cx43 deficiency results in an increased anabolic response to mechanical loading. Thus, the long-held paradigm, developed largely from in vitro studies, that Cx43 contributes to and is necessary for the anabolic response to mechanical load is not supported by recent in vivo studies. Possible reasons that the extant paradigm is not supported by in vivo studies include the fact that most in vitro experiments were completed in two- rather than three- dimensional culture; the majority of in vitro studies focused on single cell types, usually osteoblasts,(113) and did not consider the results in the context of an interconnected multicellular (osteoblast/osteoclast/osteocyte) cyto-social network; and in vitro studies focused only on one mechanical signal, usually fluid flow-induced shear stress. In any case, the newly emerging paradigm is that Cx43 deficiency increases the sensitivity of bone to mechanical load. An adjunct to this new paradigm is that Cx43-deficient bone is less sensitive to unloading.
Unloading in vivo
Mechanical unloading is experienced during spaceflight,(114) bed rest,(115) and muscle paralysis.(116) Hindlimb suspension (HLS) is a widely used method to simulate microgravity in rodents on the ground.(117) Other models, such as botulinum toxin (botox)- induced immobilization have been introduced more recently.(118) Bone loss resulting from unloading is characterized by an imbalance of bone turnover: bone formation decreases while bone resorption increases.(119) The result is a net decline in bone mass, as much as 0.5% to 1.5% per month in astronauts aboard the International Space Station.(120) Decreased bone formation is primarily mediated by the production of sclerostin by osteocytes,(97) whereas increased bone resorption is mediated by increased sclerostin and RANKL.(121,122)
In a study of botox immobilization of the hindlimbs of heterozygous Col1 cKO mice, bone loss (as measured by bone mineral content) tended to occur at a slower rate in cKOs compared with WT, although recovery of both genotypes was similar and incomplete at 12 weeks.(123) A more comprehensive follow-up by Grimston and colleagues subjected homozygous Col1-Cre cKO mice to botox immobilization.(77) This resulted in a significant loss of trabecular BV/TV along with lower trabecular number and thickness in WT mice. The degree of trabecular bone loss was similar between WT and cKO. Conversely, the enlargement of the marrow area and thinning of the cortical shell induced by disuse in WT mice was significantly attenuated in cKO mice. The investigators also documented increased endocortical osteoclast number and surface in WT mice, with no change in cKO. These initial findings suggested that, unlike loading, Cx43 deficiency results in decreased sensitivity to mechanical unloading.
A subsequent study from our laboratory subjected OCN-Cre cKO mice to mechanical unloading via HLS.(78) In contrast to the findings of Grimston and colleagues, we observed attenuated trabecular bone loss in cKO mice. And, although BFR was essentially abolished in WT mice subjected to unloading, it was maintained near baseline levels in cKO mice. In a follow-up study, we confirmed our findings of attenuated trabecular bone loss.(99) This is interesting, given the lack of trabecular phenotype in OCN-Cre mice. Furthermore, trabecular bone was not preserved in Col1-Cre cKO mice with botox immobilization, and these mice do have an osteopenic trabecular phenotype.(77) As mentioned, there was also an increased anabolic response of trabecular bone to mechanical loading in these mice,(111) suggesting that Cx43 deficiency does indeed influence the response to mechanical signals, or the lack thereof, in trabecular bone.
Our group(99) and others(79,124) have demonstrated that accumulation of apoptotic osteocytes appears to have a key role in the baseline phenotype of Cx43-deficient mice. During unloading, we have demonstrated that there is a significant increase in whole bone mRNA levels of Sost and sclerostin-positive osteocytes in WT mice subjected to HLS.(99) cKO mice, however, did not experience this same increase. Similarly, serum levels of pro-collagen type 1 N-terminal propeptide (P1NP), a marker of bone formation, declined significantly in WT but not in cKO. Considering the resorptive arm of bone remodeling, we found that unloading induces significant increases in trabecular and cortical osteoclast number and surface, with no similar increase for cKO mice. mRNA levels of Tnsfs11 (encodes RANKL) increased half as much during unloading of cKO mice compared with WT mice.
Increased osteocyte apoptosis could account for some of these findings. For example, in a series of experiments conducted by Tatsumi and colleagues, osteocytes were selectively ablated in mature mice.(125) Osteocyte ablation in this model is similar to the increased osteocyte apoptosis seen during Cx43 deficiency, albeit to a greater degree. These mice were completely resistant to the effects of HLS. They also did not exhibit increased levels of RANKL or sclerostin during unloading as seen in WT mice. In either case, the absence of mechanosensing osteocytes, which are also the primary source of sclerostin and RANKL, means that there is less potential to initially detect the unloading stimulus but also fewer cells to produce catabolic factors leading to bone loss. Thus, the reduced bone loss in unloading combined with increased bone formation with loading suggests a vertical translation of the load:response curve. Interestingly, a similar vertical translation is observed in LRP5 gain of function mice.(126) Of course, increased apoptosis does not easily explain the results in the trabecular bone compartment, where osteocyte apoptosis is not increased. An explanation for this phenotype likely requires an additional mechanism, independent of osteocyte apoptosis.
Proposed Mechanism
The mechanism by which Cx43 deficiency leads to enhanced bone formation and resorption as well as enhanced response to load and decreased response to unload is not known. However, increased periosteal bone formation and increased osteoclastic bone resorption in Cx43-deficient mice suggest the intriguing possibility that Cx43 and/or GJIC serve as a negative regulator of osteoblast and osteoclast activity. We propose that in Cx43- deficient mice, osteocytes undergo apoptosis and the subsequent release of RANKL.(84) This leads to an increase in osteoclastic bone resorption. In the remaining Cx43-deficient osteocytes, there is an increased availability of β-catenin leading to increased release of osteogenic factors from osteocytes that activate osteoblastic bone formation (Fig. 1). The increased availability of β-catenin also leads to decreased RANKL,(90–93) but this is insufficient to counteract the increased RANKL resulting from apoptosis. The net result is high-turnover bone loss. In response to mechanical load, Cx43-deficient mice experience a decrease in release of osteocytic sclerostin that leads to a further increase in availability of osteocytic β-catenin, which further increases release of osteogenic factors and an inhibition of osteocytic RANKL. The decreased RANKL resulting from load is now sufficient to counteract the increased RANKL from osteocyte apoptosis. The result is an increase in osteoblastogenesis, decrease in osteoclastogenesis, and increased bone formation, relative to WT mice, in response to load. In response to unloading, osteocytes release more sclerostin, which, acting in an autocrine fashion, decreases availability of β-catenin, thus decreasing release of osteogenic factors and increasing expression of RANKL. However, in Cx43-deficient mice, this sclerostin-induced decrease in β-catenin availability is mitigated by less β- catenin being sequestered by Cx43. Thus, the catabolic response to unloading is mitigated in Cx43-deficient mice. The model we propose can also take into account the β-catenin “threshold” proposed by Javaheri and colleagues.(95) The β-catenin threshold was proposed to explain the modest basal bone phenotype, but lack of response to mechanical load, in heterozygous β-catenin- deficient mice. Our model predicts that Cx43, through its interaction with β-catenin, adjusts this threshold. This would suggest that osteocyte selective Cx43 and β-catenin (heterozygous) deficient mice would be more responsive to mechanical load than β-catenin (heterozygous) only deficient mice (Fig. 1).
Fig. 1.
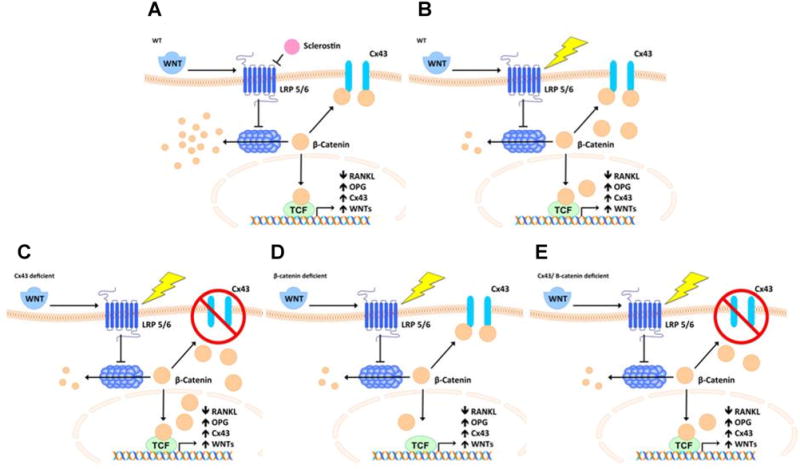
Proposed mechanism by which osteocytic Cx43, via β-catenin, regulates both bone formation and resorption in response to load. (A) WT osteocytic Cx43 binds β-catenin, decreasing its nuclear translocation. (B) Load (lightning bolt) in WT decreases sclerostin, thus increasing β-catenin, but this is governed by Cx43 functioning in a negative feedback loop. (C) In Cx43-deficient osteocytes, load-induced increases in β-catenin are amplified, resulting in further increase in expression of osteogenic genes, possibly WNTs, and decreased RANKL expression. (D) In β-catenin (heterozygous)-deficient osteocytes, β-catenin levels drop below the threshold needed for anabolic response to load. (E) In Cx43/β-catenin-deficient osteocytes, more β-catenin is made available, resulting in a modest, relative to Cx43-deficient, anabolic response to load.
It is possible that Cx43 regulates β-catenin and thereby both areas of bone remodeling, through GJIC, hemichannels, or both. However, we propose Cx43 acts in this manner independent of GJIC or hemichannels, but rather through a direct interaction with β-catenin. This concept is supported by several studies demonstrating that Cx43 is able to interact with signaling molecules such as β-arrestin,(127) kinase Src,(58,128) protein kinase C-d,(63) and zona occludens-1.(129) Cx43 itself is a target of Wnt/β-catenin,(130) which would allow for negative feedback regulation of an anabolic stimulus. Such a mechanism would mean that, in the absence of Cx43, free β-catenin is present to a greater degree and bone cells are “primed” to respond to mechanical signals.(112)
Evidence supporting this potential mechanism is provided by a study from Bivi and colleagues.(112) The investigators found that protein levels of β-catenin were greater in ulnae isolated from Dmp1-Cre cKO mice compared with WT. In vitro, they show that Cx43-deficient MLOY-4 osteocyte-like cells have increased expression of β-catenin and its targets Axin2 and Sfrp4. However, the investigators were not able to detect a difference in expression of Wnt target genes cyclin-D1 or Smad-6. Furthermore, transcription of Wnt/β-catenin target Lef1, either at baseline or with inhibition of GSK-3β by lithium chloride, is similar in WT and Cx43-deficient MLOY-4 osteocyte-like cells. These findings suggest that the accumulation of β-catenin may have an effect on increasing sensitivity to load independent of transcription of classical genes associated with Wnt/β-catenin signaling. Supporting this mechanism, mice with happloinsufficiency of β-catenin have reduced sensitivity to mechanical load.(95)
Evidence for the potential of Cx43 as part of a negative feedback system of bone formation is also found in a study from Lozupone and colleagues, which showed that loading of rat metatarsal bones increased the incidence of osteocytic GJs.(131) In a later study, our laboratory demonstrated that expression of Cx43 by osteocytes is increased in areas of bone exposed to tension relative to areas exposed to compression or to control bone.(132) Thus, loading increases Cx43, which in turn sequesters β-catenin, which leads to a decrease in β-catenin stimulation of Cx43 expression, thus closing the negative feedback loop.
Cx43 in Bone Repair
Although the mechanism described above is attractive, it does not explain the effect of Cx43 deficiency in different contexts. For instance, recent work from our group highlights the possibility for divergent interaction of Cx43 with the Wnt/β-catenin/sclerostin signaling axis in the context of loading/unloading versus fracture healing. We have shown that osteoblast/osteocyte-specific Cx43-deficient mice heal with impaired bone formation, mechanical properties,(133,134) and attenuated β-catenin expression.(134) In both OCN-Cre (Fig. 2) and Col1-Cre cKO fractures, there is an increase in the number of sclerostin-positive osteocytes in the fracture callus relative to WT. A transient increase in Sost mRNA is also observed, concurrent with attenuated β-catenin expression.(134) Additionally, increases in GSK-3β mRNA and the active form of GSK-3β protein (phospho-Tyr216)(135) occur during the time period in which β-catenin expression is decreased. Increased GSK-3β activity in Cx43cKO fractures suggests that loss of Cx43 may negatively regulate bone formation during fracture repair by increasing activating phosphorylation events of GSK-3β through an as yet determined mechanism. These data are consistent with a preliminary study that suggests that loss of Cx43 decreases inactivating phosphorylation of GSK-3β (Ser9), thus increasing GSK-3β activity.(136) We have also shown that inhibition of GSK-3β activity with lithium chloride can rescue the impairments in bone formation and mechanical properties associated with Cx43 deficiency during fracture healing. This suggests that Cx43 may promote osteogenesis by inactivating GSK-3β and therefore stimulating β-catenin expression during fracture healing.
Fig. 2.
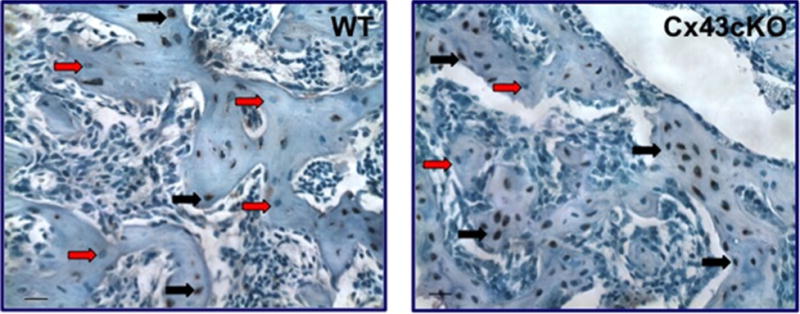
Sclerostin immunohistochemistry. An increase in the number of sclerostin+ osteocytes is observed in the fracture callus of OCN-Cre Cx43-deficient mice relative to WT at 21 days. Black arrows indicate sclerostin+ cells, red arrows indicate sclerostin− cells. Scale bar = 100 microns.
The disparate effects of Cx43 in loading/unloading and fracture repair are not completely surprising given that fracture repair is a recapitulation of development with healing occurring through endochondral ossification rather than the uncoupling of bone formation and resorption that occurs during unloading. Furthermore, vastly different cell populations including inflammatory cells and chondrocytes are involved in the fracture healing response relative to loading and unloading. Findings of impaired bone formation in Cx43-deficient fractures are supported by work from Grimston and colleagues, in which high-strain (4665 με) mechanical loading, causing an injury response, resulted in woven bone formation rather than lamellar bone.(110) This suggests that the osteogenic response may be differentially modulated by Cx43 during bone repair relative to anabolic mechanical loading. Additionally, impairments in Wnt/β-catenin signaling during fracture healing in Cx43-deficient mice raises the intriguing question of how Cx43 may modulate this important osteogenic signaling pathway, and as such is an important area of future research.
Future Studies
Future studies will need to further refine the connection between Cx43, β-catenin, and skeletal homeostasis. Findings of increased nuclear versus cytoplasmic fractions of β-catenin in primary osteocytes, MLOY-4, or IDG-SW3 osteocyte-like cells would support this hypothesis. In addition, it would be interesting to determine whether the phenotype and reduced mechanical sensitivity of β-catenin-deficient mice can be rescued by crossing with a Cx43-deficient strain. Finally, a comprehensive examination of the effect of loading and unloading on bone cell-specific Cx43-overexpressing mice would provide important insights.
Acknowledgments
This work supported by the National Space Biomedical Research Institute through NASA NCC 9-58 (MA02802), as well as R01 AG013087 from the National Institute on Aging.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: Study design: SL, AL, YZ, HD. Study conduct: SL, AL, YZ. Data collection: SL, AL, YZ. Data analysis: SL, AL, YZ, HD. Data interpretation: SL, AL, YZ, HD. Drafting manuscript: SL, AL, YZ, HD. Revising manuscript content: SL, AL, YZ, HD. Approving final version of manuscript: SL, AL, YZ, HD. HD takes responsibility for the integrity of the data.
References
- 1.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batra N, Burra S, Siller-Jackson AJ, et al. Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc Natl Acad Sci USA. 2012;109:3359–64. doi: 10.1073/pnas.1115967109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncan RL, Turner CH. Mechanotransduction and the functional response of bone to mechanical strain [Review] Calcif Tissue Int. 1995;57:344–58. doi: 10.1007/BF00302070. [DOI] [PubMed] [Google Scholar]
- 4.Civitelli R. Cell-cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys. 2008;473:188–92. doi: 10.1016/j.abb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maeda S, Tsukihara T. Structure of the gap junction channel and its implications for its biological functions. Cell Mol Life Sci. 2011;68:1115–29. doi: 10.1007/s00018-010-0551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willecke K, Eiberger J, Degen J, et al. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–37. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- 7.Stains JP, Civitelli R. Cell-to-cell interactions in bone. Biochem Biophys Res Commun. 2005;328:721–7. doi: 10.1016/j.bbrc.2004.11.078. [DOI] [PubMed] [Google Scholar]
- 8.Koval M, Harley JE, Hick E, Steinberg TH. Connexin46 is retained as monomers in a trans-Golgi compartment of osteoblastic cells. J Cell Biol. 1997;137:847–57. doi: 10.1083/jcb.137.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedner P, Steinhauser C, Theis M. Functional redundancy and compensation among members of gap junction protein families? Biochim Biophys Acta. 2012;1818:1971–84. doi: 10.1016/j.bbamem.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Taylor AF, Saunders MM, Shingle DL, Cimbala JM, Zhou Z, Donahue HJ. Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am J Physiol Cell Physiol. 2007;292:C545–52. doi: 10.1152/ajpcell.00611.2005. [DOI] [PubMed] [Google Scholar]
- 11.Ilvesaro J, Vaananen K, Tuukkanen J. Bone-resorbing osteoclasts contain gap-junctional connexin-43. J Bone Miner Res. 2000;15:919–26. doi: 10.1359/jbmr.2000.15.5.919. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Paul EM, Sathyendra V, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight MM, McGlashan SR, Garcia M, Jensen CG, Poole CA. Articular chondrocytes express connexin 43 hemichannels and P2 receptors —a putative mechanoreceptor complex involving the primary cilium? J Anat. 2009;214:275–83. doi: 10.1111/j.1469-7580.2008.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plotkin LI, Bellido T. Beyond gap junctions: connexin43 and bone cell signaling. Bone. 2013;52:157–66. doi: 10.1016/j.bone.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang JX, Siller-Jackson AJ, Burra S. Roles of gap junctions and hemichannels in bone cell functions and in signal transmission of mechanical stress. Front Biosci. 2007;12:1450–62. doi: 10.2741/2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lloyd SA, Donahue HJ. Gap junctions and biophysical regulation of bone cells. Clin Rev Bone Miner Metab. 2010;8:189–200. doi: 10.1007/s12018-011-9084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loiselle AE, Jiang JX, Donahue HJ. Gap junction and hemichannel functions in osteocytes. Bone. 2013;54:205–12. doi: 10.1016/j.bone.2012.08.132. [DOI] [PubMed] [Google Scholar]
- 18.Paznekas WA, Karczeski B, Vermeer S, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–33. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- 19.Gorlin RJ, Miskin LH, St Geme J. Oculodentodigital dysplasia. J Pediatr. 1963;63:69–75. doi: 10.1016/s0022-3476(63)80304-2. [DOI] [PubMed] [Google Scholar]
- 20.Flenniken AM, Osborne LR, Anderson N, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132:4375–86. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- 21.Dobrowolski R, Sasse P, Schrickel JW, et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17:539–54. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watkins M, Grimston SK, Norris JY, et al. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22:1240–51. doi: 10.1091/mbc.E10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewart JL, Cohen MF, Meyer RA, et al. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development. 1997;124:1281–92. doi: 10.1242/dev.124.7.1281. [DOI] [PubMed] [Google Scholar]
- 24.Sims K, Jr, Eble DM, Iovine MK. Connexin43 regulates joint location in zebrafish fins. Dev Biol. 2009;327:410–8. doi: 10.1016/j.ydbio.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziambaras K, Lecanda F, Steinberg TH, Civitelli R. Cyclic stretch enhances gap junctional communication between osteoblastic cells. J Bone Miner Res. 1998;13:218–28. doi: 10.1359/jbmr.1998.13.2.218. [DOI] [PubMed] [Google Scholar]
- 26.Alford AI, Jacobs CR, Donahue HJ. Oscillating fluid flow regulates gap junction communication in osteocytic MLO-Y4 cells by an ERK1/2 MAP kinase-dependent mechanism small star, filled. Bone. 2003;33:64–70. doi: 10.1016/s8756-3282(03)00167-4. [DOI] [PubMed] [Google Scholar]
- 27.Cherian PP, Cheng B, Gu S, Sprague E, Bonewald LF, Jiang JX. Effects of mechanical strain on the function of Gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J Biol Chem. 2003;278:43146–56. doi: 10.1074/jbc.M302993200. [DOI] [PubMed] [Google Scholar]
- 28.Cheng B, Kato Y, Zhao S, et al. PGE(2) is essential for gap junction- mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology. 2001;142:3464–73. doi: 10.1210/endo.142.8.8338. [DOI] [PubMed] [Google Scholar]
- 29.Cheng B, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells. J Bone Miner Res. 2001;16:249–59. doi: 10.1359/jbmr.2001.16.2.249. [DOI] [PubMed] [Google Scholar]
- 30.Thi MM, Kojima T, Cowin SC, Weinbaum S, Spray DC. Fluid shear stress remodels expression and function of junctional proteins in cultured bone cells. Am J Physiol Cell Physiol. 2003;284:C389–403. doi: 10.1152/ajpcell.00052.2002. [DOI] [PubMed] [Google Scholar]
- 31.Chung DJ, Castro CH, Watkins M, et al. Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J Cell Sci. 2006;119:4187–98. doi: 10.1242/jcs.03162. [DOI] [PubMed] [Google Scholar]
- 32.Saunders MM, You J, Trosko JE, et al. Gap junctions and fluid flow response in MC3T3-E1 cells. Am J Physiol Cell Physiol. 2001;281:C1917–25. doi: 10.1152/ajpcell.2001.281.6.C1917. [DOI] [PubMed] [Google Scholar]
- 33.Saunders MM, You J, Zhou Z, et al. Fluid flow-induced prostaglandin E2 response of osteoblastic ROS 17/2.8 cells is gap junction- mediated and independent of cytosolic calcium. Bone. 2003;32:350–6. doi: 10.1016/s8756-3282(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 34.Vander Molen MA, Donahue HJ, Rubin CT, McLeod KJ. Osteoblastic networks with deficient coupling: differential effects of magnetic and electric field exposure. Bone. 2000;27:227–31. doi: 10.1016/s8756-3282(00)00315-x. [DOI] [PubMed] [Google Scholar]
- 35.Yellowley CE, Li Z, Zhou Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. J Bone Miner Res. 2000;15:209–17. doi: 10.1359/jbmr.2000.15.2.209. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen NR, Henriksen Z, Brot C, et al. Human osteoblastic cells propagate intercellular calcium signals by two different mechanisms. J Bone Miner Res. 2000;15:1024–32. doi: 10.1359/jbmr.2000.15.6.1024. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Zhou Z, Yellowley CE, Donahue HJ. Inhibiting gap junctional intercellular communication alters expression of differentiation markers in osteoblastic cells. Bone. 1999;25:661–6. doi: 10.1016/s8756-3282(99)00227-6. [DOI] [PubMed] [Google Scholar]
- 38.Donahue HJ, Li Z, Zhou Z, Yellowley CE. Differentiation of human fetal osteoblastic cells is partially dependent on gap junctional intercellular communication. Am J Physiol (Cell) 2000;278:C315–C22. doi: 10.1152/ajpcell.2000.278.2.C315. [DOI] [PubMed] [Google Scholar]
- 39.Chiba H, Sawada N, Oyamada M, et al. Relationship between the expression of the gap junction protein and osteoblast phenotype in a human osteoblastic cell line during cell proliferation. Cell Structure Funct. 1993;18:419–26. doi: 10.1247/csf.18.419. [DOI] [PubMed] [Google Scholar]
- 40.Schiller PC, D’Ippolito G, Balkan W, Roos BA, Howard GA. Gap- junctional communication is required for the maturation process of osteoblastic cells in culture. Bone. 2001;28:362–9. doi: 10.1016/s8756-3282(00)00458-0. [DOI] [PubMed] [Google Scholar]
- 41.Schiller PC, Roos BA, Howard GA. Parathyroid hormone up- regulation of connexin 43 gene expression in osteoblasts depends on cell phenotype. J Bone Miner Res. 1997;12:2005–13. doi: 10.1359/jbmr.1997.12.12.2005. [DOI] [PubMed] [Google Scholar]
- 42.Upham BL, Suzuki J, Chen G, et al. Reduced gap junctional intercellular communication and altered biological effects in mouse osteoblast and rat liver oval cell lines transfected with dominant- negative connexin 43. Mol Carcinog. 2003;37:192–201. doi: 10.1002/mc.10137. [DOI] [PubMed] [Google Scholar]
- 43.Lecanda F, Towler DA, Ziambaras K, et al. Gap junctional communication modulates gene expression in osteoblastic cells. Mol Biol Cell. 1998;9:2249–58. doi: 10.1091/mbc.9.8.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donahue HJ, Li Z, Zhou Z, Yellowley CE. Differentiation of human fetal osteoblastic cells and gap junctional intercellular communication. Am J Physiol Cell Physiol. 2000;278:C315–22. doi: 10.1152/ajpcell.2000.278.2.C315. [DOI] [PubMed] [Google Scholar]
- 45.Schiller PC, D’Ippolito G, Brambilla R, Roos BA, Howard GA. Inhibition of gap-junctional communication induces the trans-differentiation of osteoblasts to an adipocytic phenotype in vitro. J Biol Chem. 2001;276:14133–8. doi: 10.1074/jbc.M011055200. [DOI] [PubMed] [Google Scholar]
- 46.Inose H, Ochi H, Kimura A, et al. A microRNA regulatory mechanism of osteoblast differentiation. Proc Natl Acad Sci USA. 2009;106:20794–9. doi: 10.1073/pnas.0909311106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gramsch B, Gabriel HD, Wiemann M, et al. Enhancement of connexin 43 expression increases proliferation and differentiation of an osteoblast-like cell line. Exp Cell Res. 2001;264:397–407. doi: 10.1006/excr.2000.5145. [DOI] [PubMed] [Google Scholar]
- 48.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277:8648–57. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 49.Lo CW. The role of gap junction membrane channels in development. J Bioenerg Biomembr. 1996;28:379–85. doi: 10.1007/BF02110114. [DOI] [PubMed] [Google Scholar]
- 50.Minkoff R, Rundus VR, Parker SB, Hertzberg EL, Laing JG, Beyer EC. Gap junction proteins exhibit early and specific expression during intramembranous bone formation in the developing chick mandible. Anat Embryol. 1994;190:231–41. doi: 10.1007/BF00234301. [DOI] [PubMed] [Google Scholar]
- 51.Minkoff R, Bales ES, Kerr CA, Struss WE. Antisense oligonucleotide blockade of connexin expression during embryonic bone formation: evidence of functional compensation within a multigene family. Dev Genet. 1999;24:43–56. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<43::AID-DVG6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 52.Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. Connexin 43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol. 2000;151:931–44. doi: 10.1083/jcb.151.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishikawa M, Iwamoto T, Nakamura T, Doyle A, Fukumoto S, Yamada Y. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J Cell Biol. 2011;193:1257–74. doi: 10.1083/jcb.201101050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schilling AF, Filke S, Lange T, et al. Gap junctional communication in human osteoclasts in vitro and in vivo. J Cell Mol Med. 2008;12:2497–504. doi: 10.1111/j.1582-4934.2008.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ransjo M, Sahli J, Lie A. Expression of connexin 43 mRNA in microisolated murine osteoclasts and regulation of bone resorption in vitro by gap junction inhibitors. Biochem Biophys Res Commun. 2003;303:1179–85. doi: 10.1016/s0006-291x(03)00502-3. [DOI] [PubMed] [Google Scholar]
- 56.Ilvesaro J, Tavi P, Tuukkanen J. Connexin-mimetic peptide Gap 27 decreases osteoclastic activity. BMC Musculoskelet Disord. 2001;2:10. doi: 10.1186/1471-2474-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hughes DE, Wright KR, Uy HL, et al. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J Bone Miner Res. 1995;10:1478–87. doi: 10.1002/jbmr.5650101008. [DOI] [PubMed] [Google Scholar]
- 58.Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–74. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plotkin LI, Bellido T. Bisphosphonate-induced, hemichannel-mediated, anti-apoptosis through the Src/ERK pathway: a gap junction- independent action of connexin43. Cell Commun Adhes. 2001;8:377–82. doi: 10.3109/15419060109080757. [DOI] [PubMed] [Google Scholar]
- 60.Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal- regulated kinase activation. J Biol Chem. 2005;280:7317–25. doi: 10.1074/jbc.M412817200. [DOI] [PubMed] [Google Scholar]
- 61.Jeong HM, Jin Y, Choi Y, et al. Risendronate increases osteoblastic differentiation and function through connexin 43. Biochem Biophys Res Commun. 2013;432:152–6. doi: 10.1016/j.bbrc.2013.01.068. [DOI] [PubMed] [Google Scholar]
- 62.Lima F, Niger C, Hebert C, Stains JP. Connexin43 potentiates osteoblast responsiveness to fibroblast growth factor 2 via a protein kinase C-delta/Runx2-dependent mechanism. Mol Biol Cell. 2009;20:2697–708. doi: 10.1091/mbc.E08-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niger C, Hebert C, Stains JP. Interaction of connexin43 and protein kinase C-delta during FGF2 signaling. BMC Biochem. 2010;11:14. doi: 10.1186/1471-2091-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niger C, Buo AM, Hebert C, Duggan BT, Williams MS, Stains JP. ERK acts in parallel to PKCdelta to mediate the connexin43-dependent potentiation of Runx2 activity by FGF2 in MC3T3 osteoblasts. Am J Physiol Cell Physiol. 2012;302:C1035–44. doi: 10.1152/ajpcell.00262.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niger C, Luciotti MA, Buo AM, Hebert C, Ma V, Stains JP. The regulation of Runx2 by FGF2 and connexin43 requires the inositol polyphosphate/protein kinase Cdelta cascade. J Bone Miner Res. 2013;28:1468–77. doi: 10.1002/jbmr.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Riddle RC, Taylor AF, Genetos DC, Donahue HJ. MAP kinase and calcium signaling mediate fluid flow-induced human mesenchymal stem cell proliferation. Am J Physiol Cell Physiol. 2006;290:C776–84. doi: 10.1152/ajpcell.00082.2005. [DOI] [PubMed] [Google Scholar]
- 67.Lim PK, Bliss SA, Patel SA, et al. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011;71:1550–60. doi: 10.1158/0008-5472.CAN-10-2372. [DOI] [PubMed] [Google Scholar]
- 68.Cherian PP, Siller-Jackson AJ, Gu S, et al. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell. 2005;16:3100–6. doi: 10.1091/mbc.E04-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang JX, Cherian PP. Hemichannels formed by connexin 43 play an important role in the release of prostaglandin E(2) by osteocytes in response to mechanical strain. Cell Commun Adhes. 2003;10:259–64. doi: 10.1080/cac.10.4-6.259.264. [DOI] [PubMed] [Google Scholar]
- 70.Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J Cell Physiol. 2007;212:207–14. doi: 10.1002/jcp.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ren J, Wang XH, Wang GC, Wu JH. 17beta estradiol regulation of connexin 43-based gap junction and mechanosensitivity through classical estrogen receptor pathway in osteocyte-like MLO-Y4 cells. Bone. 2013;53:587–96. doi: 10.1016/j.bone.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 72.Siller-Jackson AJ, Burra S, Gu S, et al. Adaptation of connexin 43- hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283:26374–82. doi: 10.1074/jbc.M803136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang JX, Cheng B. Mechanical stimulation of gap junctions in bone osteocytes is mediated by prostaglandin E2. Cell Commun Adhes. 2001;8:283–8. doi: 10.3109/15419060109080738. [DOI] [PubMed] [Google Scholar]
- 74.Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–4. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 75.Castro CH, Stains JP, Sheikh S, et al. Development of mice with osteoblast-specific connexin43 gene deletion. Cell Commun Adhes. 2003;10:445–50. doi: 10.1080/cac.10.4-6.445.450. [DOI] [PubMed] [Google Scholar]
- 76.Heersche JN, Reimers SM, Wrana JL, Waye MM, Gupta AK. Changes in expression of alpha 1 type 1 collagen and osteocalcin mRNA in osteoblasts and odontoblasts at different stages of maturity as shown by in situ hybridization. Proc Finn Dent Soc. 1992;88(Suppl1):173–82. [PubMed] [Google Scholar]
- 77.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26:2151–60. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lloyd SA, Lewis GS, Zhang Y, Paul EM, Donahue HJ. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res. 2012;27:2359–72. doi: 10.1002/jbmr.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bivi N, Condon KW, Allen MR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. 2012;27:374–89. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goodyear SR, Gibson IR, Skakle JM, Wells RP, Aspden RM. A comparison of cortical and trabecular bone from C57 Black 6 mice using Raman spectroscopy. Bone. 2009;44:899–07. doi: 10.1016/j.bone.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 81.Macdonald HM, Nishiyama KK, Kang J, Hanley DA, Boyd SK. Age- related patterns of trabecular and cortical bone loss differ between sexes and skeletal sites: a population-based HR-pQCT study. J Bone Miner Res. 2011;26:50–62. doi: 10.1002/jbmr.171. [DOI] [PubMed] [Google Scholar]
- 82.Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–46. doi: 10.1016/j.abb.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–27. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 85.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–92. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 86.Mirando AJ, Maruyama T, Fu J, Yu HM, Hsu W. Beta-catenin/cyclin D1 mediated development of suture mesenchyme in calvarial morphogenesis. BMC Dev Biol. 2010;10:116. doi: 10.1186/1471-213X-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, et al. Wnt/beta- catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 88.Yochum GS, McWeeney S, Rajaraman V, Cleland R, Peters S, Goodman RH. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proc Natl Acad Sci USA. 2007;104:3324–9. doi: 10.1073/pnas.0611576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010;38:5735–45. doi: 10.1093/nar/gkq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2012;54:258–63. doi: 10.1016/j.bone.2012.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci. 2006;119:1283–96. doi: 10.1242/jcs.02883. [DOI] [PubMed] [Google Scholar]
- 92.Glass DA, 2nd, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 93.Holmen SL, Zylstra CR, Mukherjee A, et al. Essential role of beta- catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–8. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 94.Kramer I, Halleux C, Keller H, et al. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Javaheri B, Dallas M, Zhao H, Bonewald L, Johnson M. Beta-catenin haploinsufficiency in osteocytes abolishes the osteogenic effect of mechanical loading in vivo. J Bone Miner Res. 2011;26(Suppl 1) [Google Scholar]
- 96.Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 97.Lin C, Jiang X, Dai Z, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 98.Macias BR, Aspenberg P, Agholme F. Paradoxical Sost gene expression response to mechanical unloading in metaphyseal bone. Bone. 2013;53:515–9. doi: 10.1016/j.bone.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 99.Lloyd SA, Loiselle AE, Zhang Y, Donahue HJ. Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone. 2013;57:76–83. doi: 10.1016/j.bone.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ai Z, Fischer A, Spray DC, Brown AM, Fishman GI. Wnt-1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–71. doi: 10.1172/JCI7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Talhouk RS, Mroue R, Mokalled M, et al. Heterocellular interaction enhances recruitment of alpha and beta-catenins and ZO-2 into functional gap-junction complexes and induces gap junction- dependant differentiation of mammary epithelial cells. Exp Cell Res. 2008;314:3275–91. doi: 10.1016/j.yexcr.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 102.Dbouk HA, Mroue RM, El-Sabban ME, Talhouk RS. Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Commun Signal. 2009;7:4. doi: 10.1186/1478-811X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ahn AC, Grodzinsky AJ. Relevance of collagen piezoelectricity to “Wolff’s Law”: a critical review. Med Eng Phys. 2009;31:733–41. doi: 10.1016/j.medengphy.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kontulainen S, Sievanen H, Kannus P, Pasanen M, Vuori I. Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius of female racquet-sports players: a peripheral quantitative computed tomography study between young and old starters and controls. J Bone Miner Res. 2003;18:352–9. doi: 10.1359/jbmr.2003.18.2.352. [DOI] [PubMed] [Google Scholar]
- 105.Layne JE, Nelson ME. The effects of progressive resistance training on bone density: a review. Med Sci Sports Exerc. 1999;31:25–30. doi: 10.1097/00005768-199901000-00006. [DOI] [PubMed] [Google Scholar]
- 106.Riddle RC, Donahue HJ. From streaming-potentials to shear stress: 25 years of bone cell mechanotransduction. J Orthop Res. 2009;27:143–9. doi: 10.1002/jor.20723. [DOI] [PubMed] [Google Scholar]
- 107.Sato K, Adachi T, Ueda D, Hojo M, Tomita Y. Measurement of local strain on cell membrane at initiation point of calcium signaling response to applied mechanical stimulus in osteoblastic cells. J Biomech. 2007;40:1246–55. doi: 10.1016/j.jbiomech.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 108.Castillo AB, Alam I, Tanaka SM, et al. Low-amplitude, broad- frequency vibration effects on cortical bone formation in mice. Bone. 2006;39:1087–96. doi: 10.1016/j.bone.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 109.Chapurlat RD. Bone microdamage. Osteoporos Int. 2009;20:1033–5. doi: 10.1007/s00198-009-0859-4. [DOI] [PubMed] [Google Scholar]
- 110.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1) J Bone Miner Res. 2008;23:879–86. doi: 10.1359/JBMR.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grimston SK, Watkins MP, Brodt MD, Silva MJ, Civitelli R. Enhanced periosteal and endocortical responses to axial tibial compression loading in conditional connexin43 deficient mice. PLoS One. 2012;7:e44222. doi: 10.1371/journal.pone.0044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bivi N, Pacheco-Costa R, Brun LR, et al. Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J Orthop Res. 2013;31:1075–81. doi: 10.1002/jor.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–15. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Keyak JH, Koyama AK, LeBlanc A, Lu Y, Lang TF. Reduction in proximal femoral strength due to long-duration spaceflight. Bone. 2009;44:449–53. doi: 10.1016/j.bone.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 115.Spector ER, Smith SM, Sibonga JD. Skeletal effects of long-duration head-down bed rest. Aviat Space Environ Med. 2009;80:A23–8. doi: 10.3357/asem.br02.2009. [DOI] [PubMed] [Google Scholar]
- 116.Lee TQ, Shapiro TA, Bell DM. Biomechanical properties of human tibias in long-term spinal cord injury. J Rehabil Res Dev. 1997;34:295–302. [PubMed] [Google Scholar]
- 117.Morey-Holton ER, Globus RK. Hindlimb unloading rodent model: technical aspects. J Appl Physiol. 2002;92:1367–77. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- 118.Warner SE, Sanford DA, Becker BA, Bain SD, Srinivasan S, Gross TS. Botox induced muscle paralysis rapidly degrades bone. Bone. 2006;38:257–64. doi: 10.1016/j.bone.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bikle DD, Halloran BP. The response of bone to unloading. J Bone Miner Metab. 1999;17:233–44. doi: 10.1007/s007740050090. [DOI] [PubMed] [Google Scholar]
- 120.Lang T, LeBlanc A, Evans H, Lu Y, Genant H, Yu A. Cortical and trabecular bone mineral loss from the spine and hip in long- duration spaceflight. J Bone Miner Res. 2004;19:1006–12. doi: 10.1359/JBMR.040307. [DOI] [PubMed] [Google Scholar]
- 121.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6:e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saxena R, Pan G, Dohm ED, McDonald JM. Modeled microgravity and hindlimb unloading sensitize osteoclast precursors to RANKL- mediated osteoclastogenesis. J Bone Miner Metab. 2011;29:111–22. doi: 10.1007/s00774-010-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grimston SK, Silva MJ, Civitelli R. Bone loss after temporarily induced muscle paralysis by Botox is not fully recovered after 12 weeks. Ann NY Acad Sci. 2007;1116:444–60. doi: 10.1196/annals.1402.009. [DOI] [PubMed] [Google Scholar]
- 124.Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res. 2008;23:1712–21. doi: 10.1359/JBMR.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 126.Saxon LK, Jackson BF, Sugiyama T, Lanyon LE, Price JS. Analysis of multiple bone responses to graded strains above functional levels, and to disuse, in mice in vivo show that the human Lrp5 G171V High Bone Mass mutation increases the osteogenic response to loading but that lack of Lrp5 activity reduces it. Bone. 2011;49:184–93. doi: 10.1016/j.bone.2011.03.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bivi N, Lezcano V, Romanello M, Bellido T, Plotkin LI. Connexin43 interacts with betaarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem. 2011;112:2920–30. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Giepmans BN, Hengeveld T, Postma FR, Moolenaar WH. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J Biol Chem. 2001;276:8544–9. doi: 10.1074/jbc.M005847200. [DOI] [PubMed] [Google Scholar]
- 129.Laing JG, Chou BC, Steinberg TH. ZO-1 alters the plasma membrane localization and function of Cx43 in osteoblastic cells. J Cell Sci. 2005;118:2167–76. doi: 10.1242/jcs.02329. [DOI] [PubMed] [Google Scholar]
- 130.van der Heyden MA, Rook MB, Hermans MM, et al. Identification of connexin43 as a functional target for Wnt signalling. J Cell Sci. 1998;111(Pt 12):1741–9. doi: 10.1242/jcs.111.12.1741. [DOI] [PubMed] [Google Scholar]
- 131.Lozupone E, Palumbo C, Favia A, Ferretti M, Palazzini S, Cantatore FP. Intermittent compressive load stimulates osteogenesis and improves osteocyte viability in bones cultured “in vitro”. Clin Rheumatol. 1996;15:563–72. doi: 10.1007/BF02238545. [DOI] [PubMed] [Google Scholar]
- 132.Su M, Borke JL, Donahue HJ, Li Z, Warshawsky NM, Russell CM. Expression of connexin 43 in rat mandibular bone and PDL cells during experimental tooth movement. J Dental Res. 1997;76:1357–66. doi: 10.1177/00220345970760070501. [DOI] [PubMed] [Google Scholar]
- 133.Loiselle AE, Paul EM, Lewis GS, Donahue HJ. Osteoblast and osteocyte-specific loss of connexin43 results in delayed bone formation and healing during murine fracture healing. J Orthop Res. 2013;31:147–54. doi: 10.1002/jor.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loiselle AE, Lloyd SA, Paul EM, Lewis GS, Donahue HJ. Inhibition of GSK-3β rescues the impairments in bone formation and mechanical properties associated with fracture healing in osteo-blast selective connexin 43 deficient mice. PLoS One. 2013;8:e81399. doi: 10.1371/journal.pone.0081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 136.Karl S, Ziegelhoffer B, Rabler B, Salameh A. Connexin 43 expression is impaired in beginning heart failure in spontaneously hypertensive rats. In: Entschladen F, Friedrich K, Hass R, Janssen O, editors. 12th Joint Meeting of the Signal Transduction Society. Weimar, Germany: Cell Communication and Signaling; 2008. [Google Scholar]