

Abstract
Nucleosome dynamics, such as spontaneous DNA unwrapping, are postulated to have a critical role in regulating the access of DNA repair machinery to DNA lesions within nucleosomes. However, the specific histone domains that regulate nucleosome dynamics and the impact of such changes in intrinsic nucleosome dynamics on DNA repair are not well understood. Previous studies identified a highly conserved region in the N-terminal tail of histone H2B known as the histone H2B repression (or HBR) domain, which has a significant influence on gene expression, chromatin assembly, and DNA damage formation and repair. However, the molecular mechanism(s) that may account for these observations are limited. In this study, we characterized the stability and dynamics of ΔHBR mutant nucleosome core particles (NCPs) in vitro by restriction enzyme accessibility (REA), FRET, and temperature-induced sliding of histone octamers. Our results indicate that ΔHBR–NCPs are more dynamic, with a larger steady-state fraction of the NCP population occupying the unwrapped state than for WT-NCPs. Additionally, ΔHBR-histone octamers are more susceptible to temperature-induced sliding on DNA than WT histone octamers. Furthermore, we show that the activity of base excision repair enzymes at uracil lesions and single nucleotide gaps is enhanced in a site-specific manner in ΔHBR–NCPs. This enhanced activity correlates well with regions exhibiting increased DNA unwrapping. Finally, removal of the HBR domain is not sufficient to completely alleviate the structural constraints imposed by histone octamers on the activity of base excision repair enzymes.
Keywords: nucleosome, base excision repair (BER), DNA repair, chromatin, histone modification, glycosylase, HBR domain, histone H2B repression, polymerase beta
Introduction
Eukaryotic DNA is organized into arrays of nucleosomes, which constitute the primary level of chromatin compaction. The nucleosome core particle (NCP)3 consists of 147 bp of DNA wrapped ∼1.7 times around a histone octamer composed of a heterotypic tetramer of histones H3 and H4 flanked by two heterodimers of histones H2A and H2B (1). DNA-templated processes, including DNA repair, are dependent on protein–DNA interactions that are restricted by the histone proteins in nucleosomes (2, 3). Additionally, the DNA trajectory is significantly different when bound to the histone octamer compared with that in solution (4). These differences arise from periodic sharp deformations in the DNA structure at 14 local minima where the minor groove of DNA interacts with histone residues, such as “sprocket” arginines (5). Although these contacts can be made with most DNA sequences of sufficient length, efficient bending at the histone octamer surface is facilitated by sequence-specific dinucleotide steps (6); hence, DNA sequence is an important determinant of NCP stability and dynamics. NCP stability and dynamics is also dependent on histone content. Structural and biochemical evidence suggests that amino acid sequence variability in histone proteins, such as that found in histone variants, can regulate nucleosome dynamics, either alone or in conjunction with histone post-translational modification (PTM) (7–9). For example, H3K56 acetylation has been shown to increase nucleosomal DNA unwrapping dynamics by ∼7-fold relative to unacetylated nucleosomes, based on FRET studies of in vitro reconstituted NCPs (10). Other mechanisms can also enhance nucleosome dynamics, including ATP-dependent chromatin remodeling and certain DNA lesions, such as UV photoproducts (11).
Even subtle changes in nucleosome dynamics can have important biological implications in DNA-templated processes. This is especially true for DNA repair, because nucleosomes comprise a formidable barrier to the repair of DNA lesions (2, 12, 13). The impact of nucleosomes and nucleosome dynamics on repair is best understood for the base excision repair (BER) pathway. BER is responsible for removing chemically aberrant DNA bases, abasic sites generated from enzymatic or spontaneous hydrolytic reactions, and single-strand breaks. In a coordinated stepwise fashion, damage-specific DNA glycosylases identify and remove damaged DNA bases, cleaving the N-glycosidic bond between the sugar and the base (14). The resulting abasic site is the substrate for endonuclease APE1, which cleaves the DNA backbone 5′ to the abasic site, generating a single nucleotide gap with 5′-deoxyribose phosphate and 3′-OH termini. DNA polymerase β (Pol β) removes the blocking 5′-deoxyribose phosphate intermediate, generating a 5′-phosphate, and performs template-directed DNA synthesis using the 3′-OH (15, 16). Repair is completed with the assistance of DNA ligases that seal the nick. Importantly, each of these repair steps is inhibited to varying degrees by the packaging of DNA into nucleosomes (12, 13, 17–21); however, this inhibition is mitigated when the DNA lesion is located in more accessible or dynamic locations in the nucleosomal DNA. These include “Out” rotational settings, where the DNA minor groove faces away from the histone octamer (17, 22), or at translational positions near the DNA exit/entry site (12, 17, 21), which have a much higher rate of DNA unwrapping than translational positions near the dyad center of the nucleosome (23, 24).
The N-terminal tails of histones, which constitute ∼30% of the total histone mass, contribute to sequence-dependent nucleosome positioning (25) and have been shown to be involved in intra- and internucleosomal interactions (26). Unlike the N-terminal tails of histones H3 and H4 that are highly conserved, the sequence of the N-terminal tails of H2A and H2B are more divergent across species, and less is known about their functional roles in nucleosome stability and dynamics. Wyrick and co-workers (27) previously identified a conserved sequence cassette in the yeast histone H2B N-terminal tail, called the histone H2B Repression (or HBR) domain, that has a number of phenotypes, including de-repression of many yeast genes, enhanced DNA accessibility to micrococcal nuclease, and defects in UV resistance and repair (27, 28). The HBR domain in yeast consists of residues 30–37 in the N-terminal tail of H2B, corresponding to residues 24–31 of Xenopus laevis H2B. In X. laevis, seven of the eight residues are positively charged amino acids, arginine or lysine, which make potentially important electrostatic interactions with the negatively charged DNA backbone of both DNA gyres (29). Because of the importance of electrostatic interactions between DNA and histones in regulating NCP stability and dynamics, it was originally postulated that the HBR domain may play an important role in stabilizing octamer positioning and regulating intranucleosomal interactions (28, 30). In addition, some of the HBR phenotypes may also be due to the role of the HBR domain in regulating the binding of H2A/H2B dimers to histone chaperones FACT and Nap1 (31). However, it is unclear to what extent the HBR domain regulates intrinsic nucleosome stability and dynamics, nor is it known whether differences in nucleosome stability and dynamics contribute to HBR mutant phenotypes, such as DNA damage sensitivity and repair. To shed light on these past results, our present report uses biochemical and biophysical assays to address site-specific features of nucleosome dynamics caused by HBR deletion and their impact on BER enzyme activity.
To better understand the role of the HBR domain in regulating nucleosome stability and dynamics during BER, we characterized the stability of NCPs containing the HBR deletion (ΔHBR–NCPs) and examined its effect on specific repair steps associated with removal of rotationally and translationally positioned uracil and single nucleotide DNA gaps. Restriction enzyme accessibility (REA) assays indicate that ΔHBR–NCPs are intrinsically more dynamic in regions 13–23 bp from the DNA exit/entry sites (i.e. at the sites of REA cleavage). Analysis of nucleosome dynamics by FRET of unmodified (WT) and ΔHBR–NCPs are in agreement with these studies, showing a decreased energy transfer in ΔHBR–NCPs relative to WT, which is indicative of enhanced DNA unwrapping in ΔHBR–NCPs. Furthermore, heat-induced translational repositioning (or sliding) assays show that ΔHBR–NCPs are more prone to thermally induced sliding, suggesting a weakening of histone–DNA interactions. The increased dynamics of ΔHBR–NCPs were associated with increased uracil DNA gycosylase (UDG) and APE1 activity at an inwardly oriented uracil lesion located 24 bp from the DNA ends, while showing no enhanced activity at a more centrally located uracil lesion. The activity of Pol β was similarly dependent on the location of a DNA gap in NCPs relative to the HBR domain. Taken together, our results suggest that the HBR domain restricts DNA accessibility to BER repair enzymes in a site-specific manner, which correlates with the proximity of DNA lesions to the HBR domain in nucleosomes. However, deletion of the HBR domain does not completely overcome the steric constraints imposed on BER repair proteins by nucleosomes, particularly near the dyad center of NCPs.
Results
Deletion of the HBR domain in X. laevis histone H2B does not affect nucleosome formation on 601 DNA
To determine the effects of HBR deletion (ΔHBR, residues 24–31 in X. laevis) on nucleosome formation, nucleosome core particles (NCPs) were reconstituted with WT or ΔHBR X. laevis recombinant histone octamers (Fig. 1, A and B) and the high affinity 601 nucleosome positioning sequence (34, 36). The HBR domain is the most conserved region in the H2B N-terminal tail (Fig. 1A) and is comprised of 6–7 basic amino acids that, in Saccharomyces cerevisiae, are critical for transcriptional repression of a large set of genes (27). As seen in Fig. 1C, removal of these residues in histone H2B does not impede nucleosome formation, although the ΔHBR–NCP migrates slightly faster on native polyacrylamide gels, in accordance with a previous study (39). ΔHBR histone octamers generate well positioned NCPs as indicated by the presence of a single band on these gels. Furthermore, hydroxyl radical footprinting shows no detectable changes in the •OH cleavage pattern in WT and ΔHBR–NCPs (Fig. S1). These data suggest that 601 DNA adopts, on average, the same translational and rotational positioning on WT and ΔHBR histone octamers.
Figure 1.

The HBR domain in histone H2B. A, HBR sequence alignment from various species was adapted from Parra et al. (27). As can be seen, the HBR domain (e.g. residues 24–31 in X. laevis, 30–37 in S. cerevisiae, and 27–37 in Homo sapiens) is highly conserved among species and is predominantly comprised of positively charged amino acids (50, 51). B, in the left figure, PDB code 1KX5 was modified to highlight the structural location of the HBR domain (orange spheres) relative to BER lesions (black spheres). A different view with the HBR domain displayed as sticks is shown on the right. For the lesions (uracil or single-nucleotide gap), the number in parentheses indicates the number of nucleotides away from the pseudo 2-fold axis of symmetry called the dyad, toward the 5′ end (+) or (−) toward the 3′ end of the damaged strand. All lesions are inwardly oriented or occluded, with their phosphate backbone facing the histone octamer. Lesions at −49 and +4 located in the I strand (green) and −21 in the J strand (blue). C, representative 6% native gel illustrating reconstitution efficiency of DNA with recombinant X. laevis WT and HBR (Δ24–31) octamers.
HaeIII and MspI restriction sites are more accessible in ΔHBR–NCPs. To investigate possible changes in the structure or dynamics of ΔHBR–NCPs, we first tested the effect of HBR deletion on REA. Fig. 2A shows (schematically) the locations of MspI and HaeIII restriction cut sites relative to the dyad center of NCPs. As shown, the MspI and HaeIII restriction sites are located 13 and 23 nucleotides (nt), respectively, from the DNA end (Fig. 2A). Interestingly, ΔHBR–NCPs are cleaved much more efficiently by MspI compared with WT NCPs, regardless of whether the DNA template contains uracil at position −21, relative to the dyad axis toward the 3′ end, in the J chain of the 601 NCP (Fig. 2B, right panel) or position +4 located in the complementary I chain 4 nt from the dyad axis toward the 5′ end (Fig. 2C). Additionally, the HaeIII restriction site, located closer to the dyad center, is also more accessible in ΔHBR–NCPs (Fig. 2B, left panel). These results indicate that ΔHBR–NCP DNA is intrinsically more accessible to restriction enzyme cleavage in regions as far as 23 bp from the DNA ends.
Figure 2.

Restriction enzyme accessibility assay on nucleosomal DNA containing WT (unmodified) or ΔHBR (H2BΔ24–31 amino acids) histone octamers. A, schematic of the nucleosome core particle showing the approximate locations of two restriction sites relative to the dyad, where the number in parentheses indicates the cleavage site (base number) on the I strand toward the 5′ end from the dyad. The samples were incubated with either HaeIII or MspI, where indicated (+) for 2 h at 37 °C. In B, the substrate contained a uracil at −21 in B and at +4 in C. Error bars represent standard deviation of the means for at least three independent experiments. Two-tailed, unpaired t tests were performed, and asterisks indicate the level of significance with p values of 0.0001 and 0.004 for MspI and HaeIII, respectively, in B and 0.002 in C.
Deletion of HBR domain shifts nucleosome dynamics toward the unwrapped state
The REA results suggest that the HBR domain plays a role in regulating nucleosomal DNA accessibility and/or dynamics, particularly in regions near the DNA entry/exit sites. Therefore, FRET was employed to compare the DNA unwrapping dynamics between WT and ΔHBR nucleosomes. For these experiments, the 601 DNA template was labeled with a pair of fluorescent dyes, Cy3 and Cy5, and reconstituted with WT or ΔHBR histone octamers. We chose three different locations in the nucleosome structure to incorporate the dye pair (Fig. 3A): (1) near the nucleosome's entry/exit sites (End-label) (37); (2) at the HBR–DNA-binding domain in nucleosomes (HBR label); and (3) near the dyad center (internal label) (11). As shown previously (11), with excitation of Cy3, we observed Cy5 (acceptor) emission when the 601 DNA is reconstituted into nucleosomes (Fig. 3B, red trace), and little to no Cy5 emission with the naked 601 DNA (data not shown, but see Ref. 11). Importantly, we observe less energy transfer in the ΔHBR–NCP when the dye pair is located toward the edge (end label) or near the HBR-binding domain (HBR label; Fig. 3, B and D, compare red and blue traces). In contrast, no reduction in energy transfer is seen in the HBR nucleosome when the dye pair is located near the dyad center (Fig. 3F). These results, which are consistent with our REA data, indicate that ΔHBR nucleosomes have increased DNA unwrapping dynamics for DNA regions near the DNA entry/exit sites but not at a centrally located site near the nucleosome dyad.
Figure 3.

Nucleosome dynamics of WT and ΔHBR NCPs as assessed by FRET. A, the crystal structure PDB code 1KX5 was modified to highlight the structural location of fluorophore FRET pairs, where red indicates the positioning of cy5 (acceptor) and green the positioning of cy3 (donor). For end-labeled NCPs, a Cy3-labeled base was inserted at position 6, and a Cy5-labeled base at position 81 in the 601 sequence, as described previously (37). For HBR-label NCPs, Cy3 and Cy5 fluorophores were similarly located at base position 46 from the 5′ end of the I chain and base position 25 from the 5′ end of the J chain, respectively. Internal-label fluorophores were located at position 33 from the 5′ end of the I chain (Cy3) and position 34 from the 5′ end of the J chain (Cy5). The panels on the left show emission spectra for WT and ΔHBR NCPs with FRET pairs located at the end-label (B), HBR-label (D), and internal-label (F) positions. The panels on the right show salt-induced nucleosome disassembly/unwrapping for WT and ΔHBR NCPs monitored by FRET using end-labeled (C), HBR-label (E), and internal-label (G) NCPs. Salt concentrations were adjusted to the appropriate final concentration with 5 m NaCl and allowed to equilibrate for 30 min at room temperature. Emission spectra at the salt concentrations shown were normalized to the Cy5 signal excited at 615 nm. The FRET efficiency at each salt concentration was normalized to the corresponding low salt efficiency to allow comparison of the salt-induced FRET decrease between WT and HBR NCPs.
Nucleosome stability was examined using salt-induced dissociation of NCPs (11). As expected, the FRET efficiency decreases with increased salt concentration in both WT and ΔHBR nucleosomes (Fig. 3, C, E, and G). For end-label nucleosomes, the FRET efficiency of HBR–NCPs decreases more rapidly with increased salt concentration compared with WT, indicating that histone–DNA interactions near the DNA ends are weaker and more easily disrupted by salt (Fig. 3C). On the other hand, for HBR-label nucleosomes, the ΔHBR–NCP is more sensitive than WT at low to moderate salt concentrations (i.e. 0.3 and 0.4 m) and slightly less sensitive at higher salt concentrations (Fig. 3E). This later result may indicate that the H2A-H2B dimer, which dissociates from NCPs at lower salt concentrations (40, 41), may be more easily evicted from nucleosomes in the HBR mutant octamers. Finally, no difference is observed between WT and ΔHBR–NCP salt dissociation with internal-label nucleosomes (Fig. 3G), indicating no change in the slow unwrapping kinetics of DNA near the dyad center of NCPs. Taken together, these results indicate that deletion of the HBR domain in histone H2B generates more dynamic NCPs in which DNA near the nucleosome edges has increased unwrapping kinetics relative to WT NCPs.
Thermally induced sliding is enhanced in ΔHBR NCPs
It has long been known that nucleosomes are mobile on DNA at higher temperatures in vitro, where histone octamers are able to slide translationally along the DNA over large distances (42). Moreover, histone octamer composition and DNA sequence are known to play key roles in NCP stability (40, 43). Therefore, to determine the translational stability of ΔHBR NCPs relative to WT NCPs, WT and ΔHBR-containing octamers were reconstituted with the 601 DNA sequence in a 256-bp fragment. Because 601 DNA exhibits a strong positioning power, it generates homogeneously positioned octamers at room temperature despite the presence of linker DNA (Fig. 4A, blue oval); however, when the temperature is increased, the octamer adopts alternate positions as shown by the dashed oval in Fig. 4A (44). As shown in Fig. 4B (RT lane), positioning of the WT octamer along the 256-bp 601 DNA sequence generates primarily a centrally positioned histone octamer after a 60-min incubation at room temperature. The ΔHBR octamer, however, appears to generate a slightly more heterogeneous population of NCPs positioned centrally and at the sides of the fragment. The significance of this apparent change in population of different translational positions at room temperature is revealed by quantification of the gel patterns (Fig. 4C, RT values). This effect is amplified with increased temperature, where more repositioning of the ΔHBR octamers occurs relative to WT octamers (Fig. 4B, 58 °C and 65 °C lanes). Importantly, over 50% more ΔHBR octamers than WT octamers slide off the DNA fragments completely when incubated at 65 °C, leaving naked DNA fragments (Fig. 4B). These results indicate that histone–DNA interactions are weaker in ΔHBR–NCPs and are more easily disrupted by temperature.
Figure 4.
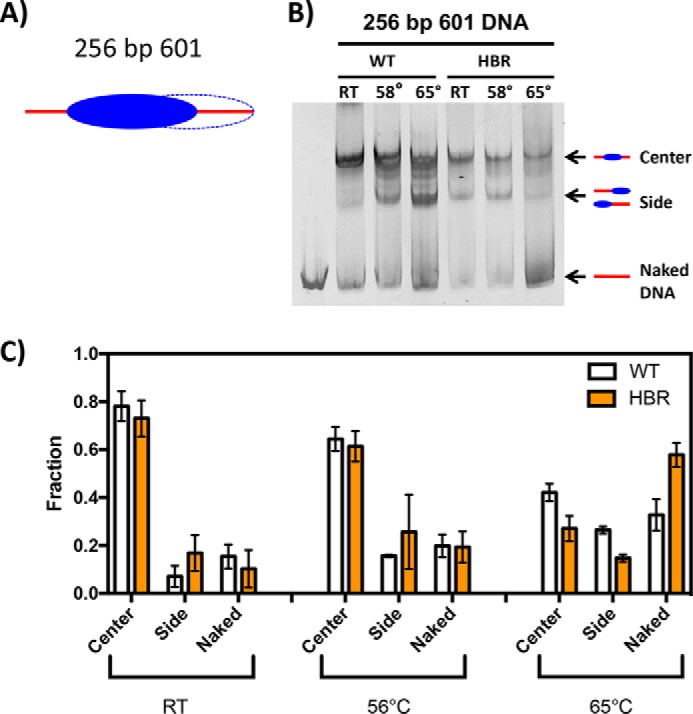
Thermal repositioning of WT and ΔHBR NCPs. A, schematic diagram of NCPs reconstituted with the 256-bp 601 DNA positioning sequence. Strongest NCP positions are indicated by solid ellipses, and weaker positions are indicated by dashed lines. B, representative 5% nondenaturing polyacrylamide gels of WT and ΔHBR NCPs reconstituted with the 256-bp 601 DNA positioning sequence. C, quantification of the fraction of DNA in each NCP structure (center or side) or as naked DNA at different temperatures. Error bars represent ± S.D. of the mean of at least three independent experiments.
HBR deletion affects uracil removal by UDG
Given that deletion of the HBR domain increases DNA unwrapping and thermally induced sliding, we wished to determine whether this increased nucleosome dynamics was sufficient to allow UDG access to occluded DNA lesions in NCPs. Surprisingly, HBR deletion has a small, but significant, negative effect on the removal of uracil at an occluded site in the DNA strand 21 nucleotides from the dyad (i.e. toward the 3′ end of the J chain; see PDB code 3LZ0) (Fig. 5A). This result suggests that if increased nucleosome dynamics extend to this location (21 nt from the dyad), they are too subtle for increased glycosylase lesion trapping during this time window. On the other hand, with a uracil located on the opposite DNA chain (i.e. the I chain) 49 nucleotides away from the dyad (toward the 3′ end), HBR deletion increases UDG activity by ∼2-fold (Fig. 5B). These data indicate that the increased nucleosome dynamics near the DNA ends increases accessibility to UDG at this site. Taken together, HBR deletion promotes uracil accessibility by UDG at sites near the DNA ends (i.e. regions that correlate with enhanced unwrapping in ΔHBR–NCPs).
Figure 5.
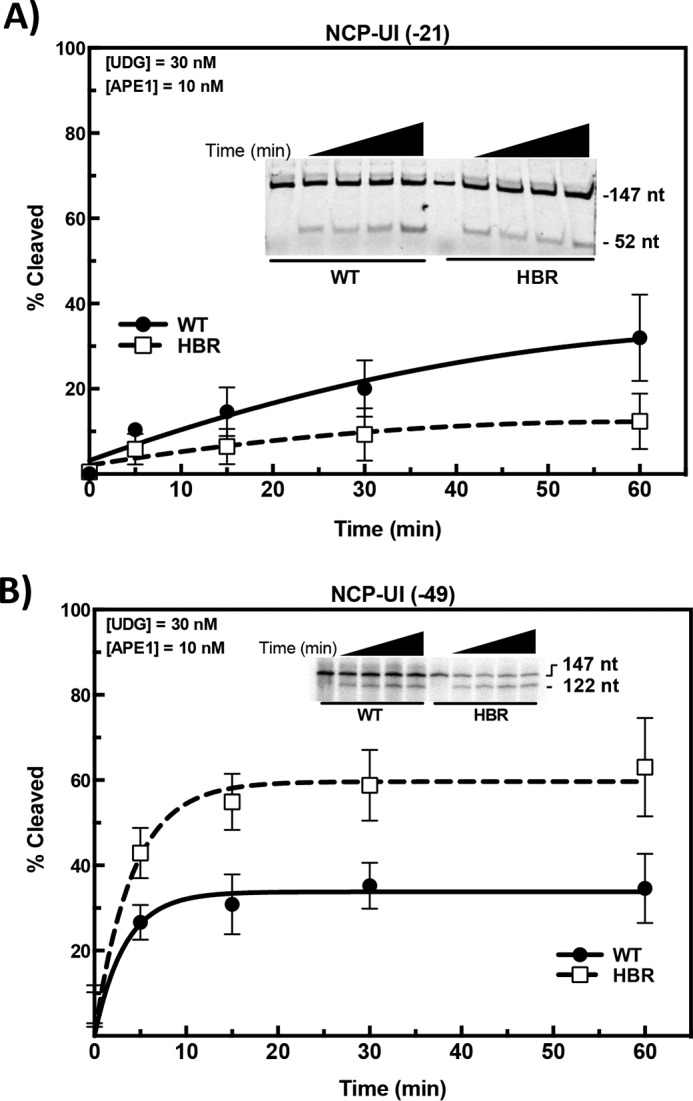
Effect of HBR deletion on uracil removal by UDG-APE1. 601 DNA, containing a single uracil at the specified locations, was 5′-end–labeled with either [γ-32P]ATP or Cy3 and annealed with the corresponding undamaged complementary strand. Substrates were reconstituted with WT or ΔHBR histone octamers, and DNA cleavage reactions were performed at 37 °C for the specified time with UDG (30 nm) and APE1 (10 nm). Cleavage activity was measured on denaturing gels (see insets), as described under “Experimental procedures.” The data points represent the means ± S.D. of at least three independent experiments.
Synthesis by DNA polymerase β in ΔHBR NCPs depends on the structural location of the DNA gap
We recently showed that the effects of histone site-specific acetylation on DNA gap-filling activity by Pol β are dependent on the structural location of the DNA gaps (45). In this study, we wished to determine whether the increased nucleosome dynamics near the DNA ends in ΔHBR–NCPs was sufficient to allow accessibility to occluded DNA gaps located near the DNA ends (−49), “midway” (−21), or near the dyad center (+4). Therefore, “gapped DNAs” were generated by pretreating 5′-end–labeled uracil-containing DNAs with UDG and APE1. The resulting NCPs containing a single-nucleotide gap were incubated with Pol β (in excess relative to the substrate). We then determined the single-nucleotide extension of substrate relative to the total substrate in the reaction. Remarkably, HBR deletion increases the initial rate of extension for NCP-gI (−21) by almost 5-fold (6.8% filled/min versus 33.1% filled/min for WT and ΔHBR, respectively; Fig. 6A). Structurally, this site lies near the region of increased DNA unwrapping as assessed by FRET (Figs. 1B and 3A). Similarly, but to a lesser extent, HBR deletion increases DNA synthesis at NCP-gI (−49), with a Ymax ∼2-fold greater in ΔHBR–NCPs (Fig. 6B). Because NCP-gI (−21) exhibits a greater effect than NCP-gI (−49), the latter being located right at the HBR domain, these data suggest that a major impediment in WT NCPs at NCP-gI (−49) is the proximity of DNA gyres (Fig. 1B, right panel). Increased DNA breathing, as well as increased local rotational flexibility of the DNA, may increase the average exposure time at NCP-gI (−21). On the other hand, at NCP-gI (−49), occlusion by the H2B N-terminal tail may lead to an overall decrease in accessibility to Pol β compared with NCP-gI (−21). Finally, at the lesion farthest away from the HBR-binding sites [NCP-gI (+4)], there is no significant difference in DNA synthesis between WT- and ΔHBR–NCPs (Fig. 6C). This result is in agreement with the FRET data and suggests that this site lies outside the dynamic range of increased DNA unwrapping resulting from HBR deletion. These results suggest that HBR deletion increases the probability of lesion recognition by Pol β, and this is sufficient to allow the repair of occluded DNA gaps located as far as 52 bp from the DNA ends (NCP-gI (−21)).
Figure 6.

Effect of HBR deletion on DNA synthesis by Pol β. WT and ΔHBR NCPs containing a single-nucleotide gap, located at positions denoted at the top of each panel, were incubated with 100 nm Pol β, a 5-fold molar excess relative to nucleosomal DNA. Extension products were separated on 8% polyacrylamide denaturing sequencing gels. Representative gels (imbedded within each chart) are shown with an arrow denoting the extension product band. Data points represent the means ± S.D. of at least three independent experiments.
Discussion
In this study, we show that the conserved HBR domain of histone H2B regulates the accessibility of repair factors to DNA lesions in nucleosomes, presumably by modulating intrinsic nucleosome dynamics. Our results indicate that NCPs reconstituted from histone octamers lacking the HBR domain (ΔHBR–NCPs) are more accessible to restriction enzyme digestion at sites as far as 23 nt from the DNA ends, consistent with the model that nucleosomal DNA unwrapping is enhanced in the ΔHBR–NCPs, particularly near the DNA entry–exit region of the NCP. Similarly, FRET studies suggest that a larger steady-state fraction of the ΔHBR–NCP population is shifted toward the unwrapped state as compared with WT-NCPs. This is in agreement with an increased susceptibility to temperature-induced octamer sliding in the ΔHBR–NCP relative to WT-NCPs. Importantly, this enhanced accessibility promotes uracil excision and single-nucleotide gap-filling by the BER enzymes UDG and Pol β, respectively, in a site-specific manner that correlates with proximity of these lesions to the HBR domain. Despite this enhanced accessibility, repair of uracil lesions is nonetheless inhibited in ΔHBR–NCPs, with only a fraction of the substrate becoming transiently accessible for repair. These findings indicate that the HBR domain represses intrinsic nucleosome dynamics, particularly near the nucleosomal DNA ends, and thus is a key factor limiting access of the BER machinery to DNA lesions in chromatin.
The HBR domain contributes to two of the strongest histone–DNA interaction sites (superhelical locations ± 4.5) as it transits between the DNA gyres (46, 47). Basic residues in the HBR domain likely contribute to favorable electrostatic interactions with the negatively charged DNA backbone, thus stabilizing histone–DNA interactions and repressing nucleosomal DNA unwrapping. This model is supported by genetic data showing that replacing the HBR domain with a series of amino acids with neutral side chains (i.e. alanine, glycine, or serine) is lethal in yeast, but replacing it with a series of lysine residues is viable, indicating that the positive charge of HBR domain residues is critical for the function of this domain in vivo (31). Locally, deletion of the HBR domain may affect the DNA trajectory, leading to an overall destabilization caused by weaker histone–DNA interactions in these regions that represent two of the most important stabilizing contacts in NCP DNA (46). Importantly, this may lead to DNA untwisting, thereby modifying distal interactions with histone H3 at the DNA entry–exit site. Indeed, a more flexible N-terminal tail of H3 is observed in the crystal structure of a tailless H2B-NCP that still contains the HBR domain (46). These findings indicate that intrinsic DNA accessibility near the HBR domain may be constrained to specific sites, which is consistent with our repair data. Our results demonstrate that sites of increased repair in ΔHBR–NCPs are generally associated with DNA regions near the HBR domain. For example, deletion of the HBR domain does not have a significant effect on DNA synthesis by Pol β at a gap-In lesion (gI (+4)) near the dyad center but does significantly increase the Pol β activity at gaps located closer to the HBR domain (i.e. gI (−21) and gI (−49)).
Previous studies have shown that the HBR domain mediates the transcriptional repression of ∼9% of yeast genes (27), and it is required to maintain normal levels of histone occupancy across the yeast genome, particularly at HBR-repressed genes (31). The HBR domain is thought to promote histone occupancy by facilitating nucleosome assembly by the histone chaperone FACT (facilitates chromatin transcription) (31). The HBR domain also regulates recruitment of the SWI/SNF ATP-dependent chromatin remodeler (28), although the HBR domain does not affect SWI/SNF remodeling activity in vitro (39). The in vitro data from this study indicate that the HBR domain may also regulate histone occupancy through a distinct mechanism, in which the HBR domain stabilizes assembled nucleosomes by repressing intrinsic nucleosome dynamics. It will be important to determine whether the role of the HBR domain in repressing intrinsic nucleosome dynamics in vitro helps promote transcriptional repression and histone occupancy in yeast.
In summary, our data indicate that the HBR domain functions to suppress intrinsic nucleosome dynamics, particularly nucleosomal DNA unwrapping, and thus restricts access of BER enzymes to DNA lesions at specific sites in nucleosomes (Fig. 7). An important implication of these findings is that BER efficiency in chromatin is likely regulated by rates of nucleosomal DNA unwrapping, which can vary widely between different nucleosome positioning sequences and at different translational positions within nucleosomes (48). This conclusion is supported by a recent genome-wide map of BER activity at single-nucleotide resolution in yeast cells (49). This study revealed that there is more efficient repair of DNA base lesions at distal translational locations within nucleosomes, where nucleosome dynamics are elevated in vitro. These findings suggest that nucleosome unwrapping dynamics regulate BER efficiency in vivo, consistent with our in vitro data. Furthermore, previous studies have identified a number of histone PTMs, including methylation, phosphorylation, and ubiquitination, occurring at histone residues within or adjacent to the HBR domain (reviewed in Ref. 50). It is interesting to speculate that these PTMs might alter HBR–DNA interactions (Fig. 7), which would be a novel mechanism to enhance nucleosome dynamics and facilitate repair in chromatin.
Figure 7.
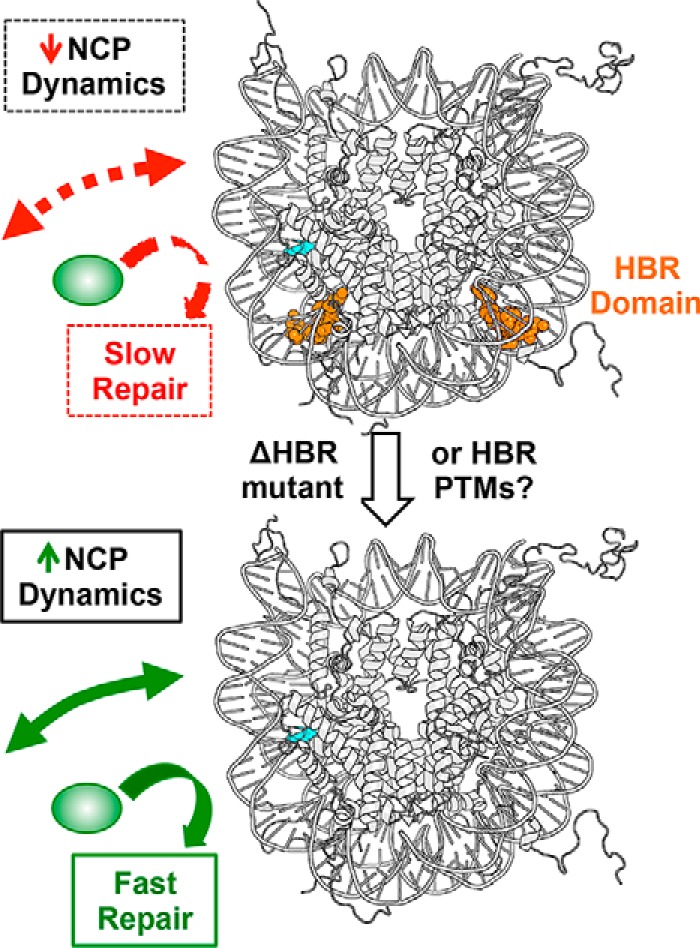
Model describing how HBR domain regulates NCP dynamics and DNA repair. In WT NCPs (upper panel), the HBR domain (depicted in orange) represses spontaneous nucleosomal DNA unwrapping, which limits access of repair enzymes (green oval) to DNA lesions (cyan) in nucleosomes. In HBR deletion mutants (lower panel), spontaneous nucleosomal DNA unwrapping is increased, which facilitates repair of DNA lesions. We hypothesize that in vivo HBR PTMs may also facilitate nucleosomal DNA unwrapping.
Experimental procedures
Preparation of DNA substrates containing uracil and single-nucleotide gaps
Uracil was inserted at distinct positions in the 147-bp 601 nucleosome positioning sequence as reported previously (32). Locations were chosen to allow for specific rotational settings when reconstituted into nucleosomes as either facing away or toward the histone octamer (12). Oligomers were purchased from Integrated DNA Technologies as ultramers (147 bp) or as primers containing a single uracil. Each of the primers or ultramers containing the uracil was either radiolabeled at the 5′ end or contained a fluorescent label as indicated in the figure legends. For the radiolabeling reactions, we used [γ-32P]ATP (PerkinElmer) and T4 polynucleotide kinase (Invitrogen) as per the manufacturers' instructions (22). After labeling, ultramers were annealed (1:1) with the complementary strand by heating to 95 °C for 10 min and slow cooling in 30 mm Tris buffer (pH 7.5) containing 100 mm potassium acetate. The resulting dsDNA fragment generated was then purified using either a QIAquick nucleotide removal kit or PCR purification kit (by Qiagen) to remove unincorporated radioactive nucleotides. We have used the nomenclature of Vasudevan et al. (33) to describe the locations of the two substrates (U(+4) and U(−49)) located in the I chain of the 601 NCP. The third substrate (UI(−21)) contained uracil in the complementary strand to the I chain (i.e. J chain) and was generated via PCR using pGEM-3z/601 as a template. The PCR product was purified using an agarose gel extraction kit (Qiagen).
Single-nucleotide gapped DNAs were generated from the uracil containing DNAs by treatment with UDG and APE1 (New England BioLabs; 30 and 10 nm, respectively) for 90 min at 37 °C to ensure complete cleavage of all uracil residues and subsequent cleavage of abasic sites. The DNA was then extracted (1:2 (v/v) ratio) with phenol:chloroform:isoamyl alcohol (PCI:25:24:1) and chloroform:isoamyl alcohol (24:1) using standard procedures, followed by ethanol precipitation overnight.
Nucleosome core particle reconstitutions
NCPs were reconstituted by salt gradient dialysis using recombinant octamer from X. laevis containing all WT core histones or core histones containing H2B histones with the HBR domain deleted. This domain consists of predominantly positively charged amino acids 24–31. WT and ΔHBR core histones were individually expressed in Escherichia coli (BL21) as previously described (34), with some modifications described in Ref. 11. After isolation, the histones were subjected to dialysis with deionized water containing 5 mm 2-mercaptoethanol and 0.2 mm phenylmethylsulfonyl fluoride. They were then lyophilized, and the histone octamer was prepared as described by Luger et al. (34). Briefly, the concentration of unfolded histone proteins was determined at A276 and equimolar ratios of all four histones were mixed and dialyzed three times in refolding buffer (2 m NaCl, 20 mm Tris-HCl, pH 7.5, 1 mm Na-EDTA, 5 mm 2-mercaptoethanol) at 4 °C for 24 h. Histone octamers were then concentrated and loaded onto a Superdex 200 column. The eluent was monitored by absorbance at 276 nm, and peak fractions were further analyzed on 16% SDS gels. Fractions containing equimolar concentrations of the histones were pooled, concentrated using an Amicon Ultra centrifugal filter, and stored in 50% glycerol at −20 °C. WT and HBR-containing histone octamers were then mixed with γ-32P–labeled DNA or fluorescence-labeled DNA in a 1.2:1 molar ratio via salt gradient dialysis to reconstitute NCPs, as described previously (11, 12).
Hydroxyl-radical footprinting
We performed hydroxyl-radical footprinting of the NCPs as described previously (22, 35). The reaction was quenched with glycerol to a final concentration of 6%. The DNA was isolated from the histones via PCI extraction followed by a chloroform:isoamyl alcohol wash. The DNA was then precipitated overnight in ethanol (2.5×, v/v), sodium acetate (1/10, v/v), pH 5.2, and glycogen (1/10, v/v). The pellet was washed twice with 70% ethanol, air dried, and suspended in 1:1 ratio of 1× Tris-EDTA (TE) and Hi-Di formamide. The samples were boiled for 10 min to denature the DNA, chilled on ice, and separated by electrophoresis in an 8% denaturing (7 m urea) polyacrylamide (19:1) gel. The gel was run at 60 W for 2.5 h, dried, exposed on a phosphor screen, and visualized on a Typhoon FLA7000 (GE Healthcare). Analysis of the image was performed using ImageQuant software (GE Healthcare). Uracil-containing DNA was treated with UDG (30 nm) and APE1 (10 nm), and incubated at 37 °C for 90 min for cleavage to take place. The cleavage reaction was stopped by PCI extraction, and the DNA was precipitated in ethanol, washed with 70% ethanol, dried, and suspended in 1× TE. A 1:1 ratio of sample and Hi-Di formamide were mixed, boiled for 10 min to denature DNA, chilled on ice, and loaded onto the sequencing gel.
Thermally induced sliding
WT and ΔHBR octamers were reconstituted with the 256-bp 601 positioning sequence (Fig. 4A). Nucleosomes were heated in a thermal cycler (Bio-Rad) at 58 or 65 °C for 60 min. The reaction was stopped by transferring the heated nucleosome samples to an ice-water bath. The samples were then analyzed by 5% PAGE in 0.25× Tris-borate-EDTA buffer (TBE). The gel was prerun at 4 °C for 1 h before running the samples for 60 min at 4 °C. Following electrophoresis, the gels were stained with SYBR Gold and imaged on a Typhoon FLA7000 imaging system (GE Healthcare). Gel images were then quantified using ImageQuant software (GE Healthcare).
FRET experiments
The 147-bp nucleosome DNA positioning sequence 601 was used for NCP reconstitution (36). In this study, three different locations were used for labeling the DNA with the fluorescence dyes. The internal-label and HBR-label DNA substrates were generated by PCR with fluorescently labeled primers. For internal-labeled DNA, a Cy3-labeled base was inserted at position 33 from the 5′ end of the I chain, and a Cy5-labeled base was inserted at position 34 from the 5′ end of the J chain. For the HBR-label, a Cy3-labeled base was inserted at position 46 from the 5′ end of the I chain, and a Cy5-labeled base was inserted at position 25 from the 5′ end of the J chain (33). The end-labeled DNA substrates were generated by inserting a Cy3-labeled base at position 6 and a Cy5-labeled base at position 81 in the 601 sequence, as described previously (37) (Fig. 3A). These fluorescently labeled DNA substrates were then used for NCP reconstitutions with WT or ΔHBR histone octamers. FRET experiments were carried out on a Photon Technology International Quantamaster UV-visible steady-state fluorometer. The samples were excited at 515 nm, and the emission spectra were collected from 550 to 700 nm. The Cy5 acceptor was excited at 615 nm for collecting “acceptor-only” emission spectra. FRET efficiencies were measured by the sensitized emission of the acceptor and calculated as described previously (11).
UDG and APE1 cleavage measurements
NCPs (20 nm) containing either WT or ΔHBR recombinant histone octamers were treated with E. coli UDG and human APE1 (New England Biolabs) at concentrations shown in the figures. All repair reactions were performed in a repair reaction buffer containing 25 mm HEPES (pH 7.5), 2 mm DTT, 100 μg/ml BSA, 10% glycerol, 5 mm MgCl2, 0.2 mm EDTA (pH 8), and 4 mm ATP. The reaction mixtures were incubated at 37 °C for the specified times (0–60 min) and terminated by the addition of PCI (1:2, sample:PCI, v/v). DNA was ethanol precipitated, resuspended in 1× TE and mixed with an equal volume of formamide-containing loading buffer. The samples were then boiled for 10 min and loaded onto a 10% polyacrylamide (19:1) 7 m urea denaturing gel in 1× TBE buffer. The gels were exposed overnight on a phosphorus-imaging screen and visualized on a Typhoon FLA7000 imaging system (GE Healthcare), and gel images were quantified using ImageQuant software (GE Healthcare).
Pol β gap-filling assays
Uracil-containing strands were radiolabeled at the 5′ end with [γ-32P]ATP using T4 polynucleotide kinase (Invitrogen). To generate single-nucleotide gaps, DNA was treated with UDG and APE1 as described above. The repair reaction was then performed by incubating radiolabeled NCPs with purified recombinant human Pol β at the specified concentrations. All extension products were separated in a DNA sequencing gel containing 8% polyacrylamide (19:1) and 7 m urea in 1× TBE buffer, and the percentage extended was calculated as described previously (12). Normalized data from either assay were fitted to a single-phase exponential curve using GraphPad Prism v.6 and the equation Y = Ymax (1 − e−kobst) as previously described (17, 38). The initial rates are reported under “Results.”
Restriction enzyme accessibility assay
Restriction enzymes HaeIII and MspI (New England Biolabs) were used to determine the accessibility of restriction sites +51 and +61, respectively, in 601 NCPs containing WT and ΔHBR histones. Gapped DNAs and NCPs were incubated with 10 units of each enzyme at 37 °C for 2 h followed by standard PCI and chloroform:isoamyl alcohol isolation of DNA. Cleavage products were separated on a 16% nondenaturing gel. Visualization and quantification of results were performed as described above.
Author contributions
Y. R. and M. D. data curation; Y. R., M. D., J. J. W., and M. J. S. formal analysis; Y. R., J. J. W., and M. J. S. supervision; Y. R. and M. D. validation; Y. R. and M. D. investigation; Y. R. and M. D. methodology; Y. R. writing-original draft; Y. R., M. D., J. J. W., and M. J. S. writing-review and editing; J. J. W. and M. J. S. conceptualization; J. J. W. and M. J. S. funding acquisition; J. J. W. and M. J. S. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Samuel H. Wilson and Rajendra Prasad (Laboratory of Structural Biology, NIEHS, National Institutes of Health) for providing human DNA polymerase β.
This work was supported by National Institutes of Health Grants ES002614 (to M. J. S. and J. J. W.), R21ES027937 (to J. J. W.), and ES004106 (to M. J. S.) from the NIEHS, National Institutes of Health, and by National Institute of General Medical Sciences Training Grant T32GM008336 (to Y. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- NCP
- nucleosome core particle
- PTM
- post-translational modification
- BER
- base excision repair
- UDG
- uracil DNA glycosylase
- Pol β
- DNA polymerase β
- nt
- nucleotide(s)
- REA
- restriction enzyme accessibility
- PDB
- Protein Data Bank
- PCI
- phenol:chloroform:isoamyl alcohol
- TE
- Tris-EDTA
- TBE
- Tris-borate-EDTA buffer.
References
- 1. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 2. Guintini L., Charton R., Peyresaubes F., Thoma F., and Conconi A. (2015) Nucleosome positioning, nucleotide excision repair and photoreactivation in Saccharomyces cerevisiae. DNA Repair (Amst.) 36, 98–104 10.1016/j.dnarep.2015.09.012 [DOI] [PubMed] [Google Scholar]
- 3. Rodriguez Y., Hinz J. M., and Smerdon M. J. (2015) Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair (Amst.) 32, 113–119 10.1016/j.dnarep.2015.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGinty R. K., and Tan S. (2015) Nucleosome structure and function. Chem. Rev. 115, 2255–2273 10.1021/cr500373h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hodges A. J., Gallegos I. J., Laughery M. F., Meas R., Tran L., and Wyrick J. J. (2015) Histone sprocket arginine residues are important for gene expression, DNA repair, and cell viability in Saccharomyces cerevisiae. Genetics 200, 795–806 10.1534/genetics.115.175885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chua E. Y., Vasudevan D., Davey G. E., Wu B., and Davey C. A. (2012) The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 10.1093/nar/gks261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bao Y., Konesky K., Park Y. J., Rosu S., Dyer P. N., Rangasamy D., Tremethick D. J., Laybourn P. J., and Luger K. (2004) Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 23, 3314–3324 10.1038/sj.emboj.7600316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugiyama M., Arimura Y., Shirayama K., Fujita R., Oba Y., Sato N., Inoue R., Oda T., Sato M., Heenan R. K., and Kurumizaka H. (2014) Distinct features of the histone core structure in nucleosomes containing the histone H2A.B variant. Biophys. J. 106, 2206–2213 10.1016/j.bpj.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowman G. D., and Poirier M. G. (2015) Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 115, 2274–2295 10.1021/cr500350x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neumann H., Hancock S. M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., and Chin J. W. (2009) A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 36, 153–163 10.1016/j.molcel.2009.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duan M. R., and Smerdon M. J. (2010) UV damage in DNA promotes nucleosome unwrapping. J. Biol. Chem. 285, 26295–26303 10.1074/jbc.M110.140087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez Y., and Smerdon M. J. (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem. 288, 13863–13875 10.1074/jbc.M112.441444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Odell I. D., Barbour J. E., Murphy D. L., Della-Maria J. A., Sweasy J. B., Tomkinson A. E., Wallace S. S., and Pederson D. S. (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632 10.1128/MCB.05715-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bauer N. C., Corbett A. H., and Doetsch P. W. (2015) The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 43, 10083–10101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hitomi K., Iwai S., and Tainer J. A. (2007) The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst.) 6, 410–428 10.1016/j.dnarep.2006.10.004 [DOI] [PubMed] [Google Scholar]
- 16. Schermerhorn K. M., and Delaney S. (2014) A chemical and kinetic perspective on base excision repair of DNA. Acc. Chem. Res. 47, 1238–1246 10.1021/ar400275a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cole H. A., Tabor-Godwin J. M., and Hayes J. J. (2010) Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem. 285, 2876–2885 10.1074/jbc.M109.073544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olmon E. D., and Delaney S. (2017) Differential ability of five DNA glycosylases to recognize and repair damage on nucleosomal DNA. ACS Chem. Biol. 12, 692–701 10.1021/acschembio.6b00921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hinz J. M. (2014) Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat. Res. 766–767, 19–24 [DOI] [PubMed] [Google Scholar]
- 20. Beard B. C., Wilson S. H., and Smerdon M. J. (2003) Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl. Acad. Sci. U.S.A. 100, 7465–7470 10.1073/pnas.1330328100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chafin D. R., Vitolo J. M., Henricksen L. A., Bambara R. A., and Hayes J. J. (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 19, 5492–5501 10.1093/emboj/19.20.5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinz J. M., Rodriguez Y., and Smerdon M. J. (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. U.S.A. 107, 4646–4651 10.1073/pnas.0914443107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tims H. S., Gurunathan K., Levitus M., and Widom J. (2011) Dynamics of nucleosome invasion by DNA binding proteins. J. Mol. Biol. 411, 430–448 10.1016/j.jmb.2011.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li G., and Widom J. (2004) Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 11, 763–769 10.1038/nsmb801 [DOI] [PubMed] [Google Scholar]
- 25. Yang Z., Zheng C., and Hayes J. J. (2007) The core histone tail domains contribute to sequence-dependent nucleosome positioning. J. Biol. Chem. 282, 7930–7938 10.1074/jbc.M610584200 [DOI] [PubMed] [Google Scholar]
- 26. Kan P. Y., Caterino T. L., and Hayes J. J. (2009) The H4 tail domain participates in intra- and internucleosome interactions with protein and DNA during folding and oligomerization of nucleosome arrays. Mol. Cell. Biol. 29, 538–546 10.1128/MCB.01343-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parra M. A., Kerr D., Fahy D., Pouchnik D. J., and Wyrick J. J. (2006) Deciphering the roles of the histone H2B N-terminal domain in genome-wide transcription. Mol. Cell. Biol. 26, 3842–3852 10.1128/MCB.26.10.3842-3852.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nag R., Kyriss M., Smerdon J. W., Wyrick J. J., and Smerdon M. J. (2010) A cassette of N-terminal amino acids of histone H2B are required for efficient cell survival, DNA repair and Swi/Snf binding in UV irradiated yeast. Nucleic Acids Res. 38, 1450–1460 10.1093/nar/gkp1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luger K., and Richmond T. J. (1998) The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8, 140–146 10.1016/S0959-437X(98)80134-2 [DOI] [PubMed] [Google Scholar]
- 30. Sivolob A., Lavelle C., and Prunell A. (2003) Sequence-dependent nucleosome structural and dynamic polymorphism: potential involvement of histone H2B N-terminal tail proximal domain. J. Mol. Biol. 326, 49–63 10.1016/S0022-2836(02)01372-4 [DOI] [PubMed] [Google Scholar]
- 31. Mao P., Kyriss M. N., Hodges A. J., Duan M., Morris R. T., Lavine M. D., Topping T. B., Gloss L. M., and Wyrick J. J. (2016) A basic domain in the histone H2B N-terminal tail is important for nucleosome assembly by FACT. Nucleic Acids Res. 44, 9142–9152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandez A. G., and Anderson J. N. (2007) Nucleosome positioning determinants. J. Mol. Biol. 371, 649–668 10.1016/j.jmb.2007.05.090 [DOI] [PubMed] [Google Scholar]
- 33. Vasudevan D., Chua E. Y., and Davey C. A. (2010) Crystal structures of nucleosome core particles containing the “601” strong positioning sequence. J. Mol. Biol. 403, 1–10 10.1016/j.jmb.2010.08.039 [DOI] [PubMed] [Google Scholar]
- 34. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 10.1016/S0076-6879(99)04003-3 [DOI] [PubMed] [Google Scholar]
- 35. Tullius T. D. (1991) DNA footprinting with the hydroxyl radical. Free Radic. Res. Commun. 12–13, 521–529 [DOI] [PubMed] [Google Scholar]
- 36. Lowary P. T., and Widom J. (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 10.1006/jmbi.1997.1494 [DOI] [PubMed] [Google Scholar]
- 37. Shim Y., Duan M. R., Chen X., Smerdon M. J., and Min J. H. (2012) Polycistronic coexpression and nondenaturing purification of histone octamers. Anal. Biochem. 427, 190–192 10.1016/j.ab.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Donigan K. A., Sun K. W., Nemec A. A., Murphy D. L., Cong X., Northrup V., Zelterman D., and Sweasy J. B. (2012) Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 287, 23830–23839 10.1074/jbc.M111.324947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng S., Crickard J. B., Srikanth A., and Reese J. C. (2014) A highly conserved region within H2B is important for FACT to act on nucleosomes. Mol. Cell. Biol. 34, 303–314 10.1128/MCB.00478-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gansen A., Valeri A., Hauger F., Felekyan S., Kalinin S., Tóth K., Langowski J., and Seidel C. A. (2009) Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 106, 15308–15313 10.1073/pnas.0903005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yager T. D., McMurray C. T., and van Holde K. E. (1989) Salt-induced release of DNA from nucleosome core particles. Biochemistry 28, 2271–2281 10.1021/bi00431a045 [DOI] [PubMed] [Google Scholar]
- 42. Pennings S., Meersseman G., and Bradbury E. M. (1991) Mobility of positioned nucleosomes on 5 S rDNA. J. Mol. Biol. 220, 101–110 10.1016/0022-2836(91)90384-I [DOI] [PubMed] [Google Scholar]
- 43. Kelbauskas L., Woodbury N., and Lohr D. (2009) DNA sequence-dependent variation in nucleosome structure, stability, and dynamics detected by a FRET-based analysis. Biochem. Cell Biol. 87, 323–335 10.1139/O08-126 [DOI] [PubMed] [Google Scholar]
- 44. Widom J. (2001) Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 34, 269–324 [DOI] [PubMed] [Google Scholar]
- 45. Rodriguez Y., Hinz J. M., Laughery M. F., Wyrick J. J., and Smerdon M. J. (2016) Site-specific acetylation of histone H3 decreases polymerase β activity on nucleosome core particles in vitro. J. Biol. Chem. 291, 11434–11445 10.1074/jbc.M116.725788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iwasaki W., Miya Y., Horikoshi N., Osakabe A., Taguchi H., Tachiwana H., Shibata T., Kagawa W., and Kurumizaka H. (2013) Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Bio. 3, 363–369 10.1016/j.fob.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hall M. A., Shundrovsky A., Bai L., Fulbright R. M., Lis J. T., and Wang M. D. (2009) High-resolution dynamic mapping of histone–DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129 10.1038/nsmb.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mao P., Wyrick J. J., Roberts S. A., and Smerdon M. J. (2017) UV-induced DNA damage and mutagenesis in chromatin. Photochem. Photobiol. 93, 216–228 10.1111/php.12646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mao P., Brown A. J., Malc E. P., Mieczkowski P. A., Smerdon M. J., Roberts S. A., and Wyrick J. J. (2017) Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Res. 27, 1674–1684 10.1101/gr.225771.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wyrick J. J., Kyriss M. N., and Davis W. B. (2012) Ascending the nucleosome face: recognition and function of structured domains in the histone H2A-H2B dimer. Biochim. Biophys. Acta 1819, 892–901 10.1016/j.bbagrm.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wyrick J. J., and Parra M. A. (2009) The role of histone H2A and H2B post-translational modifications in transcription: a genomic perspective. Biochim. Biophys. Acta 1789, 37–44 10.1016/j.bbagrm.2008.07.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.