

Abstract
Metabolic homeostasis is a fundamental property of cells that becomes dysregulated in cancer to meet the altered, often heightened, demand for metabolism for increased growth and proliferation. Oncogenic mutations can directly change cellular metabolism in a cell-intrinsic manner, priming cells for malignancy. Additionally, cell-extrinsic cues from the microenvironment, such as hypoxia, nutrient availability, oxidative stress, and crosstalk from surrounding cells can also affect cancer cell metabolism, and produce metabolic heterogeneity within the tumor. Here, we highlight recent findings revealing the complexity and adaptability of leukemia cells to coordinate metabolism.
Keywords: Leukemia, Metabolism, AMPK, Cell Intrinsic, Microenvironment
1. Introduction
Metabolic alterations have long been recognized in cancer cells. A finding made nearly a century ago by Otto Warburg that tumors consume glucose at markedly increased level has not only spurred research aimed to understand this peculiar metabolic behavior of tumors, but also has found its way into clinical application as 18F-fluorodeoxyglucose positron emission tomography for cancer diagnosis [1]. The reliance of cancer cells on glucose and its preferential consumption through glycolysis even in the presence of oxygen (aerobic glycolysis) was initially hypothesized to be due to defective mitochondria [2]. However, more recent studies have established that many cancer cells harbor and are dependent upon functional mitochondria [3–6]. The question of why cancer cells preferentially use glycolysis, an uneconomical route to produce ATP compared to mitochondrial oxidative phosphorylation, is beginning to be addressed in cancer cells. Glycolysis is not the dominant mechanism for many cancer cells to produce ATP, and ATP production is not the limiting factor for proliferation [7]. Rather, glycolysis offers cancer cells with metabolites essential for rapid division through metabolic pathways that branch away from glycolysis. For example, glucose-6-phosphate can be diverted to the pentose phosphate pathway (PPP) to produce reducing agents NADPH and ribonucleotides [8]. Additionally, 3-phospho-glycerate can enter the one-carbon cycle to generate amino acids, lipids, and reducing agents [9]. Thus, increased glycolysis meets the demand of proliferative cancer cells for anabolism, producing metabolic intermediates through multiple branching pathways. The decision of how cancer cells divert glycolysis intermediates remains elusive, but it is likely to be affected by cellular metabolic state and the environment in which the cancer cells are exposed.
The metabolic milieu of the tumor microenvironment dictates the behavior of tumors. Tumors are exposed to nutrient- and oxygen-poor conditions as they grow exponentially due to insufficient vascularization [10]. Metabolic adaptation to these stress conditions is vital for tumor survival and expansion, and as such multiple metabolic regulators that enable metabolic adaptation are dysregulated in cancer. These include the components of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling that feeds into the mechanistic target of rapamycin (mTOR) pathway, AMP-activated protein kinase (AMPK), and hypoxia inducible factor 1 (Hif1). The effects of the microenvironment on cancer cell metabolism may apply to hematological malignancies, since leukemia and lymphoma cells often expand in hypoxic areas such as the bone marrow or the thymus [11, 12]. Thus, considering cancer cell metabolism within the context of metabolically constrained tumor microenvironment is critical in understanding tumor development and progression. In this review, we will discuss mechanisms by which hematological malignancies dysregulate metabolism and how they adapt to metabolic stresses found in the native microenvironment in which they reside.
2. Increased glucose metabolism supports anabolism and redox maintenance in leukemia
Despite the fact that different hematological malignancies harbor distinct genetic lesions and reside in different microenvironment, increased glucose consumption is a conserved hallmark in hematological malignancies (Figure 1). Multiple hematological malignancies, including multiple myeloma [13], acute lymphoblastic leukemia (ALL) [14], chronic lymphatic leukemia (CLL) [15], and AML [16] exhibit increased glucose consumption and upregulated glycolysis. Pharmacological inhibition of glycolysis using 2-deoxyglucose (2-DG) [14, 17] or 3-bromopyruvate [18, 19] can arrest leukemia cell proliferation and synergize with chemotherapeutic agents. Genetic studies in mouse models also support the importance of glycolysis in initiation and maintenance of myeloid malignancies. Deletion of a pyruvate kinase isoform (PKM2) expressed specifically in embryonic tissues and cancer cells or lactate dehydrogenase A (LDHA), both of which catalyze the final steps in glycolysis, significantly attenuated the initiation and maintenance of murine CML and AML [20]. Moreover, increased glycolysis may confer increased drug resistance to leukemia cells, as has been found in patients [16], as well as in leukemia cell lines [17, 21]. As discussed above, glycolysis not only offers rapid ATP production but also gives cancer cells access to metabolic pathways that branch from glycolysis, such as the PPP that produces ribonucleotide and NADPH.
Figure 1.
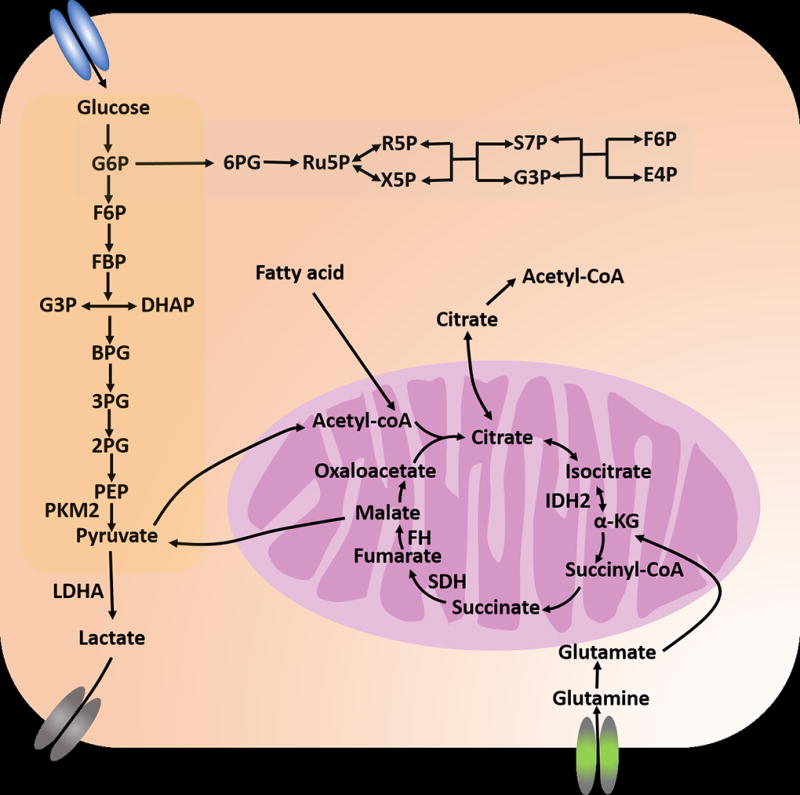
Overview of the dysregulated metabolic pathways in leukemia. Leukemia cells exhibit upregulated glycolysis rates. Perturbing key enzymes such as PKM2 and LDHA suppressed leukemogenesis. Glutamine can fuel the TCA cycle through α-KG and through reductive carboxylation. This provides a mechanism to sustain cellular energy requirement and synthesis needs from TCA metabolites when glucose metabolism is limited or TCA cycle genes are deficient.
The PPP pathway branches from glycolysis after the production of glucose 6-phosphate and is composed of two phases; the oxidative and the nonoxidative phase (Figure 1). During the oxidative phase, glucose 6-phosphate is oxidized to produce NADPH and ribose-5-phosphate. In the nonoxidative phase, the pentose phosphates produced during the oxidative phase are recycled into glucose 6-phosphate. By providing cells with ribonucleotides for nucleic acid synthesis, and NADPH for lipid synthesis and redox homeostasis, PPP plays a critical role in cancer cell metabolism and often become activated in various cancers including solid tumors [8], AML [22], multiple myeloma [23], and lymphoid leukemias [24]. Inhibition of PPP in cancer cell lines reduces NADPH levels, lipid synthesis, and also reprograms metabolism by increasing oxidative phosphorylation [22] or glycolysis. Blocking the PPP also activated AMP-activated kinase (AMPK) to suppress proliferation, pointing to the importance of this metabolic pathway in cancer cell proliferation. Increased flux through the PPP enabled by increased glucose utilization thereby supports continuous cancer proliferation.
Recent evidence supports the idea that AMPK confers cancer cells with the ability to cope with metabolic stresses by regulating glucose metabolism. AMPK is a heterotrimeric kinase complex that becomes activated upon energy stress, as sensed as an increase in AMP levels in response to ATP depletion. Activation of AMPK, either pharmacologically or by metabolic stress, suppresses the growth and proliferation of cancer cells through multiple mechanisms, including activating p53, inhibiting the growth promoting mechanistic target of rapamycin complex 1(mTORC1) pathway (Figure 2), or causing transcriptional responses [25]. AMPK activation can also promote metabolic homeostasis by activating multiple catabolic processes such as glucose uptake [26, 27], glycolysis [28], fatty acid uptake and oxidation [29], and mitochondrial biogenesis [30, 31]. Of note, AMPK activation promotes redox homeostasis by promoting NADPH production [32]. Therefore, AMPK appears to have both tumor suppressive and promoting roles, in a contextual dependent manner (Figure 3) [33–35]. In an Eμ-myc-induced lymphoma model, deletion of the α1 catalytic subunit of AMPK promoted lymphomagenesis but rendered the tumor cells sensitive to metabolic stress [36]. In mouse models of AML and CML, deletion of the two α-catalytic subunits of AMPK depleted leukemia-initiating cells and prolonged leukemia free survival [37]. AMPK deletion attenuated glucose uptake and metabolism through glycolysis and the PPP, resulting in the depletion of reducing agents NADPH and GSH. This increased oxidative stress and DNA damage accumulation in the leukemia-initiating cell population (Figure 3). Collectively, these results suggest that AMPK is a critical regulator of metabolic homeostasis in hematological malignancies, such that AMPK inhibition renders leukemia cells sensitive to metabolic stress.
Figure 2.
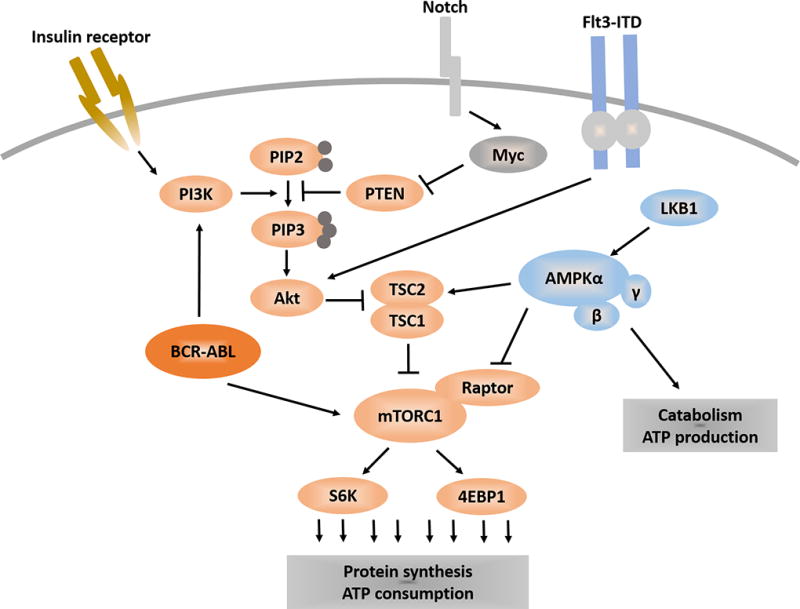
A network of metabolic pathways that regulate leukemia. PI3K catalyzes the production of PIP3, which is negatively regulated by PTEN. PIP3 recruits Akt to the plasma membrane for activation, which leads to the activation of mTORC1. mTORC1 promotes protein translation via 4EBP1 and S6P. AMPK can inhibit mTORC1 through TSC or Raptor. Leukemogenic proteins such as Notch, Flt3 and BCR-ABL can converge on the PI3K/Akt pathway.
Figure 3.
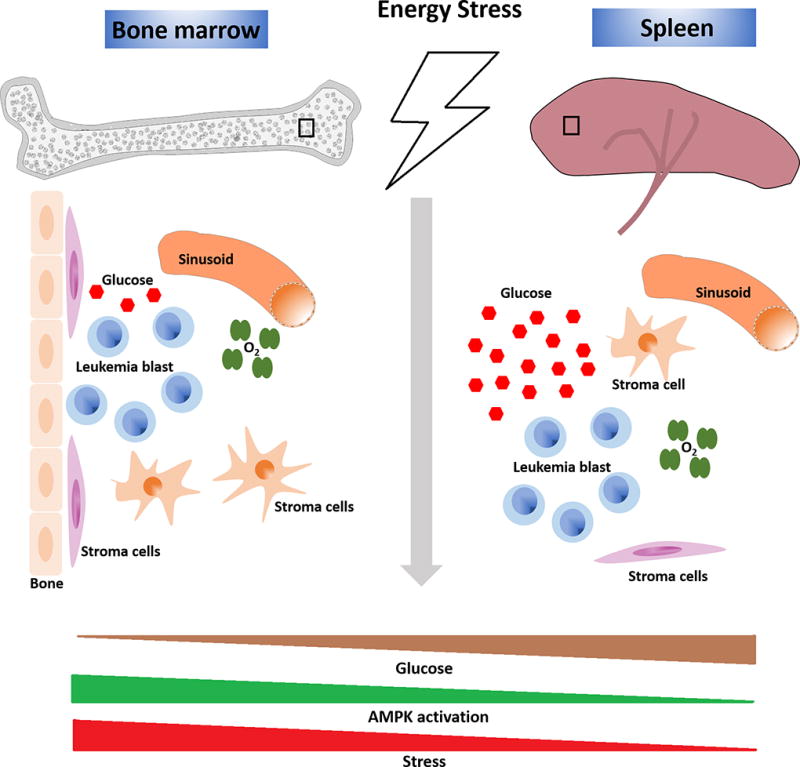
Extrinsic regulation of leukemia metabolism. Cellular and non-cellular components of the leukemia microenvironment can regulate leukemia metabolism. Signaling from the stromal cells can affect leukemia and alter its metabolism. Non-cellular components such as oxygenation, glucose concentration and perfused nutrient availability also regulates leukemia metabolism. Differences in these components between the two hematopoietic tissues, bone marrow and spleen, offer a tool to study this differences. Upon dietary restriction and possibly other metabolic stress, the glucose concentration in the bone marrow microenvironment undergoes larger decrease compared to that in the spleen. As a consequence, leukemia cells residing in the bone marrow depend upon AMPK more so that those in the spleen to maintain metabolic homeostasis.
Dietary restriction (DR) is a conserved mechanism that prolongs lifespan of multiple organisms, and reduces the incident of tumors in mammals [38–44]. One mechanism by which DR suppresses tumorigenesis appears to be through reducing insulin or insulin-like growth factor 1 (IGF1) levels, since tumors with activating mutations in the phosphoinositide 3-kinase (PI3K) pathway, which mediates the effects of insulin/IGF1 stimuli, were insensitive to DR [41, 42]. Activating mutation in the PI3K pathway is common in T-ALL [45] and while these mutations are rare in AML, activation of this pathway is commonly observed in AML [46]. In a murine model of AML induced by the MLL-AF9 oncogene, DR caused metabolic stress upon AML as indicated by the activation of AMPK but did not prolong leukemia free survival of recipient mice [37]. However, this resistance to DR was not accompanied by activation of the PI3K/Akt pathway [37, 47]. Remarkably, either genetic deletion or pharmacological inhibition of AMPK synergized with DR to suppress AML progression. This was accompanied by synergistic increase in oxidative stress and DNA damage in LICs with these two treatments, possibly caused by reduced glucose concentration in the bone marrow by DR. Thus, AMPK inhibition combined with metabolic stress, either by dietary manipulation or pharmacologically, has profound anti-leukemic effects. These results indicate that DR-resistant cancers can be rendered sensitive to the metabolic stress caused by DR by inactivating AMPK.
3. Glutamine metabolism: leukemia’s umami taste bud
Glutamine is an abundant resource of carbon and nitrogen that many cancer cells become addicted to [48]. Glutamine is a versatile metabolite that can be used to produce amino acids, fuel the mitochondria via producing α-ketoglutarate, upon which cells can use to synthesize pyruvate, NADPH, acetyl-coA, or citrate. These intermediate metabolites can then contribute in building macromolecules to support rapid cancer cell growth. Multiple cancer cell lines, such as pancreatic cancer, glioblastoma multiforme, small cell lung cancer, breast cancer, and AML, exhibit dependence on glutamine supplementation, and glutamine deprivation leads to growth arrest or apoptosis [49–52]. L-asparaginase, which suppresses ALL by depleting circulating asparagine [53], has recently been shown to suppress AML by its know deamination activity towards glutamine [50]. AML cells deprived of glutamine, either through glutamine withdrawal, pharmacological depletion of glutamine by L-asparaginase, or knockdown of glutamine transporter blocked glutamine uptake, suppressed mTORC1 pathway, and inhibited protein translation. Furthermore, blocking glutamine metabolism synergized with chemotherapy or BCL-2 inhibitors to suppress leukemia [50, 54].
A key regulator of glutaminolysis is the transcription factor Myc, one of the most frequently mutated genes in human cancer [55]. Myc regulates the expression of factors regulating glutaminolysis, such as glutamine transporters [56] and glutaminase, an enzyme that converts glutamine to glutamate for further processing in the TCA cycle [57]. Myc is a direct transcriptional target of Notch1 and supports Notch1-induced T-ALL pathogenesis [58–61]. A recent study using a Notch1 induced T-ALL model demonstrated that inhibition of Notch1 by gamma secretase inhibitor treatment attenuated glutaminolysis, making them reliant on autophagy to support metabolism and survival [62]. Pharmacological or genetic blockade of glutaminase synergized with Notch1 inhibition, highlighting the importance of glutaminolysis in T-ALL. Interestingly, deletion of Pten, a negative regulator of the PI3K pathway frequently mutated in pediatric T-ALL [63, 64], conferred T-ALL resistance against Notch1 inhibition by upregulating glycolysis. Consistently, deletion of Pten from murine hematopoietic system causes T-ALL [65, 66] through activation of the Akt pathway, a critical regulator of glucose uptake and glycolysis [67, 68]. Similar to the inhibition of AMPK rendering myeloid leukemias sensitive to lowered glucose levels [37], inhibition of Notch1 renders T-ALL cells particularly sensitive upon perturbed glutaminolysis. Combinatorial therapies aimed to expose and target the metabolic vulnerability of cancer cells may have significant therapeutic potential.
4. 1. Leukemia microenvironment and metabolism
The prevailing view in cancer biology was that oncogenic mutations confer cells with unlimited proliferative potential, making them independent from extracellular growth factors to survive and proliferate. However, it is now recognized that the non-tumor elements, such as the tumor infiltrating immune cells, the vasculature, and the stromal cells, participate in the process of tumorigenesis from the initiation, progression, and metastasis of tumors. Although much of what we know about the roles of tumor microenvironment comes from studies in solid tumors [69–71], recent evidence indicate that leukemias are no exception and are regulated by the microenvironment during leukemia initiation and progression [72–74].
The cellular components of the leukemia niche are still elusive. Recent studies on HSC niche are mostly consistent with the perivascular niche model, in which endothelial cells and the perivascular mesenchymal stromal cells retain and provide critical ligands to HSCs [75–80]. On the other hand, lymphoid progenitors reside in close proximity to the endosteum and are regulated by osteoblasts [78, 81, 82]. The niche of myeloid progenitors largely remains unknown. Since some LICs of lymphoid or myeloid leukemias share characteristics with their proposed normal progenitor counterparts (lymphoid and myeloid progenitors, respectively [83–85]), it is possible that these LICs also reside in the same microenvironment as these normal progenitors. On the other hand, it is also possible that LICs acquire a spacial preference similar to that of a more immature progenitor, such as that of HSCs. A recent study revealed that Notch1-driven murine T-ALL cells reside in close proximity with endothelial cells (where normal HSCs reside) and not in contact with osteoblasts (where normal lymphoid progenitors reside) [86]. A challenge for future investigation will be to dissect the contribution of the leukemia niche component on leukemia initiation versus maintenance, and also to determine to what extent full-blown leukemias remain niche dependent. As discussed below, leukemias may escape the constraints provided by the microenvironment and also may rearrange the microenvironment to gain growth and survival advantages.
Leukemia cells dysregulate signaling pathways to become independent of extracellular growth factors, but also become insensitive to signals produced by the microenvironment that limits their proliferation. Activating mutations in cytokine receptors such as KIT, FLT3, JAK2, IL7R are frequently found in leukemias [87]. Key signaling pathways such as the PI3K and Ras pathways function downstream of these receptors, and upregulate metabolism for growth and survival [88]. Leukemias with constitutively activating mutations in these receptors enjoy cell-autonomous activation of metabolism independent of the extracellular cues. Leukemias can also escape from differentiation-inducing signals from the microenvironment. JunB-deficient hematopoietic progenitors become insensitive to Notch and TGF-β signaling that limits myeloid differentiation, and go on to develop myeloid malignancies [89]. Consistently, deleting 3 out of 4 Notch receptors from hematopoietic cells expands myeloid progenitors and cause chronic myelomonocytic leukemia-like disorders [90]. Although mutations in the TGF-β pathway components are rare, many leukemias, such as those induced by AML-ETO or Evi-1, suppress the TGF-β pathway activation [91].
Leukemia can also remodel the microenvironment to further promote leukemogenesis. Similar to solid tumors, AML can induce angiogenesis [92, 93] and this correlates with poor prognosis [94, 95]. Primary leukemia blasts or leukemia cell lines proliferate more rapidly and exhibit increased chemotherapy resistance when co-cultured with endothelial cells [96, 97]. AML can also cause neuropathy in the bone marrow. The bone marrow is innervated by both myelinated and non-myelinated nerve fibers [98]. Non-myelinated schwann cells and the sympathetic nerves system regulate HSC mobilization and quiescence [80, 99–101]. Myeloid malignancies cause sympathetic neuropathy in the leukemic bone marrow and treatment with neuroprotective agents attenuates leukemia progression [72, 102]. Sympathetic neuropathy impairs differentiation of MSCs, decreasing stromal cells that support HSCs while expanding cells that promote leukemogenesis [72]. Taken together, leukemia induces sympathetic neuropathy in the bone marrow to create a leukemogenic environment while inhibiting HSC supportive environment. The ability of leukemias to change their responsiveness to extracellular factors that limit their expansion, and their ability to rearrange the microenvironment to promote expansion may pose a challenge in using therapies targeting extracellular signaling factors.
4.2. Metabolic effects of the leukemia microenvironment
To understand how metabolism is regulated in leukemia, we need to consider the variety of effects the leukemic microenvironment has on cells. Leukemia cell lines cultured in vitro are exposed to unphysiological levels of glucose, nutrients, and oxygen levels, all of which critically affects metabolism of cells [103]. Although the full elucidation of the metabolic milieu of leukemic microenvironment awaits further investigation, recent studies have revealed that leukemia microenvironment in the bone marrow is hypoxic, hypoglycemic, and that the cell-cell interaction of leukemia cells with stromal cells may affect cellular metabolism [20, 37, 104–109]. Thus, any perturbations to these cells, either genetic or pharmacologic, will likely have profoundly different effects when tested in culture or in vivo. Here, we will highlight some of the recent findings that are beginning to reveal metabolic interactions between leukemia cells and the microenvironment.
Hypoxia inducible factors appear to regulate adaptation of leukemia cells to different metabolic states. Studies using hypoxia indicator dyes or a nanoprobe to directly measure oxygen tension revealed that HSCs reside in hypoxic perisinusoidal regions [105, 106, 108]. These results were consistent with the findings demonstrating that HSCs have elevated levels of HIF-1α, which normally undergoes degradation by the proteasome when oxygen levels exceed 5% [105, 106]. Human AML specimen transplanted into immunocompromised mice preferentially homed to the endosteal region [110], which has been shown to be less hypoxic compared to the perisinusoidal HSC niche [108]. However, it is possible that expansion of AML cells convert the endosteal region hypoxic by disrupting the vasculature [111]. Homing to the endosteal region appears to confer chemoresistance and cell cycle quiescence to leukemia-initiating cells [112]. A hypoxia activated pro-drug called PR-104, which is reduced to a toxic agent under extremely low oxygen concentrations, was effective in suppressing leukemia cell proliferation in vitro and in vivo [111, 113] and is also under clinical trial with promising results [114]. Thus, leukemias appear to reside in hypoxic region at least at some point during their development. This may explain the mixed results regarding the role of HIF in leukemia. Deletion of Hif-1α, Hif-2α, or both accelerates the development of AML, but are dispensable for the maintenance of established AML [115]. Consistently, deletion of Hif-1α accelerated AML in three different models [116]. On the other hand, knockdown of HIF-2α from human AML decreased engraftment ability [117]. Leukemias may rely on HIF to adapt to different metabolic conditions they experience upon stimulation by transforming oncogenes or upon migration.
Residing in a hypoxic region may maintain redox homeostasis in leukemia cells. Conventional chemotherapies induces oxidative stress, and tumors in hypoxic regions are protected from chemotherapy-induced oxidative stress [118]. LICs of human AML were enriched in cells with low ROS. These cells were also metabolically dormant, exhibiting low oxidative metabolism and low glycolytic rates [119], either allowing the LICs to reside in regions with low oxygen and nutrients, or reflecting their adaptation to such environment. Interestingly, although LICs maintained low oxidative stress, they depended upon mitochondrial OXPHOS to survive. The importance of intact mitochondrial function for leukemia is also supported by the finding that inhibition of mitochondrial translation either pharmacologically or genetically eradicates human AML [119]. A subset of AML overexpress a mitochondrial protease ClpP, the inhibition of which suppressed leukemia survival by inhibiting OXPHOS [120]. In addition to supporting OXPHOS, intact mitochondrial function may be essential for leukemia survival through fatty acid oxidation (FAO) [121, 122], a mechanism by which the mitochondria produces large amount of acetyl-CoA and ATP [123]. Some leukemia cell populations, perhaps the more immature cells with leukemia-initiating capacity, may have more reliance upon mitochondrial metabolism than what has been anticipated.
Leukemia microenvironment can regulate redox homeostasis of leukemias by contributing substrates to generate antioxidants. Reduced glutathione is an important redox regulator and its synthesis is dependent upon the availability of cysteine, the unstable, reduced form of cystine [124]. Primary CLL cells have low GSH production and undergo ROS mediated cell death in vitro [125]. A recent study discovered that bone marrow stromal cells promoted GSH synthesis and alleviated oxidative stress of CLL cells in a co-culture system [126]. CLL cells had low capacity to uptake cystine for GSH production, and relied upon cysteine secreted from the stromal cells for GSH production. Pharmacological inhibition of cystine uptake, aimed to block stromal cells to produce and supply cysteine to CLL cells, reduced the disease burden in a CLL mouse model. Another amino acid provided by the leukemia microenvironment is glutamine. Adipocytes produce significant amount of glutamine, which allows ALL cells to resist the asparagine- and glutamine-lowering effects of L-asparaginase treatment [127], which may explain the association between obesity and poor outcomes in leukemia patients [128–130]. Targeting the leukemia microenvironment to disable the leukemia nurturing function therefore has important implications in cancer therapy.
Leukemia cells in different locations may encounter different levels of metabolic stress. AML is known to infiltrate various tissues, such as the spleen, liver, and the central nervous system [131], but whether distinct metabolic attributes are required to reside in different locations is unknown. We recently discovered that AMPK dependence distinguishes LICs residing in the bone marrow from those in the spleen [37]. Deletion of AMPK from MLL-AF9-induced AML increased ROS in LICs residing in the bone marrow but not those in the spleen. Since culturing AMPK-deficient AML cells in low glucose condition is sufficient to increase ROS, and that the glucose concentrations in the bone marrow were lower than those in the blood or the spleens, the simplest interpretation is that AML cells depend upon AMPK to cope with the low glucose concentration in the bone marrow to efficiently suppress ROS. A similar change in oxidative stress in tumors residing in different locations has been demonstrated in primary melanomas and distant metastases [132, 133]. These results demonstrate the capacity of cancer cells to adapt to the different metabolic requirement at different locations, and underscore the importance of conceptualizing cancer metabolism within the context of the surrounding microenvironment.
5. Conclusion
Increased uptake and assimilation of glucose and glutamine allows leukemia cells to not only generate macromolecules essential for growth and proliferation, but also to maintain redox homeostasis. Key metabolic pathways such as the AMPK pathway enable leukemia cells to adapt to metabolically harsh environment such as those found in the bone marrow. These pathways could be therapeutically exploited to reveal metabolic vulnerability of leukemia cells. With a better understanding of the molecular and cellular identity of the leukemia microenvironment, additional metabolic targets that could be exploited to disable the metabolic symbiosis of the tumor and the microenvironment are likely to emerge. As has been the case for any therapies, future challenges will be to investigate the heterogeneous behavior of leukemia cells in response to metabolic stresses, such as that caused by mutations or metabolic heterogeneity contributed by the different microenvironment in which the cancer cells reside.
Acknowledgments
This work was supported by the National Institute of Health (R01CA193235), the Cancer Prevention and Research Institute of Texas (R1201), and the Gabrielle’s Angel Foundation for Cancer Research.
References
- 1.Warburg KP O, Negelein E. Biochemische Zeitschrift. Vol. 1924. (German) Reprinted in English in the book On metabolism of tumors by O. Warburg, Publisher; Constable, London: 1930. Ueber den Stoffwechsel der Tumoren; pp. 319–344. [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 5.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 6.Burgart LJ, Zheng J, Shu Q, Strickler JG, Shibata D. Somatic mitochondrial mutation in gastric cancer. Am J Pathol. 1995;147:1105–11. [PMC free article] [PubMed] [Google Scholar]
- 7.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 8.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–54. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508:269–73. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hale LP, Braun RD, Gwinn WM, Greer PK, Dewhirst MW. Hypoxia in the thymus: role of oxygen tension in thymocyte survival. Am J Physiol Heart Circ Physiol. 2002;282:H1467–77. doi: 10.1152/ajpheart.00682.2001. [DOI] [PubMed] [Google Scholar]
- 13.Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, et al. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015;75:2071–82. doi: 10.1158/0008-5472.CAN-14-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, et al. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia. 2006;20:1731–7. doi: 10.1038/sj.leu.2404365. [DOI] [PubMed] [Google Scholar]
- 15.Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG, et al. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin Cancer Res. 2009;15:3442–50. doi: 10.1158/1078-0432.CCR-08-3291. [DOI] [PubMed] [Google Scholar]
- 16.Herst PM, Howman RA, Neeson PJ, Berridge MV, Ritchie DS. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J Leukoc Biol. 2011;89:51–5. doi: 10.1189/jlb.0710417. [DOI] [PubMed] [Google Scholar]
- 17.Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, et al. Antileukemic Activity of 2-Deoxy-d-Glucose through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia with FLT3-ITD or c-KIT Mutations. Mol Cancer Ther. 2015;14:2364–73. doi: 10.1158/1535-7163.MCT-15-0163. [DOI] [PubMed] [Google Scholar]
- 18.Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ, et al. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009;113:2014–21. doi: 10.1182/blood-2008-05-157842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvino E, Estan MC, Sanchez-Martin C, Brea R, de Blas E, Boyano-Adanez Mdel C, et al. Regulation of death induction and chemosensitizing action of 3-bromopyruvate in myeloid leukemia cells: energy depletion, oxidative stress, and protein kinase activity modulation. J Pharmacol Exp Ther. 2014;348:324–35. doi: 10.1124/jpet.113.206714. [DOI] [PubMed] [Google Scholar]
- 20.Wang YH, Israelsen WJ, Lee D, Yu VW, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–23. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen WL, Wang JH, Zhao AH, Xu X, Wang YH, Chen TL, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124:1645–54. doi: 10.1182/blood-2014-02-554204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Xu Q, Ji D, Wei Y, Chen H, Li T, et al. Inhibition of pentose phosphate pathway suppresses acute myelogenous leukemia. Tumour Biol. 2015 doi: 10.1007/s13277-015-4428-5. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Huang R, Ding J, Ji D, Song B, Yuan L, et al. Multiple myeloma acquires resistance to EGFR inhibitor via induction of pentose phosphate pathway. Sci Rep. 2015;5:9925. doi: 10.1038/srep09925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferretti A, Chen LL, Di Vito M, Barca S, Tombesi M, Cianfriglia M, et al. Pentose phosphate pathway alterations in multi-drug resistant leukemic T-cells: 31P NMR and enzymatic studies. Anticancer Res. 1993;13:867–72. [PubMed] [Google Scholar]
- 25.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, et al. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK) J Cell Sci. 2002;115:2433–42. doi: 10.1242/jcs.115.11.2433. [DOI] [PubMed] [Google Scholar]
- 27.Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5' AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–71. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 28.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 29.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito Y, Nakada D. The role of the Lkb1/AMPK pathway in hematopoietic stem cells and Leukemia. Crit Rev Oncog. 2014;19:383–97. doi: 10.1615/critrevoncog.2014011765. [DOI] [PubMed] [Google Scholar]
- 34.Jeon SM, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015;38:346–57. doi: 10.1007/s12272-015-0549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165–70. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–96. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci. 2010;31:89–98. doi: 10.1016/j.tips.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pallavi R, Giorgio M, Pelicci PG. Insights into the beneficial effect of caloric/ dietary restriction for a healthy and prolonged life. Front Physiol. 2012;3:318. doi: 10.3389/fphys.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curry NL, Mino-Kenudson M, Oliver TG, Yilmaz OH, Yilmaz VO, Moon JY, et al. Pten-null tumors cohabiting the same lung display differential AKT activation and sensitivity to dietary restriction. Cancer Discov. 2013;3:908–21. doi: 10.1158/2159-8290.CD-12-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009;458:725–31. doi: 10.1038/nature07782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–21. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mihaylova MM, Sabatini DM, Yilmaz OH. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell. 2014;14:292–305. doi: 10.1016/j.stem.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park S, Chapuis N, Tamburini J, Bardet V, Cornillet-Lefebvre P, Willems L, et al. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010;95:819–28. doi: 10.3324/haematol.2009.013797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu MC, Arimura GK, Yunis AA. Mechanism of sensitivity of cultured pancreatic carcinoma to asparaginase. Int J Cancer. 1978;22:728–33. doi: 10.1002/ijc.2910220615. [DOI] [PubMed] [Google Scholar]
- 50.Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–32. doi: 10.1182/blood-2013-03-493163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2015 doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goto M, Miwa H, Shikami M, Tsunekawa-Imai N, Suganuma K, Mizuno S, et al. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Invest. 2014;32:241–7. doi: 10.3109/07357907.2014.907419. [DOI] [PubMed] [Google Scholar]
- 53.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 54.Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–56. doi: 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–90. doi: 10.1038/nri2304. [DOI] [PubMed] [Google Scholar]
- 59.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–6. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26:8022–31. doi: 10.1128/MCB.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015;21:1182–9. doi: 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jenkinson S, Kirkwood AA, Goulden N, Vora A, Linch DC, Gale RE. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia. 2016;30:39–47. doi: 10.1038/leu.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ, et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2010;24:239–42. doi: 10.1038/leu.2009.209. [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 66.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 67.Lee JY, Nakada D, Yilmaz OH, Tothova Z, Joseph NM, Lim MS, et al. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell. 2010;7:593–605. doi: 10.1016/j.stem.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–42. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 69.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 71.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 72.Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15:365–75. doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13:285–99. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–7. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–30. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 77.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–30. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–18. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 84.Dettman EJ, Simko SJ, Ayanga B, Carofino BL, Margolin JF, Morse HC, 3rd, et al. Prdm14 initiates lymphoblastic leukemia after expanding a population of cells resembling common lymphoid progenitors. Oncogene. 2011;30:2859–73. doi: 10.1038/onc.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 86.Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell. 2015;27:755–68. doi: 10.1016/j.ccell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Etten RA. Aberrant cytokine signaling in leukemia. Oncogene. 2007;26:6738–49. doi: 10.1038/sj.onc.1210758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 89.Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, et al. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell. 2009;15:341–52. doi: 10.1016/j.ccr.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–3. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim SJ, Letterio J. Transforming growth factor-beta signaling in normal and malignant hematopoiesis. Leukemia. 2003;17:1731–7. doi: 10.1038/sj.leu.2403069. [DOI] [PubMed] [Google Scholar]
- 92.Dührsen U, Hossfeld DK. Stromal abnormalities in neoplastic bone marrow diseases. Annals of hematology. 1996;73:53–70. doi: 10.1007/s002770050203. [DOI] [PubMed] [Google Scholar]
- 93.Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000;95:309–13. [PubMed] [Google Scholar]
- 94.Shih TT, Hou HA, Liu CY, Chen BB, Tang JL, Chen HY, et al. Bone marrow angiogenesis magnetic resonance imaging in patients with acute myeloid leukemia: peak enhancement ratio is an independent predictor for overall survival. Blood. 2009;113:3161–7. doi: 10.1182/blood-2008-08-173104. [DOI] [PubMed] [Google Scholar]
- 95.Padro T, Ruiz S, Bieker R, Burger H, Steins M, Kienast J, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637–44. [PubMed] [Google Scholar]
- 96.Liesveld JL, Rosell KE, Lu C, Bechelli J, Phillips G, Lancet JE, et al. Acute myelogenous leukemia--microenvironment interactions: role of endothelial cells and proteasome inhibition. Hematology. 2005;10:483–94. doi: 10.1080/10245330500233452. [DOI] [PubMed] [Google Scholar]
- 97.Pezeshkian B, Donnelly C, Tamburo K, Geddes T, Madlambayan GJ. Leukemia Mediated Endothelial Cell Activation Modulates Leukemia Cell Susceptibility to Chemotherapy through a Positive Feedback Loop Mechanism. PLoS One. 2013;8:e60823. doi: 10.1371/journal.pone.0060823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Afan AM, Broome CS, Nicholls SE, Whetton AD, Miyan JA. Bone marrow innervation regulates cellular retention in the murine haemopoietic system. Br J Haematol. 1997;98:569–77. doi: 10.1046/j.1365-2141.1997.2733092.x. [DOI] [PubMed] [Google Scholar]
- 99.Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–7. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 100.Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–58. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 101.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 102.Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512:78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 103.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nature Cell Biology. 2012;14:276–286. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parmar K, Mauch P, Vergilio J-A, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell stem cell. 2010;7:380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 108.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508:269–73. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rovida E, Peppicelli S, Bono S, Bianchini F, Tusa I, Cheloni G, et al. The metabolically-modulated stem cell niche: a dynamic scenario regulating cancer cell phenotype and resistance to therapy. Cell Cycle. 2014;13:3169–75. doi: 10.4161/15384101.2014.964107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 111.Benito J, Shi Y, Szymanska B, Carol H, Boehm I, Lu H, et al. Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One. 2011;6:e23108. doi: 10.1371/journal.pone.0023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–80. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moradi Manesh D, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood. 2015;126:1193–202. doi: 10.1182/blood-2014-12-618900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Konopleva M, Thall PF, Yi CA, Borthakur G, Coveler A, Bueso-Ramos C, et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica. 2015;100:927–34. doi: 10.3324/haematol.2014.118455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vukovic M, Guitart AV, Sepulveda C, Villacreces A, O'Duibhir E, Panagopoulou TI, et al. Hif-1alpha and Hif-2alpha synergize to suppress AML development but are dispensable for disease maintenance. J Exp Med. 2015;212:2223–34. doi: 10.1084/jem.20150452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, Cammenga J. HIF-1alpha can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood. 2014;124:3597–607. doi: 10.1182/blood-2014-04-567065. [DOI] [PubMed] [Google Scholar]
- 117.Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, et al. HIF-2alpha protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell. 2013;13:549–63. doi: 10.1016/j.stem.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 118.Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008;8:790–7. doi: 10.2174/187152008785914798. [DOI] [PubMed] [Google Scholar]
- 119.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Larkin K, Byrd JC. Antagonizing ClpP: A New Power Play in Targeted Therapy for AML. Cancer Cell. 2015;27:747–9. doi: 10.1016/j.ccell.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 121.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–9. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 123.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–32. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830:3143–53. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Silber R, Farber CM, Papadopoulos E, Nevrla D, Liebes L, Bruck M, et al. Glutathione depletion in chronic lymphocytic leukemia B lymphocytes. Blood. 1992;80:2038–43. [PubMed] [Google Scholar]
- 126.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–86. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI, et al. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013;73:2998–3006. doi: 10.1158/0008-5472.CAN-12-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Behan JW, Yun JP, Proektor MP, Ehsanipour EA, Arutyunyan A, Moses AS, et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009;69:7867–74. doi: 10.1158/0008-5472.CAN-09-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Butturini AM, Dorey FJ, Lange BJ, Henry DW, Gaynon PS, Fu C, et al. Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol. 2007;25:2063–9. doi: 10.1200/JCO.2006.07.7792. [DOI] [PubMed] [Google Scholar]
- 130.Gelelete CB, Pereira SH, Azevedo AM, Thiago LS, Mundim M, Land MG, et al. Overweight as a prognostic factor in children with acute lymphoblastic leukemia. Obesity (Silver Spring) 2011;19:1908–11. doi: 10.1038/oby.2011.195. [DOI] [PubMed] [Google Scholar]
- 131.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–62. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 132.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–91. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re8. doi: 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]