

Abstract
Clathrin-independent endocytosis (CIE) is a form of endocytosis that lacks a defined cytoplasmic machinery. Here, we asked whether glycan interactions, acting from the outside, could be a part of that endocytic machinery. We show that the perturbation of global cellular patterns of protein glycosylation by modulation of metabolic flux affects CIE. Interestingly, these changes in glycosylation had cargo-specific effects. For example, in HeLa cells, GlcNAc treatment, which increases glycan branching, increased major histocompatibility complex class I (MHCI) internalization but inhibited CIE of the glycoprotein CD59 molecule (CD59). The effects of knocking down the expression of galectin 3, a carbohydrate-binding protein and an important player in galectin–glycan interactions, were also cargo-specific and stimulated CD59 uptake. By contrast, inhibition of all galectin–glycan interactions by lactose inhibited CIE of both MHCI and CD59. None of these treatments affected clathrin-mediated endocytosis, implying that glycosylation changes specifically affect CIE. We also found that the galectin lattice tailors membrane fluidity and cell spreading. Furthermore, changes in membrane dynamics mediated by the galectin lattice affected macropinocytosis, an altered form of CIE, in HT1080 cells. Our results suggest that glycans play an important and nuanced role in CIE, with each cargo being affected uniquely by alterations in galectin and glycan profiles and their interactions. We conclude that galectin-driven effects exist on a continuum from stimulatory to inhibitory, with distinct CIE cargo proteins having unique response landscapes and with different cell types starting at different positions on these conceptual landscapes.
Keywords: glycosylation, endocytosis, membrane trafficking, major histocompatibility complex (MHC), galectin, CD59, clathrin-independent endocytosis (CIE), galectin 3, galectin lattice, macropinocytosis
Introduction
Altered glycosylation of proteins and lipids is a hallmark of cancer (1, 2). Whereas these changes have long been used in the development of biomarkers for cancer detection (3–5), more recent studies have started to show functional roles for glycosylation in cancer (6–9). As a result, it is important to understand the cellular roles that glycans play. One of these roles that has recently received renewed focus is the mediation of clathrin-independent endocytosis (CIE).2
Unlike clathrin-mediated endocytosis (CME), which is well characterized in terms of molecular machinery (10) and regulation, CIE is still relatively poorly understood (11). CME is known to have five defined stages: initiation, cargo selection, coat assembly, scission, and uncoating. CME is initiated by proteins that create membrane curvature. Adapter proteins like the AP2 complex then select specific cargo and link the cargo to clathrin and other accessory proteins. Clathrin self-assembles into a cytoplasmic coat around the growing pit, and then dynamin facilitates vesicle scission. The endosomes are then uncoated to produce the final cargo-containing endosome (10). In stark contrast, CIE has much less known about its fundamental parameters (12). Apart from a general requirement for cholesterol and sometimes actin, the requirements for CIE vary greatly for different cargo and in different cell types (11–14). Although its molecular mechanism and regulation are still under investigation, CIE is known to play important roles in cellular signaling, plasma membrane repair, polarization, cell adhesion, and migration (12).
Galectins are a family of galactoside-binding lectins with a conserved carbohydrate-recognizing domain (CRD) (15). There are three types of galectins: proto-type (galectins 1, 2, 5, 7, 10, 11, 13, 14, and 15) have a single CRD and can homodimerize; tandem repeat-type (galectins 4, 6, 8, 9, and 12) have CRDs at either end of the protein; and chimera-type (its only member galectin 3) has a single CRD but can multimerize to give rise to a pentameric galectin (15, 16). All galectins are synthesized in the cytoplasm and are secreted by nonclassical secretory mechanisms (17). They are known to play roles both intracellularly (such as regulating RNA splicing (18)) and extracellularly (regulating cell adhesion and signaling (19)). One of the important functions galectins play extracellularly is in organizing the cell membrane via the formation of macroscale interaction networks often referred to as the galectin lattice (20). Galectin 3 is especially important in organizing the galectin lattice, as the multivalence of its pentameric form allows large networks of galectins to form (16).
Galectins and glycan interactions have been shown to play important roles in CIE. In the CLIC/GEEC (clathrin-independent carriers/GPI-enriched endocytic compartments) pathway, it was shown to stimulate internalization of the hyaluronan receptor (CD44) by promoting the formation of endocytic pits (21–23). Galectin 3 was also found to stimulate the internalization of β1 integrin by CIE (24). By contrast, in the CIE of the epidermal growth factor receptor, the galectin–glycan interactions have been demonstrated to inhibit internalization via galectin lattice–mediated cell-surface sequestration (25–30). Glycan branching has also been shown to modulate cell-surface levels of glucose transporter 2 by altering cell-surface sequestration of the receptor (31, 32). Whereas these two modes of action act in opposite directions, there have been hints that these two functions may not be mutually exclusive but could rather represent two ends of a spectrum (23), as illustrated in Fig. 1A.
Figure 1.

Schematic representation of CIE and CME and characterization of changes in glycan patterns. A, schematic representation of CIE and CME and the two modes of action that have been reported for glycan interactions on CIE: 1) stimulatory via promotion of entry into endocytic pits (23) and 2) inhibitory due to cell-surface sequestration of cargo (30). B, lectin-binding analysis by flow cytometry of HeLa cells after 48-h incubation with or without 10 mm GlcNAc demonstrates an increase in branched glycans indicated by significant increases in binding of lectin that detects terminal sugar residues (WGA and RCA) and an increase in PHA-L binding (which specifically binds to branched glycans). C, RCA binding analysis by flow cytometry of HeLa cells with or without 100 mm lactose treatment (1-h preincubation and during lectin binding) demonstrates the efficacy of lactose as an inhibitor of galactoside lectin (such as RCA and galectin) interactions. At least three independent experiments were carried out with data expressed as mean ± S.D. (error bars). *, p < 0.05.
Here, we investigate whether the well-characterized CIE cargo proteins major histocompatibility complex class I (MHCI) and CD59 are sensitive to glycan interactions. Further, as depicted in Fig. 1A, the effect of galectin–glycan interactions on cargo internalization was characterized to determine whether these two specific endogenous cargo proteins were affected via similar or different modes of action and whether these modes of action were mutually exclusive.
Results
Surface glycosylation patterns are shifted by N-acetylglucosamine treatment
Protein glycosylation is a nontemplate-driven process, and as such, glycan patterns generated on specific proteins are sensitive to the levels of the glycosyltransferases as well as the metabolic flux of their substrates (the various sugar residues) (26, 29, 33). Therefore, there are two primary approaches to altering glycan patterns: 1) altering the availability of the enzymes by altering their expression and 2) changing the metabolic flux through the glycosylation machinery by altering the availability of the sugar residues. In this study, the second approach was used to increase levels of GlcNAc in the cell by incubation with millimolar concentrations of the sugar residue. GlcNAc is essential for the branching of glycans, and hence increased availability of this sugar was expected to increase the branching of glycans (29).
After incubation with 10 mm GlcNAc for 48 h, the cell-surface glycan patterns on HeLa cells were analyzed using lectin binding and flow cytometry (Fig. 1B). The binding of wheat germ (Triticum vulgaris) agglutinin (WGA) (which detects neuraminic/sialic acid as well as GlcNAc residues), Ricinus communis agglutinin (RCA) (which detects terminal galactose residues), and Phaseolus vulgaris leucoagglutinin (PHA-L) (which binds to tri- and tetra-antennary branched complex N-glycans) was significantly increased following GlcNAc treatment, whereas the binding of Ulex europaeus agglutinin 1 (UEA1) (which binds fucose) and concanavalin A (which binds high-mannose residues) was not significantly changed by GlcNAc treatment. These results demonstrate that the increased availability of GlcNAc led to increases in glycan branching. These data also indicated that other glycan pattern characteristics were not indirectly affected.
Lactose is a competitive inhibitor of galectin–glycan interactions (23, 26), a major focus of this study. RCA is a galactoside-binding lectin that binds the same epitope as galectins and hence was used to demonstrate the efficacy of lactose competitive inhibition on galectin binding. Lactose treatment during lectin binding completely inhibited RCA binding, with lactose-treated cells having fluorescent intensity histograms very similar to cells that were not subjected to lectin binding. This demonstrates the efficacy of lactose in blocking galectin–glycan interactions (Fig. 1C).
Internalization of CIE cargo is affected by changes in glycan interactions, and increased glycan branching has cargo-specific effects
The first question we addressed was whether endocytosis of specific CIE cargo proteins is affected by glycan interactions. To do this, two well-characterized CIE cargo proteins were selected: MHCI and CD59. These two proteins, although both CIE cargo, are considerably different structurally; MHCI is a type I transmembrane protein with a single lumenal site of N-glycosylation, and CD59 is a GPI-anchored protein with two sites of N-glycosylation. These distinct cargo proteins were chosen to address a second question: whether the effects of glycan interactions were uniform for different cargo proteins. The internalization of the two cargos in HeLa cells was measured using an antibody internalization assay in control cells or in cells treated for 48 h with GlcNAc (Fig. 2, A and B).
Figure 2.

Internalization of CIE and not CME cargo is sensitive to changes in glycan interactions. Antibody internalization for 30 min at 37 °C in HeLa cells after 48-h incubation with or without 10 mm GlcNAc and in the presence or absence of 100 mm lactose for MHCI (A) and CD59 (B) indicates cargo-specific effects of increasing glycan branching (due to GlcNAc treatment) and an inhibition of CIE with lactose treatment. 48-h incubation with 10 mm glucose or 1-h pretreatment with 100 mm sucrose had no significant effect on the CIE of MHCI or CD59. C, flow cytometric analysis of clathrin-mediated endocytosis of fluorescently labeled transferrin internalized over 10 min at 37 °C in HeLa cells shows that CME is unaffected by altering glycan interactions. 48-h incubation with or without 2 μg/ml swainsonine led to similar changes in internalization as lactose treatment for MHCI (D), CD59 (E), and transferrin (F). At least three independent experiments were carried out with data expressed as mean ± S.D. (error bars). *, p < 0.05.
We found that GlcNAc treatment resulted in an increase in MHCI internalization (Fig. 2A). A similar treatment of cells with 10 mm glucose had no effect on MHCI internalization, indicating that the increase was mediated by an increase in glycan branching and not merely due to an increase in nutrient availability. This observation suggests that increased glycan branching enhances MHCI entry into endocytic pits, consistent with the endocytic pit entry modality of glycosylation's effect on endocytosis. Furthermore, lactose treatment during antibody internalization led to an ablation of the GlcNAc-mediated increase, whereas a similar treatment with sucrose had no effect on MHCI CIE (Fig. 2A). This indicates that the inhibition of all galectin–glycan interactions (and not nonspecific osmolarity effects) can counteract the effect of increased glycan branching on MHCI CIE, suggesting that this increase in internalization was mediated by extracellular galectin–glycan interactions.
In contrast, increased glycan branching (by GlcNAc treatment) inhibited CD59 internalization, whereas the analogous treatment with glucose had no effect (Fig. 2B). This suggests that CD59 may be more susceptible to the cell-surface sequestration mode of action.
Lactose treatment also inhibited CD59 internalization, whereas a similar sucrose treatment had no effect (ruling out possible effects of high osmolarity) (Fig. 2B). This indicates that glycan interactions are important for CD59 internalization, but if the level of interactions increases past a particular point, it starts to suppress internalization. The results in particular for CD59 suggest that proteins are not assigned a fixed glycan interaction mechanism that dictates internalization, but rather exist on a spectrum between the two and transition across the spectrum based on the level of glycan interactions. The results clearly indicate both that CIE cargo were sensitive to changes in glycosylation and, second, that changes in glycan interactions had cargo-specific effects.
The effect of altering glycan interaction on CME was also studied by measuring the internalization of fluorescently labeled transferrin, which enters bound to the transferrin receptor by CME. In sharp contrast to MHCI and CD59, transferrin receptor internalization by CME was not affected in any way by changes in glycan interactions (Fig. 2C). This is consistent with the extensive cytoplasmic machinery that is known to help coordinate and regulate CME.
Inhibition of synthesis of complex glycan patterns has a similar effect as lactose treatment
Chemical inhibition of Golgi α-mannosidases by treating cells for 48 h with 2 μg/ml swainsonine leads to an inhibition in the production of complex glycoforms. Decreasing the number of complex glycoforms would decrease the number of epitopes for galectin binding and thus inhibit galectin–glycan interactions. Similar to the effect of lactose, swainsonine treatment leads to a significant inhibition of CD59 internalization (Fig. 2E) and no significant effect on MHCI (Fig. 2D) or transferrin internalization (Fig. 2F). These results further indicate that the internalization of CIE cargo can be mediated via galectin–glycan interactions, whereas CME is unaffected.
Galectin 3 is secreted into the medium and bound to the surface of HeLa cells
A number of galectins have been reported to be present in HeLa cells, including galectin 1, 3, 8, and 9 (34–38). To better understand the role of individual galectins, we focused on galectin 3. Galectin 3 is the most unusual galectin, in that it is the only chimeric galectin and has the ability to pentamerize and thus mediate multivalent interactions especially important for large networks of galectins to form (16). In addition, galectin 3 has been implicated along with glycosphingolipids to play a role in mediating the CIE of CD44 (21–23). Galectin 3 is synthesized in the cytoplasm and then secreted by “nonclassical” secretory mechanisms (39, 40). To confirm that galectin 3 is produced and secreted from HeLa cells, we performed a human galectin 3 ELISA, which confirmed that after 48 h of incubation, conditioned medium from HeLa cells contained a significant amount of galectin 3 when compared with fresh complete medium (Fig. 3A). Further, when these cells were then treated with 100 mm lactose in serum-free medium to displace cell-bound galectin 3, an almost equivalent amount of galectin 3 was released into the medium (Fig. 3B). Treatment of these cells with 100 mm sucrose did not result in the release of surface galectin 3 into the medium (Fig. 3B). This indicates that 1) lactose is an effective inhibitor of galectin 3 interactions and 2) there is a significant amount of galectin 3 present on the surface of the cells.
Figure 3.
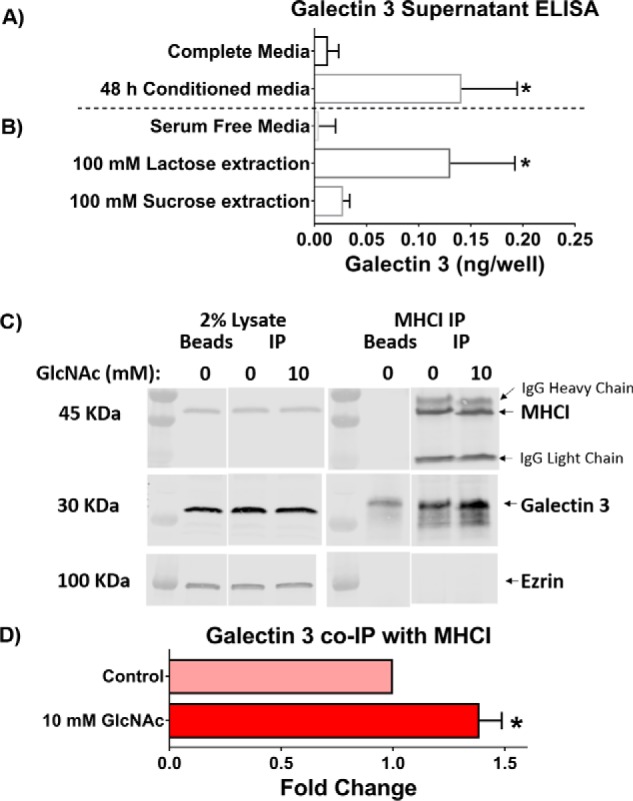
Galectin 3 is secreted and is bound to the surface and to MHCI of HeLa cells. A, endogenous galectin 3 is secreted into the medium and can be extracted from the cell surface. The galectin 3 content in complete medium, serum-free medium, and 48-h conditioned medium was measured using an ELISA. B, surface-bound galectin 3 was extracted from cells incubated with lactose-containing, but not sucrose-containing serum-free medium. C, HeLa cells were untreated or treated with 10 mm GlcNAc for 48 h before immunoprecipitation (IP) with antibodies to MHCI (w6/32). The gel on the left shows 2% lysate used as the input for immunoprecipitation, and the gel on the right shows the material pulled down with the beads. The blots were probed with antibodies to MHCI (HC110) and galectin 3, and representative Western blots are shown. D, quantification of MHCI-Gal3 co-immunoprecipitation Western blots plotting -fold change in galectin 3 normalized to MHCI pulled down for control and GlcNAc-treated conditions. At least three independent experiments were carried out with data expressed as mean ± S.D. (error bars). *, p < 0.05.
GlcNAc treatment increases galectin 3 associated with MHCI
To try to better understand whether the effects driven by GlcNAc treatment were direct or indirect, we immunoprecipitated MHCI from control cells and cells treated for 48 h with GlcNAc and blotted for galectin 3 to detect whether galectin 3 associated with the cargo protein (Fig. 3C). We found that galectin 3 pulled down along with MHCI and that GlcNAc treatment led to an increase in the amount of galectin 3 pulled down (Fig. 3D). This would suggest that for MHCI, there is a direct interaction with galectin 3 that can be stimulated by GlcNAc treatment.
Galectin 3 depletion highlights the role of the galectin lattice in the cargo-specific effects
To further explore how altering glycan interactions affected each cargo, the internalization of MHCI and CD59 was examined after siRNA-mediated knockdown (KD) of galectin 3 in HeLa cells (Fig. 4D) with or without GlcNAc treatment (Fig. 4, A and B). Once again, the results highlighted the cargo-specific effects of altering glycan interactions.
Figure 4.

MHCI and CD59 CIE have different responses to galectin 3, and CME is unaffected. Shown is antibody internalization for 30 min at 37 °C in HeLa cells after a 48-h incubation with or without 10 mm GlcNAc and with or without galectin 3 KD for MHCI (A) and CD59 (B). Galectin 3 depletion resulted in a stimulation of CD59 CIE and an ablation of GlcNAc-driven changes in MHCI CIE. C, flow cytometric analysis of the clathrin-mediated endocytosis of fluorescently labeled transferrin taken up over 10 min at 37 °C in HeLa cells shows that CME is unaffected by altering glycan interactions. D, Western blot analysis indicates that siRNA effectively depleted cells of galectin 3. E and F, the addition of exogenous recombinant galectin 3 along with the antibody in an antibody internalization assay resulted in an increase in MHCI internalization (E) and a decrease in CD59 internalization (F). At least three independent experiments were carried out with data expressed as mean ± S.D. (error bars). *, p < 0.05.
As observed previously, increased glycan branching led to an increase in MHCI internalization, but this was not observed in galectin 3–depleted cells (Fig. 4A), suggesting that this increase in endocytosis depended on galectin 3. This was again consistent with the endocytic pit entry modality of glycosylation's effect on endocytosis. It also suggests that for endocytic pit formation for MHCI, galectin 3 has a prominent role among the family of galectins, as the remaining galectins were unable to compensate for its removal.
As observed previously, increased glycan branching led to a decrease in CD59 internalization, consistent with CD59 being sequestered on the surface with extensive galectin lattice formation (Fig. 4B). However, what was most notable was that when only the galectin 3–mediated portion of the galectin lattice was disrupted by knocking down galectin 3, it led to a dramatic increase in CD59 internalization. This suggests that inhibiting only galectin 3 disrupts primarily the sequestration mode of action, allowing alternate galectins to drive an increase in internalization of CD59 via the endocytic pit entry mode, whereas, when all galectin interactions are inhibited by lactose, both the endosomal pit entry and the sequestration phases of the model are disrupted, resulting in an inhibition in CD59 CIE (Fig. 4B). On the other hand, increasing glycan branching along with galectin 3 KD caused an inhibition in CD59 internalization, potentially due to the shoring up of the galectin 3–independent portion of the galectin lattice that uses bivalent galectins from the galectin family, returning CD59 to a cell-surface sequestration modality.
The effect of altering glycan interaction and galectin lattice disruption on CME was also studied. Again, unlike CIE, CME was not affected in any way by changes in glycan interactions or the presence or absence of galectin 3 (Fig. 4C).
The addition of exogenous galectin 3 has similar effects on MHCI and CD59 CIE as GlcNAc treatment
The results from Fig. 2 using lactose as an acute extracellular inhibitor of glycan interactions suggest an extracellular role of galectins. However, galectin 3 is also known to have intracellular roles (41). To better characterize the extracellular function of galectin 3, the internalization of MHCI and CD59 was studied with the addition of exogenous recombinant human galectin 3 to the conditioned medium during antibody uptake. The addition of exogenous galectin 3 led to an increase in MHCI internalization (Fig. 4E) and a decrease in CD59 internalization (Fig. 4F), consistent with the trends observed when galectin–glycan interactions were stimulated by increasing glycan branching (using GlcNAc treatment). These data further support the idea that galectins acting on the extracellular side of the plasma membrane help mediate the CIE of MHCI and CD59. Furthermore, these data indicate that an increase in extracellular galectin–glycan interactions is responsible for the effects of increased glycan branching on the CIE of MHCI and CD59 (these effects were also reversed using lactose as an acute extracellular inhibitor of galectin–glycan interactions).
It is also important to note that even at the highest concentration of exogenous galectin 3, MHCI internalization was not inhibited. This could suggest that the presence of multiple glycosylation sites is a requisite for entry into the cell-surface sequestration modality, as MHCI, which has only one site of glycosylation, does not appear to be able to enter the cell-surface sequestration phase, whereas CD59, which has multiple sites of glycosylation, starts out in that phase. Additionally, all proteins that have been shown to have a cell-surface sequestration response to galectins have had multiple sites of glycosylation (i.e. CD44, EGFR, etc.) (23, 25, 26, 29).
The galectin lattice affects cell-surface protein mobility and cell spreading
To examine further what effect the galectin lattice has on membrane characteristics and the outcome when we disrupt these interactions, we measured the mobility of proteins in the membrane using fluorescence recovery after photobleaching (FRAP). The galectin lattice was disrupted using lactose in HeLa cells, and the FRAP of surface bound fluorescently labeled anti-CD98 antibodies (Fig. 5A) or Tac-GFP3 (Tac antigen is the IL-2 receptor α subunit) (Fig. 5C) was monitored. Both CD98 and transfected Tac-GFP3 are plasma membrane glycoproteins and CIE cargo proteins and were used as reporters to monitor changes in lateral mobility of plasma membrane proteins. For both proteins, the rate of fluorescent recovery after photobleaching was faster when the galectin lattice was disrupted by lactose, as reflected in a decrease in the half-time (t½) (time required for 50% fluorescent recovery). The diffusion coefficients were also calculated for both proteins with and without lactose treatment (Fig. 5, B and D). Control experiments with 100 mm sucrose did not have a significant effect on diffusion coefficients (data not shown), indicating that the observed effects were due to inhibition of glycan interactions and not osmolarity effects. Importantly, the diffusion coefficients were remarkably the same whether we performed the photobleaching experiment with fluorescently labeled antibody-bound CD98 or direct measurements with Tac-GFP3. Furthermore, disruption of the galectin lattice increased the diffusion coefficient of both proteins. The data suggest that disrupting the galectin lattice using lactose altered the membrane organization, thereby increasing the lateral diffusion of membrane proteins.
Figure 5.

Disruption of the galectin lattice with lactose increases the lateral diffusion of plasma membrane proteins. Fluorescence recovery after photobleaching of HeLa cells in the presence or absence of 100 mm lactose (after a 1-h pretreatment) showed that disrupting galectin–glycan interactions with lactose led to an increase in recovery rate (inversely proportionate to t½) of surface-bound Alexa Fluor–linked anti-CD98 (A) and expressed Tac-GFP3 (C). The diffusion coefficient calculated from the FRAP data highlights the increase in membrane diffusion for surface-bound anti-CD98 (B) and expressed Tac-GFP3 (D). Three independent experiments, each following FRAP regions on 30 separate cells, were carried out with data in scatter plots expressed as mean ± S.D. (error bars). FRAP curves are represented as means with goodness of fit represented by R square values. *, p < 0.05.
As another means of studying the effect of altering the galectin lattice on cell behavior, we measured the ability of cells to spread on coverslips over time with various treatments that would alter the strength of the galectin lattice. Disrupting the galectin lattice with lactose treatment significantly inhibited cell spreading at 4 h (Fig. 6, A and F). Osmolarity controls with 100 mm sucrose or 50 mm NaCl (both result in 100 mosmol/liter) had no effect on spreading (Fig. 6B), indicating that effects observed for lactose treatment were due to inhibition of glycan interactions and not nonspecific osmolarity effects. Knockdown of galectin 3 also inhibited spreading at 4 h (Fig. 6, C and F). In contrast, increasing the strength of the galectin lattice by increasing glycan branching using GlcNAc treatment led to an increase in cell spreading at 2 h, which could be abrogated with lactose treatment (Fig. 6, D and F). Increasing the strength of the galectin lattice using galectin 3 overexpression also resulted in an increase in cell spreading, and the disruption of the strengthened lattice with lactose again led to an inhibition in cell spreading (Fig. 6, E and F). To confirm that the galectin 3-GFP was being secreted and thus could mediate an extracellular effect, we also measured Gal3-GFP secretion using a fluorescent plate reader assay to measure fluorescence from conditioned medium taken from transfected cells (after a 48-h incubation). This assay confirmed that there was a significant amount of Gal3-GFP secreted into the medium and present extracellularly (Fig. 6G). These results clearly indicate that the galectin lattice plays an important role in cell spreading, with strengthening of the lattice promoting the spreading of HeLa cells, whereas disruption of the lattice inhibited their spreading.
Figure 6.

The extent of the galectin lattice modulates cell spreading. After the relevant incubations, transfections, and pretreatments, HeLa cells were allowed to spread on a coverglass and fixed after 2 or 4 h, and the area of cell spreading was measured for each cell. Disrupting the galectin lattice with lactose treatment (A) or by knocking down galectin 3 (C) leads to an inhibition in cell spreading (at 4 h). A similar increase in osmolarity using 100 mm sucrose or 50 mm NaCl did not have any effect on cell spreading (at 4 h) (B). Increasing galectin lattice strength by increasing glycan branching (using GlcNAc treatment) (at 2 h) (D) or by overexpressing galectin 3 (at 4 h) (E) leads to an increase in cell spreading, and disrupting these strengthened lattices by lactose treatment ablates these increases. F, representative images of cells for the various treatments in the cell-spreading assay. Scale bars, 20 μm. G, a secretion assay measuring GFP fluorescence in the conditioned medium after 48 h indicates that Gal3-GFP is secreted by transfected HeLa cells into the medium. At least three independent experiments were carried out with data in column graphs expressed as mean ± S.D. (error bars); scatter dot plot data are expressed as median with interquartile range; *, p < 0.05.
Different cell lines have distinct response landscapes
Having shown effects on CIE upon changing the glycan landscape in HeLa cells, we then turned to other cell types to examine their response. To characterize the effects of altering glycan interactions in different cell lines, the human bronchial epithelial cell line, Beas2b, was studied. The internalization of the MHCI and CD59 in Beas2b cells was measured using the antibody internalization assay. Unlike in HeLa cells, we found that GlcNAc treatment enhanced internalization of both MHCI (Fig. 7A) and CD59 (Fig. 7B). Thus, for this cell line, an increase in glycan branching (by GlcNAc treatment) led to an increase in endocytosis for both MHCI and CD59, suggesting that increased glycan branching promotes entry into endocytic pits for both cargos in this cell line.
Figure 7.

GlcNAc and lactose treatments stimulate CIE in Beas2b cells via alternate routes. Antibody internalization for 30 min at 37 °C in Beas2b cells after 48-h incubation with or without 10 mm GlcNAc and in the presence or absence of 100 mm lactose (after a 1-h pretreatment) for MHCI (A) and CD59 (B) shows that increased glycan branching (due to GlcNAc treatment) leads to an increase in CIE for both cargos. Surprisingly, lactose treatment also led to an increase in CIE for both cargos. C, representative images of CD59 internalization with and without lactose treatment. The increase in internalization observed with lactose corresponds to an increase in fraction of cells performing macropinocytosis (D) and an increase in the number of macropinosomes/cell (E). Scale bars, 20 μm. At least three experiments were carried out with data expressed as mean ± S.D. (error bars). *, p < 0.05.
Even more unexpectedly, lactose treatment of Beas2b cells led to an increase in internalization of both proteins, something we did not observe in any context in HeLa cells. However, we noticed that the Beas2b cells, which under control conditions performed very little macropinocytosis (a stimulated form of CIE involving actin ruffling and internalization of micron-sized vesicles (42)), started to perform a significant amount of macropinocytosis upon treatment with lactose (Fig. 7C). We discovered that lactose treatment led to increases in the fraction of cells performing macropinocytosis (Fig. 7D) and in the number of macropinosomes per cell (Fig. 7E) and that these increases correlated with the increases in internalization observed. The fact that GlcNAc treatment had no effect on macropinocytosis (Fig. 7, D and E) further emphasizes the point that its stimulation of CIE was mediated by an increase in endocytic pit entry, whereas the effect of lactose treatment was mediated via a stimulation in macropinocytosis. These results suggested that disrupting the galectin lattice and the macroscale interactions it mediates frees the cell membrane to carry out macroscale membrane movements like those required for macropinocytosis in cells that possess the requisite machinery.
Disrupting the galectin lattice has distinct effects in cells that perform macropinocytosis
Having observed a distinct effect of glycan interactions on macropinocytosis, we were interested in further characterizing these effects. To better study the effects of glycan interactions on macropinocytosis, a human fibrosarcoma cell line, HT1080, which exhibits a significant amount of macropinocytosis, was studied. As with the Beas2b cells when we disrupted the galectin lattice by treatment of the cells with lactose, we found an enhancement of macropinocytosis. The fraction of cells that were performing macropinocytosis increased (Fig. 8A), and the average number of macropinosomes per cell increased (Fig. 8B). No change in macropinocytosis was observed in experiments that used a 100 mm sucrose treatment (data not shown). This indicates that disrupting the macroscale interactions characteristic of the galectin lattice reduces the resistance to macroscale membrane movements required for macropinocytosis.
Figure 8.

Disrupting the galectin lattice increases macropinocytosis in HT1080 cells. Macropinosomes were labeled by antibody internalization for 30 min at 37 °C in HT1080 cells in the presence or absence of 100 mm lactose (after a 1-h pretreatment) for MHCI, CD59, and CD98. Disruption of the galectin lattice with lactose treatment led to an increase in fraction of cells performing macropinocytosis (A) and an increase in the number of macropinosomes/cell (B). Strengthening the galectin lattice using GlcNAc treatment led to a decrease in fraction of cells performing macropinocytosis (C) and a decrease in the number of macropinosomes/cell (D). Fluorescence recovery after photobleaching of HT1080 cells in the presence or absence of 100 mm lactose (after a 1-h pretreatment) showed that disrupting galectin–glycan interactions with lactose led to an increase in recovery rate (inversely proportionate to t½) of surface-bound Alexa Fluor-linked anti-CD98 (E) and an increase in the calculated diffusion coefficient (F). At least three independent experiments were carried out with data in scatter plots expressed as mean ± S.D. (error bars). FRAP curves are represented as means with goodness of fit represented by R2 values. Scatter dot plot data are expressed as median with interquartile range. *, p < 0.05.
Increasing the extent of the galectin lattice by increasing glycan branching and thus galectin–glycan interactions (by GlcNAc treatment) led to a small but statistically significant decrease in the fraction of cells performing macropinocytosis (Fig. 8C) and a decrease in the number of macropinosomes per cell (Fig. 8D). Consistent with the results for lactose treatment, increasing the extent of macroscale interactions by strengthening the galectin lattice resulted in an increase in resistance to macroscale membrane movements required for macropinocytosis.
Photobleaching experiments with the HT1080 cells showed that disruption of the lattice by lactose led to an increase in the membrane mobility (Fig. 8E) and diffusion coefficient (Fig. 8F) of surface-bound fluorescently labeled anti-CD98. This serves as further evidence that breaking up the galectin lattice leads to increased membrane dynamics and diffusion of proteins in the membrane for both HT1080 and HeLa cells.
Different cell behaviors explained within a conceptual interaction landscape
To better understand and discuss the results, a conceptual interaction landscape was created (Fig. 9). In this figure, changes in glycan interaction strength were represented across the horizontal axis, and changes in endocytosis and macropinocytosis were described along the vertical axis. Treatments that increased the number of glycan-mediated interactions, such as GlcNAc treatment, the addition of exogenous galectin 3, and galectin 3 overexpression, would shift the cells to the right on the horizontal axis. In contrast, treatments that inhibit galectin–glycan interactions like galectin 3 knockdown, lactose treatment, and swainsonine treatment would shift the cells to the left on the horizontal axis.
Figure 9.

A conceptual interaction landscape illustrates the unique response profiles observed for each cargo. The strength of glycan-mediated interactions is represented along the horizontal axis with GlcNAc treatment, galectin 3 overexpression and the addition of exogenous galectin 3 increasing the strength of glycan-mediated interactions and shifting the cells to the right of the landscape. By contrast, galectin 3 knockdown inhibits galectin 3–dependent glycan interactions and shifts the cells to the left of the landscape and lactose treatment, which inhibits all galectin–glycan interactions, shifts the cells to the extreme left of the landscape. Glycosylation-sensitive endocytosis is represented on the vertical axis. Response curves are depicted for CD59 (in green) and MHCI (in red). The landscape also has a macropinocytosis aspect (in dashed lines), which cell lines that possess the requisite machinery (Beas2b and HT1080) can move through. The potential starting points on this landscape are indicated for each cell line based on the experimental observations. For HeLa cells, increasing glycan-mediated interactions (moving to the right) by GlcNAc treatment or the addition of exogenous galectin 3 led to an increase in MHCI CIE and a decrease in CD59 CIE, whereas inhibiting all glycan-mediated interactions by lactose treatment (moving to the far left) led to an inhibition of CIE for CD59. Inhibition of complex glycoform synthesis by swainsonine treatment, which also leads to a dramatic inhibition of galectin–glycan interactions (moving to the far left), also led to an inhibition of CIE for CD59. A more moderate decrease in glycan-mediated interactions by galectin 3 KD led to an increase in CD59 CIE. For HT1080 cells (which perform a lot of macropinocytosis), the cells start on the macropinocytosis aspect of the landscape. Inhibiting glycan-mediated interactions by lactose treatment (shift to the left) led to an increase in macropinocytosis, whereas an increase in glycan-mediated interactions by GlcNAc treatment (shift to the right) led to a decrease in macropinocytosis. For Beas2b cells (which perform macropinocytosis to a lesser degree), the cells start at an intermediate point on the landscape. Inhibiting glycan-mediated interactions by lactose treatment (shift to the left) pushes the cells up their macropinocytosis curve and leads to an increase in CIE of both cargos. In contrast, an increase in glycan-mediated interactions by GlcNAc treatment (shift to the right) pushed both cargos up their endocytic curves and led to an increase in CIE of both cargos.
The two CIE cargos studied were depicted as having distinct response profiles, with CD59 being more sensitive to changes in glycan interactions because 1) it has no cytoplasmic domain and as a result the only interactions it “sees” were from the extracellular side, and 2) it has two sites of N-glycosylation and hence could be more susceptible to multivalence-dependent cell sequestration via the galectin lattice. Both cargos were depicted (by a dashed line) to have possible macropinocytosis aspects to their curves, depending on the availability of the necessary machinery in each cell line. The putative starting points (Fig. 9, Ctrl) on this landscape were marked based on the observed results.
The baseline in HeLa cells appears to be at a point that is on the endocytic promotion phase for MHCI (red line) and on the cell-surface sequestration phase for CD59 (green line). Thus, increasing glycan interactions with GlcNAc treatment (as demonstrated in Figs. 2 and 4) shifts the cells to the right on the landscape, leading to an increase in MHCI internalization and a decrease in CD59 internalization. Disrupting all galectin–glycan interactions with lactose treatment or incubation with swainsonine shifts the cells to the far left of the landscape (which for HeLa cells would not include the macropinocytosis curves because these cells lack the machinery to perform macropinocytosis), resulting in decreases in CD59 and an insignificant decrease in MHCI. When GlcNAc treatment was followed by lactose treatment, the cells were first shifted to the right on the landscape and then to the extreme left, corresponding to the ablation of the increase in MHCI internalization and a decrease in CD59 internalization. Inhibiting all galectin–glycan interactions by incubation with swainsonine shifts the cells to the far left of the landscape, which results in no significant effect on MHCI internalization and an inhibition of CD59 internalization.
Disrupting the galectin lattice by knocking down galectin 3 (as depicted in Fig. 4) would correspond to a moderate shift to the left on the landscape, leading to an increase in CD59 internalization and no significant change in MHCI internalization. In the case of galectin 3 KD in combination with GlcNAc treatment, increased branching by GlcNAc treatment would shift the cells to the right, and the galectin 3 KD would result in a shift back to the left, which would correspond to an ablation of the increase in MHCI (due to its less sensitive, “flatter” curve) and a decrease in CD59 internalization (due to its more sensitive, “steeper” curve). The addition of exogenous galectin 3 (shown in Fig. 4) resulted in the cells shifting to the right along the axis, leading to an increase in internalization of MHCI and a decrease in internalization of CD59.
The HT1080 cells, which perform macropinocytosis, have their baseline placed on the macropinocytosis aspect of the curves. Disrupting the galectin lattice with lactose (as shown in Fig. 8) would shift the cells to the left, resulting in the observed increase in macropinocytosis, whereas increasing the strength of glycan-mediated interactions by GlcNAc treatment (as seen in Fig. 8) shifted the cells to the right of the axis, corresponding to a decrease in macropinocytosis.
Finally, Beas2b cells, which are capable of macropinocytosis but do not perform a significant amount of it under control conditions, had their baseline placed at an intermediate position. An increase in glycan branching driven by GlcNAc treatment (as shown in Fig. 7) shifts the cells to the right, leading to increases in internalization of both cargos, whereas disrupting all galectin–glycan interactions with lactose pushes the cells to the extreme left of the landscape and up onto their macropinocytosis curves, leading to the observed increases in macropinocytosis.
Discussion
Characteristic changes in protein and lipid glycosylation are observed in almost every disease condition; however, these changes are largely viewed as passive. With an increasing number of studies highlighting numerous roles for glycosylation in various contexts, it is important to understand the functional roles these complex post-translational modifications play. CIE is a still poorly understood form of internalization and is an area in which roles for glycans are starting to emerge. The Johannes laboratory (21–23) described an important role that galectin 3 and glycosphingolipids play in stimulating the CIE of CD44 by aiding in membrane bending and endocytic pit formation. Galectin 3 has also been shown to stimulate CIE of β1 integrin (24). In contrast, the Dennis laboratory and others (25–30) have reported that CIE of EGFR was suppressed by galectin–glycan interactions, resulting in the cell-surface sequestration of the protein by the galectin lattice. Whereas these two modes of action act in opposite directions, there have been hints that these two functions may be two ends of a spectrum (23). Here, we provide evidence that this is the case. We show that CIE cargo, such as CD59 and MHCI, are sensitive to changes in glycan interactions and that modest changes in global glycan patterns caused by metabolic flux engineering can have significant effects on CIE of MHCI and CD59. The results presented here demonstrate that the two contrasting modes of action that had previously been reported can potentially use similar machinery and exist at two ends of a spectrum, with proteins shifting along the spectrum based on the shift in glycan interactions that was induced (Fig. 9).
The GPI-anchored protein CD59 was a particularly good candidate to examine this spectrum of activity. The absence of cytoplasmic inputs for GPI-anchored proteins like CD59 may be a reason that they were observed to be more sensitive to changes in extracellular inputs from galectin glycan interactions. CD59 also has more sites of glycosylation than MHCI, and the galectin-binding epitope multivalence could be a factor in determining why CD59 was more susceptible to cell-surface sequestration than endocytic pit formation. By following the changes in CIE of two distinct cargos, we highlighted how within the same cell line, different classes of proteins can exist at different positions on this glycan interaction spectrum, and their response to changes in glycan interactions depends on where on the spectrum they lie. When we caused an increase in glycan branching, we saw a decrease in CD59 CIE (as it was on the cell-surface sequestration side of the spectrum), whereas MHCI CIE was stimulated (as it was on the endocytic pit entry end of the spectrum). When all galectin–glycan interactions were inhibited by treating with lactose, both the cell-surface sequestration and the endocytic pit entry were inhibited, and hence both cargos' CIE was inhibited. When knocking down galectin 3, CD59's internalization trends clearly illustrated how cargo can transition from one mode of activity to another. CD59, which begins on the galectin lattice-dependent cell-surface sequestration end of the spectrum, transitions toward endocytic pit entry and CIE stimulation with the targeted disruption of the galectin lattice by galectin 3 knockdown.
Because galectins are also known to have intracellular functions, we also show that endogenous galectin 3 is secreted and is bound to the surface of HeLa cells. Furthermore, the addition of recombinant galectin 3 to cells is able to drive changes in the CIE of both MHCI and CD59 consistent with the trends observed when galectin–glycan interactions were stimulated using GlcNAc to increase glycan branching. This establishes that galectins can function outside the cell to mediate the CIE of both MHCI and CD59.
The data also suggest the involvement of other galectins in influencing the galectin–glycan interaction landscape. The knockdown of galectin 3 led to an increase in CD59 CIE, suggesting that alternate galectins (most likely a combination of tandem galectins like galectin 4, 6, 8, 9, or 12) could drive endocytic pit entry and thus led to an increase in CIE, whereas disrupting all galectin–glycan interactions using lactose inhibited endocytic pit entry as well, which led to a suppression in CD59 internalization. Further, when galectin 3 is knocked down, increasing the multivalency of epitopes to galectins by increasing glycan branching allowed these alternate galectins to reconstitute a lattice, once again sequestering CD59 at the cell surface. Increasing the multivalence of the CD59 by increasing glycan branching may provide the increase in multivalence required for bivalent galectins to reconstitute a galectin lattice.
Although we focus primarily on galectins, which are the predominant class of human lectin known to bind to galactoside residues on the cell surface, it is also possible that other cell-surface proteins with lectin-like domains whose functions are currently unknown could play complementary roles. The contribution of these proteins, however, is beyond the scope of this study.
The disruption of the galectin lattice leads to an inhibition of macroscale interactions at the cell surface, which in turn led to an increase in membrane fluidity and an increase in the lateral diffusion of proteins. Changes in lateral mobility have previously been reported for EGFR when the galectin lattice is disrupted (25, 26). While providing further evidence that the galectin lattice plays an important role in cell membrane organization, these data also support the intuitive prediction that disrupting the galectin lattice has widespread effects on the organization and mobility of numerous proteins at the cell surface. The disruption of macroscale interactions at the cell surface also led to an increase in macroscale movements of the cell membrane, increasing macropinocytosis in cells with the necessary machinery.
These changes in membrane dynamics and endocytosis are reflected in changes in cell spreading, with increasing galectin lattice strength increasing cell spreading and lattice disruptions inhibiting cell spreading. Cell spreading is an important facet of cancer cell metastasis (43, 44), and it is interesting that in HeLa cells, cell spreading appeared to be influenced by galectin lattice strength. From a broader point of view, increased glycan branching is one of the hallmark changes in cancer (1). Furthermore, elevated galectin 3 levels have been associated with poorer prognosis and metastasis (45, 46). In our case, this resulted in an increase in HeLa cell spreading. Our results in this broader context suggest that galectin lattice modulation of cell spreading may be one of the functional ways in which observed changes in glycosylation and galectin levels contribute toward cancer progression.
Studying the internalization of MHCI and CD59 in these alternate cell lines helps to highlight an important aspect of this study. Whereas each cargo has an individual response landscape, different cell lines can exist at different locations along these landscapes and can even have cell line–specific aspects to these landscapes (Fig. 9). This complexity in how glycosylation and galectin–glycan interactions interface with CIE is consistent with the high degree of heterogeneity in reports on both CIE and glycosylation's effect on CIE (23–27, 29). This study helps to describe in more detail the link that exists between two previously reported contrasting modes of action and helps explain why different studies observe contradictory results when looking at the effect of similar changes in glycan interactions on different proteins (often in different cell lines). This study may also shed light on the heterogeneity observed in the field of CIE for each cargo between different cell lines.
As a caveat, we were unable to effectively measure changes in glycosylation and galectin 3 binding of CD59 specifically, and whereas we believe that the changes in glycan patterns observed at a global level are reflected in the glycan patterns on both cargo proteins (based on the metabolic flux–based approach used (33)), it is also possible that the behavior of these proteins is affected indirectly by changes in glycosylation of proteins with which they interact or by global changes in membrane organization and dynamics.
Glycosylation is the most complex post-translational modification; hence, it is not surprising that a broad study of the part it plays in CIE reveals a complex, nuanced, and intricate role. Although the intracellular machinery behind CIE has yet to be fully characterized, galectins and glycan interactions are emerging as important extracellular drivers and regulators of CIE.
Finally, CIE is often viewed as a bulk membrane phenomenon that is largely passive and unregulated. However, our results highlight that galectin–glycan interactions can regulate CIE both negatively and positively in a cargo-specific manner. Global changes in glycosylation patterns (that can be induced by nutrient conditions or changes in expression of glycosyltransferases) and galectin expression levels can be decoded into cargo-specific shifts in endocytic behavior based on the intrinsic properties of the cargo protein, cell type, and glycosylation status.
Experimental procedures
Cell culture, reagents, and antibodies
HeLa and Beas2b cells were grown in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 1.0% 100× glutamine solution (Lonza), and 1.0% 100× penicillin/streptomycin antibiotic solution (Lonza). HT1080 cells were grown in Eagle's minimum essential medium (Lonza) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 1.0% 100× glutamine solution (Lonza), and 1.0% 100× penicillin/streptomycin antibiotic solution (Lonza). Cells were maintained at 37 °C in a humidified, 5% CO2-containing atmosphere.
For GlcNAc treatment, a 100 mm stock solution of GlcNAc (Sigma-Aldrich) was prepared in sterile complete culture medium, cells were then seeded on coverslips or on tissue culture plastic in 60-mm culture plates in 5.0 ml of culture medium at a density of 3 × 105 cells/plate, and the appropriate volume of GlcNAc stock solution was added to each well to achieve the desired sugar concentrations. Cells were typically incubated for 48 h with the sugar. Similarly, for glucose treatment controls, a 100 mm stock solution of glucose (J.T. Baker) in sterile complete culture medium was prepared and used.
For lactose treatments, a 100 mm solution of lactose (Sigma-Aldrich) in sterile complete culture medium was prepared, and the cells were incubated in the lactose solution for 1 h before the start of the experiment and during the experiment. Similarly, for sucrose treatment controls, a 100 mm solution of sucrose (Sigma-Aldrich) in sterile complete culture medium was prepared and used.
For swainsonine treatments, a 2 μg/ml solution of swainsonine (Sigma-Aldrich) in sterile complete culture medium was prepared, and the cells were incubated in the swainsonine solution for 48 h before the start of the experiment and during the experiment. Conditioned complete medium was the medium in which cells had been grown for 48 h.
For internalization assays with exogenous recombinant human galectin 3, a 50 μg/ml stock solution of galectin 3 (R&D Systems, catalog no. 8259-GA-050) was prepared in sterile PBS. Stock solution was added to conditioned complete medium to make up the desired concentrations before the addition of primary antibodies, and controls were made up to the highest concentration (10 μg/ml) with a similar stock solution of 50 μg/ml BSA (Sigma-Aldrich).
Monoclonal antibodies directed toward MHCI (clone w6/32), CD59 (clone p282/H19), and CD98 (clone MEM-108) were from Biolegend. A monoclonal antibody directed toward galectin 3(clone A3A12) was from ThermoFisher. A polyclonal antibody that detects Ezrin (catalog no. 3145S) was from Cell Signaling. Alexa 594–conjugated transferrin and all secondary antibodies conjugated to Alexa Fluor 594, 488, 647, 680, and 800 were purchased from Molecular Probes.
Lectin-binding assay
After a 48-h incubation with or without GlcNAc, cells were rinsed with PBS. The cells were then incubated with one of the following fluorescein labeled lectins (Vector Laboratories): WGA, RCA, UEA1, concanavalin A, or PHA-L at a concentration of 20 μg/ml in culture medium on ice for 1 h. The cells were then rinsed three times in medium and then detached by incubation with 0.25% trypsin for 1 min. The cells were collected, spun down, and resuspended in 200 μl of PBS and fixed by adding formaldehyde. After 10 min of fixation, the cells were rinsed three times in PBS and analyzed by flow cytometry using a BD Fortessa LSR2. The cell population of interest was gated appropriately, and 104 cells were used to determine mean fluorescence.
Antibody internalization assay
After the indicated incubations, transfections, and pretreatments, cells on coverslips were placed face up on parafilm, and 50 μl of conditioned complete medium containing primary antibody at concentrations of 5 μg/ml for CD59 (Biolegend) and 10 μg/ml for MHCI (Biolegend) or 10 μg/ml Alexa Fluor 488–linked transferrin (Molecular Probes) was added. Coverslips were then incubated either on ice or at 37 °C for 30 min. 10-min incubations were used for transferrin-labeled coverslips. After this, one set of coverslips that was incubated at 37 °C was fixed in 2% formaldehyde (representing total antibody bound both internally and to the surface, Ttot), and the other set of coverslips that was held at 37 °C (representing antibody internalized, Tint) as well as the set that was kept on ice (null control, T0) were acid-washed for 40 s using a solution of 0.5% acetic acid, 0.5 m NaCl, pH 3.0, after which these sets of coverslips were fixed in 2% formaldehyde. The cells were blocked for 30 min using a blocking solution of 10% FBS, 0.02% sodium azide in PBS. The coverslips were then labeled at room temperature for 1 h with Alexa Fluor 488–conjugated goat anti-mouse secondary antibody (Molecular Probes), 0.2% saponin, and 2 μg/ml HCS cell mask deep red stain (Molecular Probes) in blocking solution. The coverslips were then rinsed three times in blocking solution and a final time with PBS, and they were then mounted on glass slides.
Coverslips were imaged at room temperature using a confocal microscope (LSM 780 FCS, Carl Zeiss) with a 40× PlanApo oil immersion objective and 488- and 633-nm laser excitation. For each coverslip, six positions were imaged with 2 × 2 tiling with the pinhole kept completely open. For each condition across the six tiled images, at least 100 cells were imaged. All images for each antibody were taken with identical acquisition parameters, which are set based on the control's Ttot coverslip and tuned such that the signal was within the dynamic range.
The MetaMorph application (Molecular Devices) was used to quantify the percentage of antibody internalized. Confocal images were separated into the two different channel colors. An antibody channel threshold was set for each condition using its T0 coverslip images. The Tint and Ttot conditions were then thresholded, and the integrated signal intensity was measured for the antibody channel. The cell mask channel was autothresholded, and the threshold area was measured. The integrated signal intensity for the antibody was then normalized to the threshold area for each image. The percentage internalized is then calculated with the equation, % internal = 100 ×Ttot/area/Tint/area. Finally, the internal percentages were normalized to the untreated control.
Transferrin internalization assay
After incubations, transfections, and pretreatments, cells in a 24-well plate were incubated with 10 μg/ml Alexa Fluor 546–linked transferrin (Molecular Probes) in complete medium for 10 min either on ice (TransferrinSurface) or at 37 °C (TransferrinTotal). The cells were then washed three times in cold PBS and detached using 100 μl of 0.05% trypsin on ice for 10 min. The cells were then collected, spun down, and resuspended in 200 μl of PBS and fixed by adding formaldehyde. After 10 min of fixation, the cells were rinsed three times in PBS and analyzed by flow cytometry using a BD Fortessa LSR2. The cell population of interest was gated appropriately, and 104 cells were used to determine mean fluorescence. The percentage internalized was calculated using the equation, % internal = 100 × (TransferrinTotal − TransferrinSurface)/TransferrinTotal. Finally, the internal percentages were normalized to the untreated control.
Galectin 3 ELISA secretion assay
Cells were seeded on a 6-well plate at a density of 3 × 105 cells/well in 1 ml of complete medium in quadruplicate. After 48 h in culture, the supernatants (i.e. conditioned medium) were collected and spun down at 3000 × g for 10 min to remove any cell debris.
For lactose and sucrose extraction conditions, after the supernatant was removed, the cells were rinsed with complete medium and were then incubated for 1 h with 1 ml of 100 mm lactose (Sigma-Aldrich) or sucrose (Sigma-Aldrich) in serum-free medium. After the incubation, the lactose or sucrose extract was then collected and spun down at 3000 × g for 10 min to remove any cell debris.
Samples of complete medium, serum-free medium, conditioned medium, and the lactose and sucrose extracts were then analyzed for galectin 3 content using a human galectin 3 ELISA kit (Abeam, ab 188394) as recommended by the manufacturer.
siRNA transfection
Cells were seeded in antibiotic-free complete medium and transfected with a 50 nm final concentration of galectin 3 siRNA (Dharma on, ON-TARGET plus human LGALS2 (3958) siRNA-smartpool) or nontargeting siRNA (Dharmacon, ON-TARGETplus nontargeting pool) using Lipofectamine RNAiMAX (Invitrogen) as recommended by the manufacturer. Cells were used in experiments 48 h after transfection.
Western blot analysis
Proteins obtained from cells were analyzed by Western blotting. Briefly, after the appropriate treatment (transfection, siRNA treatment, GlcNAc incubation, etc.), cells were washed three times in cold PBS, harvested by using a tissue culture cell scraper, and pelleted by centrifugation at 4 °C (1200 × g for 10 min). Pellets were solubilized in 4× sample buffer (200 mm Tris-HCl, pH 6.8, 4% β-mercaptoethanol, 8% SDS, 0.4% bromphenol blue, and 40% glycerol), and proteins were separated by 10–20% SDS-PAGE, transferred to nitrocellulose paper, and probed with the following commercial antibodies: anti-Ezrin (Cell Signaling) and anti-galectin 3 (ThermoFisher). Membranes were imaged using LI-COR Odyssey IR imager. Protein bands were quantified using the Image Studio Lite software.
Galectin 3-MHCI co-immunoprecipitation assay
After a 48-h incubation with or without GlcNAc treatment, cells were rinsed with ice-cold PBS and lysed in 1 ml of ice-cold RIPA buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0) with protease inhibitor mixture (Sigma-Aldrich). Lysates were centrifuged at 13,300 rpm for 10 min. Supernatants were transferred to fresh Eppendorf tubes, and 0.01 μg/ml exogenous recombinant galectin 3 (R&D Systems) was added. After 1 h of incubation on ice, the samples were immunoprecipitated using 2 μg/ml anti-MHCI (Biolegend) and protein G–Sepharose beads (GE Healthcare) or beads only. The Sepharose beads were preblocked for 1 h on ice with 3% BSA (Sigma-Aldrich) in RIPA buffer. After immunoprecipitation, beads were rinsed three times with RIPA buffer and boiled for 10 min in 1× sample-loading buffer, and proteins were separated by 10–20% SDS-PAGE, transferred to nitrocellulose paper, and probed with the following antibodies: anti-Ezrin (Cell Signaling), anti-galectin 3 (ThermoFisher), and anti-MHCI (HC110). Membranes were imaged using a LI-COR Odyssey IR imager. Protein bands were quantified using the Image Studio Lite software. Protein levels measured for galectin 3 pulldown were normalized to protein levels for MHCI immunoprecipitated.
Plasmids and transient transfection
Cells were seeded in complete medium. After a 24-h incubation, cells were transfected with GFP-tagged galectin 3 (pEGFP-hGal3 (Addgene, plasmid no. 73080); pEGFP-N3 (Clontech, catalogue no. 6080-1)) or Tac-GFP3 (pEGFPx3-N1 construct with Tac cDNA from Tac in pXS inserted using the BglII and HindIII restriction sites at the 5′- and 3′-ends, respectively, a gift from Dr. J. Lipincott-Scwartz) at 2.5 μg of DNA/60-mm well using Xtremegene9 (Roche Diagnostics) as recommended by the manufacturer. Experiments were performed 48 h after transfection.
FRAP assays
Cells were cultured in 4-well Lab-Tek chambered coverglass (Thermo Scientific). Cells were either transfected with Tac-GFP3 or after incubation were labeled with anti-CD98 (BioLegend), which was Zenon-labeled with Alexa Fluor 488 (Molecular Probes) following the manufacturer's instructions. After transfections, incubations, and lactose pretreatments, cells in complete medium with 25 mm HEPES buffer were then analyzed for FRAP (47, 48). Cells were incubated at 37 °C for the duration of the FRAP experiments using an incubation chamber. Using a Zeiss 780 FCS confocal microscope together with a 488-nm argon ion laser for excitation of Alexa Fluor 488 or GFP, we monitored emissions at 525 nm. The bleaching laser intensity and number of bleaching scan iterations were adjusted to obtain a 75% loss in fluorescence in a circular 2-μm diameter photobleached region on the apical, medial, or basal focal planes of the cell membrane. Multiple regions were imaged pre- and post- photobleaching using low laser intensities, and recovery fluorescence in the selected regions was tracked over time. Fluorescence in unbleached regions on a cell (Iref) and on the coverglass (Ibackground) were also monitored over time. First, Ibackground(t) was subtracted from the measured fluorescence intensity at each time point. Next, Iref(t) was used to account for photobleaching over time as follows.
| (1) |
These fluorescence intensities (Inorm(t)) were then converted to normalized fluorescence intensities (NFI(t)) using the following equation.
| (2) |
The NFI was then plotted against time and fit to a one-phase exponential association curve using the GraphPad Prism version 6 software (GraphPad Software, Inc., La Jolla, CA). From the fit of the curves, time constants for half-recovery were derived (t½). Finally, the diffusion coefficients were calculated using the Soumpasis equation as follows: D = 0.224 × (r2/t½), where D is the diffusion coefficient, r is the radius of the region, and t½ is the time constant for half-recovery. For each condition, the FRAP was measured for at least 20 regions on separate cells in each focal plane, and the entire experiment was done in biological triplicate.
Cell-spreading assay
After incubations and transfections cells were trypsinized, collected, pelleted at 300 × g, and resuspended in complete medium. The cells were counted, and equal numbers of cells were split, pelleted, and resuspended in complete medium with or without lactose. The cells were incubated in suspension at 37 °C for 1 h and then seeded at equal density on coverslips and allowed to attach and spread for 2 or 4 h at 37 °C. The coverslips were then rinsed with PBS and fixed in 2% formaldehyde. The cells were blocked for 30 min using a blocking solution of 10% FBS, 0.02% sodium azide in PBS. The coverslips were then labeled at room temperature for 1 h with 0.67 units/ml Alexa Fluor 594–linked phalloidin (Molecular Probes) and 0.2% saponin in blocking solution. The coverslips were then rinsed three times in blocking solution and a final time with PBS. They were then mounted on glass slides.
Coverslips were imaged at room temperature using a confocal microscope (LSM 780 FCS, Carl Zeiss) with a ×40 PlanApo oil immersion objective and 488- and 594-nm laser excitation. For each coverslip, 10 positions were imaged. For each condition across the 10 images, at least 100 cells were imaged. All images were taken with identical acquisition parameters. The MetaMorph application (Molecular Devices) was used to quantify the amount of cell spreading. A region of interest was drawn around each individual cell, and an automatic threshold for light objects was set. The threshold area in each region (which corresponds to the individual cell area) was then measured. In the case of transient plasmid expression, only cells that were expressing GFP were outlined and measured.
GFP-tagged galectin 3 secretion assay
Cells were seeded on a 12-well plate at a density of 5 × 104 cells/well in 1 ml of complete medium in quadruplicate. After 24 h, the medium was replaced with 500 μl of FluoroBrite Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 1.0% 100× glutamine solution (Lonza), and 1.0% 100× penicillin/streptomycin antibiotic solution (Lonza). Cells were transfected with GFP-tagged galectin 3 (Addgene) or GFP (Clontech). 48 h post-transfection, the supernatants were collected and spun down at 3000 × g for 10 min to remove any cell debris, and 200 μl of the supernatant was transferred to a black 96-well plate in duplicate. The cells were also detached by incubating with 0.25% trypsin for 1 min at 37 °C. Cells were collected, spun down, and resuspended in 500 μl of PBS. 200 μl of the cell suspensions were also transferred to a black 96-well plate in duplicate. Bulk fluorescence in each well was measured using a Synergy H1 multimode microplate reader (BioTek). After background-subtracting any fluorescence from the medium, PBS, and cell autofluorescence, the fluorescence from the supernatant wells was normalized to the fluorescence in the cell suspension wells.
Macropinosome analysis
After incubations and pretreatments, cells on coverslips were placed face-up on parafilm, and 50 μl of medium with primary antibody in complete medium at concentrations of 5 μg/ml for CD59 (Biolegend) or CD98 (BioLegend), or 10 μg/ml for MHCI (Biolegend) was added. Coverslips were then incubated at 37 °C for 30 min. After this, the coverslips were acid-washed for 40 s using a solution of 0.5% acetic acid, 0.5 m NaCl, pH 3.0, after which these sets of coverslips were fixed in 2% formaldehyde. The cells were blocked for 30 min using a blocking solution of 10% FBS, 0.02% sodium azide in PBS. The coverslips were then labeled at room temperature for 1 h with Alexa Fluor 488–conjugated secondary antibody (Molecular Probes) and 0.2% saponin in blocking solution. The coverslips were then rinsed three times in blocking solution and a final time with PBS. They were then mounted on glass slides.
Coverslips were imaged at room temperature using a confocal microscope (LSM 780 FCS, Carl Zeiss) with a ×40 PlanApo oil immersion objective and 488- or 594-nm laser excitation. For each coverslip, five positions were imaged. For each condition across the five images, at least 100 cells were imaged. All images for each antibody were taken with identical acquisition parameters. The ImageJ software was then used to measure the size of the individual macropinosomes.
Each coverslip was also studied using an epifluorescent upright microscope (Carl Zeiss) to record the number of cells, the number of cells performing macropinocytosis, and the number of macropinosomes for numerous fields of view, with a minimum of 100 cells counted for each coverslip. The fraction of cells performing micropinocytosis and the number of macropinosomes/cell were then calculated for each condition.
Statistical analysis
At least three independent biological replicates (independent experiments) were carried out for each experiment, and data were expressed as mean ± S.D. In experiments with multiple comparisons, statistical significance was determined using a one-way ANOVA with a Dunnett's post-test to compare means of different samples with the control or a Bonferroni post-test to compare specific pairs of columns. In experiments with only two conditions, an unpaired Student's t test was used to determine statistical significance. The null hypothesis was rejected in cases where p values were <0.05.
Author contributions
M. P. M. and J. G. D. designed the study and wrote the paper. M. P. M. designed, performed, and analyzed all of the experiments. M. P. M. and J. G. D. reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Christopher Saeui, Kevin Yarema, Lois Greene, Jessica Wayt, and Sayantanee Niyogi for comments and critiques on this work. The microscopes used were in the NHLBI Light Microscopy Core facility. The flow cytometer used was in the NHLBI Flow Cytometry Core facility.
This work was supported by NHLBI, National Institutes of Health, Intramural Research Program Grant HL006130. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CIE
- clathrin-independent endocytosis
- CME
- clathrin-mediated endocytosis
- CRD
- carbohydrate-recognizing domain
- MHCI
- major histocompatibility complex class I
- WGA
- wheat germ agglutinin
- RCA
- R. communis agglutinin
- PHA-L
- P. vulgaris leucoagglutinin
- KD
- knockdown
- FRAP
- fluorescence recovery after photobleaching
- GPI
- glycosylphosphatidylinositol
- RIPA
- radioimmune precipitation assay
- UEA1
- U. europaeus agglutinin 1.
References
- 1. Hakomori S. (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U.S.A. 99, 10231–10233 10.1073/pnas.172380699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hakomori S. (1996) Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 56, 5309–5318 [PubMed] [Google Scholar]
- 3. Adamczyk B., Tharmalingam T., and Rudd P. M. (2012) Glycans as cancer biomarkers. Biochim. Biophys. Acta 1820, 1347–1353 10.1016/j.bbagen.2011.12.001 [DOI] [PubMed] [Google Scholar]
- 4. Mechref Y., Hu Y., Garcia A., Zhou S., Desantos-Garcia J. L., and Hussein A. (2012) Defining putative glycan cancer biomarkers by MS. Bioanalysis 4, 2457–2469 10.4155/bio.12.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reis C. A., Osorio H., Silva L., Gomes C., and David L. (2010) Alterations in glycosylation as biomarkers for cancer detection. J. Clin. Pathol. 63, 322–329 10.1136/jcp.2009.071035 [DOI] [PubMed] [Google Scholar]
- 6. Bellis S. L. (2004) Variant glycosylation: an underappreciated regulatory mechanism for β1 integrins. Biochim. Biophys. Acta 1663, 52–60 10.1016/j.bbamem.2004.03.012 [DOI] [PubMed] [Google Scholar]
- 7. Dennis J. W., Granovsky M., and Warren C. E. (1999) Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta 1473, 21–34 10.1016/S0304-4165(99)00167-1 [DOI] [PubMed] [Google Scholar]
- 8. Kim Y. J., and Varki A. (1997) Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj. J. 14, 569–576 10.1023/A:1018580324971 [DOI] [PubMed] [Google Scholar]
- 9. Kannagi R., Izawa M., Koike T., Miyazaki K., and Kimura N. (2004) Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 95, 377–384 10.1111/j.1349-7006.2004.tb03219.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McMahon H. T., and Boucrot E. (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517–533 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- 11. Mayor S., and Pagano R. E. (2007) Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8, 603–612 10.1038/nrm2216 [DOI] [PubMed] [Google Scholar]
- 12. Howes M. T., Mayor S., and Parton R. G. (2010) Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr. Opin. Cell Biol. 22, 519–527 10.1016/j.ceb.2010.04.001 [DOI] [PubMed] [Google Scholar]
- 13. Donaldson J. G., Porat-Shliom N., and Cohen L. A. (2009) Clathrin-independent endocytosis: a unique platform for cell signaling and PM remodeling. Cell. Signal. 21, 1–6 10.1016/j.cellsig.2008.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Radhakrishna H., and Donaldson J. G. (1997) ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 139, 49–61 10.1083/jcb.139.1.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leffler H., Carlsson S., Hedlund M., Qian Y., and Poirier F. (2002) Introduction to galectins. Glycoconj. J. 19, 433–440 10.1023/B:GLYC.0000014072.34840.04 [DOI] [PubMed] [Google Scholar]
- 16. Rabinovich G. A., Toscano M. A., Jackson S. S., and Vasta G. R. (2007) Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 17, 513–520 10.1016/j.sbi.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hughes R. C. (1999) Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta 1473, 172–185 10.1016/S0304-4165(99)00177-4 [DOI] [PubMed] [Google Scholar]
- 18. Patterson R. J., Wang W., and Wang J. L. (2002) Understanding the biochemical activities of galectin-1 and galectin-3 in the nucleus. Glycoconj. J. 19, 499–506 10.1023/B:GLYC.0000014079.87862.c7 [DOI] [PubMed] [Google Scholar]
- 19. Ochieng J., Furtak V., and Lukyanov P. (2002) Extracellular functions of galectin-3. Glycoconj. J. 19, 527–535 10.1023/B:GLYC.0000014082.99675.2f [DOI] [PubMed] [Google Scholar]
- 20. Nabi I. R., Shankar J., and Dennis J. W. (2015) The galectin lattice at a glance. J. Cell Sci. 128, 2213–2219 10.1242/jcs.151159 [DOI] [PubMed] [Google Scholar]
- 21. Johannes L., Parton R. G., Bassereau P., and Mayor S. (2015) Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 16, 311–321 10.1038/nrm3968 [DOI] [PubMed] [Google Scholar]
- 22. Johannes L., Wunder C., and Shafaq-Zadah M. (2016) Glycolipids and lectins in endocytic uptake processes. J. Mol. Biol. 428, 4792–4818 10.1016/j.jmb.2016.10.027 [DOI] [PubMed] [Google Scholar]
- 23. Lakshminarayan R., Wunder C., Becken U., Howes M. T., Benzing C., Arumugam S., Sales S., Ariotti N., Chambon V., Lamaze C., Loew D., Shevchenko A., Gaus K., Parton R. G., and Johannes L. (2014) Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol. 16, 595 [DOI] [PubMed] [Google Scholar]
- 24. Furtak V., Hatcher F., and Ochieng J. (2001) Galectin-3 mediates the endocytosis of β-1 integrins by breast carcinoma cells. Biochem. Biophys. Res. Commun. 289, 845–850 10.1006/bbrc.2001.6064 [DOI] [PubMed] [Google Scholar]
- 25. Lajoie P., Partridge E. A., Guay G., Goetz J. G., Pawling J., Lagana A., Joshi B., Dennis J. W., and Nabi I. R. (2007) Plasma membrane domain organization regulates EGFR signaling in tumor cells. J. Cell Biol. 179, 341–356 10.1083/jcb.200611106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathew M. P., Tan E., Saeui C. T., Bovonratwet P., Sklar S., Bhattacharya R., and Yarema K. J. (2016) Metabolic flux-driven sialylation alters internalization, recycling, and drug sensitivity of the epidermal growth factor receptor (EGFR) in SW1990 pancreatic cancer cells. Oncotarget 7, 66491–66511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Partridge E. A., Le Roy C., Di Guglielmo G. M., Pawling J., Cheung P., Granovsky M., Nabi I. R., Wrana J. L., and Dennis J. W. (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306, 120–124 10.1126/science.1102109 [DOI] [PubMed] [Google Scholar]
- 28. Lau K. S., Partridge E. A., Grigorian A., Silvescu C. I., Reinhold V. N., Demetriou M., and Dennis J. W. (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 10.1016/j.cell.2007.01.049 [DOI] [PubMed] [Google Scholar]
- 29. Dennis J. W., Lau K. S., Demetriou M., and Nabi I. R. (2009) Adaptive regulation at the cell surface by N-glycosylation. Traffic 10, 1569–1578 10.1111/j.1600-0854.2009.00981.x [DOI] [PubMed] [Google Scholar]
- 30. Lajoie P., Goetz J. G., Dennis J. W., and Nabi I. R. (2009) Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J. Cell Biol. 185, 381–385 10.1083/jcb.200811059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohtsubo K., and Marth J. D. (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 10.1016/j.cell.2006.08.019 [DOI] [PubMed] [Google Scholar]
- 32. Ohtsubo K., Takamatsu S., Minowa M. T., Yoshida A., Takeuchi M., and Marth J. D. (2005) Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123, 1307–1321 10.1016/j.cell.2005.09.041 [DOI] [PubMed] [Google Scholar]
- 33. Almaraz R. T., Tian Y., Bhattarcharya R., Tan E., Chen S.-H., Dallas M. R., Chen L., Zhang Z., Zhang H., Konstantopoulos K., and Yarema K. J. (2012) Metabolic flux increases glycoprotein sialylation: implications for cell adhesion and cancer metastasis. Mol. Cell. Proteomics 11, M112.01755 10.1074/mcp.M112.017558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang E.-Y., Chen Y. F., Chen Y. M., Lin I. H., Wang C. C., Su W.-H., Chuang P. C., and Yang K. D. (2012) A novel radioresistant mechanism of galectin-1 mediated by H-Ras-dependent pathways in cervical cancer cells. Cell Death Dis. 3, e251 10.1038/cddis.2011.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thurston T. L. M., Wandel M. P., von Muhlinen N., Foeglein A., and Randow F. (2012) Galectin-8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Santiago-Gómez A., Barrasa J. I., Olmo N., Lecona E., Burghardt H., Palacín M., Lizarbe M. A., and Turnay J. (2013) 4F2hc-silencing impairs tumorigenicity of HeLa cells via modulation of galectin-3 and β-catenin signaling, and MMP-2 expression. Biochim. Biophys. Acta 1833, 2045–2056 10.1016/j.bbamcr.2013.04.017 [DOI] [PubMed] [Google Scholar]
- 37. Kim H.-J., Do I.-G., Jeon H.-K., Cho Y. J., Park Y. A., Choi J.-J., Sung C. O., Lee Y.-Y., Choi C. H., Kim T.-J., Kim B.-G., Lee J.-W., and Bae D.-S. (2013) Galectin 1 expression is associated with tumor invasion and metastasis in stage IB to IIA cervical cancer. Hum. Pathol. 44, 62–68 10.1016/j.humpath.2012.04.010 [DOI] [PubMed] [Google Scholar]
- 38. Fritsch K., Mernberger M., Nist A., Stiewe T., Brehm A., and Jacob R. (2016) Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer 16, 502 10.1186/s12885-016-2546-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henderson N. C., Mackinnon A. C., Farnworth S. L., Kipari T., Haslett C., Iredale J. P., Liu F.-T., Hughes J., and Sethi T. (2008) Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 172, 288–298 10.2353/ajpath.2008.070726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Stijn C. M. W., van den Broek M., van de Weerd R., Visser M., Taşdelen I., Tefsen B., and van Die I. (2009) Regulation of expression and secretion of galectin-3 in human monocyte-derived dendritic cells. Mol. Immunol. 46, 3292–3299 10.1016/j.molimm.2009.07.026 [DOI] [PubMed] [Google Scholar]
- 41. Sano H., Hsu D. K., Apgar J. R., Yu L., Sharma B. B., Kuwabara I., Izui S., and Liu F.-T. (2003) Critical role of galectin-3 in phagocytosis by macrophages. J. Clin. Invest. 112, 389–397 10.1172/JCI200317592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Porat-Shliom N., Kloog Y., and Donaldson J. G. (2008) A unique platform for H-Ras signaling involving clathrin-independent endocytosis. Mol. Biol. Cell 19, 765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arthur W. T., and Burridge K. (2001) RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol. Biol. Cell 12, 2711–2720 10.1091/mbc.12.9.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K. M. (1998) Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 280, 1614–1617 10.1126/science.280.5369.1614 [DOI] [PubMed] [Google Scholar]
- 45. Bresalier R. S., Mazurek N., Sternberg L. R., Byrd J. C., Yunker C. K., Nangia-Makker P., and Raz A. (1998) Metastasis of human colon cancer is altered by modifying expression of the β-galactoside-binding protein galectin 3. Gastroenterology 115, 287–296 10.1016/S0016-5085(98)70195-7 [DOI] [PubMed] [Google Scholar]
- 46. Takenaka Y., Fukumori T., and Raz A. (2002) Galectin-3 and metastasis. Glycoconj. J. 19, 543–549 10.1023/B:GLYC.0000014084.01324.15 [DOI] [PubMed] [Google Scholar]
- 47. Day C. A., Kraft L. J., Kang M., and Kenworthy A. K. (2001) Analysis of protein and lipid dynamics using confocal fluorescence recovery after photobleaching (FRAP). Curr. Protoc. Cytom. Chapter 2, Unit 2.19 10.1002/0471142956.cy0219s62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Axelrod D., Koppel D. E., Schlessinger J., Elson E., and Webb W. W. (1976) Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16, 1055–1069 10.1016/S0006-3495(76)85755-4 [DOI] [PMC free article] [PubMed] [Google Scholar]