

Abstract
First step of gene expression is transcribing the genetic information stored in DNA to RNA by the transcription machinery including RNA polymerase (RNAP). In Escherichia coli, a primary σ70 factor forms the RNAP holoenzyme to express housekeeping genes. The σ70 contains a large insertion between the conserved regions 1.2 and 2.1, the σ non-conserved region (σNCR), but its function remains to be elucidated. In this study, we determined the cryo-EM structures of the E. coli RNAP σ70 holoenzyme and its complex with promoter DNA (open complex, RPo) at 4.2 and 5.75 Å resolutions, respectively, to reveal native conformations of RNAP and DNA. The RPo structure presented here found an interaction between the σNCR and promoter DNA just upstream of the −10 element, which was not observed in a previously determined E. coli RNAP transcription initiation complex (RPo plus short RNA) structure by X-ray crystallography because of restraint of crystal packing effects. Disruption of the σNCR and DNA interaction by the amino acid substitutions (R157A/R157E) influences the DNA opening around the transcription start site and therefore decreases the transcription activity of RNAP. We propose that the σNCR and DNA interaction is conserved in proteobacteria, and RNAP in other bacteria replaces its role with a transcription factor.
Keywords: cryo-electron microscopy, RNA polymerase, structural biology, promoter, transcription, cryo-EM, σ non-conserved region, promoter DNA, open complex
Introduction
In the event of formation of the transcription-ready open complex (RPo) in Escherichia coli, the conserved regions 2.4 and 4.2 of the primary σ70 factor in RNA polymerase (RNAP)4 holoenzyme recognize the −10 and −35 promoter elements, respectively, to form the closed complex (RPc). This step is followed by DNA strand separation that is stabilized by the σ region 2.3, completing the formation of the transcription bubble from −11 to +2 positions (1). E. coli RNAP forms RPo with at least two intermediates starting from the RPc. Although a series of large conformation changes of RNAP and DNA during the RPo formation have been characterized by kinetic and biophysical studies (2), the structures of intermediates have not been determined yet because of their transient and heterogeneous nature that makes them difficult to be captured by X-ray crystallography approach.
A series of technical advances such as development of a direct electron detector and also image processing techniques has moved cryo-electron microscopy (cryo-EM) to the forefront of structural biology to analyze large macromolecule complexes in unprecedented details (3). Several structures of E. coli RNAP in complex with nucleic acids (DNA/RNA and 6S RNA) (4, 5) and transcription activators (NtrC and CAP) (6, 7) have been determined by cryo-EM, demonstrating the feasibility of using this experimental approach to investigate the structure and function of E. coli RNAP. There are a couple of advantages of the cryo-EM over X-ray crystallography: 1) the 3D classification of individual particles from a single cryo-EM grid makes it possible to reveal the heterogeneity of macromolecules and determine their structures individually and 2) a cryo-EM grid preparation takes less than 10 s instead of taking days or longer for macromolecular crystallization, increasing our chances to capture elusive and unstable intermediates for cryo-EM structure determination.
The structural study of bacterial RNAP began in 1996 with the high-resolution X-ray crystal structure of the E. coli σ70, which provided insight into the DNA sequence recognition and double-strand DNA melting by the σ factor conserved regions 2.4 and 2.3, respectively (8). The structure also revealed the σ non-conserved region (σNCR) between the conserved regions 1.2 and 2.1.
In this work, we determined the cryo-EM structures of the E. coli RNAP σ70 holoenzyme and the holoenzyme–promoter DNA complex from a single cryo-EM grid. Comparing these structures revealed that the promoter DNA binding to RNAP not only triggers conformational changes of RNAP around the domains for binding the −35 and −10 elements, but also establishes the direct interaction between the σNCR and the DNA upstream of the −10 element. Our structural and biochemical data introduced a function of the σNCR during the formation of transcription ready RPo.
Results
Sample preparation and cryo-EM structure determination
In this study, we aimed to determine the structure of E. coli RNAP σ70 holoenzyme and promoter DNA complex with KdpE (KDP operon transcriptional regulatory protein) to obtain structural basis of the transcription activation by the response regulator OmpR/PhoB family. We assembled a ternary complex (Fig. S1) containing E. coli RNAP, a constitutively active variant of KdpE (KdpE-E216A) (9) and the E. coli kdpFABC promoter DNA derivative containing both a RNAP-binding site (−35 and consensus −10 elements; synthetic bubble from −6 to +1) and tandem KdpE-binding sites (from −67 to −62 and from −56 to −51) (pFABC-1, Fig. S2) for the cryo-EM grid preparation. We used the kdpFABC promoter derivative to form homogeneous RPo for the cryo-EM structure determination.
Of many grid types on which the sample was vitrified, lacey carbon grids coated with a single layer of graphene oxide followed by a layer of pyrene-nitrilotriacetic acid had images with more widely oriented particles, better suited for single-particle 3D reconstruction. From 1037 usable movie stacks, 286,424 particles were picked and used for single-particle reconstruction. The 54 classes with clear structural features representing 257,539 particles after 2D classification were selected for 3D classification. This resulted in two major 3D classes, one representing an E. coli RNAP σ70 holoenzyme–promoter DNA complex (RPo) and the other of E. coli RNAP σ70 holoenzyme by itself (Fig. S3) indicating the dissociation of DNA from some fractions of the RPo during the cryo-EM grid preparation. We failed to obtain a density map representing a ternary complex (RPo with KdpE), suggesting the dissociation of KdpE from the RPo. Determination of the cryo-EM structures of the RPo and the apo-form RNAP from a single cryo-EM grid allows us to reveal conformational changes of RNAP upon the promoter DNA binding in the identical experimental condition in solution.
Cryo-EM structure of the E. coli RNAP open complex
One of the 3D classes represents the E. coli RPo determined at 5.75 Å resolution (Fig. 1 and Fig. S3). In addition to density for RNAP holoenzyme, strong densities are observed for dsDNA (from −45 to −12 bases) and single-stranded non-template DNA (nt-DNA) in the transcription bubble (from −11 to −3 bases). However, density for the template DNA (t-DNA) in the transcription bubble (from −9 to +2 bases) is not traceable and density for downstream DNA duplex is weak and scattered, indicating their mobility within the DNA-binding main channel of RNAP (Fig. 1C). Positions of each DNA elements (−35, −10, transcription bubble, and downstream DNA) are nearly identical to a previously determined X-ray crystal structure of E. coli transcription initiation complex (TIC) (10) (PDB ID: 4YLN) (Fig. S4A). Although the overall resolution was 5.7 Å, local resolution calculations indicate the central part of the structure, including the N-terminal domain of α subunit (αNTD) as well as β and β′ subunits around the active site of RNAP, was determined close to 5 Å resolution, with peripheral areas of the structure such as σNCR (Asn-128 to Arg-373, 246 residues), β′ insertion 6 (β′i6, Ala-944 to Gly-1129, 186 residues), and ω subunit at 7–10 Å resolution (Fig. 1D). Density for the C-terminal domains of α subunit (αCTD) and σ region 1.1 domain (σ1.1) could not be traced because of the flexible nature of these domains. The distance between two pincers of RNAP (Thr-212 in the β′ clamp domain and His-165 in the β gate loop) is 28 Å, indicating the closed conformation of the RNAP clamp as observed in the E. coli TIC (27.7 Å). However, the position of σNCR in the RPo structure determined by the cryo-EM in this study is different from the one in the TIC determined by the X-ray crystallography (Fig. S4A). Compared with the structure of TIC, the loop comprising residues 151–158 of σNCR moved ∼10 Å, facing the sugar-phosphate backbone of template DNA in the cryo-EM structure. Particularly, the σNCR contacts the t-DNA at −16/−17 position via Arg-157 residue, whereas this interaction was not observed in the TIC because of possible restraint of crystal packing effect (Fig. S4B).
Figure 1.

The cryo-EM structure of the E. coli RPo. A, sequence of the promoter DNA visible in the cryo-EM structure of RPo (from −45 to +13) is depicted (full-length DNA sequence used for the RPo preparation is shown in Fig. S2). −35 and −10 elements (highlighted in yellow), transcription bubble, and transcription start site (+1) are indicated. Bases shown in gray are disordered in the RPo structures. B, cryo-EM structure of RPo. Ribbon model of RNAP and stick model of DNA are superimposed with the cryo-EM density map (white and transparent). Each subunit of RNAP, the σNCR (light blue) and β′i6 (green) are indicated in color. Template (t-DNA) and non-template DNA strands (nt-DNA) are depicted as dark gray and light gray, respectively and the −35 and −10 elements are shown in yellow. C, promoter DNA in the RPo. Stick model of promoter DNA with its cryo-EM density map are shown as the same orientation as in B. Dashed line represents the disordered t-DNA in the transcription bubble. The active site Mg2+ ion is shown as magenta sphere. Nucleotide bases at −11 and +3 (representing boundaries of the transcription bubble) and transcription start site (+1) are indicated. D, cryo-EM map of RPo colored by local resolution oriented as the same orientation as in B. E, a zoomed-in view of B shows the interaction between Arg-157 residue and t-DNA at −16/−17 positions. RNAP and DNA models are superimposed with the cryo-EM density map (white and transparent).
Cryo-EM structure of the E. coli RNAP σ70 holoenzyme
The second 3D class represents the apo-form E. coli RNAP σ70 holoenzyme with an overall resolution of 4.2 Å (Fig. 2A and Fig. S3). Local resolution calculations indicate that the center part of the structure is determined at a resolution of 4 Å, whereas the peripheral areas of the structure are determined around 6–10 Å (Fig. 2B). Densities for C-terminal domains of α subunit and σ1.1 were not traceable. Densities for σ4, σNCR, and β flap tip helix are sparse and weaker than their counterparts in the RPo, indicating their flexible nature before binding to promoter DNA. The distance between the two pincers around the main channel is 26 Å, indicating that the RNAP clamp adopts a closed conformation without DNA binding.
Figure 2.

The cryo-EM structure of the E. coli RNAP σ70 holoenzyme. A, cryo-EM structure of RNAP is shown with the cryo-EM density map (white and transparent). Subunits and domains of RNAP are shown as in Fig. 1B. A position of the RNAP main channel (MC) is indicated. B, cryo-EM map of RNAP colored by local resolution. C, rifampin-binding pocket of the β subunit is shown with the cryo-EM map. Ser-531 and His-526 residues are labeled. D, comparison of the cryo-EM structures of the holoenzyme (left, this study), RPo (middle, this study), and the crystal structure of TIC (right, PDB ID: 4YLN). Structures are shown as surface models with σ70 (orange), βlobe/i4 (blue), rim helix (yellow), β′i6 (dark green), β jaw (pale green), and β′clamp (magenta). Closure of the gap between the β′i6 domain and the βlobe/i4 domain observed in the TIC structure compared with others is shown by a black arrow.
Density around the active site located at the center of the RNAP molecule showed not only main chains but also side chains. For example, the density map of the rifampin-binding pocket of β subunit shows main chain as well as the side chains of His-526 and Ser-531 residues that play key roles in the RNAP and rifampin interaction (11) (Fig. 2C). The cryo-EM map shown here is the same quality as the X-ray crystal structure of E. coli RNAP determined at 3.6 Å resolution (PDB: 4YG2) (12), indicating that cryo-EM is able to provide the structural information for the RNAP and inhibitor interaction.
In the cryo-EM structures of both apo-form RNAP and RPo, a gap between the β′i6 domain and β′ rim helix is widely opened. In comparison, the β′i6 shifts toward the β′ rim helix about 31 Å in the X-ray crystal structure of TIC, showing the complete closure of the downstream DNA-binding cleft of RNAP (Fig. 2D and Movie S1). Significance of the movement of β′i6 will be discussed below.
Cryo-EM structure reveals a novel interaction between σNCR and DNA upstream of the −10 element
Although densities of the σ4 and σNCR domains are blurred in the apo-form RNAP, these domains are well ordered in the RPo structure. Interestingly, the σNCR is involved in the binding of upstream DNA of the −10 element. Arg-157 residue of the σNCR establishes a long-range electrostatic interaction with a phosphate between the −16/−17 DNA positions (Fig. 1E).
To investigate a role of the interaction between the σNCR and promoter DNA, we prepared E. coli RNAPs containing the σ70-R157A or -R157E substitution and tested their transcription activities. We used the WT kdpFABC promoter DNA without synthetic transcription bubble (pFABC-3) (Fig. S2) to evaluate the effects of amino acid substitution not only in DNA binding but also DNA unwinding during the transcription process. The E. coli RNAP derivatives showed major defect in transcription, the R157A and R157E variants expressing 50 and 30% activities compared with the WT enzyme (Fig. 3, A and B). To identify the step of transcription influenced by the Arg-157 substitutions, we investigated the promoter DNA binding of RNAP by using electrophoretic mobility shift assay (EMSA) and found no effect by these substitutions (Fig. 3C). To test the effect of the Arg-157 substitutions in DNA unwinding, we used fluorescence signal of 2-aminopurine (2-AP) substitutions at −7 and −1 positions in double-strand promoter DNA (pFABC_2AP-1 and pFABC_2AP-7) (Fig. S2). Fluorescence signal of 2-AP is quenched in dsDNA, whereas the increase of 2-AP fluorescence intensity is observed when RNAP unwinds dsDNA (13). Both WT and derivative RNAPs show similar increase of the 2-AP fluorescence intensity at −7 position, suggesting that the R157A/R157E substitutions do not influence the early step of DNA opening. However, 2-AP fluorescence at −1 position was 70 and 40% in the R157A and R157E derivatives, respectively, compared with the WT (Fig. 3D), indicating that the σNCR plays a role in the DNA unwinding around the transcription start site, the final step of transcription-ready RPo formation. Consistent with this functional role, the Arg-157 substitutions had no effect (Fig. 3E) on the transcription of a premelted DNA (TIS) (Fig. S2).
Figure 3.

Characterization of the σNCR and promoter DNA interaction for RNAP transcription activity. A, Arg-157 substitutions of σ70 decreases the transcription activity of RNAP. Transcription activities of the WT RNAP (lanes 4-6), the σ70-R157A (lanes 1-3), and the σ70-R157E substituted RNAP (lanes 7-9) on a double-strand promoter DNA (pFABC-3) (Fig. S2) at different time intervals as indicated. Position of the run-off transcription product (RO) is shown. B, plot comparing the rate of transcription activity between the WT and the σ70-R157E substituted RNAPs. The run-off transcript levels from A were quantified using the software ImageJ. C, EMSA showing binding of WT (lanes 2-4), the σ70-R157A (R157A, lanes 5–7) and the σ70-R157E substituted RNAPs (R157E, lanes 8-10) to pFABC-3 promoter DNA. Lane 1 is free DNA. Positions of free DNA and RNAP-DNA complex are indicated. The concentrations of heparin used as competitors are also indicated. D, fluorescence signal from the 2-aminopurines substituted at −1 and −7 positions of the template strand promoter DNA (pFABC_2AP-1 and pFABC_2AP-7) (Fig. S2) by the WT RNAP, the σ70-R157A substituted, and the σ70-R157E substituted RNAPs. Fluorescence data were normalized considering the difference in the fluorescence values of ssDNA (dotted line at the top) and dsDNA (dotted line at the bottom) as 100%. Average values from three experiments are plotted using the software GraphPad Prism. Error bars represent S.E. p values were calculated using Student's t test. E, transcription activities of the WT, the σ70-R157A substituted, and the σ70-R157E substituted RNAPs on a premelted DNA scaffold (TIC scaffold) (Fig. S2).
Discussion
Using single-particle cryo-EM reconstruction, we determined the structures of the apo-form E. coli RNAP σ70 holoenzyme and RPo at 4.2 Å and 5.7 Å resolution, respectively. Although the overall resolution of the apo-form RNAP is better than RPo, the density maps of RNAP involved in the −35 element recognition such as σ4 and β flap tip helix are better resolved in the RPo (Figs. 1B and 2A), suggesting that RNAP needs its flexibility of the −35 element–binding domain to recognize promoters with different lengths of spacer (16 to 18 bases in most case) between the −10 and −35 elements.
The density map of σNCR in the apo-form holoenzyme is weak and sparse, whereas the σ2, which directly links to the σNCR via two α helixes, is well resolved (Fig. 2A). The promoter DNA binding to RNAP enhances the rigidity of σNCR (Fig. 1B) and establishes an electrostatic interaction between Arg-157 residue and t-DNA strand at −16/−17 position (Fig. 1E). Electrostatic nature of this interaction is verified when RNAP derivatives with the side chain charge reversal (R157E) showed more quantitative effect on the DNA unwinding and the transcription than the charge deletion (R157A) compared with WT (Fig. 3). This interaction was not observed in a previously determined E. coli RNAP TIC determined by the X-ray crystallography (10) likely because of restraint of protein and DNA packings in the crystal (Fig. S4B) whereas the cryo-EM structure represents the structure of molecule in solution without any effects of crystal packing. It is also possible that TIC structure represents a conformational stage where the σNCR and t-DNA interaction do not occur.
The σNCR and t-DNA interaction presented here is distinct from the σ3 and DNA interaction, which is formed with the nt-DNA strand upstream of the −10 element (14). Our structure-based biochemical experiments revealed a role of the σNCR and t-DNA interaction, which facilitates the DNA opening of the transcription start site and formation of the transcription-ready RPo (Fig. 3). Arg-157 residue is widely conserved in Betaproteobacteria, Gammaproteobacteria, Epsilonproteobacteria, Deltaproteobacteria, and Chlamydiae (Fig. 4A), further supporting the importance of the σNCR and DNA interaction.
Figure 4.

A, alignment of the σNCR region shows the conservation of Arg-157 residue in bacteria from the classes of proteobacteria and Chlamydiae. B, the structures of E. coli RPo (top left), Thermus aquaticus RPo (top right), and Mycobacterium smegmatis (RPo with RbpA, bottom) as transparent surface models with corresponding σ factors (orange) and promoter DNA (t-DNA, dark gray; nt-DNA, light gray) shown as ribbon models. σNCR is in purple and RbpA of M. smegmatis RPo (bottom) is in green. −10 elements are colored yellow. Positively charged residues interacting with bases upstream to −10 promoter elements (Arg-157 of E. coli σNCR, Arg-79 and Lys-76 of RbpA) are shown as sphere model (red). Right bottom panel is a magnified view showing the RbpA and DNA interaction.
The size of σNCR found in the primary σ factor (σ70 in E. coli and SigA in other bacteria) depends on bacterial phyla. For example, E. coli σ70 contains 247 residues, whereas Thermus aquaticus, Mycobacterium smegmatis, and M. tuberculosis (MTB) SigA contain 72, 29, and 32 residues, respectively (Fig. 4B). There is a good correlation between the size of σNCR and the stability of RPo. Compared with MTB RNAP that forms very unstable RPo with two promoters tested, E. coli RNAP formed highly stable and irreversible complexes with the same promoters (15). Stable RPo formation of MTB RNAP requires accessory proteins RbpA and CarD (16). Because of its small size, the σNCR of MTB and M. smegmatis SigA cannot touch DNA in the RPo (16, 17). However, in the presence of RbpA, the basic linker (BL) and σ interacting domain (SID) of RbpA occupies a space between the promoter DNA and σNCR, acting like an extension of σNCR to reach DNA upstream of the −10 element (16). Basic residues (Lys-76 and Arg-79) of the RbpA-BL form salt bridges with phosphates of the nt-DNA upstream of the −10 elements (Fig. 4B) and particularly, Arg-79 and DNA interaction is critical for the stable RPo formation (16). Thus, it is likely that the RpbA in MTB is a functional counterpart of the σNCR in the E. coli RNAP transcription system.
The β′i6 is a lineage-specific insertion found between the middle of the trigger loop and changes its position at different stages in the transcription cycle. The β′i6 domain is in the open conformation in the cryo-EM structures of RNAP presented here, whereas it is in the closed conformation in the X-ray crystal structure of TIC (Fig. 2D and Movie S1). The movement of the β′i6 domain toward the βlobe/i4 domain results in closure of the gap between the β′jaw and βlobe/i4 domains. The β′jaw and β′i6 form the downstream mobile element (DME) of RNAP, and the kinetic studies of E. coli RNAP transcription proposed the conformational change of DME during the stable RPo formation (2, 18). Deletion of β′i6 drastically reduced the stability of the RPo (19), which is consistent with the idea that the β′i6 may tighten the grip of RNAP on the downstream DNA at the late stage of RPo formation. Although both the cryo-EM and X-ray crystallographic studies used the same sequence and length of downstream DNA to prepare the RNAP-DNA complex, the downstream DNA in crystal forms longer DNA as a result of head-to-tail binding with the upstream DNA of adjacent symmetrically related molecule (Fig. S4C). It is tempting to speculate that certain length of downstream DNA accommodated in the DNA-binding main channel triggers the conformational change of the DME for the establishment of the stable RPo formation.
Conclusion
The cryo-EM structure of RNAP holoenzyme was determined at 4.2 Å resolution and its density map quality is equivalent to the X-ray crystal structure of E. coli RNAP holoenzyme determined at 3.6 Å resolution, indicating that single-particle cryo-EM is an alternative and promising method for structural studies of RNAP inhibitors because of eliminating the crystallization step. The cryo-EM structure of RPo presented here revealed a novel interaction between σNCR and DNA upstream of the −10 element, which facilitates the formation of stable RPo. RNAP derivatives containing σ70-R157A/R157E accumulate intermediate species between the closed and open complexes; therefore, it, along with the cryo-EM, could be a useful tool to capture elusive intermediates during the RPo formation. Such experiments are underway.
Experimental procedures
Protein expression and purification
E. coli RNAP core enzyme and σ70 proteins were prepared and RNAP holoenzyme was reconstituted as described (20). R157E mutation in the E. coli rpoD gene was obtained by site-directed mutagenesis of plasmid pGEMD, and σ70 derivative was prepared and holoenzyme containing σ70 derivative was reconstituted as described (20). Response regulator KdpE-E216A was expressed and purified as described (9).
Sample preparation for cryo-EM
E. coli RNAP σ70 holoenzyme, response regulator KdpE-E216A, and synthetic DNA with an artificial transcription bubble (pFABC-1) (Fig. S2) were mixed at 1:2:3 molar ratio in sample buffer (10 mm Hepes, pH 8, 100 mm NaCl, 5% glycerol, 10 mm MgCl2) and incubated at room temperature for 30 min. The ternary complex was purified using size-exclusion Superose 6 column chromatography (Fig. S1) equilibrated with buffer containing 10 mm Hepes, pH 8, 100 mm NaCl, 5% glycerol, 1 mm MgCl2. The ternary complex in peak fractions was pooled and cross-linked with 0.1 mm glutaraldehyde for 30 min at room temperature. The cross-linked sample showed that the RNAP subunits formed a single band representing a large cross-linked complex, whereas the mobility of KdpE-E216A remains the same as the non–cross-linked sample (Fig. S1D), suggesting that KdpE-E216A does not directly interact with RNAP. After cross-linking, buffer was exchanged to reduce glycerol concentration to <1% and the sample was applied to lacey carbon grids coated with a layer of graphene oxide followed by a layer of pyrene nitrilotriacetic acid.
Grid preparation for cryo-EM
Lacey carbon grids (Ted Pella, Inc.) were glow discharged for 90 s, and graphene oxide solution (0.2 mg/ml, 3 μl) was applied and incubated for 1 min at room temperature. Grids were then washed with water (25 μl droplets) to remove excess graphene oxide. Pyrene nitrilotriacetic acid solution (1.91 mm, 3 μl) was applied to the graphene oxide–coated grid and incubated at room temperature for 5 min. Grids were further washed with water (50 μl droplets) before applying the ternary complex (250 μg/ml, 3 μl) followed by blotting for 6 s and immediately plunge frozen in liquid ethane with the Cryoplunge 3 (Cp3, Gatan, Inc.).
Cryo-EM data collection and image processing
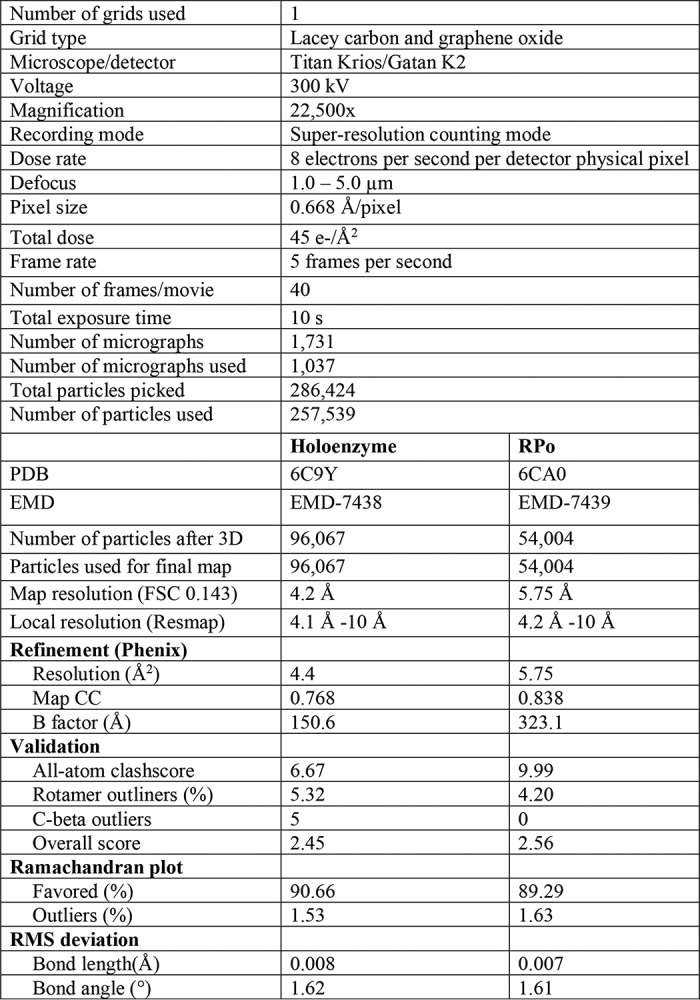
Data were collected using the Titan Krios (Thermo Fisher) microscope equipped with a K2 Summit direct electron detector (Gatan) at Purdue Cryo-EM Facility (Table 1 and Fig. S5). Sample grids were imaged at 300 kV, with an intended defocus range of 1.0–5.0 μm, a nominal magnification of 22,500× in super-resolution mode (0.668 Å per pixel), and at a dose rate of ∼8 electrons per pixel per second. Movies were collected with a total dose of ∼45 electrons per Å2 (∼1.12 electrons per frame per Å2) at 5 frames per s for 8 s. Of the total 1731 movies collected, 1037 usable movies were aligned and dose weighted using MotionCor2 (21). Contrast transfer function fitting was performed with Gctf (22). A total of 286,424 particles were picked using Gautomatch (Dr. Kai Zhang, Medical Research Council, UK). Subsequent 2D and 3D classifications, 3D refinement, post-processing, particle polishing, and local resolution estimation were performed in a beta release of Relion 2.0 (23). De novo initial model was constructed using EMAN2 (24).
Table 1.
Cryo-EM data collection and refinement statistics
Structure refinement
To refine the apo-form RNAP structure, E. coli RNAP holoenzyme crystal structure (PDB ID: 4YG2) was manually fit into the cryo-EM density map using Chimera (25) and real-space refined using Phenix (26). In the real-space refinement, domains of RNAP were rigid-body refined, then subsequently refined with secondary structure, Ramachandran, rotamer, and reference model restraints. To refine the structure of RPo, E. coli RNAP TIC crystal structure (PDB ID: 4YLN) without RNA was manually fit into the cryo-EM density map using Chimera. Upstream DNA from the −35 element was manually built by using Coot (27). The structure was refined the same as in the apo-form RNAP.
Electrophoretic mobility shift assay
For testing the promoter-specific RNAP and DNA complex formation, 10 pmol RNAP was mixed with 20 pmol DNA (pFABC-3) (Fig. S2) in binding buffer (10 mm Tris, pH 8, 50 mm NaCl, 10 mm MgCl2, 5% glycerol, 0.01% Triton X-100, and 1 mm DTT) and incubated at 37 °C for 10 min. Heparin was added to final concentrations of 10 or 50 μg/ml and incubated at 37 °C for 5 min. RNAP-DNA complex was separated from free DNA by using 6% polyacrylamide-Tris borate-EDTA gel electrophoresis and DNA was visualized by ethidium bromide staining. The experiments were conducted twice.
In vitro transcription assay
In vitro transcription assays were performed in 10 μl volume containing 100 nm RNAP and 50 nm DNA (pFABC-3 or TI scaffold) (Fig. S2) in transcription buffer (40 mm Tris-HCl (pH 8 at 25 °C), 30 mm KCl, 10 mm MgCl2, 15 μm acetylated BSA, 1 mm DTT) with nucleotide mixture (400 nm ATP, GTP, and CTP plus 100 nm UTP and 1 μCi [γ-32P]UTP). The samples were incubated at 37 °C and the reactions were stopped by adding 10 μl of 2× stop buffer (90% formamide, 50 mm EDTA, xylene cyanol, and bromophenol blue). The reaction products were electrophoretically separated on a denaturing 24% polyacrylamide/7 m urea gel and visualized with a phosphorimager (Typhoon 9410; GE Healthcare). All experiments were conducted twice.
2-Aminopurine fluorescence assay
Synthetic DNA oligos (Integrated DNA Technologies) were obtained with 2-AP substituted at −7 or −1 positions of template strand DNA and annealed to the complementary strand forming double-strand labeled DNA (pFABC_2AP-1 and pFABC_2AP-7) (Fig. S2). 100 nm ssDNA, dsDNA, and dsDNA mixed with 200 nm of purified WT RNAP or its σ70-R157A/R157E variants were incubated at 37 °C for 10 min, in a total of 100 μl reaction volume. The buffer composition of the mixture was kept as 10 mm Hepes, pH 8, 50 mm NaCl, 5% glycerol, 1 mm MgCl2. Fluorescence signal from the samples were measured in a SpectraMax M5 spectrophotometer (Molecular Devices) at excitation wavelength 320 nm and emission wavelength 380 nm.
Author contributions
A. N., D. Y., W. J., and K. S. M. conceptualization; A. N., F. S. V., and K. L. data curation; A. N., F. S. V., K. L., and M. Z. Q. investigation; A. N. writing-original draft; F. S. V., M. Z. Q., and K. S. M. formal analysis; F. S. V. and K. L. methodology; F. S. V., M. Z. Q., W. J., and K. S. M. writing-review and editing; D. Y., W. J., and K. S. M. supervision; W. J. and K. S. M. resources; W. J. and K. S. M. funding acquisition.
Supplementary Material
Acknowledgments
We thank the Purdue Cryo-EM Facility (http://cryoem.bio.purdue.edu) for the use of the Titan Krios and CM200 microscopes. We thank Shoko Murakami for critically reading the manuscript.
This work was supported by National Institutes of Health Grants GM087350 (to K. S. M.) and GM093142 (to D. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Movie S1.
The atomic coordinates and structure factors (codes 6C9Y and 6CA0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RNAP
- RNA polymerase
- σNCR
- σ non-conserved region
- nt-DNA
- non-template DNA
- t-DNA
- template DNA
- TIC
- transcription initiation complex
- DME
- downstream mobile element
- 2-AP
- 2-aminopurine
- MTB
- Mycobacterium tuberculosis.
References
- 1. Murakami K. S., and Darst S. A. (2003) Bacterial RNA polymerases: The wholo story. Curr. Opin. Struct. Biol. 13, 31–39 10.1016/S0959-440X(02)00005-2 [DOI] [PubMed] [Google Scholar]
- 2. Ruff E. F., Record M. T. Jr., and Artsimovitch I. (2015) Initial events in bacterial transcription initiation. Biomolecules 5, 1035–1062 10.3390/biom5021035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kühlbrandt W. (2014) Biochemistry. The resolution revolution. Science 343, 1443–1444 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- 4. Chen J., Wassarman K. M., Feng S., Leon K., Feklistov A., Winkelman J. T., Li Z., Walz T., Campbell E. A., and Darst S. A. (2017) 6S RNA mimics B-form DNA to regulate Escherichia coli RNA polymerase. Mol. Cell 68, 388–397.e386 10.1016/j.molcel.2017.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kang J. Y., Olinares P. D., Chen J., Campbell E. A., Mustaev A., Chait B. T., Gottesman M. E., and Darst S. A. (2017) Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. Elife 6, e25478 10.7554/eLife.25478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu B., Hong C., Huang R. K., Yu Z., and Steitz T. A. (2017) Structural basis of bacterial transcription activation. Science 358, 947–951 10.1126/science.aao1923 [DOI] [PubMed] [Google Scholar]
- 7. Glyde R., Ye F., Darbari V. C., Zhang N., Buck M., and Zhang X. (2017) Structures of RNA polymerase closed and intermediate complexes reveal mechanisms of DNA opening and transcription initiation. Mol. Cell 67, 106–116.e4 10.1016/j.molcel.2017.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malhotra A., Severinova E., and Darst S. A. (1996) Crystal structure of a σ70 subunit fragment from E. coli RNA polymerase. Cell 87, 127–136 10.1016/S0092-8674(00)81329-X [DOI] [PubMed] [Google Scholar]
- 9. Narayanan A., Kumar S., Evrard A. N., Paul L. N., and Yernool D. A. (2014) An asymmetric heterodomain interface stabilizes a response regulator-DNA complex. Nat. Commun. 5, 3282 10.1038/ncomms4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zuo Y., and Steitz T. A. (2015) Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol. Cell 58, 534–540 10.1016/j.molcel.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Molodtsov V., Scharf N. T., Stefan M. A., Garcia G. A., and Murakami K. S. (2017) Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol. Microbiol. 103, 1034–1045 10.1111/mmi.13606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Molodtsov V., Fleming P. R., Eyermann C. J., Ferguson A. D., Foulk M. A., McKinney D. C., Masse C. E., Buurman E. T., and Murakami K. S. (2015) X-ray crystal structures of Escherichia coli RNA polymerase with switch region binding inhibitors enable rational design of squaramides with an improved fraction unbound to human plasma protein. J. Med. Chem. 58, 3156–3171 10.1021/acs.jmedchem.5b00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bandwar R. P., and Patel S. S. (2001) Peculiar 2-aminopurine fluorescence monitors the dynamics of open complex formation by bacteriophage T7 RNA polymerase. J. Biol. Chem. 276, 14075–14082 10.1074/jbc.M011289200 [DOI] [PubMed] [Google Scholar]
- 14. Bae B., Feklistov A., Lass-Napiorkowska A., Landick R., and Darst S. A. (2015) Structure of a bacterial RNA polymerase holoenzyme open promoter complex. Elife 4 10.7554/eLife.08504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis E., Chen J., Leon K., Darst S. A., and Campbell E. A. (2015) Mycobacterial RNA polymerase forms unstable open promoter complexes that are stabilized by CarD. Nucleic Acids Res. 43, 433–445 10.1093/nar/gku1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hubin E. A., Fay A., Xu C., Bean J. M., Saecker R. M., Glickman M. S., Darst S. A., and Campbell E. A. (2017) Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife 6, e22520 10.7554/eLife.22520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin W., Mandal S., Degen D., Liu Y., Ebright Y. W., Li S., Feng Y., Zhang Y., Mandal S., Jiang Y., Liu S., Gigliotti M., Talaue M., Connell N., Das K., Arnold E., and Ebright R. H. (2017) Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol. Cell 66, 169–179.e168 10.1016/j.molcel.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruff E. F., Drennan A. C., Capp M. W., Poulos M. A., Artsimovitch I., and Record M. T. Jr. (2015) E. coli RNA polymerase determinants of open complex lifetime and structure. J. Mol. Biol. 427, 2435–2450 10.1016/j.jmb.2015.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Artsimovitch I., Svetlov V., Murakami K. S., and Landick R. (2003) Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 278, 12344–12355 10.1074/jbc.M211214200 [DOI] [PubMed] [Google Scholar]
- 20. Murakami K. S. (2013) X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J. Biol. Chem. 288, 9126–9134 10.1074/jbc.M112.430900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Meth. 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang K. (2016) Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheres S. H. (2016) Processing of structurally heterogeneous Cryo-EM data in RELION. Methods Enzymol. 579, 125–157 10.1016/bs.mie.2016.04.012 [DOI] [PubMed] [Google Scholar]
- 24. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 25. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 26. Afonine P. V., Mustyakimov M., Grosse-Kunstleve R. W., Moriarty N. W., Langan P., and Adams P. D. (2010) Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 66, 1153–1163 10.1107/S0907444910026582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., and Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.