

Abstract
Lafora disease (LD) is a fatal, autosomal recessive, glycogen-storage disorder that manifests as severe epilepsy. LD results from mutations in the gene encoding either the glycogen phosphatase laforin or the E3 ubiquitin ligase malin. Individuals with LD develop cytoplasmic, aberrant glycogen inclusions in nearly all tissues that more closely resemble plant starch than human glycogen. This Minireview discusses the unique window into glycogen metabolism that LD research offers. It also highlights recent discoveries, including that glycogen contains covalently bound phosphate and that neurons synthesize glycogen and express both glycogen synthase and glycogen phosphorylase.
Keywords: E3 ubiquitin ligase; epilepsy; glycogen; glycogen storage disease; Lafora disease (Lafora progressive myoclonic epilepsy, MELF); phosphatase; phosphorylation; carbohydrate binding module
Lafora disease: History
In 1911, Gonzalo Rodriguez-Lafora, one of Ramon y Cajal's (often considered the father of modern neuroscience) last great Spanish disciples, reported autopsy results from an 18-year-old patient with stimulus-sensitive resting and -active “myoclonus epilepsy with dementia” and described “amyloid bodies in the protoplasm of the ganglion cells” (1, 2). Virchow originally defined the term “amyloid” as any substance that stained in a manner similar to starch (3). The deposits were later shown to be glycogen-derived accumulations of water-insoluble, starch-like polyglucosans and were named Lafora bodies (LBs)2 (4, 5). Gabriel Schwarz named the disease Lafora disease (LD, OMIM 254780), established it as an autosomal recessive disease, and was the first to demonstrate the EEG 4–6-Hz polyspike waves in LD and to use muscle biopsies to diagnose patients (6).
LD is an invariably fatal epilepsy that equally affects both sexes (7–9). Onset is in adolescence, in apparently healthy teenagers, with absence seizures and/or visual auras. Patients then typically experience generalized tonic–clonic seizures and insidious decline in cognitive function. LD at onset is difficult to separate from the idiopathic generalized epilepsies. Myoclonic seizures, staring spells, and generalized convulsions follow, and all escalate over time. LD patients also develop highly frightening epileptic and nonepileptic visual hallucinations. Initial response to antiepileptic drugs is lost within 3 years and a constant myoclonus ensues. The young person develops severe dementia, seizes with increased frequency, becomes bedridden, and death comes in the form of a particularly massive seizure or aspiration pneumonitis (10–12).
Lafora disease: Molecular causes
LD research was revolutionized in 1995 when Serratosa et al. (13) used linkage analysis and homozygosity mapping to locate the first chromosome locus, 6q23-25, for Lafora disease. Positional cloning by the Minassian and Serratosa labs (14, 15) resulted in the discovery of the Epilepsy, Progressive Myoclonus 2A (EPM2A) gene encoding the phosphatase laforin. Mutations in EPM2A account for ∼50% of LD cases; hence, a quest for the second LD locus ensued. Scherer and co-workers (16) identified Epilepsy, Progressive Myoclonus 2B (EPM2B/NHLRC1) in 6p22 as the second LD locus encoding the protein malin.
Laforin is a 331-amino acid bimodular protein that contains a carbohydrate-binding module followed by a dual-specificity phosphatase domain (Fig. 1A). Multiple groups demonstrated that laforin dephosphorylates glycogen and phosphorylated glucans and proposed that loss of laforin results in hyperphosphorylated glycogen that disrupts glycogen branching or debranching and leads to longer glucose chains that develop into LBs (Fig. 1B) (17–21). This hypothesis was corroborated by Roach and co-workers (19, 20, 22) using a laforin-deficient mouse model that forms LBs and develops epilepsy showing that laforin dephosphorylates glycogen both in vitro and in vivo. Additionally, the recent X-ray crystal structure of laforin elucidated how laforin binds and dephosphorylates glycogen (23).
Figure 1.
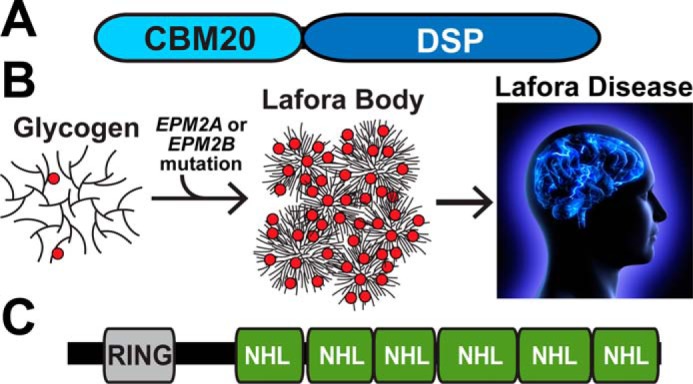
A, schematic of laforin. CBM, carbohydrate-binding module; DSP, dual-specificity phosphatase. B, glycogen is a soluble branched glucose polymer. The EPM2A gene encodes the glycogen phosphatase laforin, and EPM2B encodes the E3 ubiquitin ligase malin. Mutations in EPM2A or EPM2B result in glycogen that is hyperphosphorylated (red circles) and has disrupted glucose chain branching. This abnormal, less-soluble glycogen aggregates to form the Lafora bodies that cause Lafora disease. C, schematic of malin. RING, E2 interacting domain; NHL, protein interaction domain.
Although laforin is a glycogen phosphatase, malin contains a RING domain and six NHL repeats characteristic of a class of E3 ubiquitin ligases (Fig. 1C) (24). Dixon and co-workers (25) demonstrated that malin is an E3 ubiquitin ligase. Using cell culture systems as well as purified proteins, multiple groups reported that malin uses laforin as an adaptor protein or scaffold to polyubiquitinate protein targeting to glycogen (PTG) (26–30). PTG is a targeting subunit of protein phosphatase 1 (PP1) that dephosphorylates and activates glycogen synthase (31). Thus, it was proposed that malin forms a complex with laforin and regulates proteins involved in glycogen metabolism through ubiquitin-dependent proteasomal degradation. Additional cell culture work also identified glycogen-debranching enzyme and glycogen synthase as targets for malin ubiquitination (26, 28). However, the protein levels of these presumptive malin targets were not up-regulated in tissue from malin knockout (KO) mice with the exception of glycogen synthase (32–34). Yet for glycogen synthase, the total activity is unchanged in WT versus malin KO tissues, and the increased protein corresponds to an inactive form that accumulates in LBs. Collectively, the data demonstrate that malin does not target these proteins for proteasomal degradation, and the definitive function of malin in glycogen metabolism is unresolved (32). Nonetheless, malin is a ubiquitin ligase, and it likely modulates some aspect of glycogen metabolism.
After identification of the genes encoding laforin and malin, essentially all the Lafora research groups embarked on the elucidation of possible mechanisms to link protein function with the disease. From this work, several results and conclusions have become accepted: laforin is a glycogen phosphatase in vivo; neurons express both glycogen synthase and glycogen phosphorylase; neurons contain low levels of glycogen; neuronal glycogen provides protection during oxygen deprivation; and overaccumulation of neuronal glycogen leads to neuronal loss, locomotion defects, and reduced life span. These results also demonstrated that LD is a glycogen storage disease (GSD) and that suppressing glycogen synthesis and LB formation is a promising approach to attack Lafora disease.
Glycogen: Structure and metabolism
This topic is discussed in detail in this Minireview series by Prats et al. (99), so we only briefly describe glycogen structure and metabolism. Glycogen is the major mammalian storage carbohydrate and plays an important role in energy metabolism. Glycogen is composed of glucose units joined by α-1,4-glycosidic linkages with branches occurring every 12–14 units via α-1,6-glycosidic branches (Fig. 2A). In mammals, the two major deposits of glycogen are in the liver and skeletal muscle, but many organs, notably the brain, also synthesize the polysaccharide. Glycogen is synthesized through the cooperative action of three enzymes, namely glycogen synthase (GS), glycogenin, and glycogen-branching enzyme, using UDP-glucose (UDP-Glc) as the glucose donor. A glycogenin dimer initiates the glycogen polymer by autoglucosylation of a conserved tyrosine residue leading to an α-1,4-linked chain of 8–12 glucose units (35). This oligosaccharide remains attached to glycogenin and forms a primer that is converted into a full-size glycogen particle by the combined actions of glycogen synthase and branching enzyme (35). The coordinated action of these enzymes results in a properly branched glycogen molecule that is soluble and can be readily degraded by glycogen phosphorylase (GP) and debranching enzyme. Branches within glycogen are evenly distributed, resulting in a spherical-like structure with exposed nonreducing chain ends. This unique organization allows cells to store up to ≈55,000 glucose units/molecule in a water-soluble form that can be rapidly degraded when the cell or the body requires energy. Additionally, glycogen contains trace amounts of covalently attached phosphate, and recent work has demonstrated the importance of these phosphate moieties in both Lafora body generation and LD (19, 20, 33).
Figure 2.

A, glucose is linked by α-1,4- and α-1,6-glycosidic bonds, and it is phosphorylated at the C2-, C3-, and C6-positions (red circles). B, glucose chains of plant amylopectin and LBs are longer than glucose chains in glycogen and are proposed to form helices. C, stoichiometry of covalently attached phosphate to different glucans. D, concentration of glycogen in different cell types.
Glycogen synthesis and degradation are highly regulated processes that contribute to glucose homeostasis. Several mechanisms allow for control of GS activity, including, but not limited to, phosphorylation, allosteric activation, and intracellular localization. Regulated breakdown of glycogen is mediated by a cytosolic pathway involving glycogen phosphorylase and glycogen-debranching enzyme. Glycogen is also transported to lysosomes where it is directly hydrolyzed to glucose by a lysosomal α-glucosidase (35). Glycogen is degraded to combat ischemia in the brain, to fuel muscle contraction, and to generate free glucose in the liver to oppose hypoglycemia. One might then wonder whether the abnormal glycogen of LD disturbs glucose homeostasis, as it does in some GSDs. An initial study suggested this might be the case, but the most recent study indicates no change in whole-body glucose metabolism in LD mouse models (36, 37). Furthermore, there is no clinical evidence for hypoglycemia in LD patients.
Glycogen: Phosphorylation and LBs
Glycogen, LBs, and plant starch are all composed of α-1,4-linked glucose residues with α-1,6-glycosidic branches (Fig. 2A). A major difference is water solubility with glycogen being water-soluble and both LBs and plant starch being water-insoluble (5, 38, 39). Elegant studies in the 1960s demonstrated that there are two sizes of LBs (type I and type II) and proposed that LBs are more similar to plant starch than to human glycogen (4, 5). Starch is the major repository of glucose units in plants. It comprises two polymers: amylose that is essentially a linear polymer of α-1,4-linked glucose residues and amylopectin that contains α-1,4-linked glucose residues with α-1,6-glycosidic branches. Starch is water-insoluble due to the longer 15–25+ glucose unit chains within amylopectin and an uneven branching pattern that together allow glucose chains to form helices (Fig. 2B) (40). These helices exclude water so as to pack in a semi-crystalline structure and render starch water-insoluble (41). Reversible phosphorylation of starch has a clearly defined function in transitory plant starch where plants utilize a cycle of starch phosphorylation, degradation, and dephosphorylation (42, 43). Plants synthesize transitory starch in their leaves during the day and then release the energy cache at night. However, the semi-crystalline lattice of glucose chain helices prevents efficient amylase degradation of the starch granule. Phosphorylation of glucose at the C6 and C3 hydroxyls by glucan water dikinase and phospho-glucan water dikinase, respectively, sterically disrupts the helices and allows efficient degradation of the outer glucose layer by amylases. The amylases can release glucose down to the phosphate moiety but do not efficiently release glucose units past the phosphate. The glucan phosphatases starch excess 4 (SEX4) and like sex four 2 (LSF2) remove the phosphate, thereby resetting the cycle so that the next layer of helices can be phosphorylated and the cycle repeated (41, 43–45). The amount of phosphate within starch depends on the source, ranging from 0.034 phosphate per thousand glucose residues to 2.73 in potato tuber (Fig. 2C) (41, 46, 47). Additionally, starch phosphorylation is a dynamic process, and the starch surface has higher amounts of phosphate during the night versus the day.
Studies from the 1980s and 1990s demonstrated that glycogen contains trace amounts of phosphate (48–50). Whelan and co-workers (48) postulated that the glycogen phosphate content may relate to the age of the glycogen granule with increased phosphate being a metabolic marker to promote lysosomal degradation of glycogen. As discussed above, glycogen is degraded via the action of GP and glycogen-debranching enzyme, but it is also degraded via the lysosome in a process recently termed “glycophagy” (51). Phosphate might be a marker to distinguish these two pathways. Alternatively, a recent study (52) reported that laforin and glycogen phosphate play a role in glycogen remodeling following exhausting exercise. They showed that phosphate content remained low in WT mice following exercise-induced glycogen depletion even after glycogen levels and branching were restored. However, re-establishment of the initial glycogen-branching pattern was delayed in laforin KO mice.
Roach and co-workers (22, 54, 55) and Minassian and co-workers (53) have demonstrated that LBs contain an increased proportion of phosphate monoester groups covalently attached to hydroxyls at the C2-, C3-, and C6-positions, and they possess longer glucose chains than glycogen (Fig. 2A). They also showed that the physicochemical properties of overaccumulated muscle glycogen were significantly altered in laforin KO mice, the polysaccharide becoming less water-soluble and more aggregated, consistent with a greater tendency to precipitate in LBs (19). Although LBs possess longer glucose chains, there is no glycogen synthase hyperactivity nor branching enzyme deficiency in LD that could directly explain the change in branching or LB formation (56). An initial hypothesis was that phosphorylation at the C6-position might physically block chain formation. More recent data demonstrate that the stoichiometry of glycogen phosphate is low with only 1 phosphate per 300 glucoses to 1 phosphate per 3000 (Fig. 2C), depending on the source (22, 53, 54). Thus, no more than 1% of the potential branch points are blocked. Even with the increased phosphorylation in LD, the level of C6 phosphorylation is likely too low to directly impact branch formation. Another hypothesis is that phosphorylation may disrupt the complex hydrogen bonding and other interactions that stabilize glucose chains within glycogen, similar to what is seen in plants. This hypothesis may account for the gross chemical differences between glycogen and LBs, but the exact mechanism by which branching is decreased is unresolved.
The mechanism responsible for the introduction of phosphate into glycogen remains an active area of study. There is no biochemical or bioinformatic evidence for the existence of glycogen dikinases akin to the enzymes that phosphorylate plant starch. Roach and co-workers (54, 55) reported that glycogen synthase can transfer the β-phosphate of UDP-glucose into glycogen as a rare (1 in ∼10,000 catalytic cycles) side reaction. They proposed the formation of a cyclic glucose phosphodiester intermediate in the active site that could account for phosphate at the C2-position and possibly at the C3-position, but they could not explain the phosphorylation at C6. Therefore, other mechanisms must also be operative (57). Validating the hypothesis of a cyclic glucose phosphodiester intermediate is especially challenging, but a crystal structure of yeast glycogen synthase with glucose-1,2-cyclic phosphate bound showed that the catalytic site could accommodate the cyclic phosphate in a manner consistent with the proposed mechanism (58). In addition, incubation of glycogen synthase with UDP-Glc results in the generation of glucose-1,2-cyclic phosphate. Because of the results with glycogen synthase, Roach (59) went so far as to propose that the phosphate had no biological function and was perhaps simply the result of a minor side reaction, in which case laforin could be viewed as a repair or damage control enzyme by keeping the phosphorylation level within tolerable limits. Though excessive phosphate in glycogen may be detrimental, whether the phosphate normally has a biological role is somewhat of a mystery.
Less controversial is the idea that laforin dephosphorylates glycogen, and some studies have begun to address the site specificity. Analysis of glycogen from laforin and malin KO mice indicated similar proportional increases in C6 and C2 + C3 phosphate in glycogen, indicative of broad laforin specificity (22). Kooi and co-workers (60) explicitly examined this issue in vitro, showing that laforin can dephosphorylate both the C3- and C6-position of glycogen, but preferentially dephosphorylates the C3 position. Some LD patient mutations in laforin alter the C3/C6 specificity, suggesting that phosphorylation of different positions may have differential effects on glycogen, as is known to be the case with starch (60). Future studies are necessary to define this additional dimension of glycogen metabolism.
Glycogen in neurons
Glycogen functions as an energy reserve that is predominately found in the liver and skeletal muscle, but the brain is the organ most susceptible to decreases in glucose availability (61, 62). In fact, the brain is partly protected from hypoglycemia via elaborate blood glucose homeostatic mechanisms whereby the liver supplies glucose, derived from glycogen or gluconeogenesis, to the bloodstream as dictated by nutritional status. Additionally, the brain possesses glycogen reserves that were initially thought to simply be emergency energy stockpiles, but recent data suggest that this glycogen participates in multiple aspects of neural activity. However, the metabolism of brain glycogen is a complex process that is incompletely understood, and opposing opinions exist regarding its function and the mechanisms of energy substrate utilization. Many of these differences arise due to the difficulty of studying glycogen in the brain because of its rapid degradation ex vivo during purification, its sensitivity to stimuli via multiple signaling mechanisms, and the lack of temporal-spatial resolution in vivo. These technical hurdles have contributed to controversies regarding both the fate of glucose once it enters the brain both in neurons and glia and differing opinions regarding metabolic shuttling of glucose metabolites between cells in the brain. Most notably among these controversies is the astrocyte–neuron lactate shuttle hypothesis (ANLSH). Multiple recent reviews capture these opinions and controversies in great detail (62–66). Thus, we will not address these issues, but rather highlight the unique insights obtained regarding neuronal glycogen from studying LD.
Total brain glycogen ranges from 15 to 100 nmol of Glc/mg of protein (Fig. 2D) with astrocytes containing substantial amounts of glycogen (60–200 nmol of Glc/mg protein; Fig. 2D), especially in areas with high synaptic density (67–70). During the past 20 years, the perceived role of brain glycogen has shifted from an emergency energy supply to a dynamic participant in brain metabolism (62, 64). For example, (i) both mice and young chickens lacking glycogen in the brain show an impairment in long-term memory formation and in synaptic plasticity (71, 72). (ii) Clearance of extracellular K+ ions appears to be fueled by glucose 6-phosphate generated from glycogen (73, 74). In the absence of K+ ion clearance, neuronal hypersynchronization burst firing and seizures occur. (iii) The metabolism of glutamate, the most important excitatory neurotransmitter in the CNS, is linked with glycogen metabolism (75, 76). These studies suggest that excess glutamate is shuttled into glycogen, whereas the decreased glycogen breakdown leads to glutamate deficiency. (iv) One of the most potent proconvulsant drugs, l-methionine-SR-sulfoximine, promotes formation of masses of abnormally structured glycogen (62). Mice that resist formation of these malstructured glycogen particles resist the proconvulsant effect of the drug through unknown mechanisms (62). Conversely, mice devoid of brain glycogen show an increased susceptibility to kainate-induced epilepsy (77). (v) In cases of common refractory epilepsy, mesial temporal lobe epilepsy, up to 50% of cases show abundant, sometimes profuse, polyglucosan bodies, indistinguishable from LBs, in surgically resected tissue (78, 79). These and other data are establishing an as yet poorly understood link between glycogen metabolism and epileptogenesis. The surprisingly severe epilepsy of LD renders this disorder, and its further elucidation, an entryway to understanding this poorly explored but possibly critical facet of epilepsy and epilepsy intractability.
Although many cell types in LD patients develop LBs, neurons are especially susceptible to energy perturbations; thus, LBs trigger neuronal apoptosis, LD, and ultimately early death (38). The CNS is an interesting case in relation to glycogen metabolism. In embryonic stages, glycogen appears both in glial and neuronal cells, but in adults glycogen is almost exclusively found in astrocytes (80). However, paradoxically, adult neurons express glycogen phosphorylase (81, 82). Guinovart and co-workers (28, 83) demonstrated that neurons also express glycogen synthase and glycogen phosphorylase, thereby conferring them the capacity to synthesize and degrade glycogen. They also revealed that cultured neurons contain low levels of glycogen (6 nmol of Glc/mg of protein) (Fig. 2D) (83). Furthermore, glycogen metabolism protects both cultured primary neurons and in vivo fly neurons during oxygen deprivation, thus demonstrating a functional role for neuronal glycogen (83). These discoveries were remarkable because until then it had been widely accepted that neurons do not accumulate glycogen.
Although glycogen synthesis occurs in neurons, it must be tightly controlled because overaccumulation (≥400 nmol of Glc/mg of protein) induces neuronal apoptosis (28). Strikingly, when primary neurons are forced to overaccumulate glycogen, they produce a poorly branched glucose polymer reminiscent of an LB that induces neuronal apoptosis (28). This finding was confirmed in vivo by overexpressing an active form of GS in neurons of mice and flies, independent of manipulating laforin or malin (84). Guinovart and co-workers (28, 83) demonstrated that progressive glycogen accumulation in mouse and Drosophila neurons leads to neuronal loss, locomotion defects, and reduced life span, supporting the hypothesis that aberrant glycogen accumulation is the etiological cause of autophagy impairment, neuronal apoptosis, and LD.
Cumulatively, these data contradict the widely held view that neurons do not contain glycogen. Additionally, these results settle the apparent paradox of how a cell type that was thought to have no capacity to synthesize glycogen accumulates Lafora bodies.
LD mouse models establish disease mechanisms and therapeutic options
Several mouse models of LD have been generated (32–34, 85–88). Yamakawa and co-workers (86) generated the first laforin LD mouse model and demonstrated that whole body, laforin-deficient mice form LBs and are more prone to epileptic activity. Multiple laboratories independently generated malin-deficient mouse models that recapitulate LD (Fig. 3A) (32–34). The Minassian, Roach, DePaoli-Roach, Guinovart, and Sanz laboratories have all generated KO LD mouse models as well as double KO models. Glycogen content in the brains of laforin or malin KO animals is more than double that of WT animals (32, 34, 87–89). This increase was accompanied by progressive loss of neuronal cells and the consequent neurophysiological alterations, namely changes in the electrophysiological properties of hippocampal synapses and an increased susceptibility to kainate-induced epilepsy (34). Thus, each LD model to date replicates LB formation, spontaneous myoclonus, and reduced threshold for convulsions.
Figure 3.
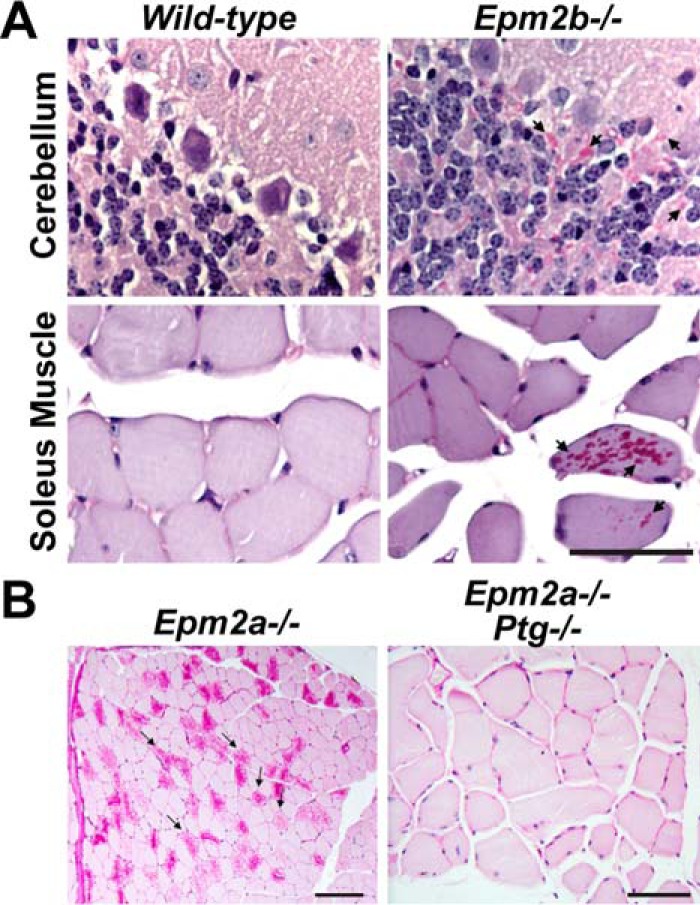
A, periodic acid-Schiff (PAS) staining of cerebellum and soleus sections from WT mice show few stainable structures. Conversely, Epm2b−/− mice exhibit dramatic numbers of stainable polyglucosans, i.e. LBs. Arrows indicate examples of areas rich in LBs. Data are modified from Ref. 32. B, periodic acid-Schiff staining of Epm2a−/− skeletal muscle reveals dramatic accumulations of LBs. Mice lacking both Epm2a and Ptg display minimal to no LBs. Data are modified from Ref. 89.
Despite the long-recognized accumulation of aberrant glycogen in LD, there was still no direct evidence whether the accumulation of glycogen is the cause of LD. The LD mouse models and cell culture models exhibit perturbations in more than just glycogen metabolism. Mice and cells lacking laforin and/or malin display increased endoplasmic reticulum stress, autophagy impairment, and reduced clearance of misfolded toxic proteins through the ubiquitin proteasome system (85, 90–92). Each of these pathways was proposed as an alternative underlying cause of LD instead of LBs. Some hypothesized that laforin and malin directly regulate these cellular processes. One hypothesis postulated that the loss of malin or laforin results in an autophagy defect, and autophagy was proposed as the primary cause of LD neurodegeneration. Under these circumstances, the accumulation of LBs could be a mere epiphenomenon.
However, several groups independently and definitively demonstrated that LBs are the cause of LD myoclonus epilepsy and neuronal apoptosis using multiple mouse models as follows. 1) Laforin KO mice lacking Ptg exhibit reduced LB accumulation, resolved neurodegeneration, and resolved myoclonic epilepsy (Fig. 3B) (88). 2) The lack of Ptg in malin-deficient mice nearly completely eliminates LBs and rescues the neurodegeneration, myoclonus, seizure susceptibility, and behavioral abnormality (87). 3) Malin-deficient mice lacking glycogen synthase (Gys1) are devoid of LBs, exhibit normal electrophysiological properties, show decreased susceptibility to kainate-induced epilepsy, and do not exhibit increased neurodegeneration (89). 4) Malin-deficient mice lacking one Gys1 allele, and thus showing reduced GS expression, partially rescue the phenotype (89).
Results from these mouse models demonstrate that decreased or complete absence of the glycogen synthesis machinery ablates LB formation, neurodegeneration, and epilepsy. Additionally, it is important to highlight that malin-deficient mice lacking Gys1 also exhibit normal autophagy, strongly suggesting that malin does not regulate autophagy but rather LBs trigger autophagy impairment (89). These results demonstrate that other cellular perturbations are consequences of aberrant glycogen accumulation, i.e. LBs, rather than being directly regulated by malin or laforin.
Thus, excessive glycogen accumulation appears to be responsible for the observed defects in malin and laforin KO mice, including their epilepsy and neurodegeneration. Also, the finding that deletion of one Gys1 allele partially rescues the pathological phenotype and wholly rescues the neurological phenotype strongly suggests that partial inhibition of glycogen synthase activity prevents disease progression. This hypothesis is further strengthened by the finding that the absence of Ptg by itself, which decreases glycogen accumulation by 50%, also resolves the murine disease. Cumulatively, these groups have shown in the LD mouse models that reducing glycogen synthesis by 50–100% through knockouts of the glycogen synthase gene or the glycogen synthase activator Ptg gene rescue murine LD. These findings suggest that glycogen accumulation is responsible for the epilepsy and functional consequences seen in the LD mouse models, defining LD as a glycogenosis. More importantly, they demonstrate that down-regulation of glycogen synthesis eliminates LBs and rescues LD.
An emerging link between glycogen metabolism and epilepsy
LD offers a unique window into both normal neuronal glycogen metabolism and epileptic disease when this metabolism is perturbed. Although much effort focuses on defining the basic mechanisms of LD and translating this work into therapeutics, it may reveal pathogenic mechanisms related to common epilepsies. These insights may be particularly informative to the most daunting aspect of epilepsy, namely the intractability that afflicts over 30% of patients. The mechanisms by which LD glycogenosis affects neurons and neuronal networks to generate an intractable seizure disorder are intriguing. There are several lines of evidence that implicate abnormal glycogen in the generation of seizures and epilepsy.
First, a recent study and accompanying review suggested that epilepsy may be as much a disease of energy metabolism as of neuronal discharge (93, 94). Sada et al. (94) demonstrated that blocking lactate dehydrogenase (LDH), the metabolic enzyme proposed by the ANLSH to provide astrocyte–neuron cross-talk, ablated neuronal excitation both in vitro and in an epilepsy animal model. Furthermore, they demonstrated that the antiseizure drug (ASD) stiripentol inhibits LDH (94). The authors surmise the mechanism of action is to block lactate generated from astrocytic glycogen from being shuttled to neurons and being utilized as a key energy source when energy consumption is increased, i.e. during seizures. A subsequent study by Samokhina et al. (95) studying epileptogenesis showed neuronal release of lactate rather than uptake. They concluded that blocking LDH instead causes energy deprivation via pyruvate-induced inhibition of glycolysis. These results are similar to those of Hall et al. (96) reporting that inhibition of LDH did not interfere with neuronal metabolism, and lactate shuttling is inconsistent with their data. Given these opposing conclusions, there is clearly more work to do in this arena. Regardless of the mechanism, the physiologically relevant targets of other ASDs are currently unknown, with many only modestly affecting ion channels; thus, other ASDs could also target key metabolic enzymes.
Other studies have demonstrated that normal glycogen is needed to clear extracellular K+, because the astrocytic Na+/K+-ATPase uses ATP generated from glucose 6-phosphate originating from glycogen breakdown and only from glycogen breakdown (73–75). Nonclearance of extracellular K+ results in neuronal hypersynchronization and burst firing, the critical mechanism of seizure generation and propagation. Therefore, normal glycogen is essential to controlling hyperexcitability and seizures (62, 77). In addition, normal glycogen synthesis and breakdown are critical to the homeostasis of glutamate, the most important excitatory neurotransmitter in the brain (62). Furthermore, mesial temporal lobe epilepsy is one of the most common types of refractory epilepsy. In tissue from refractory cases going to surgery, up to 50% of patients show abundant, sometimes profuse, polyglucosan bodies, indistinguishable from LBs, indicating an important yet poorly studied link between the accumulation of an abnormal glycogen and refractory epilepsy (78, 79). Recent work has demonstrated that neuronal death in LD could be influenced by secondary cellular insults via impairment in proteostasis mechanisms (97, 98). Although these data are convincing, there is clearly much more work to elucidate links between proteostasis, aberrant glycogen metabolism, and epilepsy.
Defining the basic mechanisms of LD and translating these findings into therapeutic options may provide insight into the cause of intractability in epilepsy and may lead to rational approaches to new therapies.
Conclusions
The central theme of LD revolves around the overaccumulation of aberrantly structured glycogen as follows: LBs largely consist of aberrant glycogen resulting from mutations in two critical enzymes; laforin binds and dephosphorylates glycogen; and the E3 ubiquitin ligase malin ubiquitinates enzymes involved in glycogen metabolism. LD is thus one of the family of glycogen storage diseases and offers a unique window into neuronal glycogen metabolism and disease. Despite glycogen metabolism being a mainstay in textbooks, work on LD has yielded a number of novel advances in our understanding of glycogen metabolism: 1) glycogen contains covalently bound phosphate; 2) laforin binds and dephosphorylates glycogen; 3) neurons express both glycogen synthase and glycogen phosphorylase; 4) neurons synthesize glycogen; and 5) aberrant glycogen accumulation causes autophagy impairment.
The LD field has generated a number of in vitro biochemical and structural techniques, in situ cell culture models, and in vivo mouse models. The quest now begins to utilize these systems to translate the cellular, structural, biochemical, and physiological insights into putative treatments. Although these hypotheses are directly applicable to LD, future findings may impact other epilepsies by targeting key cellular processes as a basis for treatment. Additionally, these findings may assist in resolving some of the controversies surrounding the relationship and mechanisms of energy metabolism in neurons and astrocytes.
This work was supported by the National Institutes of Health under Award Numbers R01NS070899 (to M. S. G.), P01NS097197 (to M. S. G.), R01NS056454 (to P. J. R.), and R01DK037221 (to P. J. R.); a Mitzutani Foundation for Glycoscience award 130095 (to M. S. G.); a National Science Foundation CAREER award MCB-1252345 (to M. S. G.); and Ministerio de Economia de Spain, Industria y Competitividad Grants SAF2014-59594-R (to J. M. S.) and SAF2014-55525-P (to J. J. G.). This is the fourth article in the Thematic Minireview series “Brain glycogen metabolism.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, Mitzutani Foundation, National Science Foundation, and Ministerio de Economia de Spain.
- LB
- Lafora bodies
- LD
- Lafora disease
- PTG
- protein targeting to glycogen
- GSD
- glycogen storage disease
- GS
- glycogen synthase
- GP
- glycogen phosphorylase
- ANLSH
- astrocyte-neuron lactate shuttle hypothesis
- LDH
- lactate dehydrogenase
- ASD
- antiseizure drug
- CNS
- central nervous system.
References
- 1. Lafora G. R. (1911) Uber des Vorkommen amyloider KJrperchen im innern der Ganglienzellen. Virchows Arch. Path. Anat. 205, 295 10.1007/BF01989438 [DOI] [Google Scholar]
- 2. Lafora G. R., and Gluck B. (1911) Beitrag zur histopathologie der myoklonischen epilepsie. Z Ges Neurol. Psychiatr. 6, 1–14 10.1007/BF02863929 [DOI] [Google Scholar]
- 3. Virchow R. L. (1858) Die Cellularpathologie Inihrer Begründung auf Physiologische and Pathologische Gewebelehre, Hirschwald, Berlin [Google Scholar]
- 4. Yokoi S., Austin J., and Witmer F. (1967) Isolation and characterization of Lafora bodies in two cases of myoclonus epilepsy. J. Neuropathol. Exp. Neurol. 26, 125–127 [PubMed] [Google Scholar]
- 5. Yokoi S., Austin J., Witmer F., and Sakai M. (1968) Studies in myoclonus epilepsy (Lafora body form). I. Isolation and preliminary characterization of Lafora bodies in two cases. Arch. Neurol. 19, 15–33 10.1001/archneur.1968.00480010033002 [DOI] [PubMed] [Google Scholar]
- 6. Schwarz G. A., and Yanoff M. (1965) Lafora's disease. Distinct clinico-pathologic form of Unverricht's syndrome. Arch. Neurol. 12, 172–188 10.1001/archneur.1965.00460260062008 [DOI] [PubMed] [Google Scholar]
- 7. Berkovic S. F., So N. K., and Andermann F. (1991) Progressive myoclonus epilepsies: clinical and neurophysiological diagnosis. J. Clin. Neurophysiol. 8, 261–274 10.1097/00004691-199107010-00003 [DOI] [PubMed] [Google Scholar]
- 8. Minassian B. A., Andrade D. M., Ianzano L., Young E. J., Chan E., Ackerley C. A., and Scherer S. W. (2001) Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann. Neurol. 49, 271–275 10.1002/1531-8249(20010201)49:2%3C271::AID-ANA52%3E3.0.CO%3B2-D [DOI] [PubMed] [Google Scholar]
- 9. Van Heycop Ten Ham M. W. (1975) in Handbook of Clinical Neurology (Vinken P. J., and Bruyn G. W., eds) pp. 382–422, North Holland Publishing Co., Holland, Amsterdam [Google Scholar]
- 10. Minassian B. A. (2001) Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 25, 21–29 10.1016/S0887-8994(00)00276-9 [DOI] [PubMed] [Google Scholar]
- 11. Serratosa J. M. (1999) Idiopathic epilepsies with a complex mode of inheritance. Epilepsia 40, Suppl. 3, 12–16 [DOI] [PubMed] [Google Scholar]
- 12. Turnbull J., Tiberia E., Striano P., Genton P., Carpenter S., Ackerley C. A., and Minassian B. A. (2016) Lafora disease. Epileptic Disord. 18, 38–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Serratosa J. M., Delgado-Escueta A. V., Posada I., Shih S., Drury I., Berciano J., Zabala J. A., Antúnez M. C., and Sparkes R. S. (1995) The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum. Mol. Genet. 4, 1657–1663 10.1093/hmg/4.9.1657 [DOI] [PubMed] [Google Scholar]
- 14. Minassian B. A., Lee J. R., Herbrick J. A., Huizenga J., Soder S., Mungall A. J., Dunham I., Gardner R., Fong C. Y., Carpenter S., Jardim L., Satishchandra P., Andermann E., Snead O. C. 3rd., Lopes-Cendes I., et al. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 20, 171–174 10.1038/2470 [DOI] [PubMed] [Google Scholar]
- 15. Serratosa J. M., Gómez-Garre P., Gallardo M. E., Anta B., de Bernabé D. B., Lindhout D., Augustijn P. B., Tassinari C. A., Malafosse R. M., Topcu M., Grid D., Dravet C., Berkovic S. F., and de Córdoba S. R. (1999) A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet. 8, 345–352 10.1093/hmg/8.2.345 [DOI] [PubMed] [Google Scholar]
- 16. Chan E. M., Young E. J., Ianzano L., Munteanu I., Zhao X., Christopoulos C. C., Avanzini G., Elia M., Ackerley C. A., Jovic N. J., Bohlega S., Andermann E., Rouleau G. A., Delgado-Escueta A. V., Minassian B. A., and Scherer S. W. (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127 10.1038/ng1238 [DOI] [PubMed] [Google Scholar]
- 17. Gentry M. S., Dowen R. H 3rd, Worby C. A., Mattoo S., Ecker J. R., and Dixon J. E. (2007) The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J. Cell Biol. 178, 477–488 10.1083/jcb.200704094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Worby C. A., Gentry M. S., and Dixon J. E. (2006) Laforin: A dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 281, 30412–30418 10.1074/jbc.M606117200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tagliabracci V. S., Girard J. M., Segvich D., Meyer C., Turnbull J., Zhao X., Minassian B. A., Depaoli-Roach A. A., and Roach P. J. (2008) Abnormal metabolism of glycogen phosphate as a cause for lafora disease. J. Biol. Chem. 283, 33816–33825 10.1074/jbc.M807428200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tagliabracci V. S., Turnbull J., Wang W., Girard J. M., Zhao X., Skurat A. V., Delgado-Escueta A. V., Minassian B. A., Depaoli-Roach A. A., and Roach P. J. (2007) Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 19262–19266 10.1073/pnas.0707952104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gentry M. S., Romá-Mateo C., and Sanz P. (2013) Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii. FEBS J. 280, 525–537 10.1111/j.1742-4658.2012.08549.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DePaoli-Roach A. A., Contreras C. J., Segvich D. M., Heiss C., Ishihara M., Azadi P., and Roach P. J. (2015) Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J. Biol. Chem. 290, 841–850 10.1074/jbc.M114.607796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raththagala M., Brewer M. K., Parker M. W., Sherwood A. R., Wong B. K., Hsu S., Bridges T. M., Paasch B. C., Hellman L. M., Husodo S., Meekins D. A., Taylor A. O., Turner B. D., Auger K. D., Dukhande V. V., et al. (2015) Structural mechanism of laforin function in glycogen dephosphorylation and Lafora disease. Mol. Cell 57, 261–272 10.1016/j.molcel.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Borden K. L., and Freemont P. S. (1996) The RING finger domain: a recent example of a sequence-structure family. Curr. Opin. Struct. Biol. 6, 395–401 10.1016/S0959-440X(96)80060-1 [DOI] [PubMed] [Google Scholar]
- 25. Gentry M. S., Worby C. A., and Dixon J. E. (2005) Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. U.S.A. 102, 8501–8506 10.1073/pnas.0503285102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng A., Zhang M., Gentry M. S., Worby C. A., Dixon J. E., and Saltiel A. R. (2007) A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease. Genes Dev. 21, 2399–2409 10.1101/gad.1553207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Ros S., Fernandez-Sanchez M. E., Garcia-Fojeda B., Criado Garcia O., Vilchez D., Dominguez J., Garcia-Rocha M., Sanchez-Piris M., Aguado C., Knecht E., Serratosa J., Guinovart J. J., Sanz P., and Rodriguez de Córdoba S. (2008) Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 17, 667–678 10.1093/hmg/ddm339 [DOI] [PubMed] [Google Scholar]
- 28. Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medraño-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez de Córdoba S., and Guinovart J. J. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413 10.1038/nn1998 [DOI] [PubMed] [Google Scholar]
- 29. Worby C. A., Gentry M. S., and Dixon J. E. (2008) Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076 10.1074/jbc.M708712200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Romá-Mateo C., Sanz P., and Gentry M. S. (2012) Deciphering the role of malin in the lafora progressive myoclonus epilepsy. IUBMB Life 64, 801–808 10.1002/iub.1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Printen J. A., Brady M. J., and Saltiel A. R. (1997) PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science 275, 1475–1478 10.1126/science.275.5305.1475 [DOI] [PubMed] [Google Scholar]
- 32. DePaoli-Roach A. A., Tagliabracci V. S., Segvich D. M., Meyer C. M., Irimia J. M., and Roach P. J. (2010) Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 285, 25372–25381 10.1074/jbc.M110.148668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turnbull J., Wang P., Girard J. M., Ruggieri A., Wang T. J., Draginov A. G., Kameka A. P., Pencea N., Zhao X., Ackerley C. A., and Minassian B. A. (2010) Glycogen hyperphosphorylation underlies lafora body formation. Ann. Neurol. 68, 925–933 10.1002/ana.22156 [DOI] [PubMed] [Google Scholar]
- 34. Valles-Ortega J., Duran J., Garcia-Rocha M., Bosch C., Saez I., Pujadas L., Serafin A., Cañas X., Soriano E., Delgado-García J. M., Gruart A., and Guinovart J. J. (2011) Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol. Med. 3, 667–681 10.1002/emmm.201100174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roach P. J. (2002) Glycogen and its metabolism. Curr. Mol. Med. 2, 101–120 10.2174/1566524024605761 [DOI] [PubMed] [Google Scholar]
- 36. DePaoli-Roach A. A., Segvich D. M., Meyer C. M., Rahimi Y., Worby C. A., Gentry M. S., and Roach P. J. (2012) Laforin and malin knockout mice have normal glucose disposal and insulin sensitivity. Hum. Mol. Genet. 21, 1604–1610 10.1093/hmg/ddr598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vernia S., Heredia M., Criado O., Rodriguez de Cordoba S., Garcia-Roves P. M., Cansell C., Denis R., Luquet S., Foufelle F., Ferre P., and Sanz P. (2011) Laforin, a dual specificity phosphatase involved in Lafora disease, regulates insulin response and whole-body energy balance in mice. Hum Mol. Genet. 20, 2571–2584 10.1093/hmg/ddr157 [DOI] [PubMed] [Google Scholar]
- 38. Gentry M. S., Dixon J. E., and Worby C. A. (2009) Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem. Sci. 34, 628–639 10.1016/j.tibs.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S. (2012) Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 10.1042/BJ20111416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blennow A., and Engelsen S. B. (2010) Helix-breaking news: fighting crystalline starch energy deposits in the cell. Trends Plant Sci. 15, 236–240 10.1016/j.tplants.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 41. Blennow A., Bay-Smidt A. M., Olsen C. E., and Møller B. L. (2000) The distribution of covalently bound phosphate in the starch granule in relation to starch crystallinity. Int. J. Biol. Macromol. 27, 211–218 10.1016/S0141-8130(00)00121-5 [DOI] [PubMed] [Google Scholar]
- 42. Blennow A. (2015) in Starch (Nakamura Y., ed) pp. 399–424, Springer, Tokyo, Japan [Google Scholar]
- 43. Smirnova J., Fernie A. R., and Steup M. (2015) in Starch (Nakamura Y., ed) pp. 239–290, Springer, Tokyo, Japan [Google Scholar]
- 44. Gentry M. S., Brewer M. K., and Vander Kooi C. W. (2016) Structural biology of glucan phosphatases from humans to plants. Curr. Opin. Struct. Biol. 40, 62–69 10.1016/j.sbi.2016.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pfister B., and Zeeman S. C. (2016) Formation of starch in plant cells. Cell. Mol. Life Sci. 73, 2781–2807 10.1007/s00018-016-2250-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kozlov S. S., Blennow A., Krivandin A. V., and Yuryev V. P. (2007) Structural and thermodynamic properties of starches extracted from GBSS and GWD suppressed potato lines. Int. J. Biol. Macromol. 40, 449–460 10.1016/j.ijbiomac.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 47. Verbeke J., Penverne C., D'Hulst C., Rolando C., and Szydlowski N. (2016) Rapid and sensitive quantification of C3- and C6-phosphoesters in starch by fluorescence-assisted capillary electrophoresis. Carbohydr. Polym. 152, 784–791 10.1016/j.carbpol.2016.07.057 [DOI] [PubMed] [Google Scholar]
- 48. Lomako J., Lomako W. M., Kirkman B. R., and Whelan W. J. (1994) The role of phosphate in muscle glycogen. Biofactors 4, 167–171 [PubMed] [Google Scholar]
- 49. Lomako J., Lomako W. M., Whelan W. J., and Marchase R. B. (1993) Glycogen contains phosphodiester groups that can be introduced by UDPglucose: glycogen glucose 1-phosphotransferase. FEBS Lett. 329, 263–267 10.1016/0014-5793(93)80234-L [DOI] [PubMed] [Google Scholar]
- 50. Fontana J. D. (1980) The presence of phosphate in glycogen. FEBS Lett. 109, 85–92 10.1016/0014-5793(80)81317-2 [DOI] [PubMed] [Google Scholar]
- 51. Jiang S., Wells C. D., and Roach P. J. (2011) Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem. Biophys. Res. Commun. 413, 420–425 10.1016/j.bbrc.2011.08.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Irimia J. M., Tagliabracci V. S., Meyer C. M., Segvich D. M., DePaoli-Roach A. A., and Roach P. J. (2015) Muscle glycogen remodeling and glycogen phosphate metabolism following exhaustive exercise of wild type and laforin knockout mice. J. Biol. Chem. 290, 22686–22698 10.1074/jbc.M115.673897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nitschke F., Wang P., Schmieder P., Girard J. M., Awrey D. E., Wang T., Israelian J., Zhao X., Turnbull J., Heydenreich M., Kleinpeter E., Steup M., and Minassian B. A. (2013) Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab. 17, 756–767 10.1016/j.cmet.2013.04.006 [DOI] [PubMed] [Google Scholar]
- 54. Tagliabracci V. S., Heiss C., Karthik C., Contreras C. J., Glushka J., Ishihara M., Azadi P., Hurley T. D., DePaoli-Roach A. A., and Roach P. J. (2011) Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab. 13, 274–282 10.1016/j.cmet.2011.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Contreras C. J., Segvich D. M., Mahalingan K., Chikwana V. M., Kirley T. L., Hurley T. D., DePaoli-Roach A. A., and Roach P. J. (2016) Incorporation of phosphate into glycogen by glycogen synthase. Arch. Biochem. Biophys. 597, 21–29 10.1016/j.abb.2016.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang W., Lohi H., Skurat A. V., DePaoli-Roach A. A., Minassian B. A., and Roach P. J. (2007) Glycogen metabolism in tissues from a mouse model of Lafora disease. Arch. Biochem. Biophys. 457, 264–269 10.1016/j.abb.2006.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roach P. J. (2015) Glycogen phosphorylation and Lafora disease. Mol. Aspects Med. 46, 78–84 10.1016/j.mam.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chikwana V. M., Khanna M., Baskaran S., Tagliabracci V. S., Contreras C. J., DePaoli-Roach A., Roach P. J., and Hurley T. D. (2013) Structural basis for 2′-phosphate incorporation into glycogen by glycogen synthase. Proc. Natl. Acad. Sci. U.S.A. 110, 20976–20981 10.1073/pnas.1310106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roach P. J. (2011) Are there errors in glycogen biosynthesis and is laforin a repair enzyme? FEBS Lett. 585, 3216–3218 10.1016/j.febslet.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meekins D. A., Raththagala M., Auger K. D., Turner B. D., Santelia D., Kötting O., Gentry M. S., and Vander Kooi C. W. (2015) Mechanistic insights into glucan phosphatase activity against polyglucan substrates. J. Biol. Chem. 290, 23361–23370 10.1074/jbc.M115.658203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fryer K. L., and Brown A. M. (2015) Pluralistic roles for glycogen in the central and peripheral nervous systems. Metab. Brain Dis. 30, 299–306 [DOI] [PubMed] [Google Scholar]
- 62. Dinuzzo M., Mangia S., Maraviglia B., and Giove F. (2015) Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab. Brain Dis. 30, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brown A. M., and Ransom B. R. (2015) Astrocyte glycogen as an emergency fuel under conditions of glucose deprivation or intense neural activity. Metab. Brain Dis. 30, 233–239 10.1007/s11011-014-9588-2 [DOI] [PubMed] [Google Scholar]
- 64. Dienel G. A., and Cruz N. F. (2015) Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 30, 281–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mergenthaler P., Lindauer U., Dienel G. A., and Meisel A. (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36, 587–597 10.1016/j.tins.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dienel G. A. (2017) Lack of appropriate stoichiometry: strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 95, 2103–2125 10.1002/jnr.24015 [DOI] [PubMed] [Google Scholar]
- 67. Brown A. M., and Ransom B. R. (2007) Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271 10.1002/glia.20557 [DOI] [PubMed] [Google Scholar]
- 68. Cummins C. J., Lust W. D., and Passonneau J. V. (1983) Regulation of glycogen metabolism in primary and transformed astrocytes in vitro. J. Neurochem. 40, 128–136 10.1111/j.1471-4159.1983.tb12662.x [DOI] [PubMed] [Google Scholar]
- 69. Dringen R., and Hamprecht B. (1993) Differences in glycogen metabolism in astroglia-rich primary cultures and sorbitol-selected astroglial cultures derived from mouse brain. Glia 8, 143–149 10.1002/glia.440080302 [DOI] [PubMed] [Google Scholar]
- 70. Cruz N. F., and Dienel G. A. (2002) High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J. Cereb. Blood Flow Metab. 22, 1476–1489 10.1097/01.WCB.0000034362.37277.C0 [DOI] [PubMed] [Google Scholar]
- 71. Duran J., Saez I., Gruart A., Guinovart J. J., and Delgado-García J. M. (2013) Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J. Cereb. Blood Flow Metab. 33, 550–556 10.1038/jcbfm.2012.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gibbs M. E., and Hutchinson D. S. (2012) Rapid turnover of glycogen in memory formation. Neurochem. Res. 37, 2456–2463 10.1007/s11064-012-0805-2 [DOI] [PubMed] [Google Scholar]
- 73. Choi H. B., Gordon G. R., Zhou N., Tai C., Rungta R. L., Martinez J., Milner T. A., Ryu J. K., McLarnon J. G., Tresguerres M., Levin L. R., Buck J., and MacVicar B. A. (2012) Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75, 1094–1104 10.1016/j.neuron.2012.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xu J., Song D., Xue Z., Gu L., Hertz L., and Peng L. (2013) Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res. 38, 472–485 10.1007/s11064-012-0938-3 [DOI] [PubMed] [Google Scholar]
- 75. Obel L. F., Müller M. S., Walls A. B., Sickmann H. M., Bak L. K., Waagepetersen H. S., and Schousboe A. (2012) Brain glycogen-new perspectives on its metabolic function and regulation at the subcellular level. Front. Neuroenergetics 4, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schousboe A., Sickmann H. M., Walls A. B., Bak L. K., and Waagepetersen H. S. (2010) Functional importance of the astrocytic glycogen-shunt and glycolysis for maintenance of an intact intra/extracellular glutamate gradient. Neurotox. Res. 18, 94–99 10.1007/s12640-010-9171-5 [DOI] [PubMed] [Google Scholar]
- 77. López-Ramos J. C., Duran J., Gruart A., Guinovart J. J., and Delgado-García J. M. (2015) Role of brain glycogen in the response to hypoxia and in susceptibility to epilepsy. Front. Cell. Neurosci. 9, 431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Streichenberger N., Ryvlin P., Guénot M., Sindou M., Kopp N., and Mauguière F. (2001) Polyglucosan bodies and temporal lobe epilepsy: an incidental finding or more? Clin. Neuropathol. 20, 172–175 [PubMed] [Google Scholar]
- 79. Abubakr A., Wambacq I., Donahue J. E., and Zappulla R. (2005) The presence of polyglucosan bodies in temporal lobe epilepsy: its role and significance. J. Clin. Neurosci. 12, 911–914 10.1016/j.jocn.2004.12.007 [DOI] [PubMed] [Google Scholar]
- 80. Cataldo A. M., and Broadwell R. D. (1986) Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J. Neurocytol. 15, 511–524 10.1007/BF01611733 [DOI] [PubMed] [Google Scholar]
- 81. Pfeiffer-Guglielmi B., Fleckenstein B., Jung G., and Hamprecht B. (2003) Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J. Neurochem. 85, 73–81 10.1046/j.1471-4159.2003.01644.x [DOI] [PubMed] [Google Scholar]
- 82. Lovatt D., Sonnewald U., Waagepetersen H. S., Schousboe A., He W., Lin J. H., Han X., Takano T., Wang S., Sim F. J., Goldman S. A., and Nedergaard M. (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 27, 12255–12266 10.1523/JNEUROSCI.3404-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Saez I., Duran J., Sinadinos C., Beltran A., Yanes O., Tevy M. F., Martínez-Pons C., Milán M., and Guinovart J. J. (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow Metab. 34, 945–955 10.1038/jcbfm.2014.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Duran J., Tevy M. F., Garcia-Rocha M., Calbó J., Milán M., and Guinovart J. J. (2012) Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol. Med. 4, 719–729 10.1002/emmm.201200241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Criado O., Aguado C., Gayarre J., Duran-Trio L., Garcia-Cabrero A. M., Vernia S., San Millán B., Heredia M., Romá-Mateo C., Mouron S., Juana-López L., Domínguez M., Navarro C., Serratosa J. M., Sanchez M., et al. (2012) Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol. Genet. 21, 1521–1533 10.1093/hmg/ddr590 [DOI] [PubMed] [Google Scholar]
- 86. Ganesh S., Delgado-Escueta A. V., Sakamoto T., Avila M. R., Machado-Salas J., Hoshii Y., Akagi T., Gomi H., Suzuki T., Amano K., Agarwala K. L., Hasegawa Y., Bai D. S., Ishihara T., Hashikawa T., et al. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 11, 1251–1262 10.1093/hmg/11.11.1251 [DOI] [PubMed] [Google Scholar]
- 87. Turnbull J., Epp J. R., Goldsmith D., Zhao X., Pencea N., Wang P., Frankland P. W., Ackerley C. A., and Minassian B. A. (2014) PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann. Neurol. 75, 442–446 10.1002/ana.24104 [DOI] [PubMed] [Google Scholar]
- 88. Turnbull J., DePaoli-Roach A. A., Zhao X., Cortez M. A., Pencea N., Tiberia E., Piliguian M., Roach P. J., Wang P., Ackerley C. A., and Minassian B. A. (2011) PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 7, e1002037 10.1371/journal.pgen.1002037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Duran J., Gruart A., García-Rocha M., Delgado-García J. M., and Guinovart J. J. (2014) Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 23, 3147–3156 10.1093/hmg/ddu024 [DOI] [PubMed] [Google Scholar]
- 90. Aguado C., Sarkar S., Korolchuk V. I., Criado O., Vernia S., Boya P., Sanz P., de Córdoba S. R., Knecht E., and Rubinsztein D. C. (2010) Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 19, 2867–2876 10.1093/hmg/ddq190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vernia S., Rubio T., Heredia M., Rodríguez de Córdoba S., and Sanz P. (2009) Increased endoplasmic reticulum stress and decreased proteasomal function in Lafora disease models lacking the phosphatase laforin. PLoS ONE 4, e5907 10.1371/journal.pone.0005907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Puri R., and Ganesh S. (2010) Laforin in autophagy: a possible link between carbohydrate and protein in Lafora disease? Autophagy 6, 1229–1231 10.4161/auto.6.8.13307 [DOI] [PubMed] [Google Scholar]
- 93. Scharfman H. E. (2015) Neuroscience. Metabolic control of epilepsy. Science 347, 1312–1313 10.1126/science.aaa9607 [DOI] [PubMed] [Google Scholar]
- 94. Sada N., Lee S., Katsu T., Otsuki T., and Inoue T. (2015) Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367 10.1126/science.aaa1299 [DOI] [PubMed] [Google Scholar]
- 95. Samokhina E., Popova I., Malkov A., Ivanov A. I., Papadia D., Osypov A., Molchanov M., Paskevich S., Fisahn A., Zilberter M., and Zilberter Y. (2017) Chronic inhibition of brain glycolysis initiates epileptogenesis. J. Neurosci. Res. 95, 2195–2206 10.1002/jnr.24019 [DOI] [PubMed] [Google Scholar]
- 96. Hall C. N., Klein-Flügge M. C., Howarth C., and Attwell D. (2012) Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 32, 8940–8951 10.1523/JNEUROSCI.0026-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rao S. N., Maity R., Sharma J., Dey P., Shankar S. K., Satishchandra P., and Jana N. R. (2010) Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 19, 4726–4734 10.1093/hmg/ddq407 [DOI] [PubMed] [Google Scholar]
- 98. Sinadinos C., Valles-Ortega J., Boulan L., Solsona E., Tevy M. F., Marquez M., Duran J., Lopez-Iglesias C., Calbó J., Blasco E., Pumarola M., Milán M., and Guinovart J. J. (2014) Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 13, 935–945 10.1111/acel.12254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Prats C., Graham T. E., and Shearer J. (2018) The dynamic life of the glycogen granule. J. Biol. Chem. 293, 7089–7098 10.1074/jbc.R117.802843 [DOI] [PMC free article] [PubMed] [Google Scholar]