

Abstract
Atypical hemolytic uremic syndrome (aHUS) and bone marrow transplantation-associated thrombotic microangiopathy (TA-TMA) are associated with excessive activation of the alternative complement pathway (AP) and with severe renal, but rarely cerebral, microvascular damage. Here, we compared AP activation and regulation in human glomerular and brain microvascular endothelial cells (GMVECs and BMVECs, respectively) unstimulated or stimulated by the proinflammatory cytokine, tumor necrosis factor (TNF). Compared with GMVECs and under both experimental conditions, BMVECs had increased gene expression of the AP-related genes C3, CFB, and C5 and decreased expression of CFD. This was associated with increased expression in BMVECs (relative to GMVECs) of the genes for surface and soluble regulatory molecules (CD46, THBD, CD55, CFI, and CFH) suppressing formation of the AP C3 and C5 convertases. Of note, unlike GMVECs, BMVECs generated extremely low levels of C3a and C5a and displayed decreased activation of the AP (as measured by a lower percentage of Ba generation than GMVECs). Moreover, BMVECs exhibited increased function of CD141, mediating activation of the natural anticoagulant protein C, compared with GMVECs. We also found that the C3a receptor (C3aR) is present on both cell types and that TNF greatly increases C3AR1 expression in GMVECs, but only slightly in BMVECs. Higher AP activation and C3a generation in GMVECs than in BMVECs, coupled with an increase in C3aR production in TNF-stimulated GMVECs, provides a possible explanation for the predominance of renal damage, and the absence of cerebral injury, in individuals with episodes of aHUS and TA-TMA.
Keywords: tumor necrosis factor (TNF), endothelial cell, inflammation, kidney, brain, alternative complement pathway, anaphylatoxin receptors, anaphylatoxins, C3a, C3aR
Introduction
Thrombotic microangiopathies (TMAs)2 are disorders characterized by microvascular thrombosis, microangiopathic hemolytic anemia, thrombocytopenia, and microvascular endothelial injury (1–3). Atypical hemolytic uremic syndrome (aHUS) and bone marrow transplantation-associated TMA (TA-TMA) are TMAs associated with abnormalities in the alternative complement pathway (AP) as well as severe renal damage (3–6).
The AP is one of three pathways of the innate immune complement system responsible for protecting the host from unwanted pathogens or foreign invaders. It is initiated when C3b is cleaved from complement component C3 and attaches to an activating surface, releasing a soluble C3a fragment (7, 8). Factor B (FB) then binds to C3b (9, 10), and factor D (FD) cleaves FB in this complex to form C3bBb (the active C3 convertase of the AP) (11), releasing the activation product Ba. The C3 convertase is stabilized by factor P (properdin) (12–14). The Bb in C3bBb cleaves C3 to generate additional C3b and C3a in an activation amplification loop (15, 16); as the ratio of C3b to Bb increases, C3bBbC3b (the C5 convertase of the AP) forms and cleaves C5 to C5b, releasing the soluble C5a activation fragment (11, 17). AP activation is likely to occur on ultra-large von Willebrand factor (ULVWF) multimeric strings secreted by, and attached to, human endothelial cells. The attachment of AP components to the ULVWF multimeric strings and AP activation has been previously demonstrated on human umbilical vein endothelial cells (HUVECs) (18–20). The classical complement pathway (CP) component, C4, does not attach to the strings; therefore, this pathway is not activated by ULVWF (18). The potent inflammatory cytokine, tumor necrosis factor (TNF), likely promotes AP activation by inducing ULVWF secretion (21).
C3a and C5a function as pro-inflammatory polypeptides (anaphylatoxins) that cause neutrophil chemotaxis, histamine release from basophils and mast cells, and increased vascular permeability (22). C3a and C5a are generated by activation in each complement pathway: the AP, the lectin pathway, or the CP. The activation product, Ba, is unique to the AP and is not generated with activation of the lectin pathway or CP. No receptor for the Ba protein has been identified. Receptors for C3a (C3aR) and C5a (C5aR) are reported to be present on myeloid and lymphoid cells (23, 24), platelets (25), HUVECs, and dermal microvascular endothelial cells (26).
Regulatory molecules suppress excessive AP activation and potential host cell damage. Factor H (FH) and factor I (FI) are soluble inhibitory regulators: FH suppresses the formation or persistence of C3bBb by displacing Bb (27, 28), and FI, with cofactor FH, promotes the cleavage/inactivation of C3b (29). CD46, CD141, and CD55 are cell surface-bound membrane regulatory receptors that protect the host cell from AP-mediated injury; CD46 and CD141 function as cofactors for FI-mediated proteolysis of C3b (30, 31), and CD55 accelerates the decay of both the C3 convertase and the C5 convertase (32). CD141, also known as thrombomodulin (and gene name of THBD), has an additional function as a natural anticoagulant. CD141 binds and diverts thrombin substrate specificity to the activation of protein C (PC). Activated PC, with bound protein S, cleaves and inactivates coagulation factors Va and VIIIa and thereby regulates thrombosis (33).
Atypical HUS and TA-TMA are associated with excessive activation or defective regulation of the AP. aHUS is predominantly caused by mutations in complement genes or the development of antibodies against FH (Table 1) (30, 34–37). Whereas variants in complement genes have been reported in patients with TA-TMA (38), post-bone marrow transplant risk factors more likely predispose to TA-TMA development in this group of patients (Table 1) (39–47). Excessive AP activation in aHUS and TA-TMA contributes to microvascular endothelial injury; the kidney is the organ that is the most commonly damaged, and in contrast, the brain is rarely involved (6, 48, 49).
Table 1.
Etiology of abnormalities in the alternative complement pathway in TMA
aHUS is caused by mutations in complement protein genes or from the development of autoantibodies directed against FH. TA-TMA is associated with post-bone marrow transplant environmental stressors. CFHR1/CFHR3 indicate the deletions of complement factor H-related genes 1 and 3.
| aHUS, mutations in complement genes | TA-TMA, environmental risk factors |
|---|---|
| Gain of function | Immunosuppressive drugs |
| C3 | Total body irradiation |
| CFB | High-dose chemotherapy–conditioning regimens |
| Loss of function | Graft versus host disease |
| CD46 | Infection |
| THBD | ABO incompatibility |
| CFI | Donor type |
| CFH | |
| CFHR1/CFHR3 (deletion; plus autoantibody to FH) |
Infectious/inflammatory events have been demonstrated to trigger initial or recurrent episodes of aHUS and TA-TMA (6, 47, 48, 50–52). Furthermore, TNF has been demonstrated to be elevated in aHUS and TA-TMA patients clinically (53, 54). We have previously shown that TNF contributes to AP activation in human glomerular microvascular endothelial cells (GMVECs), as demonstrated by higher levels of activation products C3a, Ba, and C5a in these cells compared with levels in HUVECs after TNF stimulation (55). We also found that TNF caused substantial down-regulation of GMVEC THBD expression, resulting in diminished PC activation by CD141-bound thrombin (55).
The vulnerability of the kidney to AP-mediated injury in aHUS and TA-TMA led us to hypothesize that there is a difference in AP activation and regulation in GMVECs (the cell type that is predominantly involved in these two types of TMA) compared with brain microvascular endothelial cells (BMVECs), a microvascular endothelial cell type that is not affected. To achieve our objectives, we compared AP activation and regulation in GMVECs and in BMVECs that were either unstimulated or, as a model for inflammation/infection, stimulated by TNF. These MVECs serve as ideal models for our studies as they produce and secrete all AP components and regulators, as well as VWF (18, 55).
Results
Gene expression of AP components in unstimulated and TNF-stimulated BMVECs relative to GMVECs
We compared in BMVECs and GMVECs the expression of genes that encode essential proteins in the activation of the AP: C3, CFB, C5, CFP, and CFD. Although the classical pathway is not activated on ULVWF strings, we included the gene expression of C4 as a control. Unstimulated BMVECs had 14-fold higher mRNA levels for C3, ∼3-fold higher levels for both CFB and C5, and ∼5-fold higher mRNA levels for C4 compared with levels in unstimulated GMVECs. Unstimulated BMVECs had ∼4-fold lower mRNA levels for both CFP and CFD compared with unstimulated GMVECs (Fig. 1A). Expression levels of C3, CFB, C5, and C4 were ∼7-, ∼4-, ∼5-, and ∼8-fold higher, respectively, and CFD expression levels were 3-fold lower in TNF-stimulated BMVECs compared with TNF-stimulated GMVECs (Fig. 1B).
Figure 1.
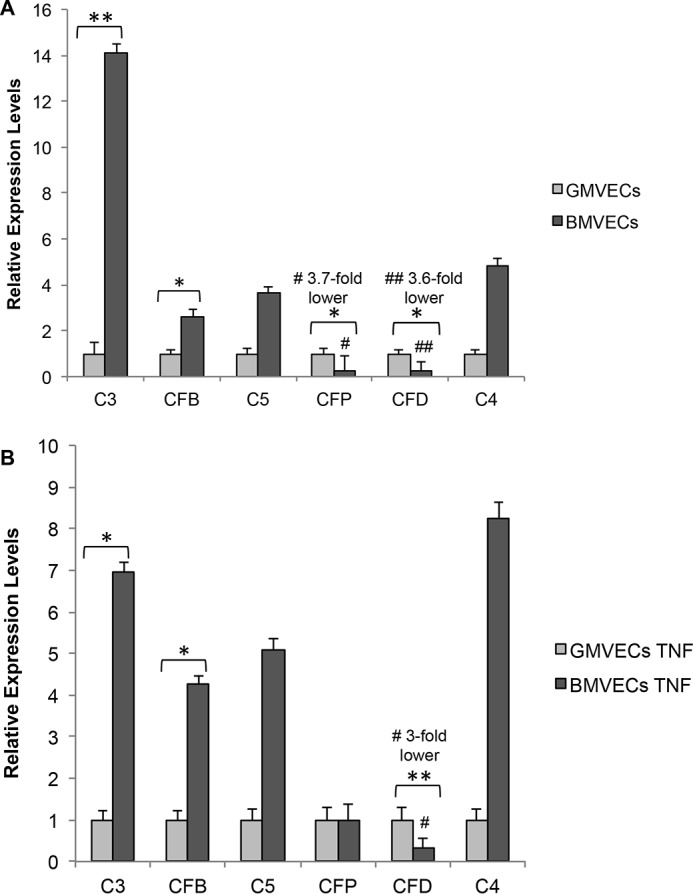
Gene expression of AP components in unstimulated and TNF-stimulated BMVECs relative to GMVECs. The mRNA levels of the genes for AP components C3, CFB, C5, CFP, and CFD and the classical complement component C4 in unstimulated BMVECs and GMVECs (A) and in TNF-stimulated BMVECs and GMVECs (B) were evaluated by real time (RT)-qPCR. RNA was extracted from unstimulated BMVECs and GMVECs that were maintained in serum-free medium for 24 h and in these cells after incubation with 10 ng/ml TNF for 48 h (24 h in complete medium and 24 h in serum-free medium). Fold-changes in BMVEC mRNA levels were calculated relative to the mRNA levels in GMVECs. RNA was extracted in 4–6 separate experiments from each cell type. Data are means plus standard deviations (S.D.) from RT-qPCR runs with triplicate measurements. GAPDH was used for normalization. *, p < 0.05; **, p < 0.001.
Quantitative gene expression of AP components by TNF-stimulated GMVECs and BMVECs
We additionally quantified changes in gene expression of each AP component in BMVECs and GMVECs after exposure to TNF from unstimulated gene levels in each MVEC type (Fig. 2). AP component expression of both cell types changed with TNF stimulation in a similar pattern (but not in magnitude). Both GMVECs and BMVECs had increased gene expression of C3 (150- and 50-fold, respectively) and of CFB (60- and 80-fold, respectively) and reduced expression of CFP (10- and 2-fold respectively). CFD expression changed minimally in GMVECs and BMVECs with TNF stimulation (<2-fold increases), and C5 and C4 mRNA levels did not change substantially in either cell type with TNF.
Figure 2.
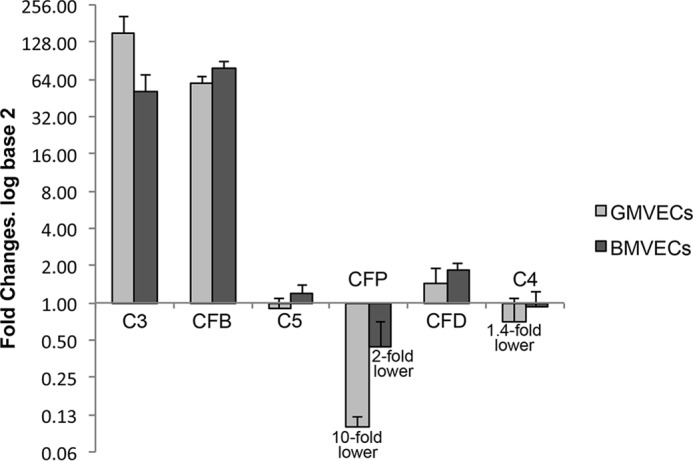
Quantitative gene expression of AP components by TNF-stimulated GMVECs and BMVECs. The mRNA levels of the genes for the AP components C3, CFB, C5, CFP, and CFD and the classical complement component C4 in TNF-stimulated BMVECs and GMVECs were quantified relative to the same genes in unstimulated BMVECs and GMVECs. RNA was extracted from unstimulated BMVECs and GMVECs that were maintained in serum-free medium for 24 h and in these cells after incubation with 10 ng/ml TNF for 48 h (24 h in complete medium and 24 h in serum-free medium). Fold changes in TNF-stimulated BMVEC and GMVEC mRNA levels were calculated relative to levels in unstimulated MVECs. RNA was extracted in 4–6 separate experiments from each cell type. Data are means plus standard deviations (S.D.) from RT-qPCR runs with triplicate measurements. GAPDH was used for normalization.
Gene expression of AP surface and soluble regulatory protein genes in unstimulated and TNF-stimulated BMVECs relative to GMVECs
Gene expression levels of surface (CD46, THBD, and CD55) and soluble (CFI and CFH) AP regulatory protein genes in BMVECs were calculated relative to levels in GMVECs. BMVECs had significantly higher levels of mRNA encoding each of these five AP regulatory proteins studied, compared with GMVECs. The largest relative differences were in CD46, CD55, and CFI, where expression levels were 6-, 14-, and 18-fold higher, respectively (Fig. 3A and Table 2). TNF-stimulated BMVECs also had higher expression levels of all five AP regulatory protein genes studied relative to TNF-stimulated GMVECs, and TNF magnified the relative differences for THBD, CD55, CFI and CFH, which were 10-, 18-, 23-, and 7-fold higher, respectively (Fig. 3B and Table 2).
Figure 3.

Gene expression of AP surface and soluble regulatory protein genes in unstimulated and TNF-stimulated BMVECs relative to GMVECs. The mRNA levels of the genes for three membrane-bound regulatory receptors (CD46, THBD, and CD55) and two soluble regulatory proteins (CFH and CFI) were evaluated by RT-qPCR in unstimulated BMVECs and GMVECs (A) and in TNF-stimulated BMVECs and GMVECs (B). RNA was extracted from unstimulated BMVECs and GMVECs that were maintained in serum-free medium for 24 h and from TNF-stimulated cells after incubation with 10 ng/ml TNF for 48 h (24 h in complete medium and 24 h in serum-free medium). Fold-changes in BMVEC mRNA levels were calculated relative to the mRNA levels in GMVECs. RNA was extracted in 4–6 separate experiments from each cell type. Data are means plus S.D. from RT-qPCR runs with triplicate measurements. GAPDH was used for normalization. *, p < 0.05; **, p < 0.001.
Table 2.
BMVEC complement regulatory gene expression levels relative to GMVECs
Summary of regulatory gene expression levels in unstimulated and TNF-stimulated BMVECs relative to levels in unstimulated and TNF-stimulated GMVECs.
| Unstimulated BMVECs | TNF-stimulated BMVECs | |
|---|---|---|
| CD46 | 6-Fold ↑ | 4-Fold ↑ |
| THBD | 2.5-Fold ↑ | 10-Fold ↑ |
| CD55 | 14-Fold ↑ | 18-Fold ↑ |
| CFI | 18-Fold ↑ | 23-Fold ↑ |
| CFH | 3.5-Fold ↑ | 7-Fold ↑ |
Surface AP regulatory proteins in unstimulated and TNF-stimulated BMVECs and GMVECs
Protein levels of the surface AP regulators, CD46 and CD55, on unstimulated and TNF-stimulated BMVECs were determined by flow cytometry and compared with levels on GMVECs (55). Under both conditions CD55 receptors were ∼2-fold higher on BMVECs compared with GMVECs (Fig. 4A). CD46 protein presence on BMVEC and GMVEC surfaces was similar (Fig. 4A), even though CD46 gene expression was higher in BMVECs compared with GMVECs (Fig. 3). A possible explanation for this enigma is that the mAb used to detect surface CD46 does not bind and detect the complete array of BMVEC CD46 isoforms. CD46 has multiple isoforms as a result of alternate gene splicing, and identical CD46 isoforms are not present in cerebral and renal tissue (56). TNF resulted in statistically significant increases of detectable CD46 on the surface of both cell types (1.6-fold on GMVECs and 2.1-fold on BMVECs) but did not result in significant changes in CD55 on either cell type (Fig. 4A).
Figure 4.
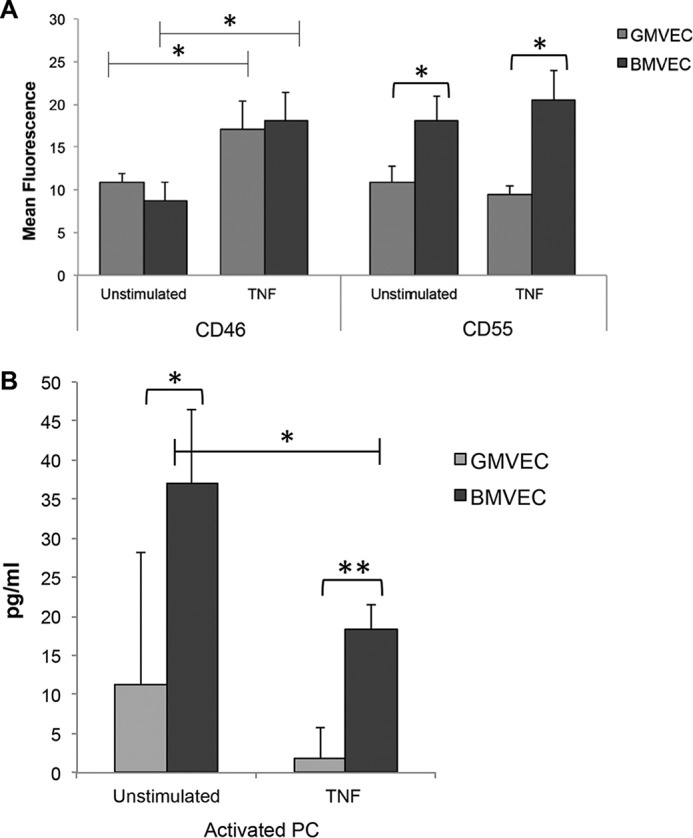
CD46 and CD55 on the surface of and activated PC in the supernatant of unstimulated and TNF-stimulated BMVECs and GMVECs. A, surface complement regulatory receptors were measured by flow cytometry. BMVECs and GMVECs were incubated with or without TNF (10 ng/ml)-supplemented media for 48 h (without TNF n = 4 for BMVECs and n = 6 for GMVECs; with TNF n = 4 for BMVECs and n = 8 for GMVECs). Samples of 2 × 104 unstimulated or TNF-stimulated BMVECs and GMVECs were labeled with saturating amounts of FITC-conjugated mAbs to CD46 and CD55 or with FITC-conjugated isotype control antibodies. Values are mean fluorescence intensities plus S.D. B, levels of activated PC were measured in BMVEC (n = 4) and GMVEC (n = 5) supernatant after the addition of 0.2 μm PC and 10 nm thrombin over 60 min. The BMVECs and GMVECs in T-75 flasks were either unstimulated or incubated with 10 ng/ml TNF for 48 h before the experiment. *, p < 0.05; **, p < 0.001.
Our previous study demonstrating a 6.7-fold down-regulation of GMVEC THBD expression by TNF (55) led us to compare CD141 receptor function for thrombin binding and protein C activation on both cell types under nonstimulating and TNF-stimulating conditions. TNF stimulation resulted in decreased activation of PC in both GMVECs and BMVECs. Without TNF stimulation, BMVECs generated 3.3-fold more activated PC than unstimulated GMVECs, and with TNF stimulation, BMVECs generated 10-fold more activated PC than TNF-stimulated GMVECs (Fig. 4B).
Soluble AP regulatory protein, FH, in BMVEC and GMVEC supernatant with and without TNF stimulation
FH levels measured from the supernatant of both unstimulated and TNF-stimulated BMVECs were ∼1.5-fold higher than levels measured from the supernatant of unstimulated and TNF-stimulated GMVECs, respectively (Fig. 5).
Figure 5.
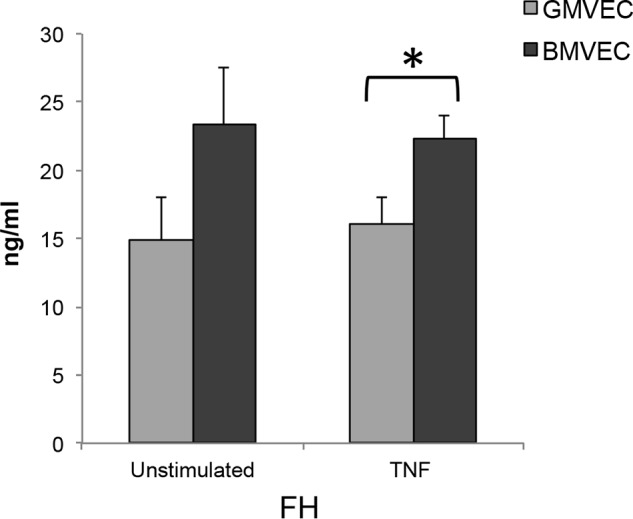
Levels of the soluble AP regulatory protein, FH, in the supernatant of unstimulated and TNF-stimulated GMVECs and BMVECs. Supernatant from unstimulated GMVECs (n = 4) and BMVECs (n = 4) in T-75 flasks was collected after 24 h in serum-free media and assayed for FH by ELISA. The same flasks were then stimulated for 48 h with 10 ng/ml TNF (24 h in complete media and 24 h in serum-free media), and the 24-h supernatant was again collected and assayed for FH. *, p < 0.05.
Levels of complement proteins and activation products in unstimulated and TNF-stimulated BMVEC and GMVEC supernatants
C3 protein released from unstimulated and TNF-stimulated BMVECs were ∼5- and ∼4-fold higher, respectively, than C3 released from unstimulated and TNF-stimulated GMVECs (Fig. 6A). FB released from unstimulated BMVECs and GMVECs was undetectable, and with TNF stimulation, BMVECs released substantially more FB (∼4-fold) relative to TNF-stimulated GMVECs (Fig. 6A).
Figure 6.

Levels of complement proteins, C3 and FB, and complement activation products, C3a, C5a, and Ba, in the supernatant of unstimulated and TNF-stimulated BMVECs and GMVECs. Supernatant from unstimulated BMVECs and GMVECs in T-75 flasks was collected and assayed by ELISA for complement components C3 and FB (A) and AP activation products C3a, C5a, and Ba (B and C). The same flasks were then stimulated for 48 h with 10 ng/ml TNF (24 h in complete media and 24 h in serum-free media), and the 24-h supernatant was again collected and assayed for the same proteins. A, n = 4 for BMVECs and n = 6 for GMVECs for C3, and n = 4 for both GMVECs and BMVECs for FB; FB levels in unstimulated BMVECs and GMVECs were below detection limits (bdl) of the assay. B, n = 6 for GMVECs and n = 4 for BMVECs for C3a experiments, n = 6 for both BMVECs and GMVECs for C5a experiments, and n = 4 for both BMVECs and GMVECs for Ba experiments; C3a levels in unstimulated and TNF-stimulated BMVECs were below detection limits of the assay. C, percentage of Ba generated from FB under TNF-stimulating conditions was calculated from Ba levels in B and FB levels in A; n = 4 for both BMVECs and GMVECs. *, p < 0.05; **, p < 0.001.
We studied differences in AP activation between GMVECs and BMVECs with and without TNF stimulation by measuring complement activation products, C3a, C5a, and Ba, in the cell supernatant. Without or with TNF stimulation, GMVECs produced much higher concentrations of C3a and C5a than did BMVECs. Unstimulated and TNF-stimulated BMVECs did not generate detectable C3a and produced only minute amounts of C5a, demonstrating that GMVECs have increased complement activation compared with BMVECs under both experimental conditions (Fig. 6B). In contrast to C3a and C5a, Ba was generated by both cell types under both experimental conditions and increased significantly in the supernatant of both cell types with TNF stimulation (consistent with TNF-mediated AP activation as demonstrated previously (55)). Ba is an AP-specific activation product that is released from C3b-bound FB after cleavage by FD to form C3bBb (11). The Bb in this convertase can be displaced by FH or CD46 (without changing released Ba levels), followed by inactivation by co-factor FI to iC3b (57).
As shown in Fig. 6A, TNF-stimulated BMVECs released about 4-fold more FB than TNF-stimulated GMVECs. Thus, under conditions of uncontrolled TNF-mediated AP activation and 100% conversion, BMVECs would be capable of generating 4-fold more Ba than GMVECs. However, we found that only ∼2% of the FB produced by TNF-stimulated BMVECs was cleaved/activated to Ba. In contrast, in TNF-stimulated GMVECs, ∼4% of the FB produced was cleaved/activated to Ba (Fig. 6C). Therefore, with TNF stimulation, BMVECs converted half as much Ba from FB than GMVECs. This indicates that although TNF-stimulated BMVECs released higher concentrations of both C3 and FB than TNF-stimulated GMVECs, a smaller percentage of possible C3 convertases was formed in BMVECs. The undetectable levels of C3a in BMVECs (Fig. 6B) also may indicate that the C3 convertases that did form were subsequently inactivated (by FH and CD46). Consequently, further AP activation (C5 convertase formation) was suppressed.
Gene expression of C3a and C5a receptors, C3AR1 and C5AR1, in unstimulated and TNF-stimulated BMVECs and GMVECs
BMVEC mRNA levels of C3AR1 and C5AR1, the genes encoding C3aR and C5aR, were calculated relative to levels in GMVECs. Under both unstimulated and TNF-stimulated conditions, GMVECs expressed considerably higher mRNA levels of C3AR1 than BMVECs. Unstimulated BMVECs had 4.8-fold fewer C3AR1 mRNA copies relative to unstimulated GMVECs (Fig. 7A). TNF-stimulated BMVECs had C3AR1 mRNA levels that were 23-fold lower relative to levels in TNF-stimulated GMVECs (Fig. 7B). TNF stimulation induced a 5.8-fold increase in C3AR1 expression in GMVECs and only a 1.4-fold increase in BMVECs (Fig. 8), accounting for the 23-fold relative difference in the TNF-stimulated cell types. Expression levels for C5AR1 were undetectable in both BMVECs and GMVECs without and with TNF stimulation (Table S1).
Figure 7.
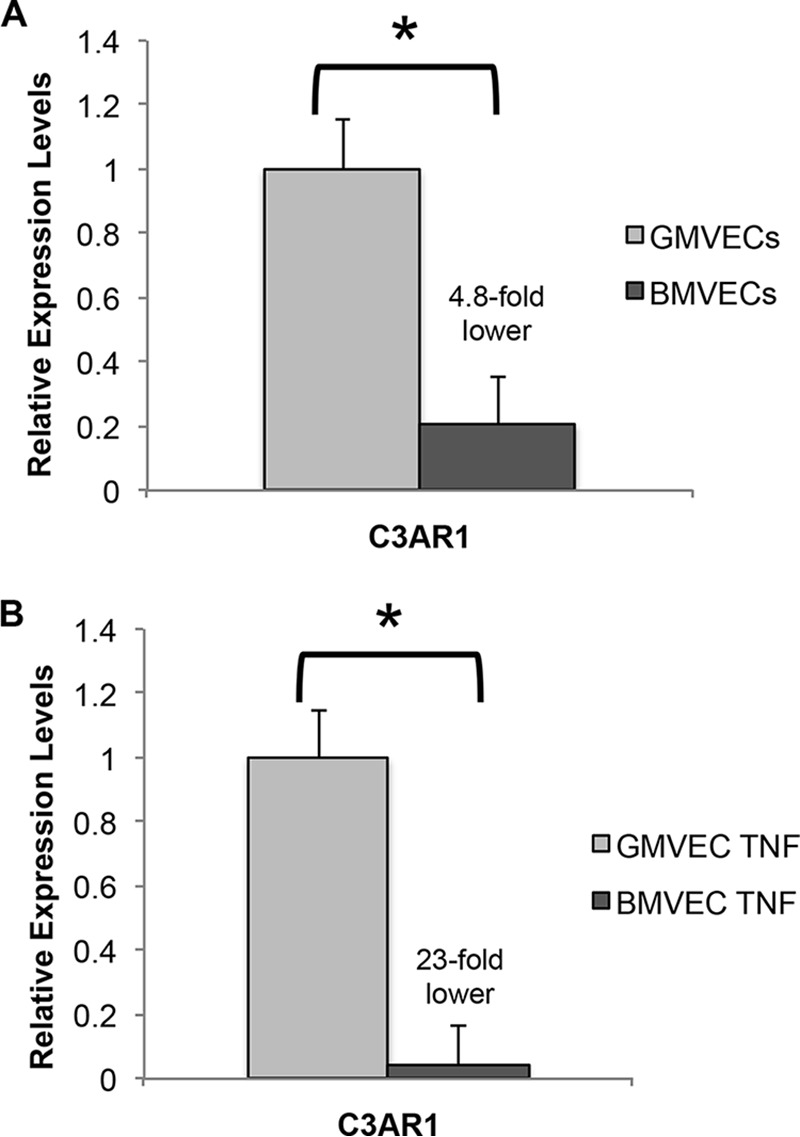
Gene expression levels of C3AR1 in unstimulated and TNF-stimulated BMVECs and GMVECs. Expression levels of C3AR1, the gene encoding the C3a receptor, were measured by RT-qPCR in unstimulated (A) and in TNF-stimulated (B) BMVECs and GMVECs. BMVEC values were calculated relative to levels in GMVECs. Unstimulated cells were maintained in serum-free medium for 24 h, and TNF-stimulated BMVECs and GMVECs were treated with 10 ng/ml TNF for 48 h (24 h in complete medium and 24 h in serum-free medium). RNA was extracted in 4–6 separate experiments from BMVECs and GMVECs with and without TNF. Data are means plus S.D. from RT-qPCR runs with triplicate measurements. GAPDH was used for normalization. *, p < 0.05.
Figure 8.
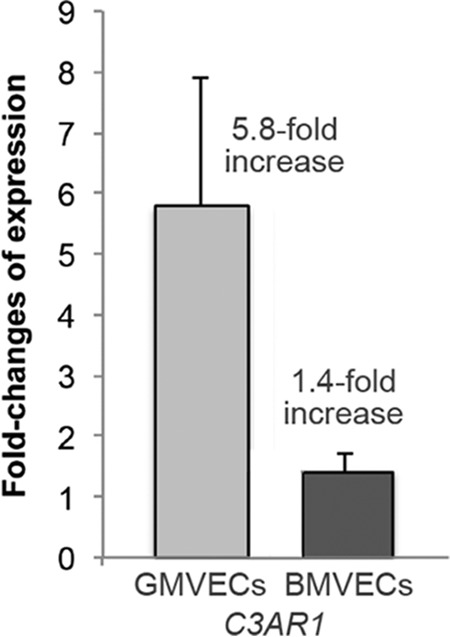
Quantitative gene expression of C3AR1 in TNF-stimulated BMVECs and GMVECs. The fold-changes in mRNA levels in BMVECs and GMVECs after TNF stimulation were measured by RT-qPCR for the C3a receptor gene C3AR1. RNA was extracted in 4–6 separate experiments from unstimulated BMVECs and GMVECs that were maintained in serum-free medium for 24 h and in cells stimulated with 10 ng/ml TNF for 48 h (24 h in complete medium and 24 h in serum-free medium). Changes in gene expression in GMVECs and BMVECs in the presence of TNF were calculated using the method developed by Pfaffl et al. (78). Data are means plus standard deviations from 3 to 5 real-time qPCR runs with triplicate measurements. GAPDH was used for normalization.
C3aR and C5aR proteins on the surface of unstimulated and TNF-stimulated BMVECs and GMVECs
C3aR was ∼2-fold higher on BMVECs under both unstimulated and TNF-stimulated conditions. TNF stimulation increased C3aR detection by ∼1.5-fold on the surface of both cell types, although this was only statistically significant in GMVECs (Fig. 9A). This detection of more C3aR on BMVEC compared with GMVEC surfaces, along with the finding that the receptor only increased by 1.5-fold on TNF-stimulated GMVEC surfaces, was unexpectedly different from the gene expression results (Figs. 7 and 8). Analysis of the single-gated cell populations by flow cytometry showed that a considerable percentage of both BMVECs (∼73%) and GMVECs (∼85%) were negative for surface C3aR, although saturating concentrations of the detection antibody were used (Fig. 9B). Similar results showing a reduction in C3a receptors have been previously reported by others to be caused by receptor complex (C3a-bound C3aR) internalization (58, 59).
Figure 9.

C3aR protein on surfaces of unstimulated and TNF-stimulated BMVECs and GMVECs. BMVECs and GMVECs were incubated with or without TNF (10 ng/ml)-supplemented media for 48 h. Samples of 2 × 104 BMVECs (n = 5) or GMVECs (n = 5) were labeled with saturating amounts of PE-conjugated mAbs to C3aR or with PE-conjugated isotype control antibodies alone (to measure background fluorescence), fixed, and analyzed by flow cytometry. A, mean fluorescence intensities (plus S.D.) of C3aR on BMVEC and GMVEC surfaces without and with TNF. B, single gated population of GMVECs (upper panel) or BMVECs (lower panel) detected by forward and side scatter was analyzed in quadrant plots of PE fluorescence (y axis) and forward scatter (FSC, x axis). Fluorescent detection of C3aR within this single gate showed two distinct cell populations, positive or negative for C3aR, in each cell type and under both experimental conditions (unstimulated and TNF-stimulated). The same quantity of isotype antibody and monoclonal mouse antibody to C3aR was used in both cell types. Quadrant plots are representative of at least five experiments on each cell type, with and without TNF stimulation. *, p < 0.05; **, p < 0.001.
C5aR was not detected by flow cytometry on the surfaces of unstimulated or TNF-stimulated BMVECs or GMVECs (Fig. S1).
Discussion
We compared AP activation and regulation in BMVECs to GMVECs under nonstimulating and TNF-stimulating conditions in an effort to elucidate relative cerebral and renal injury in aHUS and TA-TMA. Our data indicate that BMVECs are more effective in resisting TNF-mediated AP activation than GMVECs, possibly contributing to the explanation for the absence of cerebral microvascular injury and the prominence of renal damage in patients with inflammation/infection-induced episodes of aHUS and TA-TMA.
Under both experimental conditions, the AP component gene expression levels of C3, CFB, and C5 in BMVECs were much higher than the expression levels in GMVECs. Conversely, gene expression levels of CFD were ∼3-fold lower in unstimulated and TNF-stimulated BMVECs compared with GMVECs under the same conditions (Fig. 1). The relatively reduced quantity of FD produced in BMVECs may contribute to the diminished cleavage of FB into active Bb (restricting C3 convertase formation and AP activation) in BMVECs relative to GMVECs.
Compared with GMVECs, BMVECs demonstrated increased gene expression for the negative AP regulatory protein genes, CD46, THBD, CD55, CFI, and CFH (Table 2 and Fig. 3) under both unstimulated and TNF-stimulated conditions. TNF did not affect levels of CD55 protein in either cell type, but it did result in statistically significant increases of detectable CD46 receptors on both cell types (with a greater increase in BMVECs, Fig. 4A). Because BMVECs had higher levels of gene expression for all five AP regulatory proteins than GMVECs under basal conditions, control of AP activation would be predicted to be more effective in BMVECs than in GMVECs with an AP-activating agent, such as TNF. To assess this functionally, we measured AP activation products C3a, C5a, and Ba in the supernatant of unstimulated and TNF-stimulated BMVECs and GMVECs and found that control of AP activation was, in fact, more effective in BMVECs. With provocation of AP activation by TNF, BMVECs displayed significantly decreased AP activation by converting half as much Ba from FB than GMVECs (2% in BMVECs and 4% in GMVECs, Fig. 6C). In this in vitro system, this is evidence that GMVECs are less able to control AP activation than BMVECs. In aHUS and TA-TMA patients with underlying susceptibilities to AP activation, even this seemingly inconsequential inability to control excessive AP activation may lead to deleterious effects.
Previously, we found that GMVECs generated ∼5-fold higher levels of C3a than HUVECs with and without TNF stimulation (55). In contrast to GMVECs and HUVECs, BMVECs in this study did not generate detectable levels of C3a under either condition. This is likely because of the following two reasons: 1) the ample quantities of soluble and surface BMVEC-negative regulatory molecules available to suppress AP initiation and active C3 convertase formation (Figs. 3–5 and Table 2); and 2) binding of C3a to C3aR, preventing detection in the cell supernatant (which likely also occurs in GMVECs). Finally, BMVECs generated barely detectable levels of C5a without (<0.1 pg/ml) and with (∼2 pg/ml) TNF stimulation. In contrast, C5a levels generated by GMVECs were about 50 pg/ml under both experimental conditions (Fig. 6B). The low level of C5a generated by BMVECs is compatible with minimal formation of C3 and C5 convertases by this cell type because of ample AP regulatory molecules to suppress AP activation. Conversely, the vigorous generation of C3a and C5a by GMVECs is likely the result of abundant formation of these convertases due to insufficient amounts of AP regulatory molecules (compared with BMVECs).
Because of the higher production of FH and CD46, and the nearly negligible production of C5a by TNF-stimulated BMVECs, the elevated Ba generated by BMVECs during TNF stimulation (Fig. 6B) likely does not indicate increased AP activation compared with GMVECs. FB that is bound to C3b can be cleaved/activated by FD to produce Bb and release Ba during the formation of the AP C3 convertase (C3bBb). The cleaved Bb can be displaced by FH (57) or CD46 (60), which function as co-factors of FI to generate iC3b, to reduce further amplification of C3b. Without enough active C3 convertase formation to increase the affinity of C5 binding, the C5 convertase will not be generated (61). Therefore, C5 convertase generation in BMVECs is suppressed without affecting the quantity of Ba released. Additionally, the higher amounts of Ba in TNF-stimulated BMVEC supernatant are unlikely to have much effect on BMVEC membranes without a known receptor or function for Ba.
Functionally, BMVECs, compared with GMVECs, generated ∼3- and 10-fold higher levels of activated PC under unstimulated and TNF-stimulated conditions, respectively, indicating a higher amount of CD141 on BMVEC surfaces under both conditions (Fig. 4B). CD141, as both a complement regulator and a natural anticoagulant, has the potential to play a role in host cell protection from AP-mediated injury (by acting as a co-factor for FI-mediated proteolysis of C3b (30, 31)) and from thrombotic injury (by increasing activation of PC (33)). Because of the strong relationship between low activated PC levels and thrombosis, GMVECs are likely more prone to thrombus generation, which in vivo could lead to occlusion of the microvessels and renal dysfunction.
Gene expression levels of C3AR1 were many-fold lower in both unstimulated and TNF-stimulated BMVECs than in GMVECs (Fig. 7), and were increased to a greater extent by TNF stimulation in GMVECs (Fig. 8). In contrast, BMVECs had more C3aR surface protein under both experimental conditions compared with GMVECs (Fig. 9), and TNF stimulation only resulted in a 1.5-fold increase of the receptor on GMVEC surfaces (versus 5.8-fold gene level increase). The higher amounts of C3a generated by GMVECs (compared with BMVECs) likely leads to increased C3a–C3aR binding, followed by functional internalization of the C3aR–C3a complex and therefore lower detection of C3aR on GMVECs by flow cytometry under both unstimulated and TNF-stimulated conditions. This interpretation is compatible with previous observations by others demonstrating that human granulocytes internalize C3a–C3aR complexes (40, 41). C3a–C3aR internalization also likely explains why C3a levels in GMVECs did not increase in parallel with increasing Ba levels during TNF-mediated AP activation (Fig. 6B).
Expression of the C5aR gene, C5AR1, as well as presence of C5aR, was below the levels of detection for BMVECs and GMVECs under unstimulated and TNF-stimulated conditions (Table S1 and Fig. S1). The absence of C5aR confirmed that generated C5a is not partially receptor-bound, the measured C5a levels are accurate, and that the extremely low C5a levels measured in BMVECs are the result of limited AP activation.
We have shown that TNF activates the AP in GMVECs and BMVECs, evidenced by the increases in Ba protein with TNF stimulation. However, our data do not show parallel increases in C3a and C5a (Fig. 6B). C3a likely does increase with TNF stimulation, but the corresponding rise of C3aR prevents detection of the increase because of C3a binding and C3a–C3aR complex internalization. TNF stimulated GMVECs still generated enough additional C3a to exceed the saturation of their increased numbers of C3aR but not enough to detect a significant rise in C3a in their supernatant. The TNF-stimulated BMVECs, even with 20-fold fewer C3aR and 7-fold more C3 synthesis (based on gene expression data), did not generate even enough C3a to exceed C3aR saturation, and therefore, C3a was not detected in TNF-stimulated BMVEC supernatant. Because neither the gene expression nor the C5aR protein was detected in either BMVECs or GMVECs, the detected levels of generated C5a were more accurate. C5a most likely did not increase in TNF-stimulated BMVECs because the increased amounts of FH and CD46 prevented sufficient C3b amplification to form C5 convertases. Alternatively, the production of C5a in GMVECs with TNF stimulation may have been limited by the synthesis rate of C5; as shown in Fig. 2, TNF stimulation resulted in extensive increases in C3 and CFB gene expression levels, without changes in C5 gene expression.
C3a and C5a are potent anaphylatoxins implicated in many disease states, including sepsis, asthma and allergy, cancer, neurodegenerative diseases, and autoimmune diseases, among others (22, 62–73). Some studies have reported that these proteins may contribute to renal damage (74–77). These data, along with the data reported here on GMVEC generation of C3a and increased production of C3aR, suggest that C3a may be involved in the pathogenesis of renal injury in aHUS and TA-TMA. Our in vitro findings of increased alternative complement pathway activation along with vigorous C3a generation in GMVECs, compared with BMVECs, correspond strongly to the in vivo clinical observation that injury to the kidneys is much more frequently observed than injury to the brain in patients with aHUS and TA-TMA.
Plasma AP protein concentrations are much higher than the levels generated by the MVECs in this study. In vivo, circulating plasma complement proteins may promote supplementary assembly/activation of AP components on these two types of MVECs. However, these circulating plasma proteins are unlikely to alter the number or function of membrane-bound regulatory receptors (CD141, CD55, and CD46), which provide the most effective host cell defense. Additionally, in our experiments, BMVECs and GMVECs express and produce their own AP components and AP regulatory proteins. Because there was no external addition of complement proteins, our models provide an ideal system for the elucidation of basic molecular mechanisms of AP activation and regulation.
In summary, we compared AP activation and regulation in BMVECs and GMVECs and demonstrated that GMVECs produce decreased amounts of the AP regulatory proteins, activate the AP to a greater extent, and generate more C3a and C5a than BMVECs under both nonstimulating and TNF-stimulating conditions. The data may contribute to the susceptibility of the kidneys and relative resistance of the brain to AP-mediated injury in aHUS and TA-TMA.
Experimental procedures
Cells
BMVECs and GMVECs
Pooled primary human BMVECs (ACBRI-376 V) and GMVECs (ACBRI-128 V) were purchased from Cell Systems. BMVECs and GMVECs were grown in complete media (CM131, MCDB-131 medium (Sigma), supplemented with penicillin/streptomycin/l-glutamine (Life Technologies, Inc.), plus microvascular growth supplement (Life Technologies, Inc.)). These MVECs serve as an ideal model for our studies as they produce and secrete all AP components and regulators, as well as VWF (18, 55). Additionally, GMVECs are the cells that are primarily affected in TMA, whereas BMVECs serve as an optimal control, because the brain is an organ that has no pathologic association with aHUS and TA-TMA.
Seeding
GMVECs and BMVECs were seeded in T-25 flasks for flow cytometry experiments and in T-75 flasks for gene expression experiments and AP component/activated PC measurements.
VWF detection in Weibel-Palade bodies (WPBs)
Characterization of the cells as endothelial was confirmed by immunofluorescence that showed 95% of the GMVECs or BMVECs positive for VWF in Weibel-Palade bodies (Fig. S2). BMVECs and GMVECs were grown to confluence (8–12 days) on coverslips. For internal detection of VWF in Weibel-Palade bodies, the ECs were first washed with PBS, fixed for 10 min with 2% p-formaldehyde in PBS, and made permeable with 0.2% Triton X-100 for 5 min. After further PBS washes, the ECs were stained with polyclonal rabbit anti-human VWF (Ramco Laboratories) plus secondary Alexa Fluor 488 (green) chicken anti-rabbit IgG (Invitrogen catalog no. A21441) for 15 min. Cell nuclei (blue) were detected with 1 nm 4,6 diamidino-2-phenylindole (DAPI) in the mounting solution (Fluoro-gel II). Fluorescent images were acquired using IP Lab software version 3.9.4r4 (Scanalytics, Inc., Fairfax, VA) on a Nikon Diaphot TE300 microscope equipped with a CFI Plan Apo Lambda ×100 oil N.A. 1.45 objective plus ×10 projection lens (Nikon, Garden City, NY), SensiCamQE CCD camera (Cooke Corp., Romulus, MI), motorized stage and dual filter wheels (Prior) with single band excitation, and emission filters for FITC/TRITC/CY5/DAPI (Chroma, Rockingham, VT).
Reverse transcription real-time quantitative PCR (RT-qPCR)
RNA was extracted from unstimulated and TNF-stimulated BMVECs and GMVECs using TRIzol and isolated using chloroform extraction and isopropyl alcohol precipitation. RNA integrity was verified by 260:280 optical ratios and 1% agarose/formaldehyde electrophoresis. RNA samples were reverse-transcribed using SuperScript VILO MasterMix (Invitrogen), and the resulting cDNA samples (100 ng) were amplified in triplicate by real-time qPCR under the following conditions: 95 °C for 3 min, 40 cycles of 10 s at 95 °C, 10 s at 55 °C, and 30 s at 72 °C, and 95° for 10 s (CFX96, Bio-Rad). Amplified products were detected using TaqMan Gene Expression Assays (with probes that span target exon junctions) and Perfect FastMix II (Quanta).
Quantitative changes in gene expression of BMVECs and GMVECs in the presence of TNF were calculated using the method developed by Pfaffl et al. (78), which uses specific primer efficiencies to evaluate the amount of cDNA amplification per each PCR cycle. Gene efficiencies were determined by amplification of 100 to 0.01 ng of cDNA and calculating the slope of the line after plotting the quantification cycle (Cq) versus nanograms of cDNA. The relative quantification of gene expression with/without TNF stimulation in BMVECs and GMVECs was calculated as described by Livak and Schmittgen (79). The standard deviation in gene expression assays (s) was determined by Equation 1,
| (1) |
where s1 and s2 are the standard deviations of triplicate Cq measurements for the reference and target genes.
Genes studied include the complement surface regulatory proteins CD46, CD55, and THBD (gene for CD141); soluble regulatory proteins CFH and CFI; AP components C3, C5, CFB, CFD, and CFP; the classical component C4; anaphylatoxin receptors C3AR1 and C5AR1; and the reference gene GAPDH. Assay probe ID numbers and gene efficiencies are listed in Table S2. The Minimum Information for publication of Quantitative real-time PCR Experiment (MIQE) guidelines state that using a single reference gene is permissible if there is consistent quantification in the quantification cycle of the reference gene under untreated and treated experimental conditions (80). Data in Table S3 show the consistent quantification cycle of GAPDH in untreated and TNF-treated BMVECs and GMVECs, thus satisfying these criteria.
Flow cytometry
TNF stimulation of GMVECs and BMVECs
Once confluent in T-25 flasks, control cells were incubated for 24 h in serum-free media (MCDB-131 plus insulin–transferrin–selenium, Life Technologies, Inc.), and experimental cells were incubated for 24 h with CM131 plus TNF (10 ng/ml, Life Technologies, Inc.) and then incubated for an additional 24 h in serum-free media plus 10 ng/ml TNF (total TNF exposure 48 h). Both control and experimental flasks were incubated in serum-free media for a total of 24 h to eliminate any proteins derived from serum that might affect surface receptor detection. The specific concentration and duration of exposure of TNF were chosen based on our previous experimentations (55).
Cell-surface labeling of CD46, CD141, CD55, C3aR, and C5aR
Control and experimental cells were detached by a 10-min incubation with 5 mm EDTA in Ca+2/Mg+2-free PBS (to retain surface proteins) and centrifuged (10 min at 400 × g). Cells were counted using the TC10 automated cell counter (Bio-Rad) and resuspended in 1% BSA/Ca2+/Mg+2-free PBS at 106 cells/ml. Samples of 2 × 104 GMVECs or BMVECs (20 μl) were labeled individually with saturating amounts of each fluorescently conjugated mAb (see below) or labeled with saturating amounts of FITC-conjugated (CD46 and CD55) or PE-conjugated (CD141, C3aR, and C5aR) isotype controls (see below) to measure background fluorescence. Saturating amounts of antibody were as follows: 1 μl of CD46, CD141, CD55, or C5aR antibody + 1 μl of corresponding isotype control per 20-μl cell suspension, and 4 μl of C3aR antibody + 4 μl of corresponding isotype control per 20-μl cell suspension. Samples were incubated for 20 min in the dark. Cells were then fixed with 0.5 ml of 1% formaldehyde/Ca2+/Mg2+-free PBS. Cells expressing higher fluorescence than background (isotype control alone) were considered positive, and background fluorescence was subtracted from positive fluorescence. Experiments were repeated with increasing passage number (passages 5–7 for GMVECs and passages 4–6 for BMVECs) to exclude the possibility that any differences appreciated were related to passage differences.
Abs to human AP regulatory proteins (flow cytometry)
Abs include the following: CD46, clone E4.3, mouse monoclonal IgG2a,κ conjugated to FITC (BD Biosciences, 555949); CD141, clone 1A4, mouse monoclonal IgG1,κ conjugated to PE (BD Biosciences, 559781); and CD55, clone IA10, mouse monoclonal IgG2a,κ conjugated to FITC (BD Biosciences, 555693). Isotype controls included PE mouse, clone IgG1 (BD Biosciences, 349043), and FITC mouse, clone IgG2a,κ (BD Biosciences, 555573).
Abs to human C3aR and C5aR (flow cytometry)
Abs include the following: C3aR, clone hC3aRZ8, mouse monoclonal IgG2b,κ conjugated to PE (BD Biosciences, 561178), and C5aR, clone D53-1473, mouse monoclonal IgG1,κ conjugated to PE (BD Biosciences, 550494). Isotype controls included PE mouse, clone IgG1 (BD Biosciences, 349043), and FITC mouse, clone IgG2a,κ (BD Biosciences, 555573).
Acquisition and analysis
Samples were acquired using FACScan (BD Biosciences), and the data were analyzed using CellQuest software (BD Biosciences) or FlowJo Collector's Edition software (Cytec). The instrument settings of the forward scatter and side scatter profiles were log mode. MVEC samples appeared as single populations and were gated based on their forward and side-scatter profiles. Five thousand gated events were analyzed for each sample.
Measurement of AP components, regulators, and activation products from BMVEC and GMVEC supernatant
In these experiments, AP components C3 and FB, AP regulator FH, and AP activation products, C3a, C5a, and Ba, were assayed by ELISA from the supernatant of unstimulated or TNF-stimulated BMVECs. These experiments assessed nondirectional release of AP components and regulators from the surface of the two types of MVECs.
Supernatant collection and TNF stimulation
Once confluent in T-75 flasks, GMVECs and BMVECs were incubated for 24 h in 1 ml of serum-free media per T-75 flask to concentrate the components in the cell supernatant. The supernatant was collected after 24 h and immediately frozen in liquid nitrogen for 20 s (to prevent further activation of the AP) prior to storing at −80 °C until assayed. These samples were designated the unstimulated controls. After at least 24–48 h of recovery in CM131, the same flasks were then incubated for 24 h in CM131 plus 10 ng/ml TNF and incubated an additional 24 h in 1 ml per T-75 flask of serum-free media plus 10 ng/ml TNF (total TNF exposure of 48 h). The concentrated supernatant was again collected, flash-frozen, and stored at −80 °C until assayed. These samples were designated as the TNF-stimulated samples. GMVECs in these experiments were studied as passages 3 and 4 and BMVECs were used at passages 3–5.
Sample preparation
Samples were rapidly thawed and kept on ice prior to use to prevent spontaneous complement activation, and then analyzed for the various complement pathway components, regulators, or activation products.
C3 fluorescence immunoassay
Black 96-well plates were coated with 50 ng/well polyclonal rabbit anti-human C3a (detects human C3a and C3, Complement Technologies, catalog no. A218) in 100 nm bicarbonate buffer (pH 9.6) overnight at 4 °C. TBST-washed wells were blocked overnight with 1% Ig-free BSA in PBS (BSA/PBS), followed by a 50-min incubation with 100 μl/well test samples (MVEC supernatant from unstimulated or TNF-stimulated cells, no dilution) or purified C3 protein (Complement Technologies, catalog no. A113) for the standard curve (with a range of 9.4–600 ng/ml). TBST-washed wells were next incubated with 31 ng/ml goat polyclonal Ab to human C3 (Complement Technologies, catalog no. A213) for 25 min, followed by incubation with 100 ng/ml secondary donkey anti-goat IgG-HRP (Pierce, catalog no. PA 1-28664). Fluorescence was measured in a Tecan Infinite M200 Pro plate reader 25 min after the addition of HRP substrate 10-acetyl-3,7-dihydroxyphenoxazine (AnaSpec, Fremont, CA) with excitation of 530 nm and emission of 590 nm.
The high range of sensitivity of the C3 immunoassay (as well as the others detailed below) is based on the 10-acetyl-3,7-dihydroxyphenoxazine, a substrate for HRP that reacts with hydrogen peroxide to produce a highly fluorescent product. The raw fluorescent readings for the standards range from 1000 to 45,000. Reciprocal plots of standard dilutions (1/concentration) versus fluorescence intensity at 590 nm (1/590 intensity) produce linear equations that allow the interpolation of complement component concentrations from 0 to the lower limit of the standard curve, in addition to values between the lower limit and higher limit of the standard curve (55).
FB, C3a, and C5a fluorescence immunoassays
FB, C3a, and C5a levels were measured in unstimulated and TNF-stimulated GMVEC and BMVEC supernatant samples with FB, C3a, and C5a ELISAs using the same protocol as the C3 immunoassay. The Abs and standard curves used were as follows: 1) FB: capture, 100 ng/well polyclonal goat anti-human FB (Complement Technologies, catalog no. A235); standard, FB protein (Complement Technologies, catalog no. A135) with a range of 12.5–800 ng/ml; detection Abs, 0.1 μg/ml monoclonal mouse anti-factor Ba (Quidel Corp., catalog no. A225) that was generated using purified human FB as the Ag and is reactive with both FB and the Ba fragment, and 0.1 μg/ml secondary donkey anti-mouse IgG-HRP (Pierce, catalog no. PA1-28748). 2) C3a: capture, 50 ng/well polyclonal rabbit anti-human C3a (Complement Technologies, catalog no. A218); standard, purified C3a des-Arg protein (Complement Technologies, catalog no. A119) with a range of 61 pg/ml to 3.9 ng/ml; detection Abs, 100 ng/ml mouse mAb to human C3a (Pierce, Thermo Fisher Scientific, catalog no. GAU 013-16-02) and 0.25 μg/ml secondary goat anti-mouse IgG-HRP (Rockland Immunochemicals, Limerick, PA). 3) C5a: capture, 100 ng/well polyclonal rabbit anti-human C5a (Complement Technologies, catalog no. A221); standard, C5a des-Arg protein (Complement Technologies, catalog no. A145) with a range of 0.156–10 ng/ml; detection Abs, 0.1 μg/ml mouse monoclonal anti-human C5a (Pierce, Thermo Fisher Scientific, catalog no. MA 1-40162) and 0.25 μg/ml secondary goat anti-mouse IgG-HRP.
Ba and C5a ELISA kits
Ba levels were measured in unstimulated and TNF-stimulated GMVEC and BMVEC supernatant samples using an ELISA kit (Quidel, San Diego, catalog no. A033). C5a levels were measured in unstimulated and TNF-stimulated BMVEC (but not GMVEC) supernatant samples by an ELISA kit (Quidel, catalog no. A021) as a means to confirm the results from the C5a fluorescence immunoassay.
Measurement of activated PC from BMVEC and GMVEC supernatant
In these experiments, activated PC was assayed by ELISA from the supernatant of unstimulated or TNF-stimulated GMVECs and BMVECs after the addition of PC and thrombin.
PC/thrombin supplementation, supernatant collection, and TNF stimulation
GMVECs and BMVECs confluent in T-75 flasks were incubated for 24 h in serum-free media. The cells were then washed with PBS and supplemented with 0.2 μm human PC (Hematologic Technologies, catalog no. HCPC-0070) and 10 nm human α-thrombin (Hematologic Technologies, catalog no. HCT-0020) in 1 ml of activated PC buffer (0.1% BSA, 3 mm CaCl2, 0.6 mm MgCl2 in Ca+2/Mg+2-free PBS) (33) and incubated at 37 °C for 60 min (33, 81). Further activation of PC to activated PC was inhibited with the addition of 10 nm hirudin (1.5 units/ml, Sigma, catalog no. H7016) (33). Supernatants were collected and frozen in liquid nitrogen for 20 s prior to storage at −80 °C until analysis. These samples were designated as the unstimulated controls. After allowing recovery for at least 24–48 h in complete media (CM131), the same flasks were incubated for 24 h in CM131 plus 10 ng/ml TNF and then for an additional 24 h in serum-free media plus 10 ng/ml TNF. The flasks were again washed with PBS and supplemented with 0.2 μm human PC and 10 nm human α-thrombin in 1 ml of activated PC buffer and incubated at 37 °C for 60 min. Supernatant was collected, flash-frozen, and stored at −80 °C. These samples were designated as the TNF-stimulated samples. The 24–48-h recovery time in CM131 after collection of control supernatant samples allowed the MVECs to replenish nutrients after serum-free media incubation. Cell numbers were not affected by the presence of TNF (55).
Sample preparation and assay of activated PC
Samples were rapidly thawed and kept on ice prior to use. Activated PC levels were measured in unstimulated and TNF-stimulated GMVEC and BMVEC supernatant samples using an ELISA kit (Cloud-Clone, Houston, TX, catalog no. SEA738Hu).
Statistics
The p values for statistical significance for Figs. 4–6 and 9 were obtained by using two-tailed distribution and two-sample equal variance Student's t tests. A p value < 0.05 was considered statistically significant.
Author contributions
S. E. S., N. A. T., and J. L. M. conceptualization; S. E. S. and N. A. T. data curation; S. E. S. and N. A. T. formal analysis; S. E. S. and J. L. M. funding acquisition; S. E. S. investigation; S. E. S. and N. A. T. methodology; S. E. S. writing-original draft; S. E. S. and J. L. M. project administration; S. E. S., N. A. T., and J. L. M. writing-review and editing; J. L. M. supervision.
Supplementary Material
This work was supported by grants from the Baylor College of Medicine Pediatric Pilot Award, the Mary R. Gibson Foundation, and the Mabel and Everett Hinkson Memorial Fund. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2, Tables S1–S3, and supporting Refs. 1 and 2.
- TMA
- thrombotic microangiopathy
- aHUS
- atypical hemolytic uremic syndrome
- TA-TMA
- transplantation-associated thrombotic microangiopathy
- AP
- alternative complement pathway
- GMVEC
- glomerular microvascular endothelial cell
- BMVEC
- brain microvascular endothelial cell
- TNF
- tumor necrosis factor
- C3aR
- complement C3a receptor
- FB
- factor B
- FD
- factor D
- ULVWF
- ultra large von Willebrand factor
- HUVEC
- human umbilical vein endothelial cell
- CP
- classical complement pathway
- C5aR
- complement C5a receptor
- FH
- factor H
- FI
- factor I
- THBD
- thrombomodulin
- PC
- protein C
- BSA
- bovine serum albumin
- VWF
- von Willebrand factor
- Ab
- antibody
- qPCR
- quantitative PCR
- PE
- phycoerythrin.
References
- 1. Ruggenenti P., Noris M., and Remuzzi G. (2001) Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 60, 831–846 10.1046/j.1523-1755.2001.060003831.x [DOI] [PubMed] [Google Scholar]
- 2. Moake J. L. (2002) Thrombotic microangiopathies. N. Engl. J. Med. 347, 589–600 10.1056/NEJMra020528 [DOI] [PubMed] [Google Scholar]
- 3. Meri S. (2013) Complement activation in diseases presenting with thrombotic microangiopathy. Eur. J. Intern Med. 24, 496–502 10.1016/j.ejim.2013.05.009 [DOI] [PubMed] [Google Scholar]
- 4. Brodsky R. A. (2015) Complement in hemolytic anemia. Blood 126, 2459–2465 10.1182/blood-2015-06-640995 [DOI] [PubMed] [Google Scholar]
- 5. Jodele S., Licht C., Goebel J., Dixon B. P., Zhang K., Sivakumaran T. A., Davies S. M., Pluthero F. G., Lu L., and Laskin B. L. (2013) Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 122, 2003–2007 10.1182/blood-2013-05-501445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laskin B. L., Goebel J., Davies S. M., and Jodele S. (2011) Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood 118, 1452–1462 10.1182/blood-2011-02-321315 [DOI] [PubMed] [Google Scholar]
- 7. Law S. K., and Levine R. P. (1977) Interaction between the third complement protein and cell surface macromolecules. Proc. Natl. Acad. Sci. U.S.A. 74, 2701–2705 10.1073/pnas.74.7.2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pangburn M. K., Ferreira V. P., and Cortes C. (2008) Discrimination between host and pathogens by the complement system. Vaccine 26, Suppl. 8, l15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Law S. K., and Dodds A. W. (1997) The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 6, 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schreiber R. D., Pangburn M. K., Lesavre P. H., and Müller-Eberhard H. J. (1978) Initiation of the alternative pathway of complement: recognition of activators by bound C3b and assembly of the entire pathway from six isolated proteins. Proc. Natl. Acad. Sci. U.S.A. 75, 3948–3952 10.1073/pnas.75.8.3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rawal N., and Pangburn M. (2001) Formation of high-affinity C5 convertases of the alternative pathway of complement. J. Immunol. 166, 2635–2642 10.4049/jimmunol.166.4.2635 [DOI] [PubMed] [Google Scholar]
- 12. Fearon D. T., Austen K. F., and Ruddy S. (1973) Formation of a hemolytically active cellular intermediate by the interaction between properdin factors B and D and the activated third component of complement. J. Exp. Med. 138, 1305–1313 10.1084/jem.138.6.1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pillemer L., Blum L., Lepow I. H., Ross O. A., Todd E. W., and Wardlaw A. C. (1954) The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 120, 279–285 10.1126/science.120.3112.279 [DOI] [PubMed] [Google Scholar]
- 14. Weiler J. M., Daha M. R., Austen K. F., and Fearon D. T. (1976) Control of the amplification convertase of complement by the plasma protein β1H. Proc. Natl. Acad. Sci. U.S.A. 73, 3268–3272 10.1073/pnas.73.9.3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pangburn M. K. (1983) Activation of complement via the alternative pathway. Fed. Proc. 42, 139–143 [PubMed] [Google Scholar]
- 16. Lachmann P. J., and Hughes-Jones N. C. (1984) Initiation of complement activation. Springer Semin. Immunopathol. 7, 143–162 10.1007/BF01893018 [DOI] [PubMed] [Google Scholar]
- 17. Kinoshita T., Takata Y., Kozono H., Takeda J., Hong K. S., and Inoue K. (1988) C5 convertase of the alternative complement pathway: covalent linkage between two C3b molecules within the trimolecular complex enzyme. J. Immunol. 141, 3895–3901 [PubMed] [Google Scholar]
- 18. Turner N. A., and Moake J. (2013) Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PLoS ONE 8, e59372 10.1371/journal.pone.0059372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng S., Liang X., Cruz M. A., Vu H., Zhou Z., Pemmaraju N., Dong J. F., Kroll M. H., and Afshar-Kharghan V. (2013) The interaction between factor H and Von Willebrand factor. PLoS ONE 8, e73715 10.1371/journal.pone.0073715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tati R., Kristoffersson A. C., Ståhl A. L., Rebetz J., Wang L., Licht C., Motto D., and Karpman D. (2013) Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J. Immunol. 191, 2184–2193 10.4049/jimmunol.1301221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernardo A., Ball C., Nolasco L., Moake J. F., and Dong J. F. (2004) Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 104, 100–106 10.1182/blood-2004-01-0107 [DOI] [PubMed] [Google Scholar]
- 22. Klos A., Tenner A. J., Johswich K. O., Ager R. R., Reis E. S., and Köhl J. (2009) The role of the anaphylatoxins in health and disease. Mol. Immunol. 46, 2753–2766 10.1016/j.molimm.2009.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mueller-Ortiz S. L., Morales J. E., and Wetsel R. A. (2014) The receptor for the complement C3a anaphylatoxin (C3aR) provides host protection against Listeria monocytogenes-induced apoptosis. J. Immunol. 193, 1278–1289 10.4049/jimmunol.1302787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mueller-Ortiz S. L., Calame D. G., Shenoi N., Li Y. D., and Wetsel R. A. (2017) The complement anaphylatoxins C5a and C3a suppress IFN-β production in response to Listeria monocytogenes by inhibition of the cyclic dinucleotide-activated cytosolic surveillance pathway. J. Immunol. 198, 3237–3244 10.4049/jimmunol.1601420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patzelt J., Mueller K. A., Breuning S., Karathanos A., Schleicher R., Seizer P., Gawaz M., Langer H. F., and Geisler T. (2015) Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis 238, 289–295 10.1016/j.atherosclerosis.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 26. Schraufstatter I. U., Trieu K., Sikora L., Sriramarao P., and DiScipio R. (2002) Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J. Immunol. 169, 2102–2110 10.4049/jimmunol.169.4.2102 [DOI] [PubMed] [Google Scholar]
- 27. Kazatchkine M. D., Fearon D. T., and Austen K. F. (1979) Human alternative complement pathway: membrane-associated sialic acid regulates the competition between B and β1H for cell-bound C3b. J. Immunol. 122, 75–81 [PubMed] [Google Scholar]
- 28. Whaley K., and Ruddy S. (1976) Modulation of the alternative complement pathways by β1H globulin. J. Exp. Med. 144, 1147–1163 10.1084/jem.144.5.1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harrison R. A., and Lachmann P. J. (1980) The physiological breakdown of the third component of human complement. Mol. Immunol. 17, 9–20 10.1016/0161-5890(80)90119-4 [DOI] [PubMed] [Google Scholar]
- 30. Delvaeye M., Noris M., De Vriese A., Esmon C. T., Esmon N. L., Ferrell G., Del-Favero J., Plaisance S., Claes B., Lambrechts D., Zoja C., Remuzzi G., and Conway E. M. (2009) Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 361, 345–357 10.1056/NEJMoa0810739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liszewski M. K., Post T. W., and Atkinson J. P. (1991) Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 9, 431–455 10.1146/annurev.iy.09.040191.002243 [DOI] [PubMed] [Google Scholar]
- 32. Nicholson-Weller A., Burge J., Fearon D. T., Weller P. F., and Austen K. F. (1982) Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J. Immunol. 129, 184–189 [PubMed] [Google Scholar]
- 33. Stearns-Kurosawa D. J., Kurosawa S., Mollica J. S., Ferrell G. L., and Esmon C. T. (1996) The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. U.S.A. 93, 10212–10216 10.1073/pnas.93.19.10212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maga T. K., Nishimura C. J., Weaver A. E., Frees K. L., and Smith R. J. (2010) Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum. Mutat. 31, E1445–E1460 10.1002/humu.21256 [DOI] [PubMed] [Google Scholar]
- 35. Zipfel P. F., Edey M., Heinen S., Józsi M., Richter H., Misselwitz J., Hoppe B., Routledge D., Strain L., Hughes A. E., Goodship J. A., Licht C., Goodship T. H., and Skerka C. (2007) Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 3, e41 10.1371/journal.pgen.0030041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frémeaux-Bacchi V., Miller E. C., Liszewski M. K., Strain L., Blouin J., Brown A. L., Moghal N., Kaplan B. S., Weiss R. A., Lhotta K., Kapur G., Mattoo T., Nivet H., Wong W., Gie S., et al. (2008) Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112, 4948–4952 10.1182/blood-2008-01-133702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goicoechea de Jorge E., Harris C. L., Esparza-Gordillo J., Carreras L., Arranz E. A., Garrido C. A., López-Trascasa M., Sánchez-Corral P., Morgan B. P., and Rodríguez de Córdoba S. (2007) Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. U.S.A. 104, 240–245 10.1073/pnas.0603420103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jodele S., Zhang K., Zou F., Laskin B., Dandoy C. E., Myers K. C., Lane A., Meller J., Medvedovic M., Chen J., and Davies S. M. (2016) The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 127, 989–996 10.1182/blood-2015-08-663435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamae H., Yamane T., Hasegawa T., Nakamae M., Terada Y., Hagihara K., Ohta K., and Hino M. (2006) Risk factor analysis for thrombotic microangiopathy after reduced-intensity or myeloablative allogeneic hematopoietic stem cell transplantation. Am. J. Hematol. 81, 525–531 10.1002/ajh.20648 [DOI] [PubMed] [Google Scholar]
- 40. Willems E., Baron F., Seidel L., Frère P., Fillet G., and Beguin Y. (2010) Comparison of thrombotic microangiopathy after allogeneic hematopoietic cell transplantation with high-dose or nonmyeloablative conditioning. Bone Marrow Transplant. 45, 689–693 10.1038/bmt.2009.230 [DOI] [PubMed] [Google Scholar]
- 41. Uderzo C., Bonanomi S., Busca A., Renoldi M., Ferrari P., Iacobelli M., Morreale G., Lanino E., Annaloro C., Volpe A. D., Alessandrino P., Longoni D., Locatelli F., Sangalli H., and Rovelli A. (2006) Risk factors and severe outcome in thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Transplantation 82, 638–644 10.1097/01.tp.0000230373.82376.46 [DOI] [PubMed] [Google Scholar]
- 42. Hale G. A., Bowman L. C., Rochester R. J., Benaim E., Heslop H. E., Krance R. A., Horwitz E. M., Cunningham J. M., Tong X., Srivastava D. K., Handgretinger R., and Jones D. P. (2005) Hemolytic uremic syndrome after bone marrow transplantation: clinical characteristics and outcome in children. Biol. Blood Marrow Transplant. 11, 912–920 10.1016/j.bbmt.2005.07.012 [DOI] [PubMed] [Google Scholar]
- 43. Worel N., Greinix H. T., Leitner G., Mitterbauer M., Rabitsch W., Rosenmayr A., Höcker P., and Kalhs P. (2007) ABO-incompatible allogeneic hematopoietic stem cell transplantation following reduced-intensity conditioning: close association with transplant-associated microangiopathy. Transfus. Apher. Sci. 36, 297–304 10.1016/j.transci.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 44. Batts E. D., and Lazarus H. M. (2007) Diagnosis and treatment of transplantation-associated thrombotic microangiopathy: real progress or are we still waiting? Bone Marrow Transplant. 40, 709–719 10.1038/sj.bmt.1705758 [DOI] [PubMed] [Google Scholar]
- 45. Rosenthal J., Pawlowska A., Bolotin E., Cervantes C., Maroongroge S., Thomas S. H., and Forman S. J. (2011) Transplant-associated thrombotic microangiopathy in pediatric patients treated with sirolimus and tacrolimus. Pediatr. Blood Cancer 57, 142–146 10.1002/pbc.22861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Changsirikulchai S., Myerson D., Guthrie K. A., McDonald G. B., Alpers C. E., and Hingorani S. R. (2009) Renal thrombotic microangiopathy after hematopoietic cell transplant: role of GVHD in pathogenesis. Clin. J. Am. Soc. Nephrol. 4, 345–353 10.2215/CJN.02070508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loirat C., and Frémeaux-Bacchi V. (2011) Atypical hemolytic uremic syndrome. Orphanet J. Rare Dis. 6, 60 10.1186/1750-1172-6-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jodele S., Laskin B. L., Dandoy C. E., Myers K. C., El-Bietar J., Davies S. M., Goebel J., and Dixon B. P. (2015) A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 29, 191–204 10.1016/j.blre.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siami K., Kojouri K., Swisher K. K., Selby G. B., George J. N., and Laszik Z. G. (2008) Thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation: an autopsy study. Transplantation 85, 22–28 10.1097/01.tp.0000297998.33418.7e [DOI] [PubMed] [Google Scholar]
- 50. Sellier-Leclerc A. L., Fremeaux-Bacchi V., Dragon-Durey M. A., Macher M. A., Niaudet P., Guest G., Boudailliez B., Bouissou F., Deschenes G., Gie S., Tsimaratos M., Fischbach M., Morin D., Nivet H., Alberti C., et al. (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 18, 2392–2400 10.1681/ASN.2006080811 [DOI] [PubMed] [Google Scholar]
- 51. Loirat C., Noris M., and Fremeaux-Bacchi V. (2008) Complement and the atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 23, 1957–1972 10.1007/s00467-008-0872-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fakhouri F., Delmas Y., Provot F., Barbet C., Karras A., Makdassi R., Courivaud C., Rifard K., Servais A., Allard C., Besson V., Cousin M., Châtelet V., Goujon J. M., Coindre J. P., et al. (2014) Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. Am. J. Kidney Dis. 63, 40–48 10.1053/j.ajkd.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 53. Cofiell R., Kukreja A., Bedard K., Yan Y., Mickle A. P., Ogawa M., Bedrosian C. L., and Faas S. J. (2015) Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 125, 3253–3262 10.1182/blood-2014-09-600411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seeber C., Hiller E., Holler E., and Kolb H. J. (1992) Increased levels of tissue plasminogen activator (t-PA) and tissue plasminogen activator inhibitor (PAI) correlate with tumor necrosis factor α (TNFα)-release in patients suffering from microangiopathy following allogeneic bone marrow transplantation (BMT). Thromb. Res. 66, 373–383 10.1016/0049-3848(92)90286-J [DOI] [PubMed] [Google Scholar]
- 55. Sartain S. E., Turner N. A., and Moake J. L. (2016) TNF regulates essential alternative complement pathway components and impairs activation of protein C in human glomerular endothelial cells. J. Immunol. 196, 832–845 10.4049/jimmunol.1500960 [DOI] [PubMed] [Google Scholar]
- 56. Tang S. J., Luo S., Ho J. X., Ly P. T., Goh E., and Roca X. (2016) Characterization of the regulation of CD46 RNA alternative splicing. J. Biol. Chem. 291, 14311–14323 10.1074/jbc.M115.710350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pangburn M. K. (1986) Differences between the binding sites of the complement regulatory proteins DAF, CR1, and factor H on C3 convertases. J. Immunol. 136, 2216–2221 [PubMed] [Google Scholar]
- 58. Settmacher B., Bock D., Saad H., Gärtner S., Rheinheimer C., Köhl J., Bautsch W., and Klos A. (1999) Modulation of C3a activity: internalization of the human C3a receptor and its inhibition by C5a. J. Immunol. 162, 7409–7416 [PubMed] [Google Scholar]
- 59. Settmacher B., Rheinheimer C., Hamacher H., Ames R. S., Wise A., Jenkinson L., Bock D., Schaefer M., Köhl J., and Klos A. (2003) Structure-function studies of the C3a-receptor: C-terminal serine and threonine residues which influence receptor internalization and signaling. Eur. J. Immunol. 33, 920–927 10.1002/eji.200323293 [DOI] [PubMed] [Google Scholar]
- 60. Seya T., and Atkinson J. P. (1989) Functional properties of membrane cofactor protein of complement. Biochem. J. 264, 581–588 10.1042/bj2640581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pangburn M. K., and Rawal N. (2002) Structure and function of complement C5 convertase enzymes. Biochem. Soc. Trans. 30, 1006–1010 10.1042/bst0301006 [DOI] [PubMed] [Google Scholar]
- 62. Bengtson A., and Heideman M. (1988) Anaphylatoxin formation in sepsis. Arch. Surg. 123, 645–649 10.1001/archsurg.1988.01400290131023 [DOI] [PubMed] [Google Scholar]
- 63. Hack C. E., Nuijens J. H., Felt-Bersma R. J., Schreuder W. O., Eerenberg-Belmer A. J., Paardekooper J., Bronsveld W., and Thijs L. G. (1989) Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am. J. Med. 86, 20–26 10.1016/0002-9343(89)90224-6 [DOI] [PubMed] [Google Scholar]
- 64. Selberg O., Hecker H., Martin M., Klos A., Bautsch W., and Köhl J. (2000) Discrimination of sepsis and systemic inflammatory response syndrome by determination of circulating plasma concentrations of procalcitonin, protein complement 3a, and interleukin-6. Crit. Care Med. 28, 2793–2798 10.1097/00003246-200008000-00019 [DOI] [PubMed] [Google Scholar]
- 65. Köhl J., Baelder R., Lewkowich I. P., Pandey M. K., Hawlisch H., Wang L., Best J., Herman N. S., Sproles A. A., Zwirner J., Whitsett J. A., Gerard C., Sfyroera G., Lambris J. D., and Wills-Karp M. (2006) A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J. Clin. Invest. 116, 783–796 10.1172/JCI26582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Drouin S. M., Corry D. B., Hollman T. J., Kildsgaard J., and Wetsel R. A. (2002) Absence of the complement anaphylatoxin C3a receptor suppresses Th2 effector functions in a murine model of pulmonary allergy. J. Immunol. 169, 5926–5933 10.4049/jimmunol.169.10.5926 [DOI] [PubMed] [Google Scholar]
- 67. Bautsch W., Hoymann H. G., Zhang Q., Meier-Wiedenbach I., Raschke U., Ames R. S., Sohns B., Flemme N., Meyer zu Vilsendorf A., Grove M., Klos A., and Köhl J. (2000) Cutting edge: guinea pigs with a natural C3a-receptor defect exhibit decreased bronchoconstriction in allergic airway disease: evidence for an involvement of the C3a anaphylatoxin in the pathogenesis of asthma. J. Immunol. 165, 5401–5405 10.4049/jimmunol.165.10.5401 [DOI] [PubMed] [Google Scholar]
- 68. Ji H., Ohmura K., Mahmood U., Lee D. M., Hofhuis F. M., Boackle S. A., Takahashi K., Holers V. M., Walport M., Gerard C., Ezekowitz A., Carroll M. C., Brenner M., Weissleder R., Verbeek J. S., et al. (2002) Arthritis critically dependent on innate immune system players. Immunity 16, 157–168 10.1016/S1074-7613(02)00275-3 [DOI] [PubMed] [Google Scholar]
- 69. Trcka J., Moroi Y., Clynes R. A., Goldberg S. M., Bergtold A., Perales M. A., Ma M., Ferrone C. R., Carroll M. C., Ravetch J. V., and Houghton A. N. (2002) Redundant and alternative roles for activating Fc receptors and complement in an antibody-dependent model of autoimmune vitiligo. Immunity 16, 861–868 10.1016/S1074-7613(02)00327-8 [DOI] [PubMed] [Google Scholar]
- 70. Kumar V., Ali S. R., Konrad S., Zwirner J., Verbeek J. S., Schmidt R. E., and Gessner J. E. (2006) Cell-derived anaphylatoxins as key mediators of antibody-dependent type II autoimmunity in mice. J. Clin. Invest. 116, 512–520 10.1172/JCI25536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rogers J., Cooper N. R., Webster S., Schultz J., McGeer P. L., Styren S. D., Civin W. H., Brachova L., Bradt B., and Ward P. (1992) Complement activation by β-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 89, 10016–10020 10.1073/pnas.89.21.10016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McGeer P. L., and McGeer E. G. (2004) Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat. Disord. 10, Suppl. 1, S3–S7 [DOI] [PubMed] [Google Scholar]
- 73. Singhrao S. K., Neal J. W., Morgan B. P., and Gasque P. (1999) Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp. Neurol. 159, 362–376 10.1006/exnr.1999.7170 [DOI] [PubMed] [Google Scholar]
- 74. Li L., Yin Q., Tang X., Bai L., Zhang J., Gou S., Zhu H., Cheng J., Fu P., and Liu F. (2014) C3a receptor antagonist ameliorates inflammatory and fibrotic signals in type 2 diabetic nephropathy by suppressing the activation of TGF-β/smad3 and IKBα pathway. PLoS one 9, e113639 10.1371/journal.pone.0113639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bao L., Osawe I., Haas M., and Quigg R. J. (2005) Signaling through up-regulated C3a receptor is key to the development of experimental lupus nephritis. J. Immunol. 175, 1947–1955 10.4049/jimmunol.175.3.1947 [DOI] [PubMed] [Google Scholar]
- 76. Liu L., Zhang Y., Duan X., Peng Q., Liu Q., Zhou Y., Quan S., and Xing G. (2014) C3a, C5a renal expression and their receptors are correlated to severity of IgA nephropathy. J. Clin. Immunol. 34, 224–232 10.1007/s10875-013-9970-6 [DOI] [PubMed] [Google Scholar]
- 77. Peng Q., Li K., Smyth L. A., Xing G., Wang N., Meader L., Lu B., Sacks S. H., and Zhou W. (2012) C3a and C5a promote renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 23, 1474–1485 10.1681/ASN.2011111072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 80. Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M. W., Shipley G. L., Vandesompele J., and Wittwer C. T. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 81. Esmon C. T., and Owen W. G. (1981) Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc. Natl. Acad. Sci. U.S.A. 78, 2249–2252 10.1073/pnas.78.4.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.