

Abstract
Increased myeloperoxidase (MPO) levels and activity are associated with increased cardiovascular risk among individuals with chronic kidney disease (CKD). However, a lack of good animal models for examining the presence and catalytic activity of MPO in vascular lesions has impeded mechanistic studies into CKD-associated cardiovascular diseases. Here, we show for the first time that exaggerated atherosclerosis in a pathophysiologically relevant CKD mouse model is associated with increased macrophage-derived MPO activity. Male 7-week-old LDL receptor–deficient mice underwent sham (control mice) or 5/6 nephrectomy and were fed either a low-fat or high-fat, high-cholesterol diet for 24 weeks, and the extents of atherosclerosis and vascular reactivity were assessed. MPO expression and oxidation products—protein-bound oxidized tyrosine moieties 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine—were examined with immunoassays and confirmed with mass spectrometry (MS). As anticipated, the CKD mice had significantly higher plasma creatinine, urea nitrogen, and intact parathyroid hormone along with lower hematocrit and body weight. On both the diet regimens, CKD mice did not have hypertension but had lower cholesterol and triglyceride levels than the control mice. Despite the lower cholesterol levels, CKD mice had increased aortic plaque areas, fibrosis, and luminal narrowing. They also exhibited increased MPO expression and activity (i.e. increased oxidized tyrosines) that co-localized with infiltrating lesional macrophages and diminished vascular reactivity. In summary, unlike non-CKD mouse models of atherosclerosis, CKD mice exhibit increased MPO expression and catalytic activity in atherosclerotic lesions, which co-localize with lesional macrophages. These results implicate macrophage-derived MPO in CKD-accelerated atherosclerosis.
Keywords: myeloperoxidase, 3-chlorotyrosine, atherosclerosis, mass spectrometry (MS), macrophage, oxidative stress, 3-nitrotyrosine, Chronic Kidney Disease, oxidized amino acids
Introduction
Chronic kidney disease (CKD)3 affects 15% of Americans, and cardiovascular disease (CVD) continues to be the leading cause of mortality in these patients (>10-fold mortality compared with the general population) (1–6). An increased risk for CVD is evident even in milder and nonalbuminuric forms of CKD and cannot be fully explained by traditional risk factors (7–10). Furthermore, control of established risk factors such as hyperlipidemia results in substantially lower risk reduction in CKD patients compared with the general population (11). Recent evidence supports the key role of nontraditional risk factors (e.g. oxidative stress) in the pathogenesis of CVD in CKD (12–17), suggesting that CKD-specific mechanisms are responsible for CVD in kidney patients.
Atherosclerosis, the major cause of CVD, is a chronic inflammatory disease characterized by the infiltration of lipids and inflammatory cells, such as monocyte-derived macrophages and T-lymphocytes, into the artery wall (18). Although elevated levels of low-density lipoprotein (LDL) are known to increase the risk of atherosclerosis greatly (19), in vitro studies suggest that LDL by itself is not atherogenic but must instead be oxidized to initiate atherosclerotic disease (20, 21). Oxidized LDL is taken up by scavenger receptors of macrophages, which then become lipid-laden foam cells, the pathologic hallmark of early atherosclerotic lesions (22). Oxidized LDL has been isolated from human and animal atherosclerotic tissue, and immunohistochemical studies have detected oxidized lipids in atherosclerotic lesions, further substantiating the role of oxidative stress in atherosclerosis (23–25).
A primary source of oxidative stress is myeloperoxidase (MPO), a heme peroxidase enzyme that is highly expressed in neutrophils, monocytes, and macrophages (26). Although essential for bacterial killing, unrestrained MPO can induce surrounding tissue damage. Tyrosine residues in proteins are susceptible to covalent oxidative modifications, including the formation of 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine (27). Because these modifications are stable covalent interactions, protein tyrosine modifications serve as a metric of oxidative injury. MPO catalyzes the conversion of chloride ions in the presence of hydrogen peroxide to generate hypochlorous acid, a potent oxidant. Hypochlorous acid causes the post-translational modification 3-chlorotyrosine, a highly sensitive and specific marker of MPO activity (28). Similarly, MPO can generate nitrogen dioxide radical (NO2·) to form 3-nitrotyrosine and produce tyrosyl radicals from free tyrosine that can subsequently oxidize protein tyrosyl residues to form o,o′-dityrosine (29–31). By consuming nitric oxide, MPO action can also reduce vasodilator bioavailability, leading to vascular dysfunction (32).
The presence and activity of MPO in human atherosclerosis are well-established. MPO co-localizes with macrophages in human atherosclerotic lesions (33), and MPO oxidation products are present in human atherosclerotic lesions demonstrated by immunohistochemistry and quantification using MS (28, 33–36). More importantly, elevated plasma MPO levels predict the risk of cardiovascular events and mortality in patients with unstable angina, chest pain (37), acute coronary syndrome, or peripheral arterial disease (37–40). Systemic levels of 3-nitrotyrosine have also been found to be higher among patients with CVD compared with those with healthy arteries (41–43). We have recently demonstrated that plasma MPO levels and MPO oxidation products are elevated in autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematous, both of which carry an increased risk of CVD (44, 45).
Although the role of MPO-related oxidative processes is well-characterized in atherosclerosis, the specific role of MPO in CKD-accelerated atherosclerosis remains unclear. In general, studies in human patients with CKD have suggested that MPO plays an important role. For example, MPO-derived protein carbamylation is associated with CVD in the CKD population (46–49). Plasma MPO levels increase with each stage of CKD and are linked to CVD mortality in patients with end-stage renal disease (ESRD) who are undergoing peritoneal dialysis (50–53). In a recent study, our group reported that both MPO and 3-chlorotyrosine levels rise in the presence of coronary artery disease at various stages of CKD (54). It is important to note, however, that most of these studies are associative; hence, causality remains unclear.
Lack of good animal models to study the role of MPO in CKD-associated atherosclerosis makes the determination of causality a challenge. Despite strong evidence of MPO activity in human atherosclerotic lesions (28, 38, 55), mouse models of non-CKD atherosclerosis have undetectable MPO oxidation products in mouse atheroma (56). Paradoxically, MPO-deficient mice exhibit exaggerated atherosclerosis, although a study of mice overexpressing the human MPO transgene in macrophages showed increased atherosclerosis (57). This lack of MPO activity in mouse atheroma of non-CKD mice further highlights the need for an appropriate CKD-atherosclerosis model that demonstrates MPO activity in atheromatous lesions and lends itself to the systematic study of MPO action in CKD. In this study, using a pathophysiologically relevant 5/6 nephrectomy model of CKD in the LDL receptor knockout (LDLr−/−) mouse, we tested the presence of MPO and its activity in aortic tissue. This model has been extensively studied and reported by our group and others as a model that exhibits accelerated atherosclerosis with CKD (58–61). These CKD mice exhibit all the biochemical features of CKD and demonstrate accelerated atherosclerosis without elevated blood pressure, diabetes, or worsened hyperlipidemia compared with non-CKD mice. Thus, the CKD LDLr−/− model is an excellent animal model for studying the effects of CKD on atherosclerosis without the concomitant added risk factors. In contrast to non-CKD models, we report robust MPO expression and catalytic activity as demonstrated by elevated MPO oxidation products that co-localize with macrophages in atherosclerotic lesions. This implicates macrophage-derived MPO as a source of oxidants in the artery wall, which might propagate CKD-accelerated atherosclerosis.
Results
CKD LDLr−/− mice exhibit biochemical features of moderate CKD
C57BL/6 LDLr−/− mice were subjected either to sham operation (CTL, n = 20) or to 5/6 nephrectomy (CKD, n = 21). At 9 weeks of age, the mice in each group were further randomly divided into two subgroups and fed a low-fat diet (LFD) or high-fat, high-cholesterol diet (HFD). The mice in each group, CTL-LFD, CTL-HFD, CKD-LFD, or CKD-HFD, were maintained on these diets for either 12 or 24 weeks.
As shown in Table 1, serum blood urea nitrogen (BUN) and creatinine from CKD-LFD and CKD-HFD mice were elevated compared with their CTL counterparts, confirming the renal insufficiency of CKD mice. Serum BUN was 1.5 times and serum creatinine was ∼3 times higher in the CKD mice compared with their CTL counterparts indicating moderate CKD. There was a consistent decrease in renal function over time in all four groups. Hematocrit in CKD mice on either diet was decreased at 12 weeks (p < 0.05) and decreased further at 24 weeks (p < 0.05). Despite the increased intact parathyroid hormone (iPTH), there was no change in the serum calcium or phosphorous levels. Unlike human CKD, the CKD mice tended to have lower blood pressure compared with controls over time, although this trend did not reach statistical significance. There were no differences in the hemoglobin A1c levels among all four groups.
Table 1.
Biological characteristics of the four groups of mice
Male mice (n = 6–12/group) were utilized for this study; CTL, control LDLr−/− mice; CKD, 5/6 nephrectomized LDLr−/− mice; LFD, low-fat diet; HFD, high-fat, high-cholesterol diet; BUN, blood urea nitrogen; iPTH, intact parathyroid hormone.
| Biological parameters(units) | Weeks | CTL-LFD | CKD-LFD | CTL-HFD | CKD-HFD |
|---|---|---|---|---|---|
| Body weight (g) | 0 | 22.6 ± 0.4 | 22.2 ± 0.3 | 22.6 ± 0.3 | 22.2 ± 0.3 |
| 12 | 32.1 ± 0.9a | 27.7 ± 0.4a.b | 34.6 ± 0.9a | 28.7 ± 0.5a,b | |
| 24 | 34.5 ± 0.9a,b | 30.3 ± 0.9a,b | 39.3 ± 1.4a | 33.1 ± 1.0a,b,c | |
| Systolic blood pressure (mm Hg) | 0 | 103.0 ± 4.9 | 110.4 ± 4.8 | 110.4 ± 3.9 | 111.7 ± 5.4 |
| 12 | 102.7 ± 4.1 | 99.7 ± 2.4 | 101.8 ± 4.6 | 104.0 ± 6.2 | |
| 24 | 108.9 ± 4.4 | 89.70 ± 3.2 | 108.2 ± 3.4 | 91.8 ± 4.4 | |
| Glycohemoglobin (%) | 0 | 7.1 ± 0.4 | 6.7 ± 0.2 | 6.6 ± 0.2 | 7.0 ± 0.5 |
| 24 | 6.9 ± 0.2 | 6.5 ± 0.2 | 6.4 ± 0.1 | 7.0 ± 0.3 | |
| Hematocrit (%) | 0 | 60.8 ± 0.9 | 57.4 ± 0.7 | 61.7 ± 0.5 | 57.1 ± 0.8 |
| 12 | 61.7 ± 0.4 | 54.0 ± 0.4a,b | 59.8 ± 0.5 | 53.5 ± 0.7a,b | |
| 24 | 59.6 ± 0.7 | 51.6 ± 0.8a,b | 59.0 ± 0.6 | 51.9 ± 0.8a,b | |
| BUN(mg/dl) | 24 | 29.5 ± 1.5 | 43.7 ± 1.4b | 27.0 ± 1.7 | 44.7 ± 3.4b |
| Creatinine (mg/dl) | 0 | 0.06 ± 0.00 | 0.21 ± 0.01b | 0.08 ± 0.00 | 0.20 ± 0.01b |
| 12 | 0.08 ± 0.00 | 0.20 ± 0.01b | 0.08 ± 0.03 | 0.20 ± 0.01b | |
| 24 | 0.12 ± 0.01 | 0.24 ± 0.02b | 0.11 ± 0.01 | 0.22 ± 0.05b | |
| Calcium (mg/dl) | 0 | 10.4 ± 0.4 | 9.9 ± 1.1 | 9.8 ± 0.6 | 10.1 ± 0.7 |
| 24 | 9.8 ± 0.1 | 10.5 ± 0.3 | 10.3 ± 1.0 | 10.1 ± 0.5 | |
| Phosphorus (mg/dl) | 0 | 10.3±.2.5 | 11.6 ± 3.2 | 11.7 ± 1.5 | 11.1 ± 1.0 |
| 24 | 9.7 ± 1.6 | 9.6 ± 1.1 | 9.7 ± 1.1 | 10.1 ± 1.0 | |
| Plasma iPTH (pg/ml) | 0 | 336 ± 65 | 341 ± 124 | 366 ± 98 | 368 ± 136 |
| 24 | 314 ± 7 | 594 ± 8b | 329 ± 7 | 700 ± 6b | |
| Plasma cholesterol (mg/dl) | 0 | 249.2 ± 8.1 | 281.4 ± 25.5 | 278.2 ± 11.7 | 290.1 ± 27.2 |
| 12 | 389.9 ± 212.0 | 282.8 ± 49.0b | 1712 ± 937.4a,c | 1478 ± 599.6a,b,c | |
| 24 | 857.7 ± 93.4a | 471 ± 53.1a,b | 2432 ± 207.8a,c | 1532 ± 244.1a,b,c | |
| Plasma triglyceride (mg/dl) | 0 | 198.5 ± 9.8 | 263.6 ± 17.2 | 235.4 ± 35.6 | 216.6 ± 21.9 |
| 12 | 186.4 ± 25.7 | 412.6 ± 239.2 | 231.2 ± 79.0 | 368.3 ± 135.1 | |
| 24 | 389.4 ± 66.2a | 308 ± 16.8b | 1094 ± 147.5a,c | 668.2 ± 77.2a,b,c |
a p value <0.05 was compared with mice in the same group of different ages.
b p value <0.05 of CKD mice was compared with controls within same diet group.
c p value <0.05 was compared with mice fed low fat diet with similar renal function.
Mice from all four groups gained significant body weight over time (p < 0.001). Compared with the CTL mice, the CKD mice gained less weight at 12 weeks, and this difference persisted at 24 weeks. Although there was no significant difference at 12 weeks, the 24-week HFD-fed mice in the CKD groups gained much more than their LFD mice (p < 0.01).
Effects of CKD and HFD on plasma lipid levels and lipoprotein profiles
With the exception of triglycerides in CKD-LFD mice, plasma cholesterol and triglyceride concentrations were increased at 24 weeks in all diet categories, especially with HFD (p < 0.0001; Table 1). CKD mice fed HFD had decreased levels of cholesterol and triglycerides when compared with CTL mice fed HFD at 24 weeks (p < 0.05). As expected, the HFD mice in each renal function category had higher cholesterol and triglycerides compared with LFD mice with similar renal function.
Lipid distribution among the various lipoprotein fractions was analyzed by FPLC in pooled plasma samples from each group at 24 weeks. As shown in Fig. 1A, mice on LFD had lower cholesterol in the very-low-density (VLDL) and LDL fractions than mice on the HFD. The CKD mice had considerably lower VLDL and LDL cholesterol, and unchanged high-density lipoprotein (HDL) cholesterol compared with the CTL mice. Similarly, as shown in Fig. 1B, CKD mice, compared with CTL mice in the same diet category, had lower triglyceride content in the VLDL and LDL fractions. Mice on HFD had higher triglyceride content in the VLDL and LDL fractions compared with mice on LFD. Therefore, overall CKD mice had a more favorable atherogenic lipid profile compared with CTL mice.
Figure 1.
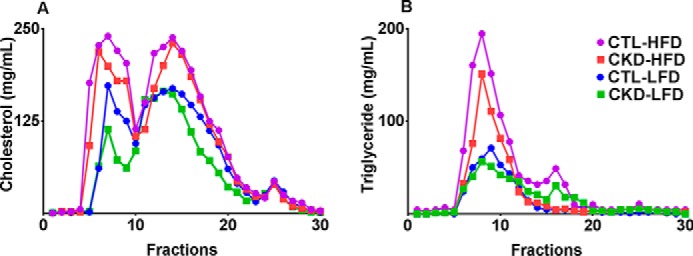
5/6 nephrectomized mice have lower cholesterol and triglyceride content in the VLDL and LDL fractions. FPLC fractions of pooled plasma from the four groups of male LDLr−/− control (CTL) and 5/6 nephrectomized (CKD) mice on high-fat, high-cholesterol diet (HFD) and low-fat diet (LFD) for 24 weeks reveal that CKD mice have lower VLDL and LDL cholesterol levels (A) with similar HDL cholesterol levels compared with CTL mice of the same diet. Similarly, the VLDL and LDL triglyceride levels (B) are lower in CKD mice when compared with CTL mice of the same diet. On the whole, mice on LFD had lower VLDL and LDL triglyceride and cholesterol content than mice on the HFD.
Effects of CKD and HFD on aortic atherosclerotic lesions
The en face analysis of aortic tree lesion area and cross-sectional analysis at the root of the aorta were performed to assess atherosclerosis. As shown in Fig. 2, Oil Red staining of the aortic tree and subsequent comparative morphometry revealed that all groups demonstrated increasing atherosclerotic lesions with age. At 12 weeks, HFD diet accelerated atherosclerosis in both CTL and CKD mice. At 24 weeks, CKD HFD mice had greater atherosclerosis than CKD-LFD and CTL-HFD (p value <0.05) mice. The CKD-HFD mice had more advanced atherosclerotic lesions in the aortic root (Fig. 3), aortic arch, brachiocephalic arteries, thoracic aorta, and abdominal bifurcations as shown in cross-sections of aortic root stained with hematoxylin and eosin (H&E) (Fig. 3, A and B) and Masson Trichrome (Fig. 3, C and D). Specifically, at 24 weeks the CKD-HFD mice (Fig. 3, B and D) had significantly elevated aortic plaque area fraction, luminal narrowing, and cellular filtration with significant fibrosis when compared with CTL-HFD mice (Fig. 3, A and C).
Figure 2.

5/6 nephrectomized (CKD) mice on high-fat, high-cholesterol diet (HFD) demonstrate increased atherosclerosis compared with control mice on HFD and CKD mice on low-fat diet. Representative Oil Red O staining and morphometry of aorta and main branches from male LDLr−/− mice are shown. A and F, control (CTL) mice on low-fat diet (LFD) for 12 and 24 weeks. B and G, 5/6 nephrectomized CKD mice on LFD for 12 and 24 weeks. C and H, CTL mice on HFD for 12 and 24 weeks. D and I, CKD mice on HFD for 12 and 24 weeks. E and J, analysis of total atherosclerotic lesion area ratio between the four groups at 12 and 24 weeks reveals increases in lesional ratio (ratio of Oil Red O-stained area to the total surface area of en face section of the aortic tree expressed as a percentage) in the CKD and HFD groups. The CKD HFD group has the most extensive lesional area among the four groups that is significantly higher when compared with CTL HFD group. (*, p < 0.05; number of mice is shown in parentheses.)
Figure 3.

5/6 nephrectomized mice on high-fat, high-cholesterol diet (HFD) have advanced atherosclerotic lesions. Representative cross-sections at aorta root from male LDLr−/− mice were stained with hematoxylin and eosin (H&E; A and B) and Masson Trichrome (C and D). A and C, control mice on HFD for 24 weeks. B and D, 5/6 nephrectomized (CKD) mice on HFD for 24 weeks. Magnification ×400. The atherosclerotic lesions from CKD mice on HFD had elevated aortic plaque area fraction, luminal narrowing, macrophage filtration, and fibrosis.
Effects of CKD and HFD on the vasodilatory response
Given the intimate link between atherosclerosis and vascular function, we assessed the cholinergic responsiveness of mouse aortic rings from the four mouse groups. CKD-HFD mice differed significantly in response to acetylcholine from that observed in rings from mice of the other three groups. The rings of CKD-HFD mice initially relaxed with acetylcholine. However, they then re-contracted moderately and maintained a similar tension at higher concentrations of acetylcholine. This phenomenon is illustrated by the data points (3 × 10−7 m and 10−6 m acetylcholine) that lie above the interpolated relaxation curve for the CKD-HFD mice (Fig. 4). The maximum vasodilatory response (Emax) for the aortic rings of CKD-HFD mice was significantly different compared with those observed for the other three groups (p < 0.01 versus CKD-LFD/CTL-HFD; p < 0.0001 versus CTL-LFD). There were no differences in the Emax to acetylcholine exhibited between rings from the CTL-LFD, CKD-LFD, or CTL-HFD mice (5.80 ± 3.30, 13.39 ± 2.94, and 13.01 ± 2.30%, respectively; Fig. 4). There was no difference in relaxation in response to sodium nitroprusside in any of the groups (data not shown).
Figure 4.
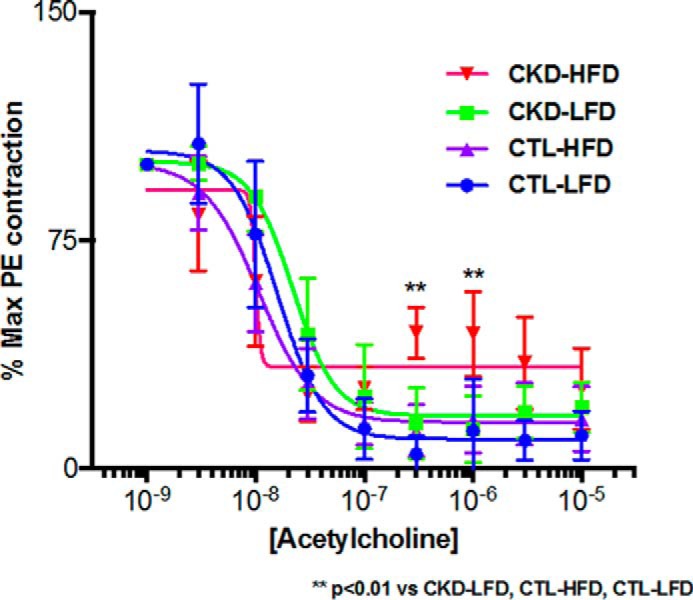
5/6 nephrectomized mice on high-fat, high-cholesterol diet (HFD) demonstrate decreased response to acetylcholine in aortic rings. Responses to acetylcholine in pre-contracted (phenylephrine) aortic rings from male LDLr−/− control (CTL) and 5/6 nephrectomized (CKD) mice on 24 weeks of a low-fat diet (LFD) and a HFD: CTL-LFD (Emax: 5.804 ± 3.296%), CKD-LFD (Emax: 13.39 ± 2.941%), and CTL-HFD (Emax: 13.01 ± 2.295%) mice were similar. In contrast, cholinergic responsiveness was significantly decreased in aortic rings from CKD-HFD (32.48 ± 3.126%) mice (Emax: **, p < 0.01 versus CKD-LFD/CTL-HFD/CTL-LFD; n = 5/group).
Immunochemical staining of MPO and MS assessment of MPO oxidation products in atherosclerotic lesions
MPO can oxidize tyrosine moieties in proteins to generate oxidized tyrosines. Although 3-chlorotyrosine is a specific marker for MPO, MPO can also form 3-nitrotyrosine and o,o′-dityrosine. We examined MPO expression and the presence of oxidized tyrosines with immunohistochemistry. Fig. 5 shows representative immunostaining of atherosclerotic lesions in aortic cross-sections from LDLr−/− mice at 24 weeks. MPO expression was observed in atherosclerotic lesions in CKD-HFD mice (Fig. 5B) when compared with CTL-HFD mice (Fig. 5A). Similarly, immunohistochemical staining for macrophage marker F4/80 protein and MPO-derived tyrosine modifications using specific antibodies for 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine demonstrated robust staining in the CKD-HFD mice at 24 weeks compared with the CTL HFD (data not shown).
Figure 5.
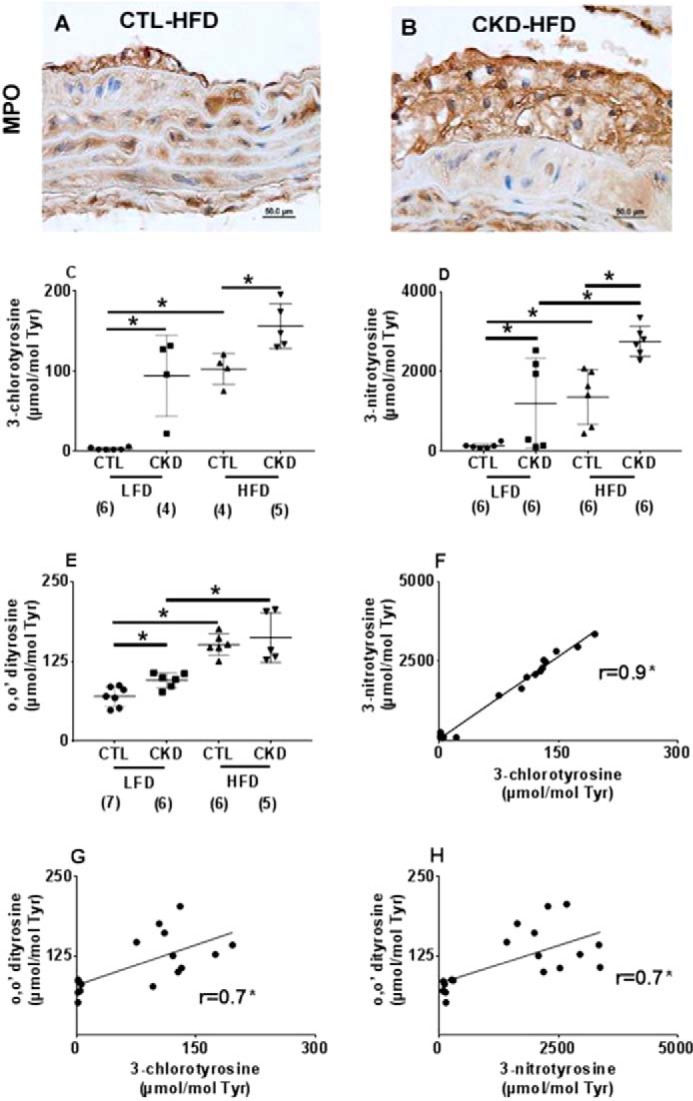
Increased myeloperoxidase and its products present in atherosclerotic lesions of 5/6 nephrectomized mice on high-fat, high-cholesterol diet (HFD). Representative immunohistologic staining for myeloperoxidase (MPO) in the aortic cross-sections from male LDLr−/− mice after 24 weeks of HFD is shown. Magnification ×1000. When compared with control (CTL-HFD) mice (A), intense staining for MPO was observed in the intimal lesions of 5/6 nephrectomized (CKD-HFD) mice (B) atherosclerotic lesions. Mass spectrometric quantification of oxidized amino acids in aortic proteins in male LDLr−/− mice on 24 weeks of HFD or LFD. C, 3-chlorotyrosine; D, 3-nitrotyrosine; and E, o,o′-dityrosine expressed as ratios to precursor amino acid tyrosine in μmol/mol (n = 4–7 each); F–H show the correlation using Pearson's correlation between the three oxidation products (*, p < 0.05).
To confirm the activity of MPO in these lesions, we utilized stable isotope-dilution mass spectrometry (MS), a highly sensitive and specific method to quantify the levels of protein tyrosine oxidative modifications: 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine in the aortic lesions. In contrast to immunoassays, which are qualitative, MS provides accurate quantitative data that are essential to ascribe enzyme activity. All results were normalized for the precursor amino acid tyrosine. As shown in Fig. 5C, at 24 weeks the average 3-chlorotyrosine levels (the specific MPO product) were markedly increased (156.3 ± 28.1 versus 102.5 ± 19.6 μmol/mol tyrosine; p < 0.05) in CKD versus CTL mice fed HFD in the aortic tissue. Similarly, CKD LFD mice showed increased lesional 3-chlorotyrosine compared with CTL-LFD mice (94.1 ± 50.9 versus 2.6 ± 1.7 μmol/mol tyrosine; p < 0.0001). Hence, CKD status, independent of the type of diet, augmented the presence of the MPO product 3-chlorotyrosine in atherosclerotic lesions.
The average 3-nitrotyrosine levels approximately doubled (2757 ± 376.1 versus 1365 ± 693.8 μmol/mol tyrosine, p < 0.01) in CKD compared with control aortic tissue. 3-Nitrotyrosine was higher in CKD-LFD compared with CTL-LFD (1204 ± 1133 versus 137.3 ± 65.3 μmol/mol tyrosine; p < 0.05, Fig. 5D).
The o,o′-dityrosine levels were 1.36-fold higher in CKD-LFD compared with CTL-LFD mice (95.67 ± 11.7 versus 70.2 ± 15.5 μmol/mol tyrosine; p < 0.05), whereas the levels were unchanged between CKD-HFD mice and CTL-HFD mice (148.4 ± 39.9 versus 151.6 ± 16.8 μmol/mol tyrosine; p = 0.70). o,o′-Dityrosine in lesions of CKD-HFD were increased compared with the CKD-LFD mice (n = 8, p < 0.01, Fig. 5E). These data suggest that tyrosine oxidation products are increased in aortic tissues of CKD mice and accentuated with HFD.
In addition to their increased presence, these oxidation products correlate with each other. Importantly, 3-chlorotyrosine, a specific product of MPO, correlated strikingly with 3-nitrotyrosine (r = 0.93; p value <0.0001) suggesting that MPO is the source of both of these products. The other correlations were less striking (3-chlorotyrosine versus o,o′-dityrosine, r = 0.70; p value < 0.01; o,o′-dityrosine versus 3-nitrotyrosine, r = 0.70; p value < 0.005) suggesting that in addition to MPO other pathways might contribute to their formation (Fig. 5, F–H).
MPO expression and its tyrosine oxidation products in atherosclerotic lesions in CKD-HFD mice
Using a double-labeling immunofluorescence technique, we found that Mac-2 (a macrophage marker), MPO, o,o′-dityrosine, 3-nitrotyrosine, and 3-chlorotyrosine co-localized intensely in atherosclerotic plaques in the CKD-HFD mice. The labeling was concentrated at the luminal surface of the plaque areas (Fig. 6, I–L).
Figure 6.
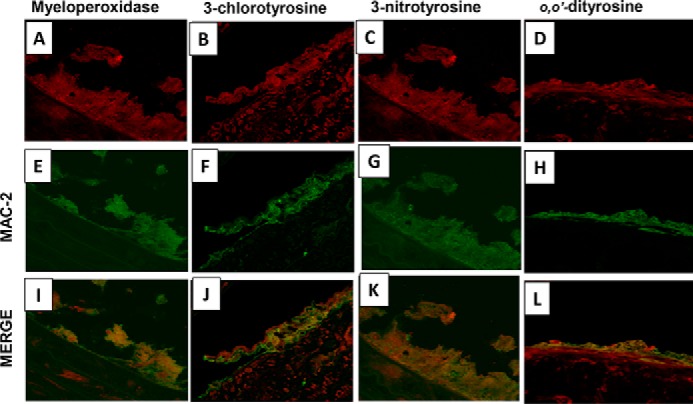
Macrophages, myeloperoxidase, and myeloperoxidase products co-localize in the arterial wall of 5/6 nephrectomized mice on high-fat, high-cholesterol diet. Representative immunofluorescence and double labeling in the aortic cross-sections from male LDLr−/− 5/6 nephrectomized mice on 24 weeks of high-fat, high-cholesterol diet for Mac-2 (macrophage marker), myeloperoxidase, 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine are shown. Shown is a section view of the aortic wall double-labeled for Mac-2 (green; E–H) and MPO (red, A), 3-chlorotyrosine (red, B), 3-nitrotyrosine (red, C), and o,o′-dityrosine (red, D). In all sections, the signals of MPO, o,o′-dityrosine, 3-nitrotyrosine, and 3-chlorotyrosine co-localized with Mac-2 (yellow, I–L).
To ascertain the specificity and cross-reactivity of these antibodies, we generated oxidized bovine serum albumin (BSA) in vitro with conditions favoring chlorination (MPO-peroxide-chloride with a resultant increase in 3-chlorotyrosine), nitration (MPO-nitrite-peroxide system with an increase in 3-nitrotyrosine), and hydroxyl radical formation (copper-peroxide; with o,o′-dityrosine, ortho-tyrosine (o-tyrosine), and meta-tyrosine (m-tyrosine) generation) as described previously and elaborated in the supporting Methods (55, 62–64). The formation of the anticipated oxidized amino acids following the reactions was confirmed by MS. We then pooled the reaction mixtures to form a mixture of BSA containing all oxidized tyrosine modifications (o,o′-dityrosine, o-tyrosine, m-tyrosine, 3-nitrotyrosine, and 3-chlorotyrosine). We subsequently subjected the mixture to immunoprecipitation with the anti-3-chlorotyrosine, anti-3-nitrotyrosine, and anti-o,o′-dityrosine antibodies. The pre- and post-immunoprecipitation mixtures were tested for enrichment of modified tyrosines by MS following acid hydrolysis. Immunoprecipitation with anti-3-nitrotyrosine antibody showed ∼3-fold enrichment of 3-nitrotyrosine but not of the other tyrosine moieties. Immunoprecipitation with anti-o,o′-dityrosine antibody resulted in ∼18-fold enrichment of o,o′-dityrosine compared with pre-immunoprecipitation levels but not of the other modified tyrosines, confirming the specificity of these two antibodies (Table S1). In contrast, anti-3-chlorotyrosine antibody (utilizing the only commercially available antibody) did not result in enrichment of 3-chlorotyrosine (nor any other oxidized amino acid; Table S1). This could be potentially because (a) the anti-3-chlorotyrosine antibody is not specific, (b) is not sensitive as 3-chlorotyrosine generation was lower than the other modifications, or (c) the antibody is more conducive to immunochemistry studies but may not work well in the immunoprecipitation reactions.
These results suggest that MPO derived from lesional macrophages co-localizes with 3-nitrotyrosine and o,o′-dityrosine residues in atherosclerotic plaques of CKD-HFD mice. However, given the nonspecificity of the 3-chlorotyrosine antibody, we cannot ascertain the same for 3-chlorotyrosine, the specific marker for MPO. Taken in conjunction with the quantitative MS data, which show a strong correlation of 3-chlorotyrosine with other oxidative tyrosine moieties, the data are highly suggestive that MPO is derived from lesional macrophages as the source of all three oxidatively modified tyrosines in atherosclerotic lesions.
Discussion
MPO-mediated oxidative stress is associated with elevated CVD risk in CKD patients (65, 66); however, testing whether MPO is pathogenically important in atherosclerosis in CKD patients is difficult given a lack of strong animal models of atherosclerosis that also demonstrate significant MPO activity in atherosclerotic lesions. Our CKD atherosclerosis mouse model is unique and is the first to demonstrate increased MPO expression and activity in atherosclerotic lesions. The atherosclerotic lesions in this model clearly exhibit elevated 3-chlorotyrosine, a specific marker for MPO activity, in addition to 3-nitrotyrosine and o,o′-dityrosine (two modified tyrosines that can be derived from MPO or alternative sources). Importantly, the oxidative tyrosine modifications show a high degree of correlation among each other, supporting the notion that MPO mediates all three oxidative modifications in this model. Furthermore, these oxidative MPO products co-localize with macrophages in the lesions, confirming the presence of catalytically active macrophage-derived MPO in the lesions in this model.
The mouse model of atherosclerosis and CKD bears some similarities to human CKD and atherosclerosis, including elevated LDL cholesterol and biochemical features of moderate CKD (e.g. tripling of serum creatinine, mild anemia, and elevated iPTH levels with normal calcium and phosphorous levels consistent with mild secondary hyperparathyroidism). These mice do not develop diabetes and hypertension even when exposed to HFD, making this an ideal model for the study of cardiovascular complications of moderate CKD alone. The CKD mice also exhibit a more favorable lipid profile compared with the CTL mice when exposed to an HFD; however, they develop enhanced atherosclerosis, suggesting that CKD significantly accelerates atherosclerosis in this model beyond the influence of hyperlipidemia. To our knowledge, this is the first mouse model that demonstrates increased lesional MPO expression and catalytic activity likely derived from macrophages.
Macrophages are closely associated with atherogenesis and represent a major source of oxidative stress in CKD (51, 67). However, the role of macrophage-derived MPO in CKD-associated atherosclerosis is yet to be explored using mechanistic models, as non-CKD animal models of atherosclerosis that demonstrate clear MPO involvement are not established. LDLr−/− mice of C57BL/6 background, similar to our model on HFD, did not demonstrate remarkable MPO presence or activity in atherosclerotic lesions. This finding is dissimilar from human lesions, which have increased 3-chlorotyrosine, a specific product of MPO activity (56). This calls into question the appropriateness of mouse models for studying MPO-related atherosclerosis, as there appear to be notable differences between murine and human atherosclerosis. Moreover, MPO-deficient mice with both macrophage-specific and whole-body knockouts paradoxically demonstrated a 50% increase in atherosclerosis after 14 weeks of HFD, suggesting that alternative pathways are involved in the process. This inability to replicate the MPO activity evident in human atherosclerosis in mouse models has deterred further mechanistic studies. Thus, our CKD animal model that demonstrates robust MPO involvement in vascular lesions after 24 weeks of HFD is invaluable in developing a better understanding of atherosclerosis in CKD.
MPO expression is restricted to hematopoietic cells and is controlled by promoter, enhancer, and repressor elements in addition to transcription and growth factors that influence these elements (68). −463G/A polymorphism at one of the upstream Alu MPO promoter elements has been linked to increased incidence of coronary artery disease in the general population (69, 70) and CVD incidence in CKD (71) and ESRD patients (72). This primate-specific promoter is competitively inhibited by estrogen (68) and contains binding site for nuclear transcription factors, which promote MPO expression, namely SP1-thyroid hormone–retinoic acid-response element (73), peroxisome proliferator-activated receptor α and γ, retinoid X receptors (74), statins (75), and the liver X receptor (76). This promotor is absent in mice, a finding that may partially explain the marked absence of MPO or MPO activity in the mouse model and the failure of MPO-deficient mice to ameliorate atherosclerosis. Repopulating the bone marrow of LDLr−/− mice with bone marrow from transgenic mice expressing human myeloperoxidase resulted in increased atherosclerosis (57). Another study, utilizing the overexpression of different human MPO allele polymorphisms in the promoter region, likewise demonstrated increased aortic lesions in male mice only, thus highlighting the role of estrogen in ameliorating MPO expression (77). Yet MPO polymorphism and its effect on MPO expression are much more complicated (78); although some studies implicate the GG phenotype with an increased risk of CVD (72, 78), other studies have not clearly replicated this finding (79, 80). Our model of CKD atherosclerosis is the first of its kind to demonstrate marked up-regulation of mouse MPO and its specific oxidation products in response to reduced renal function and HFD in murine atherosclerosis. Similar to our work, apolipoprotein E-deficient (apoE−/−) mice with chronic renal failure demonstrate enhanced atherosclerosis with increased macrophage infiltration and 3-nitrotyrosine expression in their atherosclerotic lesions, suggesting a similar process is plausible (81). However, this study did not specifically investigate whether MPO or MPO-specific oxidation products are elevated. It is unclear how reduced renal function influenced MPO expression in these mice who have such low baseline MPO expression with functioning kidneys. One possible reason is the prolonged exposure to HFD for 24 weeks in our model, in addition to other factors altered by decreased renal function. Our demonstration using the quantitative measurement of MPO oxidation products co-localizing with macrophage markers suggests that macrophage-derived MPO may propagate enhanced atherosclerosis in this model. The correlation between these oxidized moieties strongly implicates MPO as the source of all of these modifications.
MPO scavenges the reactive nitrogen species generated in the vascular wall to modulate vascular reactivity by consuming vasodilator nitric oxide. Nitric oxide can also combine with NADPH oxidase-generated superoxide (O2˙̄) to produce the reactive nitrogen species peroxynitrite (ONOO−), which has a very short half-life and causes nitration of proteins. In addition to decreased availability, nitric oxide production by itself could be the issue in this model as suggested by our earlier work demonstrating high levels of circulating methylated arginines that inhibit NO synthase (58). Thus, extensive atherosclerotic burden, modulation from the reactive nitrogen species, elevated methylated arginines, and nitric oxide consumption contribute to endothelial dysfunction that is associated with an increased risk of atherosclerosis disease in CKD (82). We report that this dysfunction is manifested as decreased vasodilation in response to cholinergic stimulation in CKD-HFD mice only. This is an interesting observation, as it clearly demonstrates the important interaction between diet and renal function necessary to induce endothelial dysfunction. Prior reports of endothelial dysfunction are limited to the double knockout of LDLr−/− with apoE−/− mice (83, 84). These mice, whether on HFD or not, fail to exhibit reduced cholinergic vasodilation (85–87). These results are consistent with our findings for CTL-LFD and CTL-HFD mice. Furthermore, although there are reports of 5/6 nephrectomy inducing a reduction in cholinergic responsiveness, such research seems to have been conducted exclusively in rats (88, 89). In these cases, the reduction in response was demonstrated with renal insufficiency but was associated with related hypertension and disruption in the angiotensin-dependent pathway. Clearly, our results show that reduction in cholinergic response is not dependent on kidney function alone but is rather a response to extensive atherosclerosis caused by the combination of decreased renal function and HFD in the absence of hypertension. Thus, our findings indicate that our model provides a foundation from which to explore the interaction of diet and renal function without the confounding influence of hypertension.
Our 5/6 nephrectomized LDLr−/− model achieved by surgical means, in contrast to uni-nephrectomy and 5/6 nephrectomy achieved via renal ablation, demonstrates sufficient and consistent loss of renal function to mimic moderate CKD as confirmed by creatinine and BUN measurements. As with humans, these CKD mice develop anemia and iPTH elevation, which confirms secondary hyperparathyroidism and effects consistent with an advanced degree of renal function. Although both LDLr−/− and apoE−/− mice are well established atherosclerotic models, LDLr−/− mice exhibit more modestly elevated LDL cholesterol levels compared with apoE−/− mice. Our study demonstrates the development of atherosclerosis with HFD exposure in LDLr−/− mice at 12 weeks that becomes more pronounced at 24 weeks. This elevated atherosclerotic plaque burden was observed despite reduced plasma cholesterol and triglycerides compared with controls on the same diet. The LDLr−/− CKD mice also show lower triglyceride and cholesterol levels in the VLDL and LDL fractions compared with controls irrespective of diet. These findings differ from the prior literature on other uremic atherosclerosis models that use both LDLr−/− and apoE−/− mice with an exaggerated aortic lesional area independent of hypertension but, unlike our observations, exhibit marked hypercholesterolemia compared with controls on the same diet (60, 90–94). In a study by Bro et al. (94), uremic apoE−/− mice that were fed a regular diet for 22 weeks had 50% higher total plasma cholesterol concentrations than normal apoE−/− mice. Plasma triglyceride concentrations did not differ between uremic and normal apoE−/− mice (81, 94). This is in contrast to our trend of decreased cholesterol and triglycerides in CKD LDLr−/− mice when compared with CTL mice regardless of diet. Despite decreased cholesterol and triglyceride levels in the VLDL and LDL fractions of CKD-HFD mice, these mice exhibited markedly accelerated atherosclerosis compared with CTL mice beginning at 12 weeks. Our CKD model is thus able to produce an increased atherosclerotic burden even in the absence of exaggerated hypercholesterolemia, hyperlipidemia, hypertension, and insulin resistance.
In our study, the aortic root and the abdominal aorta and its branches in CKD-HFD mice appear laden with a plaque that is more mature and thick, including stained sections that are more fibrotic and necrotic. Similar to our findings, Massy et al. (96) discovered that the relative proportion of atherosclerotic lesions to lesion-free vascular tissue is increased in the aortic root of uremic apoE−/− mice when compared with controls. Our model did not show any changes in serum calcium and phosphate levels but did indicate increased iPTH. Plaque composition in uremic apoE−/− mice demonstrates macrophage infiltration, increased cholesterol, collagen, and calcium content than classical atherosclerosis (96). Although we did not observe more than minimal changes in serum calcium and phosphate, this model and the apoE−/− model nonetheless exhibit significantly increased vascular calcification demonstrated in prior work by other groups (59, 60, 96–99).
In conclusion, our study demonstrates for the first time that increased MPO levels and activity co-localize with lesional macrophages in the artery wall in a mouse model of CKD atherosclerosis. In addition to decreased cholinergic response in the vessel, our work suggests that MPO expression and activity may play an important role in the propagation of atherosclerotic lesions in CKD mice. However, the key limitation of this work is that the evidence is associative and does not unequivocally demonstrate the causal role of macrophage-derived MPO in CKD-accelerated atherosclerosis. Studies examining the effects of MPO knockdown and overexpression in macrophages in chimeric CKD mice (using MPO knock-out and transgenic animals) are warranted to provide more definitive evidence on the role of macrophage-derived MPO in CKD-accelerated atherosclerosis.
Experimental procedures
Mouse model of CKD accelerated atherosclerosis
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals. Six-week-old male C57BL/6 LDLr−/− mice (The Jackson Laboratory, Bar Harbor, ME) were maintained with water ad libitum and on a standard rodent diet (Lab Diet® Hudson, NH) containing 28.5% protein, 13.5% fat, 58.0% carbohydrates by calories, and 200 ppm cholesterol. These mice were housed in a climate-controlled, light-regulated facility with a 12:12 h light/dark cycle. At age 7 weeks, the mice were subjected either to sham operation (CTL, n = 20) or to 5/6 nephrectomy by removing the whole right kidney in a first procedure followed by 2/3 left kidney by renal artery ligation (CKD, n = 21) after a week. At 9 weeks of age, the mice in each group were further randomly divided into two subgroups and fed on LFD containing 19.6% protein, 10.7% fat, and 69.7% carbohydrates by calories or HFD containing 19.5% protein, 40.5% fat, and 40.0% carbohydrates (Harlan Teklad Laboratory, Winfield, IA). The LFD contained 0% cholesterol, and the HFD contained 0.5% cholesterol by weight. The mice in each group, CTL-LFD, CTL-HFD, CKD-LFD, or CKD-HFD, were maintained on these diets for either 12 or 24 weeks. Evaluation of these mice was done in a blinded manner.
Murine systolic blood pressure was measured by the IITC Life Science blood pressure (tail cuff) system (Woodland Hills, CA) as described previously (100). Hematocrit was measured by CritSpin® Micro-Hematocrit centrifuge with Digital Hematocrit Reader (StatSpin® Co.). HgBA1c was measured by the Helena GLYCO-Tek Affinity column method (Beaumont, Texas). Plasma iPTH was measured by ELISA kit bought from ALPICO Diagnostics (Salem, NH).
Lipoprotein analysis
Plasma lipoprotein profiles were generated by FPLC (Bio-Rad) using two Superose 6 PC 3.2/30 columns in series (GE Healthcare) as discussed previously (95).
Determination of kidney function
Plasma creatinine levels were measured by highly-specific LC electron spray ionization and tandem MS (LC-ESI-MS/MS) as described previously (58). BUN was measured directly on IDExx Vettest 8008 Chemistry Analyzer (Westbrook, ME) using dry slide technology.
Atherosclerosis assessment
Each mouse was anesthetized and perfused with PBS through the left ventricle followed by 3 ml of 10% buffered formalin for fixing the vascular tree. The aortic tree was removed, microdissected to remove adventitial fat, cut longitudinally, stained with Oil Red O (Sigma) to visualize neutral lipids, and then pinned onto wax plates. The images of the ∼10-mm ascending and abdominal aorta were captured on a digital camera, and en face plaque quantification of total and lesional surface area was performed with computerized image analysis program (Image Pro software, Media Cybernetics, Bethesda) (56). The aortic lesion ratio is derived from the ratio of Oil Red O-stained area to the total surface area of en face section of the aortic tree expressed as a percentage. Aortic root cross-sections were cut from paraffin blocks of the aortic root with Leica RM 2155 Microtome and collected on glass slides. Sections were stained with H&E and Masson Trichrome for general tissue morphology and photographed with Olympus BX-51 microscope and DP-70 high resolution digital camera.
Immunohistochemistry
Immunohistochemistry was performed on paraffin sections with anti-mouse MPO antibody (1:500; Abcam, UK) and negative controls.
Immunofluorescence and confocal imaging
Snap-frozen aortic sections were incubated with rabbit antibodies for anti-mouse Mac-2 (macrophage marker; 1:500; Cedarlane, NC), 3-chlorotyrosine (1:1000; Cell Sciences, MA), 3-nitrotyrosine (1:50; Abcam, UK), and o,o′-dityrosine (1:200; Cosmo Bio, CA) at 4 °C overnight for immunofluorescence experiments. Appropriate negative controls were simultaneously processed to examine for background autofluorescence. Dual immunofluorescence labeling was simultaneously scanned by an Olympus FV500 confocal laser-scanning microscope, equipped with complete integrated image analysis software system (Olympus America Inc., Melville, NY).
Oxidized amino acid quantification by MS
Aortic samples were analyzed as outlined previously (27) using known concentrations of isotopically labeled internal standards [13C6]tyrosine, 3-[13C6] nitrotyrosine, o,o′-[13C12]dityrosine, or 3-[13C6]chlorotyrosine. Oxidized amino acids were quantified by LC-ESI-MS/MS with multiple reaction monitoring MS/MS-positive ion acquisition mode utilizing an Agilent 6410 triple quadrupole MS system equipped with an Agilent 1200 LC system. Labeled precursor amino acid, [13C9,15N1]tyrosine, was added to monitor potential internal artifact formation of 3-chlorotyrosine, 3-nitrotyrosine, and o,o′-dityrosine and was noted to be negligible.
Vascular reactivity experiments
Mouse aortic rings were mounted in a myograph system (Danish Myo Technology A/S, Aarhus, Denmark), and reactivity was measured as reported previously (100). Force was expressed as a percent of that achieved with 80% of maximal force with phenylephrine.
Statistical analysis
Results are presented as the mean ± S.D. Differences between the groups at different time periods were considered significant at p <0.05 using the ANOVA and Tukey-Kramer tests. For vascular reactivity studies, data were plotted using sigmoidal interpolation. Maximal steady-state vasodilation values were obtained using nonlinear regression and comparison between groups achieved by one-way ANOVA with Bonferroni post hoc analysis. All analyses were made using Graph Pad Prism 7.0 (La Jolla, CA).
Author contributions
L. Z., A. V. M., J. B., K. B. A., F. C. B., and S. P. data curation; L. Z., A. V. M., K. B. A., F. C. B., and S. P. formal analysis; L. Z., A. V. M., and S. P. visualization; L. Z., A. V. M., K. B. A., F. C. B., and S. P. writing-review and editing; A. V. M. writing-original draft; J. B. methodology; F. C. B. and S. P. project administration; S. P. conceptualization; S. P. supervision; S. P. funding acquisition.
Supplementary Material
This work was supported National Institutes of Health Grants P30DK081943 and P30DK089503 from NIDDK (to S. P.) and Grant K08HL130944 from NHLBI (to A. V. M.), and a grant from the Renal Research Institute (to K. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1, supporting Methods, and supporting Refs. 1–5.
- CKD
- chronic kidney disease
- MPO
- myeloperoxidase
- CVD
- cardiovascular disease
- CTL
- control
- LFD
- low-fat diet
- HFD
- high-fat, high-cholesterol diet
- LDL
- low-density lipoprotein
- VLDL
- very-low-density lipoprotein
- HDL
- high density lipoprotein
- LDLr−/−
- LDL receptor deficient mice
- apoE−/−
- apolipoprotein E-deficient mice
- ESRD
- end-stage renal disease
- BUN
- serum blood urea nitrogen
- iPTH
- intact parathyroid hormone
- H&E
- hematoxylin and eosin
- BSA
- bovine serum albumin
- LC-ESI-MS/MS
- LC electron spray ionization and tandem MS.
References
- 1. Go A. S., Chertow G. M., Fan D., McCulloch C. E., and Hsu C. Y. (2004) Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 351, 1296–1305 10.1056/NEJMoa041031 [DOI] [PubMed] [Google Scholar]
- 2. Foley R. N., Parfrey P. S., and Sarnak M. J. (1998) Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 32, S112–S119 10.1053/ajkd.1998.v32.pm9820470 [DOI] [PubMed] [Google Scholar]
- 3. Cheung A. K., Sarnak M. J., Yan G., Dwyer J. T., Heyka R. J., Rocco M. V., Teehan B. P., and Levey A. S. (2000) Atherosclerotic cardiovascular disease risks in chronic hemodialysis patients. Kidney Int. 58, 353–362 10.1046/j.1523-1755.2000.00173.x [DOI] [PubMed] [Google Scholar]
- 4. Sarnak M. J., Coronado B. E., Greene T., Wang S. R., Kusek J. W., Beck G. J., and Levey A. S. (2002) Cardiovascular disease risk factors in chronic renal insufficiency. Clin. Nephrol. 57, 327–335 10.5414/CNP57327 [DOI] [PubMed] [Google Scholar]
- 5. Sarnak M. J., Levey A. S., Schoolwerth A. C., Coresh J., Culleton B., Hamm L. L., McCullough P. A., Kasiske B. L., Kelepouris E., Klag M. J., Parfrey P., Pfeffer M., Raij L., Spinosa D. J., Wilson P. W., et al. (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 42, 1050–1065 10.1161/01.HYP.0000102971.85504.7c [DOI] [PubMed] [Google Scholar]
- 6. United States Renal Data System (USRDS) (2017) Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. NIDDK, National Institutes of Health, Bethesda, MD [Google Scholar]
- 7. Hostetter T. H. (2004) Chronic kidney disease predicts cardiovascular disease. N. Engl. J. Med. 351, 1344–1346 10.1056/NEJMe048236 [DOI] [PubMed] [Google Scholar]
- 8. Wattanakit K., Folsom A. R., Selvin E., Coresh J., Hirsch A. T., and Weatherley B. D. (2007) Kidney function and risk of peripheral arterial disease: results from the Atherosclerosis Risk in Communities (ARIC) Study. J. Am. Soc. Nephrol. 18, 629–636 10.1681/ASN.2005111204 [DOI] [PubMed] [Google Scholar]
- 9. Drüeke T. B., and Massy Z. A. (2010) Atherosclerosis in CKD: differences from the general population. Nat. Rev. Nephrol. 6, 723–735 10.1038/nrneph.2010.143 [DOI] [PubMed] [Google Scholar]
- 10. Penno G., Solini A., Bonora E., Fondelli C., Orsi E., Zerbini G., Trevisan R., Vedovato M., Gruden G., Cavalot F., Cignarelli M., Laviola L., Morano S., Nicolucci A., Pugliese G., et al. (2011) Clinical significance of nonalbuminuric renal impairment in type 2 diabetes. J. Hypertens. 29, 1802–1809 10.1097/HJH.0b013e3283495cd6 [DOI] [PubMed] [Google Scholar]
- 11. Kon V., Yang H., and Fazio S. (2015) Residual cardiovascular risk in chronic kidney disease: role of high-density lipoprotein. Arch. Med. Res. 46, 379–391 10.1016/j.arcmed.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Locatelli F., Canaud B., Eckardt K. U., Stenvinkel P., Wanner C., and Zoccali C. (2003) Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrol. Dial. Transplant. 18, 1272–1280 10.1093/ndt/gfg074 [DOI] [PubMed] [Google Scholar]
- 13. Guilgen G., Werneck M. L., de Noronha L., Martins A. P., Varela A. M., Nakao L. S., and Pecoits-Filho R. (2011) Increased calcification and protein nitration in arteries of chronic kidney disease patients. Blood Purif. 32, 296–302 10.1159/000330327 [DOI] [PubMed] [Google Scholar]
- 14. Vaziri N. D. (2004) Oxidative stress in uremia: Nature, mechanisms, and potential consequences. Semin. Nephrol. 24, 469–473 10.1016/j.semnephrol.2004.06.026 [DOI] [PubMed] [Google Scholar]
- 15. Vaziri N. D. (2004) Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr. Opin. Nephrol. Hypertens. 13, 93–99 10.1097/00041552-200401000-00013 [DOI] [PubMed] [Google Scholar]
- 16. Landmesser U., and Drexler H. (2003) Oxidative stress, the renin-angiotensin system, and atherosclerosis. Eur. Heart J. Suppl. 5, A3–A7 10.1016/S1520-765X(03)90057-5 [DOI] [Google Scholar]
- 17. Klebanoff S. J. (1980) Oxygen metabolism and the toxic properties of phagocytes. Ann. Intern. Med. 93, 480–489 10.7326/0003-4819-93-3-480 [DOI] [PubMed] [Google Scholar]
- 18. Ross R. (1999) Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340, 115–126 10.1056/NEJM199901143400207 [DOI] [PubMed] [Google Scholar]
- 19. Brown M. S., and Goldstein J. L. (1992) Koch's postulates for cholesterol. Cell 71, 187–188 10.1016/0092-8674(92)90346-E [DOI] [PubMed] [Google Scholar]
- 20. Goldstein J. L., Ho Y. K., Basu S. K., and Brown M. S. (1979) Binding site on macrophages that mediates uptake and degradation of acetylated low-density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. U.S.A. 76, 333–337 10.1073/pnas.76.1.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Witztum J. L., and Steinberg D. (1991) Role of oxidized low-density lipoprotein in atherogenesis. J. Clin. Invest. 88, 1785–1792 10.1172/JCI115499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinberg D. (2002) Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat. Med. 8, 1211–1217 10.1038/nm1102-1211 [DOI] [PubMed] [Google Scholar]
- 23. Haberland M. E., Fong D., and Cheng L. (1988) Malondialdehyde-altered protein occurs in atheroma of Watanabe heritable hyperlipidemic rabbits. Science 241, 215–218 10.1126/science.2455346 [DOI] [PubMed] [Google Scholar]
- 24. Ylä-Herttuala S., Palinski W., Butler S. W., Picard S., Steinberg D., and Witztum J. L. (1994) Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler. Thromb. 14, 32–40 10.1161/01.ATV.14.1.32 [DOI] [PubMed] [Google Scholar]
- 25. Ylä-Herttuala S., Palinski W., Rosenfeld M. E., Parthasarathy S., Carew T. E., Butler S., Witztum J. L., and Steinberg D. (1989) Evidence for the presence of oxidatively modified low-density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 84, 1086–1095 10.1172/JCI114271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klebanoff S. J., and Rosen H. (1978) The role of myeloperoxidase in the microbicidal activity of polymorphonuclear leukocytes. Ciba Found. Symp. 263–284 [DOI] [PubMed] [Google Scholar]
- 27. Vivekanandan-Giri A., Byun J., and Pennathur S. (2011) Quantitative analysis of amino acid oxidation markers by tandem mass spectrometry. Methods Enzymol. 491, 73–89 10.1016/B978-0-12-385928-0.00005-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hazen S. L., and Heinecke J. W. (1997) 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low-density lipoprotein isolated from human atherosclerotic intima. J. Clin. Invest. 99, 2075–2081 10.1172/JCI119379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eiserich J. P., Hristova M., Cross C. E., Jones A. D., Freeman B. A., Halliwell B., and van der Vliet A. (1998) Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 391, 393–397 10.1038/34923 [DOI] [PubMed] [Google Scholar]
- 30. McCormick M. L., Gaut J. P., Lin T. S., Britigan B. E., Buettner G. R., and Heinecke J. W. (1998) Electron paramagnetic resonance detection of free tyrosyl radical generated by myeloperoxidase, lactoperoxidase, and horseradish peroxidase. J. Biol. Chem. 273, 32030–32037 10.1074/jbc.273.48.32030 [DOI] [PubMed] [Google Scholar]
- 31. Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., Lusis A. J., Lee J. J., Lee N. A., Abu-Soud H. M., Ischiropoulos H., and Hazen S. L. (2002) A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277, 17415–17427 10.1074/jbc.M112400200 [DOI] [PubMed] [Google Scholar]
- 32. Abu-Soud H. M., and Hazen S. L. (2000) Nitric oxide is a physiological substrate for mammalian peroxidases. J. Biol. Chem. 275, 37524–37532 10.1074/jbc.275.48.37524 [DOI] [PubMed] [Google Scholar]
- 33. Daugherty A., Dunn J. L., Rateri D. L., and Heinecke J. W. (1994) Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94, 437–444 10.1172/JCI117342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hazell L. J., Arnold L., Flowers D., Waeg G., Malle E., and Stocker R. (1996) Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Invest. 97, 1535–1544 10.1172/JCI118576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leeuwenburgh C., Hardy M. M., Hazen S. L., Wagner P., Oh-ishi S., Steinbrecher U. P., and Heinecke J. W. (1997) Reactive nitrogen intermediates promote low-density lipoprotein oxidation in human atherosclerotic intima. J. Biol. Chem. 272, 1433–1436 10.1074/jbc.272.3.1433 [DOI] [PubMed] [Google Scholar]
- 36. Heller J. I., Crowley J. R., Hazen S. L., Salvay D. M., Wagner P., Pennathur S., and Heinecke J. W. (2000) p-Hydroxyphenylacetaldehyde, an aldehyde generated by myeloperoxidase, modifies phospholipid amino groups of low-density lipoprotein in human atherosclerotic intima. J. Biol. Chem. 275, 9957–9962 10.1074/jbc.275.14.9957 [DOI] [PubMed] [Google Scholar]
- 37. Brennan M. L., Penn M. S., Van Lente F., Nambi V., Shishehbor M. H., Aviles R. J., Goormastic M., Pepoy M. L., McErlean E. S., Topol E. J., Nissen S. E., and Hazen S. L. (2003) Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 349, 1595–1604 10.1056/NEJMoa035003 [DOI] [PubMed] [Google Scholar]
- 38. Pennathur S., Wagner J. D., Leeuwenburgh C., Litwak K. N., and Heinecke J. W. (2001) A hydroxyl radical-like species oxidizes cynomolgus monkey artery wall proteins in early diabetic vascular disease. J. Clin. Invest. 107, 853–860 10.1172/JCI11194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baldus S., Eiserich J. P., Brennan M. L., Jackson R. M., Alexander C. B., and Freeman B. A. (2002) Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic. Biol. Med. 33, 1010 10.1016/S0891-5849(02)00993-0 [DOI] [PubMed] [Google Scholar]
- 40. Düzgünçinar O., Yavuz B., Hazirolan T., Deniz A., Tokgözoğlu S. L., Akata D., and Demirpençe E. (2008) Plasma myeloperoxidase is related to the severity of coronary artery disease. Acta Cardiol. 63, 147–152 10.2143/AC.63.2.2029520 [DOI] [PubMed] [Google Scholar]
- 41. Shishehbor M. H., Aviles R. J., Brennan M. L., Fu X., Goormastic M., Pearce G. L., Gokce N., Keaney J. F. Jr., Penn M. S., Sprecher D. L., Vita J. A., and Hazen S. L. (2003) Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 289, 1675–1680 10.1001/jama.289.13.1675 [DOI] [PubMed] [Google Scholar]
- 42. Shishehbor M. H., Brennan M. L., Aviles R. J., Fu X., Penn M. S., Sprecher D. L., and Hazen S. L. (2003) Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 108, 426–431 10.1161/01.CIR.0000080895.05158.8B [DOI] [PubMed] [Google Scholar]
- 43. Shishehbor M. H., and Hazen S. L. (2004) Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr. Atheroscler. Rep. 6, 243–250 10.1007/s11883-004-0038-1 [DOI] [PubMed] [Google Scholar]
- 44. Vivekanandan-Giri A., Slocum J. L., Byun J., Tang C., Sands R. L., Gillespie B. W., Heinecke J. W., Saran R., Kaplan M. J., and Pennathur S. (2013) High density lipoprotein is targeted for oxidation by myeloperoxidase in rheumatoid arthritis. Ann. Rheum. Dis. 72, 1725–1731 10.1136/annrheumdis-2012-202033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith C. K., Vivekanandan-Giri A., Tang C., Knight J. S., Mathew A., Padilla R. L., Gillespie B. W., Carmona-Rivera C., Liu X., Subramanian V., Hasni S., Thompson P. R., Heinecke J. W., Saran R., Pennathur S., and Kaplan M. J. (2014) Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 66, 2532–2544 10.1002/art.38703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Z., Nicholls S. J., Rodriguez E. R., Kummu O., Hörkkö S., Barnard J., Reynolds W. F., Topol E. J., DiDonato J. A., and Hazen S. L. (2007) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 13, 1176–1184 10.1038/nm1637 [DOI] [PubMed] [Google Scholar]
- 47. Jaisson S., Pietrement C., and Gillery P. (2011) Carbamylation-derived products: bioactive compounds and potential biomarkers in chronic renal failure and atherosclerosis. Clin. Chem. 57, 1499–1505 10.1373/clinchem.2011.163188 [DOI] [PubMed] [Google Scholar]
- 48. Tang W. H., Shrestha K., Wang Z., Borowski A. G., Troughton R. W., Klein A. L., and Hazen S. L. (2013) Protein carbamylation in chronic systolic heart failure: relationship with renal impairment and adverse long-term outcomes. J. Card. Fail. 19, 219–224 10.1016/j.cardfail.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koeth R. A., Kalantar-Zadeh K., Wang Z., Fu X., Tang W. H., and Hazen S. L. (2013) Protein carbamylation predicts mortality in ESRD. J. Am. Soc. Nephrol. 24, 853–861 10.1681/ASN.2012030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang A. Y., Lam C. W., Chan I. H., Wang M., Lui S. F., and Sanderson J. E. (2010) Prognostic value of plasma myeloperoxidase in ESRD patients. Am. J. Kidney Dis. 56, 937–946 10.1053/j.ajkd.2010.05.008 [DOI] [PubMed] [Google Scholar]
- 51. Kisic B., Miric D., Dragojevic I., Rasic J., and Popovic L. (2016) Role of myeloperoxidase in patients with chronic kidney disease. Oxid. Med. Cell. Longev. 2016, 1069743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bansal V., Cunanan J., Hoppensteadt D., Jeske W., and Fareed J. (2007) Hemodialysis mediated upregulation of myeloperoxidase in end-stage renal disease: pathophysiologic implications. FASEB J. 21, A438–A439 [Google Scholar]
- 53. Madhusudhana Rao A., Anand U., and Anand C. V. (2011) Myeloperoxidase in chronic kidney disease. Indian J. Clin. Biochem. 26, 28–31 10.1007/s12291-010-0075-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Afshinnia F., Zeng L., Byun J., Gadegbeku C. A., Magnone M. C., Whatling C., Valastro B., Kretzler M., Pennathur S., and Michigan Kidney Translational Core, CPROBE Investigator Group. (2017) Myeloperoxidase levels and its product 3-chlorotyrosine predict chronic kidney disease severity and associated coronary artery disease. Am. J. Nephrol. 46, 73–81 10.1159/000477766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pennathur S., Jackson-Lewis V., Przedborski S., and Heinecke J. W. (1999) Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o′-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J. Biol. Chem. 274, 34621–34628 10.1074/jbc.274.49.34621 [DOI] [PubMed] [Google Scholar]
- 56. Brennan M.-L., Anderson M. M., Shih D. M., Qu X.-D., Wang X., Mehta A. C., Lim L. L., Shi W., Hazen S. L., Jacob J. S., Crowley J. R., Heinecke J. W., and Lusis A. J. (2001) Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Invest. 107, 419–430 10.1172/JCI8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McMillen T. S., Heinecke J. W., and LeBoeuf R. C. (2005) Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation 111, 2798–2804 10.1161/CIRCULATIONAHA.104.516278 [DOI] [PubMed] [Google Scholar]
- 58. Mathew A. V., Zeng L., Byun J., and Pennathur S. (2015) Metabolomic profiling of arginine metabolome links altered methylation to chronic kidney disease–accelerated atherosclerosis. J. Proteomics Bioinform. Suppl. 14, pii 001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davies M. R., and Hruska K. A. (2001) Pathophysiological mechanisms of vascular calcification in end-stage renal disease. Kidney Int. 60, 472–479 10.1046/j.1523-1755.2001.060002472.x [DOI] [PubMed] [Google Scholar]
- 60. Davies M. R., Lund R. J., and Hruska K. A. (2003) BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J. Am. Soc. Nephrol. 14, 1559–1567 10.1097/01.ASN.0000068404.57780.DD [DOI] [PubMed] [Google Scholar]
- 61. Bisgaard L. S., Bosteen M. H., Fink L. N., Sørensen C. M., Rosendahl A., Mogensen C. K., Rasmussen S. E., Rolin B., Nielsen L. B., and Pedersen T. X. (2016) Liraglutide reduces both atherosclerosis and kidney inflammation in moderately uremic LDLr−/− mice. PLoS ONE 11, e0168396 10.1371/journal.pone.0168396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pennathur S., Vivekanandan-Giri A., Locy M. L., Kulkarni T., Zhi D., Zeng L., Byun J., de Andrade J. A., and Thannickal V. J. (2016) Oxidative modifications of protein tyrosyl residues are increased in plasma of human subjects with interstitial lung disease. Am. J. Respir. Crit. Care Med. 193, 861–868 10.1164/rccm.201505-0992OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leeuwenburgh C., Rasmussen J. E., Hsu F. F., Mueller D. M., Pennathur S., and Heinecke J. W. (1997) Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low-density lipoprotein isolated from human atherosclerotic plaques. J. Biol. Chem. 272, 3520–3526 10.1074/jbc.272.6.3520 [DOI] [PubMed] [Google Scholar]
- 64. Zheng L., Nukuna B., Brennan M. L., Sun M., Goormastic M., Settle M., Schmitt D., Fu X., Thomson L., Fox P. L., Ischiropoulos H., Smith J. D., Kinter M., and Hazen S. L. (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Invest. 114, 529–541 10.1172/JCI200421109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zalba G., Fortuño A., and Díez J. (2006) Oxidative stress and atherosclerosis in early chronic kidney disease. Nephrol. Dial. Transplant. 21, 2686–2690 10.1093/ndt/gfl398 [DOI] [PubMed] [Google Scholar]
- 66. Gluba-Brzózka A., Michalska-Kasiczak M., Franczyk B., Nocuń M., Toth P., Banach M., and Rysz J. (2016) Markers of increased atherosclerotic risk in patients with chronic kidney disease: a preliminary study. Lipids Health Dis. 15, 22 10.1186/s12944-016-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kon V., Linton M. F., and Fazio S. (2011) Atherosclerosis in chronic kidney disease: the role of macrophages. Nat. Rev. Nephrol. 7, 45–54 10.1038/nrneph.2010.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chumakov A. M., Chumakova E. A., Chih D., and Koeffler H. P. (2000) Molecular analysis of the human myeloperoxidase promoter region. Int. J. Oncol. 16, 401–411 [PubMed] [Google Scholar]
- 69. Asselbergs F. W., Reynolds W. F., Cohen-Tervaert J. W., Jessurun G. A., and Tio R. A. (2004) Myeloperoxidase polymorphism related to cardiovascular events in coronary artery disease. Am. J. Med. 116, 429–430 10.1016/j.amjmed.2003.10.025 [DOI] [PubMed] [Google Scholar]
- 70. Nikpoor B., Turecki G., Fournier C., Théroux P., and Rouleau G. A. (2001) A functional myeloperoxidase polymorphic variant is associated with coronary artery disease in French-Canadians. Am. Heart J. 142, 336–339 10.1067/mhj.2001.116769 [DOI] [PubMed] [Google Scholar]
- 71. Grahl D. A., Axelsson J., Nordfors L., Heimburger O., Bárány P., Gao Y. Z., Qureshi A. R., Kato S., Watanabe M., Suliman M., Riella M. C., Lindholm B., Stenvinkel P., and Pecoits-Filho R. (2007) Associations between the CYBA 242C/T and the MPO −463G/A polymorphisms, oxidative stress and cardiovascular disease in chronic kidney disease patients. Blood Purif. 25, 210–218 10.1159/000100419 [DOI] [PubMed] [Google Scholar]
- 72. Pecoits-Filho R., Stenvinkel P., Marchlewska A., Heimburger O., Barany P., Hoff C. M., Holmes C. J., Suliman M., Lindholm B., Schalling M., and Nordfors L. (2003) A functional variant of the myeloperoxidase gene is associated with cardiovascular disease in end-stage renal disease patients. Kidney Int. Suppl. 2003, 172–176 [DOI] [PubMed] [Google Scholar]
- 73. Piedrafita F. J., Molander R. B., Vansant G., Orlova E. A., Pfahl M., and Reynolds W. F. (1996) An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 271, 14412–14420 10.1074/jbc.271.24.14412 [DOI] [PubMed] [Google Scholar]
- 74. Kumar A. P., Piedrafita F. J., and Reynolds W. F. (2004) Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the −463GA promoter polymorphism. J. Biol. Chem. 279, 8300–8315 10.1074/jbc.M311625200 [DOI] [PubMed] [Google Scholar]
- 75. Kumar A. P., and Reynolds W. F. (2005) Statins downregulate myeloperoxidase gene expression in macrophages. Biochem. Biophys. Res. Commun. 331, 442–451 10.1016/j.bbrc.2005.03.204 [DOI] [PubMed] [Google Scholar]
- 76. Reynolds W. F., Kumar A. P., and Piedrafita F. J. (2006) The human myeloperoxidase gene is regulated by LXR and PPARα ligands. Biochem. Biophys. Res. Commun. 349, 846–854 10.1016/j.bbrc.2006.08.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Castellani L. W., Chang J. J., Wang X., Lusis A. J., and Reynolds W. F. (2006) Transgenic mice express human MPO −463G/A alleles at atherosclerotic lesions, developing hyperlipidemia and obesity in −463G males. J. Lipid Res. 47, 1366–1377 10.1194/jlr.M600005-JLR200 [DOI] [PubMed] [Google Scholar]
- 78. Cayley W. E., Jr. (2004) Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 350, 516–518 10.1056/NEJM200401293500519 [DOI] [PubMed] [Google Scholar]
- 79. Doi K., Noiri E., Maeda R., Nakao A., Kobayashi S., Tokunaga K., and Fujita T. (2007) Functional polymorphism of the myeloperoxidase gene in hypertensive nephrosclerosis dialysis patients. Hypertens. Res. 30, 1193–1198 10.1291/hypres.30.1193 [DOI] [PubMed] [Google Scholar]
- 80. Bouali H., Nietert P., Nowling T. M., Pandey J., Dooley M. A., Cooper G., Harley J., Kamen D. L., Oates J., and Gilkeson G. (2007) Association of the G-463A myeloperoxidase gene polymorphism with renal disease in African Americans with systemic lupus erythematosus. J. Rheumatol. 34, 2028–2034 [PMC free article] [PubMed] [Google Scholar]
- 81. Bro S., Bentzon J. F., Falk E., Andersen C. B., Olgaard K., and Nielsen L. B. (2003) Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J. Am. Soc. Nephrol. 14, 2466–2474 10.1097/01.ASN.0000088024.72216.2E [DOI] [PubMed] [Google Scholar]
- 82. Malyszko J. (2010) Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta 411, 1412–1420 10.1016/j.cca.2010.06.019 [DOI] [PubMed] [Google Scholar]
- 83. Yamamoto Y., Yamashita T., Kitagawa F., Sakamoto K., Giddings J. C., and Yamamoto J. (2010) The effect of the long term aspirin administration on the progress of atherosclerosis in apoE−/− LDLR−/− double knockout mouse. Thromb. Res. 125, 246–252 10.1016/j.thromres.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 84. Csányi G., Gajda M., Franczyk-Zarow M., Kostogrys R., Gwoźdź P., Mateuszuk L., Sternak M., Wojcik L., Zalewska T., Walski M., and Chlopicki S. (2012) Functional alterations in endothelial NO, PGI(2) and EDHF pathways in aorta in ApoE/LDLR−/− mice. Prostaglandins Other Lipid Mediat. 98, 107–115 10.1016/j.prostaglandins.2012.02.002 [DOI] [PubMed] [Google Scholar]
- 85. Wölfle S. E., and de Wit C. (2005) Intact endothelium-dependent dilation and conducted responses in resistance vessels of hypercholesterolemic mice in vivo. J. Vasc. Res. 42, 475–482 10.1159/000088101 [DOI] [PubMed] [Google Scholar]
- 86. Ketonen J., and Mervaala E. (2008) Effects of dietary sodium on reactive oxygen species formation and endothelial dysfunction in low-density lipoprotein receptor-deficient mice on high-fat diet. Heart Vessels 23, 420–429 10.1007/s00380-008-1066-5 [DOI] [PubMed] [Google Scholar]
- 87. Meyrelles S. S., Peotta V. A., Pereira T. M., and Vasquez E. C. (2011) Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. 10, 211 10.1186/1476-511X-10-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wu-Wong J. R., Li X., and Chen Y. W. (2015) Different vitamin D receptor agonists exhibit differential effects on endothelial function and aortic gene expression in 5/6 nephrectomized rats. J. Steroid Biochem. Mol. Biol. 148, 202–209 10.1016/j.jsbmb.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 89. Eräranta A., Törmänen S., Kööbi P., Vehmas T. I., Lakkisto P., Tikkanen I., Moilanen E., Niemelä O., Mustonen J., and Pörsti I. (2014) Phosphate binding reduces aortic angiotensin-converting enzyme and enhances nitric oxide bioactivity in experimental renal insufficiency. Am. J. Nephrol. 39, 400–408 10.1159/000362507 [DOI] [PubMed] [Google Scholar]
- 90. Bro S., Borup R., Andersen C. B., Moeller F., Olgaard K., and Nielsen L. B. (2006) Uremia-specific effects in the arterial media during development of uremic atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 26, 570–575 [DOI] [PubMed] [Google Scholar]
- 91. Bro S., Binder C. J., Witztum J. L., Olgaard K., and Nielsen L. B. (2007) Inhibition of the renin-angiotensin system abolishes the proatherogenic effect of uremia in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 1080–1086 10.1161/ATVBAHA.107.139634 [DOI] [PubMed] [Google Scholar]
- 92. Bro S., Flyvbjerg A., Binder C. J., Bang C. A., Denner L., Olgaard K., and Nielsen L. B. (2008) A neutralizing antibody against receptor for advanced glycation end products (RAGE) reduces atherosclerosis in uremic mice. Atherosclerosis 201, 274–280 10.1016/j.atherosclerosis.2008.01.015 [DOI] [PubMed] [Google Scholar]
- 93. Bro S., Bollano E., Brüel A., Olgaard K., and Nielsen L. B. (2008) Cardiac structure and function in a mouse model of uraemia without hypertension. Scand. J. Clin. Lab. Invest. 68, 660–666 10.1080/00365510802037272 [DOI] [PubMed] [Google Scholar]
- 94. Bro S. (2009) Cardiovascular effects of uremia in apolipoprotein E-deficient mice. Dan. Med. Bull. 56, 177–192 [PubMed] [Google Scholar]
- 95. Gerdes L. U., Gerdes C., Klausen I. C., and Faergeman O. (1992) Generation of analytic plasma lipoprotein profiles using two prepacked Superose 6B columns. Clin. Chim. Acta 205, 1–9 10.1016/0009-8981(92)90348-T [DOI] [PubMed] [Google Scholar]
- 96. Massy Z. A., Ivanovski O., Nguyen-Khoa T., Angulo J., Szumilak D., Mothu N., Phan O., Daudon M., Lacour B., Drüeke T. B., and Muntzel M. S. (2005) Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J. Am. Soc. Nephrol. 16, 109–116 [DOI] [PubMed] [Google Scholar]
- 97. Phan O., Ivanovski O., Nguyen-Khoa T., Mothu N., Angulo J., Westenfeld R., Ketteler M., Meert N., Maizel J., Nikolov I. G., Vanholder R., Lacour B., Drüeke T. B., and Massy Z. A. (2005) Sevelamer prevents uremia-enhanced atherosclerosis progression in apolipoprotein E-deficient mice. Circulation 112, 2875–2882 10.1161/CIRCULATIONAHA105.541854 [DOI] [PubMed] [Google Scholar]
- 98. Phan O., Ivanovski O., Nikolov I. G., Joki N., Maizel J., Louvet L., Chasseraud M., Nguyen-Khoa T., Lacour B., Drüeke T. B., and Massy Z. A. (2008) Effect of oral calcium carbonate on aortic calcification in apolipoprotein E-deficient (apoE−/−) mice with chronic renal failure. Nephrol. Dial. Transplant. 23, 82–90 [DOI] [PubMed] [Google Scholar]
- 99. Shobeiri N., Adams M. A., and Holden R. M. (2010) Vascular calcification in animal models of CKD: a review. Am. J. Nephrol. 31, 471–481 10.1159/000299794 [DOI] [PubMed] [Google Scholar]
- 100. Atkins K. B., Seki Y., Saha J., Eichinger F., Charron M. J., and Brosius F. C. (2015) Maintenance of GLUT4 expression in smooth muscle prevents hypertension-induced changes in vascular reactivity. Physiol. Rep. 3, e12299 10.14814/phy2.12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.