

Abstract
Members of the bromodomain and extra-terminal domain (BET) family of proteins (bromodomain-containing (BRD) 2, 3, 4, and T) are widely expressed and highly conserved regulators of gene expression in eukaryotes. These proteins have been intimately linked to human disease, and more than a dozen clinical trials are currently underway to test BET-protein inhibitors as modulators of cancer. However, although it is clear that these proteins use their bromodomains to bind both histones and transcription factors bearing acetylated lysine residues, the molecular mechanisms by which BET family proteins regulate gene expression are not well defined. In particular, the functions of the other domains such as the ET domain have been less extensively studied. Here, we examine the properties of the ET domain of BRD3 as a protein/protein interaction module. Using a combination of pulldown and biophysical assays, we demonstrate that BRD3 binds to a range of chromatin-remodeling complexes, including the NuRD, BAF, and INO80 complexes, via a short linear “KIKL” motif in one of the complex subunits. NMR-based structural analysis revealed that, surprisingly, this mode of interaction is shared by the AF9 and ENL transcriptional coregulators that contain an acetyl-lysine-binding YEATS domain and regulate transcriptional elongation. This observation establishes a functional commonality between these two families of cancer-related transcriptional regulators. In summary, our data provide insight into the mechanisms by which BET family proteins might link chromatin acetylation to transcriptional outcomes and uncover an unexpected functional similarity between BET and YEATS family proteins.
Keywords: transcriptional coactivator, acetylation, nuclear magnetic resonance (NMR), protein structure, protein/protein interaction, AF9, BET family proteins, bromodomain, extraterminal domain
Introduction
The bromodomain and extra-terminal domain (BET)3 family of proteins, namely BRD2,- 3, -4 and -T, is defined by an architecture consisting of two N-terminal bromodomains and an ∼80-residue extra-terminal (ET) domain (Fig. 1E) (1). BET bromodomains specifically recognize ϵ-acetylated lysines, with targets including histone H3 acetylated at Lys-14 and histone H4 bearing either single or multiple acetylation modifications at Lys-5/Lys-8/Lys-12/Lys-15 (2–4). Transcription factors such as GATA-1, TWIST, RelA, and STAT3 are also established targets (5–8). These interactions are postulated to form the basis for recruitment of chromatin-modifying effectors to promoters and enhancers, thus underpinning the functional readout of histone and transcription factor acetylation.
Figure 1.

BRD3 can bind to a large number of gene regulatory proteins. A, reducing SDS-PAGE showing proteins pulled down from HEK293 nuclear extract by FLAG-Brd3 immobilized on FLAG-Sepharose beads. The control comprised beads alone. The gel was cut into the four indicated sections (right), and the proteins contained in each section were identified by MS. B, coimmunoprecipitation showing the ability of FLAG-Brd3 to pull CHD4 and MTA2 from HEK293 cells. Western blottings were carried out with the indicated antibodies. C, coimmunoprecipitation showing the ability of FLAG-Brd3 to pull BRG1 and BAF170 from HEK293 cells. Western blots were carried out with the indicated antibodies. D, coimmunoprecipitations mapping the BAF-binding domain of Brd3. Western blots were carried out with the indicated antibodies. E, domain architecture of BET proteins. Residue numbering is based on human proteins (Uniprot IDs: BRD2, P25440; BRD3, Q15059; BRD4, O60885; and BRDT, Q58F21). BD, bromodomain; ET, extra-terminal domain; CTD, C-terminal domain. Regions of BRD2, BRD3, and BRD4 previously identified to interact with full-length CHD4 (46), as well as BRD3 constructs used in this study, are also shown.
BET proteins play important roles in the regulation of gene transcription (9–11). In mammals, BRD2/4 are essential for normal development; homozygous deletion of either gene in mice is embryonic lethal (12–14). BRD2 and BRD3 (but not BRD4) are highly enriched at erythroid gene promoters where their direct interaction with the transcription factor GATA1 regulates the expression of many erythroid genes (15, 16). Furthermore, BRD2 and to a lesser degree BRD3 are found at CTCF-occupied sites where they contribute to genome organization and chromatin boundary function (17). As well as being targeted by several viruses during infection (18–20), BET proteins have also emerged as key players in oncogenesis. Fusions of BRD3/4 with the protein Nuclear in Testis give rise to NUT midline carcinoma, an aggressive malignancy, and BRD4 has been shown to occupy super-enhancer regions that drive oncogenic transcription programs in a range of cancers (21–25). As a consequence, BRD4 in particular has become a major target for the design of cancer therapeutics that inhibit the acetyl-lysine–binding properties of its bromodomains (26–28).
Although many studies have focused on the acetyl-lysine–binding specificity of BET bromodomains and on the design of small-molecule inhibitors of these domains, the roles of other parts of these proteins, and the biochemical mechanisms by which BET proteins actually regulate gene expression, are less well understood. For example, although the functional importance of BRD2/3 in erythropoiesis is clear and it is known that the direct interaction between GATA1 and BRD2/3 is important for GATA1 chromatin occupancy (6, 15), the biochemical mechanism underlying these effects is not well understood.
The ET domain of BRD4 has been shown to be the cellular target of the viral proteins Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen (KSHV–LANA), and the murine leukemia virus integrase (MLV-IN) (29–31). Recently, NMR structures of the BRD4 ET domain in complex with peptides derived from these viral proteins have been reported, as has the structure of a complex formed between BRD4 ET and a peptide from the histone methyltransferase NSD3 (32, 33). Overall, however, the role played by the ET domain in normal biology remains relatively under-explored.
The remodeling of chromatin, an essential aspect of genomic function, is achieved by large, multisubunit protein complexes that contain ATP-dependent DNA translocase domains (34). Several classes of these chromatin-remodeling complexes have been defined in mammals, including (i) the Nucleosome Remodeling and Deacetylation (NuRD) complex, in which CHD3, CHD4, or CHD5 is the ATP-driven DNA translocase (35, 36); (ii) the BAF and PBAF complexes, for which remodeling is driven by BRG1 or BRM (37, 38); and (iii) INO80 family complexes, which are involved in DNA damage repair and the exchange of variant histones such as H2AX into nucleosomes (39, 40).
The mechanisms by which these machines are directed to specific genomic loci are areas of active research. For example, the NuRD complex has been shown to colocalize with GATA-1 during erythroid differentiation and to interact directly with FOG-1 (41–43), which is recruited to these sites through its zinc finger-mediated interaction with GATA-1 (44, 45). Intriguingly, the NuRD subunit CHD4 was among a number of cellular interaction partners identified in pulldowns that used a BRD4 fragment containing the ET domain as bait (46). This result raises the possibility that the BRD3 ET domain might also interact with CHD4 and potentially contribute to recruitment or retention of the NuRD complex to GATA-1 sites during hematopoiesis.
In an effort to better define the biochemical mechanisms by which BET family proteins regulate gene expression, we searched for binding partners of BRD3. Among partners that we identified were multiple members of the NuRD, BAF, and INO80 chromatin-remodeling complexes. Biochemical and sequence analysis led us to demonstrate that the ET domain of BRD3 was sufficient to mediate direct interactions with the CHD4, BRG1, and INO80B components of the respective complexes. In each case BRD3-ET recognized a short linear motif comprising a KIKL-like sequence. We determined three-dimensional structures of BRD3-ET bound separately to a CHD4 and a BRG1 KIKL peptide, demonstrating that the ET domain recognizes a wide range of chromatin-remodeling complexes via a conserved mechanism. Analysis of the structures also reveals that, strikingly, the architecture of the interaction is conserved in another class of transcriptional regulators: the AF9 and ENL YEATS-domain proteins that form part of the superelongation complex (SEC) and are prominent MLL fusion partners in a subset of acute leukemias (47). Our analysis provides general insight into the molecular mechanisms by which BET family proteins might regulate gene expression and points to a remarkable functional similarity between BET and YEATS family proteins.
Results
Immobilized BRD3 can capture a number of gene-regulatory complexes
To delineate the mechanisms by which BRD3 regulates gene expression, we immobilized full-length murine FLAG-tagged Brd3 on anti-FLAG-Sepharose beads and treated the beads with nuclear extract from HEK293 cells (Fig. 1A). Among the 752 proteins retained by Brd3 that could be identified by MS, multiple members of several multiprotein complexes associated with gene regulation were observed. These complexes include NuRD, SWI/SNF, INO80, TFIID, TFIIH, Mediator, and the PAF complex, as well as a range of elongation, splicing, and DNA replication factors (Table 1 and Tables S1–S5). Of these, two of the proteins detected with the largest number of peptides were the chromatin-remodeling enzymes CHD4 and BRG1. An interaction between CHD4 and a C-terminal fragment of both BRD3 and BRD4 (that includes the ET domain) had previously been detected in a similar pulldown experiment (46).
Table 1.
Components of NuRD, BAF, and INO80 complexes identified in the Brd3 pulldown experiment
The numbers of peptides identified for each protein in the Brd3-loaded beads and the beads alone are indicated. IP, immunoprecipitation.
| Protein | No. of peptides identified |
|
|---|---|---|
| FLAG-Brd3 IP | Control IP | |
| NuRD complex | ||
| CHD4 | 77 | 2 |
| GATAD2A | 6 | 0 |
| GATAD2B | 7 | 2 |
| HDAC1 | 6 | 3 |
| HDAC2 | 6 | 2 |
| MBD3 | 9 | 1 |
| MTA1 | 4 | 0 |
| MTA2 | 15 | 0 |
| RBBP4 | 6 | 2 |
| RBBP7 | 1 | 0 |
| BAF complex | ||
| BAF200 | 5 | 0 |
| BAF180 | 40 | 0 |
| BAF170 | 18 | 0 |
| BAF155 | 21 | 1 |
| BAF60B | 3 | 0 |
| BAF57 | 6 | 0 |
| BAF53A | 17 | 1 |
| BAF47 | 8 | 0 |
| BCL7A | 4 | 0 |
| BCL7C | 4 | 0 |
| BRD7 | 4 | 0 |
| BRD9 | 10 | 0 |
| BRG1 | 44 | 0 |
| BRM | 4 | 0 |
| β-Actin | 3 | 0 |
| GLTSCR1 | 29 | 0 |
| INO80 complex | ||
| ACTR5 | 8 | 0 |
| ACTR8 | 5 | 0 |
| INO80 | 8 | 0 |
| INO80B | 4 | 0 |
| INO80C | 3 | 0 |
| INO80E | 5 | 0 |
| NFRKB | 5 | 0 |
To corroborate these interactions, we transfected HEK293 cells with a FLAG-Brd3 construct, captured the FLAG-Brd3 protein on anti-FLAG-Sepharose beads, ran it on SDS-PAGE, and probed the gel with antibodies against components of the NuRD complex (CHD4 and MTA2, Fig. 1B) or the BAF complex (BRG1 and BAF170, Fig. 1C). In both cases, the two complex subunits that were queried were clearly detected. To assess which part of Brd3 was responsible for interacting with the BAF complex, the experiment was repeated using Brd3 deletion constructs. Fig. 1D indicates that the ET domain is essential for the Brd3/BAF complex interaction.
BRD3-binding motif of CHD4 maps to a disordered region
It was shown previously that BRD4(608–671), which encompasses the ET domain but not the bromodomains or C-terminal domain (Fig. 1E), can interact with full-length CHD4 in pulldown assays (46). The same study showed that a larger region of BRD3 (residues 570–726) can likewise bind CHD4. Following these findings, we first mapped the BRD3-interacting domain of CHD4 using pulldown assays. When full-length BRD3 was coexpressed in HEK293 cells with the N-terminal, middle, or C-terminal sections of CHD4 (Fig. 2A), only the N-terminal third (residues 1–355) was able to robustly bind BRD3 (Fig. 2B). This portion of the protein contains an HMG-box–like domain (48) and is otherwise predicted to be disordered (by programs such as DISOPRED3 (49)). We next made a series of bacterially expressed GST-tagged CHD4 fragments focused on the N-terminal third of the protein (Fig. 2A), and we used these as baits to pull down HA-tagged BRD3-L expressed in mammalian cells (Fig. 2C). These pulldowns indicated that CHD4-Ne (residues 265–310), a region predicted to be largely disordered, was both necessary and sufficient for interaction with BRD3.
Figure 2.

BRD3 binds to a short motif in the N-terminal disordered region of CHD4. A, human CHD4 constructs used in this study. Domains with known structures are indicated. The presence or absence of an interaction with BRD3 is indicated by + or −, respectively. CD = chromodomain. B, BRD3 coimmunoprecipitates with the N-terminal third of CHD4. BRD3-HA (full-length, L or BD) and CHD4-FLAG (full-length, NT, M, or CT) constructs were coexpressed in HEK293 cells and applied to anti-FLAG-agarose beads. Western blottings using anti-HA or anti-FLAG antibodies are shown. C, CHD4(265–310) (fragment Ne) binds BRD3. Bacterially expressed CHD4 fragments immobilized on GSH beads were used to pull down mammalian-expressed HA-BRD3-L. Each Western blotting was visualized using α-HA and α-GST antibodies. PD = pulldown; FT = flow-through; * = unidentified band.
Sequence similarity with the BRD4 ET domain, together with the published interaction data (46), suggested that BRD3-ET alone might also interact with CHD4. To ascertain whether BRD3-ET was sufficient for interaction with CHD4, we turned to NMR spectroscopy. 15N HSQC spectra of BRD3-L and BRD3-ET are shown in Fig. 3A. The majority of peaks in the spectrum of BRD3-L (Fig. 3A, black) appear as intense, overlapping signals with amide proton shifts in the 8.0–8.6 ppm region of the spectrum, a pattern characteristic of the disordered polypeptide. A number of weaker and more dispersed signals are also observed. In contrast, peaks in the 15N HSQC of BRD3-ET (Fig. 3A, red) are well-dispersed, as expected of a small, folded protein domain. An overlay of the two 15N HSQC spectra reveals that almost all signals in the BRD3-ET spectrum are essentially unchanged in the BRD3-L spectrum, indicating that the folded ET domain takes up the same conformation in the context of the longer polypeptide.
Figure 3.

BRD3-ET binds CHD4(265–310). 15N HSQC spectra were acquired in 20 mm sodium phosphate, pH 6.5, 50 mm NaCl, 1 mm DTT at 298 K. A, 15N HSQC spectra of BRD3-L (black) and BRD3-ET (red). Most of BRD3-L outside of the core ET domain appears to be disordered. B, titration of 15N-BRD3-L with CHD4-Ne. Arrows indicate large chemical shift changes induced by CHD4-Ne. Boxed regions are shown in detail in D. C, 15N HSQC spectra of 15N-BRD3-ET alone and in the presence of 2 m eq of CHD4-Ne. Boxed regions are shown in detail in D. D, comparison of CHD4-induced peak movements in BRD3-L and BRD3-ET, showing that BRD3-ET and BRD-L bind CHD4 in a comparable manner.
Titration of 15N BRD3-L with CHD4-Ne does not appear to induce a significant disorder-to-order transition (Fig. 3B). Instead, only the signals attributable to BRD3-ET are affected. Furthermore, the pattern of intensity changes suggests the formation of a complex under an intermediate–slow exchange regime. To confirm that BRD3-ET is sufficient to mediate the interaction with CHD4, a titration was conducted with 15N-labeled BRD3-ET and unlabeled CHD4-Ne (Fig. 3, C and D). The CHD4-induced changes in peak positions were almost exactly reproduced in BRD3-ET (Fig. 3D), indicating that the ET domain is likely to be the minimal interaction domain for CHD4.
BRD3-ET interacts with a conserved linear motif found associated with many classes of chromatin-remodeling complexes
The 15N HSQC spectrum of free CHD4-Ne was partially assigned using a combination of homonuclear 2D spectra (1H,1H NOESY and TOCSY) and a 15N-separated NOESY. Because of extensive overlap of lysine resonances, complete assignment of CHD4-Ne amide resonances was not possible; however, a number of peaks affected by titration with BRD3-ET were able to be identified (Figs. 4A and 5). Comparison of CHD4-Ne with a 23-residue MLV-IN peptide known to bind the BRD4 ET domain (50) revealed a highly related “PLKI(K/R)L” sequence (Fig. 4A) in the vicinity of the BRD3-affected peaks, suggesting that this might constitute the core ET-binding motif. We therefore made a synthetic peptide corresponding to residues 290–301 of CHD4 (CHD4-Ng, Fig. 2A, Table 2), which contains the hypothesized ET-binding site. To verify that CHD4-Ng binds BRD3-ET in the same manner as the longer, recombinant CHD4-Ne peptide, we carried out a 15N HSQC titration of BRD3-ET with CHD4-Ng (Fig. 4B). The chemical shift perturbations induced by CHD4-Ne and CHD4-Ng in BRD3-ET are very similar (Fig. S1), indicating that CHD4-Ng is sufficient to recapitulate the interaction of BRD3-ET with CHD4-Ne. A fit of the titration data to an intermediate chemical exchange model, using an approach previously described (51), yielded a KD estimate of ∼2 μm. We also used surface plasmon resonance to measure this affinity, which yielded a KD of 95 ± 10 μm (Fig. 5A, Table 2). The SPR and NMR were carried out at 4 and 25 °C, respectively, which could be the source of this difference.
Figure 4.

BRD3-ET can interact in a conserved manner with motifs from a range of transcriptional coregulators. A, sequence alignment of CHD-Ne with a peptide from MLV-IN that is known to bind the ET domain of Brd4 (50), as well as related sequences found in INO80B, BRG1, NSD3, GLTSCR1, and TAF7. A region of high similarity (representing a potential ET recognition motif) is boxed. Residues that display the largest chemical shift perturbations in the presence of BRD3-ET are indicated by arrows. B, partial 15N HSQC spectra of BRD3-ET alone (red) and titrated with peptides from (top to bottom) CHD4, BRG1, INO80B, NSD3, and MLV-IN. The number of molar equivalents of each peptide added are indicated in each spectrum. C, chemical shift changes induced in BRD3 ET domain at saturation with CHD4, INO80B, and NSD3. The horizontal red line indicates 1 S.D. above mean chemical shift change. Peaks no longer visible at the titration end point are indicated by vertical dashed lines. Residues that are identical or highly similar between CHD4 and MLV integrase are indicated by asterisks and a colon, respectively.
Figure 5.
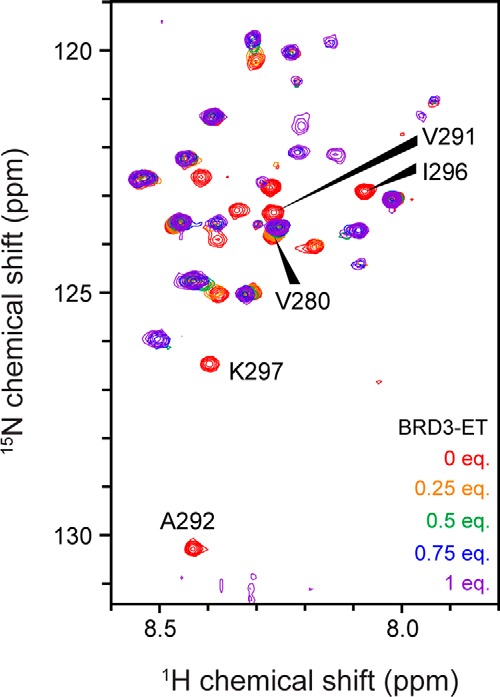
15N HSQC titration of 15N-labeled CHD4-Ne with BRD3-ET. 200 μm 15N CHD4-Ne was titrated with BRD3-ET in 20 mm sodium phosphate, pH 6.5, 50 mm NaCl, 1 mm DTT at 298 K. Several peaks that undergo large chemical shift changes are labeled with their assignments.
Table 2.
Synthetic peptides used in 15N HSQC titrations together with affinities determined by SPR
Because the PLKIKL sequence in both CHD4-Ng and MLV-IN was sufficient to bind BRD3-ET, we hypothesized that it might represent a common motif by which the ET domain interacts with partner proteins. Indeed, this motif is present in both NSD3 and GLTSCR1 (Fig. 4A), which along with CHD4 have been previously identified as potential ET domain interaction partners in a pulldown-MS screen (46). Interrogation of the human proteome using SCANSITE3 (scansite3.mit.edu (52)) revealed instances of this motif in a number of other putative BET interaction partners, including INO80B, a subunit of the INO80 complex involved in chromatin remodeling and DNA damage repair (53) and BRG1 (SMARCA4), a member of the SWI/SNF chromatin remodeling complex that has been previously found in complexes containing BRD2 (Fig. 4A) (54). For INO80B, BRG1, and NSD3, the motif was located in a region of the protein that is likely to be disordered based on disorder predictions.
To assess whether the mode of interaction is conserved between different ET-domain–binding partners, synthetic peptides derived from NSD3, BRG1, and INO80B were tested in 15N HSQC titrations (Fig. 4B and Table 2); a titration was also carried out with an MLV-IN peptide for comparison. Fig. 4C shows that the chemical perturbation shift patterns induced in BRD3-ET by each peptide are very similar, indicating that the same binding pocket on BRD3-ET mediates all of these interactions. The interaction of BRD3-ET with NSD3 appeared to be the weakest, based on the larger excess of NSD3 peptide required to reach saturation, the magnitude of the chemical shift changes induced by NSD3, and the fact that it occurs in a faster chemical exchange regime. Surface plasmon resonance measurements (Fig. 6) yielded a KD of 7 ± 1 μm for the ET/BRG1 interaction, 80 ± 10 μm for ET/INO80B, and 950 ± 100 μm for ET/NSD3.
Figure 6.

SPR-derived binding affinities for interactions between BRD3-ET and KIKL peptides. Sensograms (upper panels) and fits to equilibrium responses from the sensograms (lower panels) are shown for BRD3-ET binding to CHD4 (A), BRG1 (B), INO80B (C), and NSD3 (D) peptides. Data were fitted to a simple Langmuir 1:1 binding isotherm in the Biacore software. Measurements were made in a Biacore T200 at 4 °C in a buffer containing 20 mm HEPES and 150 mm NaCl, pH 7.5.
Three-dimensional structures of BRD3–CHD4 and BRD3–BRG1 complexes reveal a conserved interaction mode
We next determined the three-dimensional structure of the BRD3–CHD4 complex by NMR methods. Samples of the 1:1 complex were generated by titrating 15N/13C-labeled BRD3-ET to a slight excess of CHD4-Ng (∼1:1.1), and a range of standard 2D and 3D NMR spectra was recorded. A total of 1324 NOEs, which included 49 intermolecular NOEs, were obtained, and the structure is shown in Fig. 7, A–C. The geometry of the structure is good, with >99% of residues in the ordered region lying in allowed regions of the Ramachandran plot. The fold of BRD3-ET consists of three α-helices, designated α1 (residues 574–586), α2 (residues 589–602), and α3 (residues 623–637), consistent with the structure of the free BRD4 ET domain (1). The CHD4 peptide binds in a groove that is formed by residues from the α1 helix and the α2–α3 loop of BRD3-ET (Fig. 7, B and C), resulting in a buried surface area of ∼1150 Å2. A short antiparallel β-sheet is formed between CHD4 Ile-296–Leu-298 and Ile-614–Ile-616 in the α2–α3 loop of BRD3, as inferred from CO–N distances.
Figure 7.

Solution NMR structure of the BRD3-ET–CHD4 and BRD3-ET–BRG1 complexes. A, ribbon representation of the ensemble of 20 lowest energy structures for the BRD3-ET–CHD4-Ng complex. N and C termini are labeled. B, surface representation of BRD3-ET with CHD4-Ng shown as sticks. Only ordered CHD4 residues are shown and labeled. BRD3 residues with CHD4-induced chemical shift changes of >1 standard deviation above the mean are indicated in yellow. C, surface representation showing the electrostatic properties of the binding pocket. BRD3-ET residues are labeled. The electrostatic complementarity is clear. D–F, representations of the BRD3-ET·BRG1 structure, as for A–C. BRG1 is shown in magenta.
BRD3 residues with large CHD4-induced chemical shift perturbations (Fig. 4C) are shown mapped onto the structure of the complex in Fig. 7B. Hydrophobic residues (Leu-294, Ile-296, and Leu-298) alternate with charged residues (Lys-295 and Lys-297) in the CHD4 peptide, and this arrangement is mirrored by residues on the BRD3 α2–α3 loop. Leu-294 interacts with the aromatic ring of Phe-618; Ile-296 contacts Ile-616; and Leu-298 packs between Ile-614 and Leu-592 (in the α2 helix). A network of electrostatic interactions is also apparent: the side-chain amine of Lys-297 lies adjacent to the carboxyl groups of Glu-613/Glu-615 and that of Lys-295 contacts Glu-615/Asp-617 (Fig. 7C). Other residues that do not directly contact the peptide also display large chemical shift changes, which is likely due to the large movement in the α2–α3 loop and helix α3 induced by peptide binding. These residues include Val-595 and Val-596 on the α2 helix, which pack against the binding-pocket residues Leu-592 and Ile-614, respectively, and residues Arg-607–Asn-610, which form a small 310 helix at the N terminus of the α2–α3 loop that may contribute to stabilization of the loop conformation in the peptide-bound state.
To ascertain whether this interaction is important for mediating an interaction between full-length CHD4 and BRD3, we expressed human CHD4 (either WT or a mutant in which the Lys-297–Leu-298 sequence was mutated to AA) in HEK293 cells and applied cell lysate from these cells to GST–BRD3-ET bound to GSH-Sepharose beads. As shown in Fig. 8, binding of the mutant to BRD3-ET was significantly attenuated.
Figure 8.
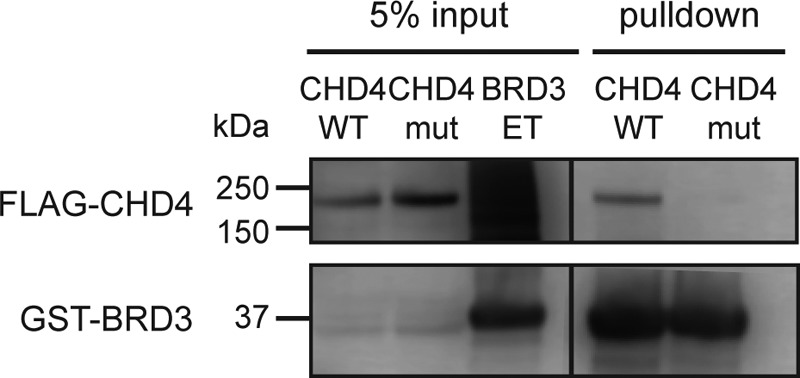
KIKL motif in CHD4 is important for the BRD3/CHD4 interaction. Bacterially-expressed GST–BRD3-ET immobilized on GSH beads was used to pull down mammalian-expressed full-length FLAG-CHD4 (WT or K297A/L298A mutant). Western blots using anti-GST and anti-FLAG antibodies are shown.
Finally, we used the same NMR-based approach to determine the three-dimensional structure of the BRD3–BRG1 complex. A total of 873 NOEs, including 71 intermolecular NOEs, were used to derive the structure shown in Fig. 7, D–F. Line broadening resulted in fewer NOEs than observed for the BRD3–CHD4 complex, and consequently a higher root mean square deviation for the overlay of the 20 conformers used to represent the structure (Table 3). Overall, however, the structure and binding mode are essentially the same as for the BRD3–CHD4 complex, with the three hydrophobic residues Val-1595, Ile-1597, and Leu-1599 packing against BRD3-ET and the three alternating residues Lys-1594, Lys-1596, and Lys-1598 making contacts with the acidic surface of the binding pocket.
Table 3.
Structural statistics for BRD3-ET complexes with CHD4 and BRG1
| BRD3·CHD4 | BRD3·BRG1 | |
|---|---|---|
| NOE-based distance restraints | ||
| Total | 1324 | 873 |
| Intraresidue (i, i) | 337 | 248 |
| Sequential (i, i + 1) | 359 | 253 |
| Medium-range (2 ≤ |i − j| ≤4) | 352 | 196 |
| Long-range (|i − j| >4) | 276 | 176 |
| Total dihedral restraints | 136 | 144 |
| Root mean square deviation from lowest energy structurea | ||
| All backbone atoms (N, Cα, C′) (Å) | 0.8 | 1.0 |
| All heavy atoms (N, C, O, S) (Å) | 1.5 | 1.5 |
| PROCHECK Ramachandran statistics | ||
| Residues in most favored regions (%) | 95.5 | 91.7 |
| Residues in additionally allowed regions (%) | 4.1 | 7.2 |
| Residues in generously allowed regions (%) | 0.3 | 0.9 |
| Residues in disallowed regions (%) | 0.2 | 0.2 |
| Deviations from idealized geometry | ||
| Bond lengths (Å) | 0.014 | 0.015 |
| Bond angles (°) | 1.3 | 1.6 |
a Deviations are over residues 295–298 and 570–637.
Discussion
ET domains are highly conserved
During our work, structures of the ET domain of BRD4 bound to peptides from the MLV-IN (33), the transcriptional coregulator NSD3 (residues 152–163 and 593–605) (32), and the KSHV–LANA protein (32) were reported. The sequences of human BRD2-, BRD3-, and BRD4-ET are conserved (>85% identity and only one conservative substitution on the peptide-binding surface; Fig. S2A), and a comparison of the structures of BRD3-ET–CHD4, BRD3-ET–BRG1, BRD4-ET–NSD3, and BRD4-ET–LANA shows that the interaction mode is also conserved (Fig. 9). All of the interactions occur in the same peptide-binding pocket of the ET domain and involve the formation of an intermolecular two-stranded β-sheet that positions alternating hydrophobic and charged residues in the peptide to contact the ET domain. In contrast, the MLV-IN peptide is longer and folds back onto itself to form a third antiparallel β-strand in the complex with BRD4-ET. Interestingly, our data show that an MLV-IN peptide containing only the core motif can bind BRD3-ET, albeit with significantly lower affinity than that reported for the BRD4/MLV-IN interaction (160 nm). Furthermore, the similarity of chemical shift perturbation data for CHD4-Ne and the shorter CHD4-Ng confirm that, for CHD4, the shorter peptide contains the full ET-binding motif.
Figure 9.
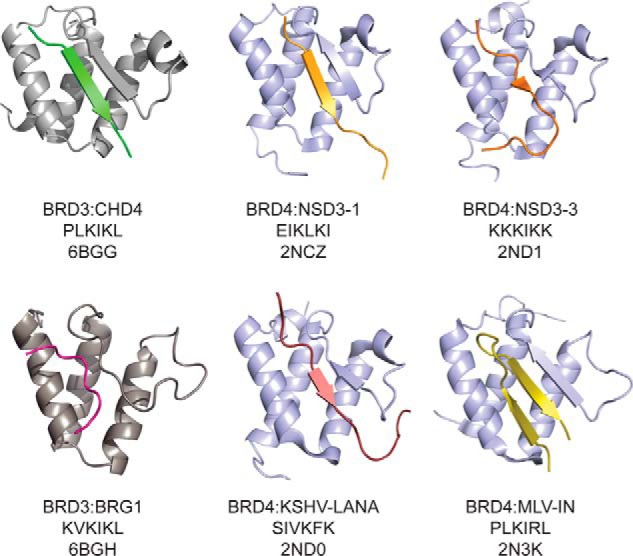
Comparison of structures of ET domain complexes. ET-binding peptides, as well as the ET domain of BRD3 (gray) or Brd4 (blue), are shown as ribbons. The BET protein, core peptide sequence, and PDB code are indicated below the structures.
In addition to the high conservation of ET domains among human BET proteins, high similarity is observed throughout evolution. For example, only one conservative substitution distinguishes human BRD3-ET from the corresponding domain in Xenopus laevis. Furthermore, biochemical data (55) and sequence analysis indicate that the yeast BET protein Bdf1 interacts with the TAF7 component of the TFIID complex via a similar binding mechanism that involves Bdf1-ET and an LKLKN motif in TAF7 (Fig. 4A).
This high sequence similarity throughout evolution argues for a critical role for ET/partner interactions in BET protein function. In contrast, the high degree of identity between family members within a species suggests that a degree of functional overlap is likely to exist between BRD2, -3, and -4, given that they have an overlapping pattern of expression across mammalian tissues.
However, genetic evidence suggests that BET proteins perform both redundant as well as nonredundant roles (10, 16, 56); whether the ET domain contributes to discrimination between interaction partners has yet to be determined. This study, together with published data (33, 57), indicates that different partners display a range of affinities for the BRD3 and BRD4 ET domains. In addition, there are residues in the α2–α3 loop of the ET domain (namely Arg-607–Asn-610 in BRD3) that exhibit large chemical shift changes upon peptide binding but are not conserved between BET family members (Fig. S2A), and these might conceivably influence binding affinity and/or specificity. Outside of the bromodomains and ET domain, BET proteins display lower sequence conservation, and these regions might play a greater role in delineating paralogue-specific in vivo interactions of each family member.
Conservation of ET-binding motifs in transcriptional regulators
The ET-interacting peptide motif is relatively short and therefore is likely to occur by chance in many proteins, especially large proteins such as CHD4. Conservation of putative binding sites, both between paralogs and across species, might be of use in distinguishing those that are functionally relevant. For example, at least one additional potential binding site for the ET domain has been identified in both NSD3 (KKKIKK, residues 596–601) and CHD4 (LKKVKL, residues 689–694) (32). However, only the first ET-binding site (EIKLKI, residues 152–157) is fully conserved between NSD2 and NSD3, with partial conservation observed in NSD1 (Fig. S2E). Consistent with this observation, mutation of the first NSD3 site has been shown to abolish the NSD3/ET interaction in pulldown experiments, whereas mutation of the second site has little effect (32). Similarly, the first site in CHD4 (PLKIKL, residues 293–298) is conserved in human and mouse CHD3/4 (Fig. S2B), both of which can participate in formation of the NuRD complex. In contrast, the second site is not conserved between any of the nine members of the CHD family of proteins in humans. Therefore, it might be predicted that the ET domain interacts with mammalian CHD3/4 primarily through the first site. The Drosophila homolog of CHD4, dMi-2β, also displays the first ET-binding site (Fig. S2B), which we predict will interact with the ET domain of the sole BET protein fs(1)h in Drosophila melanogaster. The ability of any KIKL sequence to interact with an ET domain will also depend on sequence context due to issues of folding and steric accessibility.
A counterpart for the BRG1 (SMARCA4) ET-binding site also exists in the BRG1 paralog BRM (SMARCA2) (Fig. S2C); both can serve as the ATPase subunit in the mammalian SWI/SNF remodeling complexes, consistent with the hypothesis that the ET domain plays a central role in the recruitment of ATP-dependent chromatin–remodeling complexes to gene-regulatory elements. The INO80B site is conserved from human to frog (Fig. S2D).
Curiously, BRD4 itself also contains a potential ET-binding site within the disordered C-terminal domain (DLKIKN, residues 1193–1198). Intramolecular interaction between the second BRD4 bromodomain and an adjacent phosphorylated control region has previously been proposed to regulate its interaction with acetylated chromatin (58), and it is possible that additional autoregulation might arise through this ET/CTD interaction.
AF9/ENL leukemia-associated proteins display unexpected structural and functional commonality with BET proteins
A BLAST search of the human proteome using BRD3-ET as a query does not return any hits other than BRD4-ET. However, a structure-based search using DALI (59) reveals that the ET domain fold closely resembles the structure of the ANC1 homology domain (AHD) of AF9 (Fig. 10A) (60); the two domains share 25% sequence identity (Fig. 10B, top), suggesting the idea that they are descended from a common ancestor. AF9 and its close paralog ENL (which also contains an AHD) are common translocation partners of MLL in human leukemia (61). Unexpectedly, the AF9-AHD is intrinsically disordered in isolation (62) but takes up an ET-like fold upon interacting with peptides from AF4 (62), CBX8 (PDB code 2N4Q),4 or DOT1L (60). AF4 and DOT1L (an H3K79 methyltransferase) are components of complexes associated with transcriptional elongation (the super-elongation (63) and Dot1 (47) complexes, respectively).
Figure 10.

ET interaction mechanism is conserved in AF9/ENL family proteins. A, comparison of BRD3–CHD4 (left) and AF9–DOT1L (right) complexes showing the ET/AHD as a surface and the partner peptide as cartoon/stick representation. B, top, amino acid sequences of the ET and AHD domains of human BRD3 and AF9, respectively. Identical residues are shown in orange and similar residues in yellow. The sequence that forms a β-sheet with the partner is underlined. Bottom, alignment of the ET-binding regions of CHD4 and BRG1 with the corresponding regions from the AHD-binding proteins CBX8, AF4, and BCoR. Sequences from the human proteins are shown in all cases. C, comparison of the interface residues of ET/AHD domains (left) and of the partner peptides (right); strong conservation is apparent in the former case, whereas the alternating pattern of hydrophobic and basic residues observed in ET-binding sequences is not as strongly conserved in AHD-binding sequences.
In a remarkable recapitulation of the ET/peptide interaction mechanism, the AF9/partner interactions are mediated by the formation of a short antiparallel β-sheet between the α2–α3 loop of AF9 and an amphipathic peptide from AF4/CBX8/DOT1L (Fig. 10A). There is a clear similarity between the peptide-binding sites of AHD and ET domains (Fig. 10, A and C, bottom), although the alternation between charged and hydrophobic residues (Fig. 10, B and C) is less distinct for the AHD-binding motifs than for the ET-binding sequences.
Similar to the BET proteins, AF9/ENL harbor an N-terminal domain that recognizes acetylated lysine side chains, in this case a YEATS domain (64, 65), rather than the twin bromodomains of the BET family. This architectural similarity gives rise to clear functional commonalities between the two protein families. Both BRD4 and ENL can read the histone (or transcription factor) acetylation status of target genes and promote transcriptional elongation. Both are also essential for the proliferation of a subset of aggressive leukemias (24, 66, 67); inhibition of their activity impairs proliferation through selective repression of the transcriptional program initiated by the oncoprotein c-Myc (24, 66, 67).
At the biochemical level, the signal encoded in the acetylated lysine residue(s) is interpreted by the respective epigenetic reader domains (YEATS and bromodomain) and then translated into a downstream action by the ET/AHD domain recognizing a short, linear motif in specified partner proteins. It is at this point that the activities of the two classes of proteins appear to diverge, despite utilizing the same structural mechanism for signal transmission. Thus, targets of the AF9/ENL AHD include the lysine methyltransferase DOT1L, which is the only enzyme that methylates histone H3K79. In contrast, targets of the ET domain are dominated by chromatin remodelers, including CHD4, CHD8 (indirectly through the interaction of ET with a short form of NSD3 (68)), BRG1, and the INO80 complex subunit INO80B. Thus the ET/peptide interactions represent a conserved regulatory mechanism across (at least) these two families of gene-regulatory proteins.
From an evolutionary perspective, a search of the protein sequence database using PHMMER (69) reveals that ET-related sequences are found very widely throughout Eukaryota, including in deep-branching species such as the plankton Emiliania huxleyi, the ciliate Tetrahymena thermophila, and the oomycete (water mold) Phytophthora infestans (where they are found alongside bromodomains). In contrast, AHD-related sequences appear to be restricted to Metazoa (animals, from Homo sapiens to the sponge Amphimedon queenslandica). The most parsimonious explanation for this observation is that the intrinsically disordered AHD domain has evolved from an ancestral ET domain. The selective advantage afforded by the intrinsically disordered nature of the AHD (if any) is currently not clear, but it might involve an ability to rapidly exchange binding partners (62), even high-affinity ones.
Functional implications of ET interactions with transcriptional coregulators
The first BRD3 bromodomain has been shown to bind to acetylated GATA-1, and this interaction has been proposed to stabilize chromatin occupancy of GATA-1, possibly through the simultaneous binding of the second bromodomain to acetylated histones (15). The NuRD complex, of which CHD4 is a component, is also suggested to be recruited to promoters of GATA-1 target genes via Friend of GATA-1 (FOG-1), which interacts with the RBBP4/7 and MTA1/2 subunits of NuRD (42, 70). The interaction between the BRD3 ET domain and CHD4 suggests that BRD3 may play an additional role in the recruitment or retention of the NuRD complex at GATA-1 sites. It is possible that both the BRD3/CHD4 and FOG-1/RBBP4 interactions are required for the functional recruitment of NuRD, because RBBP4/7 can also participate in other regulatory complexes (71), and yet only the NuRD complex is recruited to GATA-1-dependent promoters. An analogous arrangement might also exist for BRD4, which is also able to bind the acetylated transcription factor RelA (7). The ubiquitous expression pattern of BRD2/3/4 suggests that recognition of acetylated transcription factors by BET–protein bromodomains, followed by ET-mediated interactions with KIKL motifs, might be a general mechanism through which chromatin remodeling complexes are recruited to promoters/enhancers.
Given that recruitment of transcriptional coregulators by the ET and AHD domains appears to play an important role in oncogenesis, targeting the ET domain might represent an alternative approach in the design of therapeutics for BET-dependent malignancies. The high degree of conservation in the ET domain might, like the bromodomains, present a challenge in terms of designing small molecules that selectively inhibit individual BET family members. Nevertheless, the unusual dependence of malignant cells on BET-occupied enhancers suggests that disrupting the ET-mediated interactions of BET proteins with transcriptional regulators that drive oncogenic expression programs might be a viable therapeutic strategy (72). In this regard, our findings provide fundamental insight into the mechanisms by which BET proteins link chromatin acetylation to transcriptional outcomes in normal physiology and in human disease.
Materials and methods
Constructs
All constructs described in this work are derived from human gene sequences unless otherwise specified. The full-length murine Brd3 coding sequence was cloned into the pEF1α vector with a single FLAG tag (DYKDDDDK) at the N terminus. Brd3 deletion mutants were amplified by PCR with a single FLAG tag at the N terminus and inserted into pEF1α. BRD3-L (residues 420–726) was cloned into pcDNA3.1 with a C-terminal hemagglutinin (HA) tag (YPYDVPDYA) and both BRD3-L and BRD3-ET (577–644) were cloned into pGEX-6P (for bacterial expression as GST-fusion proteins) using BamHI/EcoRI sites. CHD4 constructs were cloned either into pcDNA3.1 (with an N-terminal FLAG tag or HA tag) or into pGEX-6P. CHD4 KL-AA (K297A/L298A) was generated by Gibson assembly of synthetic gene fragments.
Mammalian cell culture
For pulldowns in Fig. 1, HEK-293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 2% penicillin/streptomycin, 1% glutamine, and 1% sodium pyruvate. For other experiments, HEK293FT cells were cultured (at 37 °C in 5% CO2) in Dulbecco's modified Eagle's medium (Gibco) containing 10% (v/v) fetal bovine serum, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin. Cultures were grown to 70–80% confluency and then incubated in fresh medium without antibiotics for 4–6 h prior to transfection. Cells were transfected with plasmid DNA (∼17 μg) using polyethyleneimine in a 3:1 mass ratio, and growth was continued for 24–48 h before harvesting the cells.
Protein overexpression in E. coli
Escherichia coli Rosetta (DE3) pLysS cells transformed with a CHD4 or BRD3 encoding plasmid were cultured at 37 °C with shaking. Log-phase cultures (OD 0.4–0.8) were induced by addition of isopropyl β-d-thiogalactopyranoside (IPTG, 0.2–0.4 mm), and cultured for a further 16–24 h at 25 °C. For preparation of isotopically-labeled protein, log-phase cells were washed in M9 minimal media salts and equilibrated for 1 h at 25 °C in minimal media containing 15NH4Cl and (in the case of 15N/13C-labeled samples) d-[13C]glucose. The cultures were then induced with IPTG and protein expressed as described above. For production of 15N/13C-labeled BRD3-ET, cultures were grown completely in minimal media containing 15NH4Cl and d-[13C]glucose over 12–14 h at 37 °C prior to induction.
Protein purification
Cell pellets containing overexpressed GST–BRD3-ET and GST–BRD3-L were lysed in lysis buffer (50 mm Tris, pH 7.3, 500 mm NaCl, 100 μm MgCl2, 1 mm DTT, 0.5 μm PMSF, 25 μg ml−1 DNase I) and purified using GSH-affinity chromatography. The N-terminal GST tag was removed by incubation of GSH elution fractions with HRV-3C protease (produced in-house) at 4 °C. Recombinant protein was separated from GST and other contaminants using column chromatography. BRD3-L was purified by ion exchange on a UNOTM S1 column (Bio-Rad) in 50 mm Tris, pH 7.5, 50–1000 mm NaCl, 1 mm DTT. BRD3-ET was purified using size-exclusion chromatography on a Superdex® 75 preparative grade HiLoad 16/60 column (GE Healthcare) in 50 mm Tris, pH 7.3, 150 mm NaCl, 1 mm DTT. Separation of proteins was monitored using UV absorbance at 215 and/or 280 nm using a BioLogic QuadTec UV-visible detector (Bio-Rad) and SDS-PAGE.
Preparation of nuclear extracts
Cells were scraped from the plate in cold PBS. After centrifugation, the cell pellet was lysed in Buffer A (10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, protease inhibitor mixture (Roche Applied Science), 1 mm PMSF, 1 mm DTT) on ice for 30 min. After vigorous vortexing, nuclei were pelleted by centrifugation and lysed in Buffer C (20 mm HEPES-KOH, pH 7.9, 25% glycerol, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, protease inhibitor mixture, 1 mm PMSF, 1 mm DTT) on ice for 30 min. Material was then spun at maximum speed in a tabletop centrifuge for 10 min at 4 °C. The resulting supernatant was diluted in Buffer C lacking NaCl to reduce the salt concentration to 150 mm before being used for immunoprecipitations.
Immunoprecipitations
Immunoprecipitations were performed with anti-FLAG beads (Sigma). After extensive washing (150 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0.5% Igepal), elution was carried out with 3× FLAG peptide (Sigma), and bound proteins were run on SDS-PAGE and analyzed by Western blotting or MS. For MS, SDS-polyacrylamide gels were stained with Colloidal Blue and cut into four segments: >100, 95 to 50, 50 to 25, and <25 kDa. Mass spectrometry was performed at the Taplin Facility at Harvard University.
GST pulldown assays
Clarified cell lysates from 0.5 to 1 ml of E. coli Rosetta2 cells expressing GST-CHD4 proteins were incubated with GSH-Sepharose® 4B beads for 30 min at 4 °C. The beads were subsequently washed with 5 column volumes of lysis buffer, followed by equivalent washing into pulldown buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 1 mm DTT, 1× cOmplete EDTA-free protease inhibitor mixture). 1 ml of HEK293FT cells expressing HA-BRD3-L was lysed by incubation in pulldown buffer for 30 min at 4 °C. Clarified HEK293FT cell lysate was incubated with 20 μl of GST-CHD4 beads for 1 h at 4 °C. The beads were subsequently washed with 5 column volumes of pulldown buffer. Protein retained on the beads and 10% input of HEK293FT cell lysate were analyzed by SDS-PAGE followed by Western blotting.
Antibodies
Antibodies used in this study were as follows: BAF155 (A301-021A, Bethyl), BAF170 (sc-17838, Santa Cruz Biotechnology), BRG1 (sc-10768, Santa Cruz Biotechnology), CHD4 (sc-11378, Santa Cruz Biotechnology), FLAG (F1804, Sigma), and MTA2 (sc-9447, Santa Cruz Biotechnology).
Handling of synthetic peptides
Peptides derived from murine leukemia virus integrase (MLV-IN; residues 1191–1202) and from human CHD4 (residues 290–301), INO80B (residues 67–78), BRG1 (residues 1591–1602), and NSD3 (residues 149–160) were purchased as N-terminally acetylated and C-terminally amidated peptides at >80% purity from ChinaPeptides (Table 2). The lyophilized peptides were resuspended in NMR buffer (20 mm sodium phosphate, pH 6.5, 50 mm NaCl, 1 mm DTT) and the peptide solutions adjusted to pH 6.5 before making up the volume to a final concentration of 5 mm. Concentrations were determined either by mass or by UV absorbance at 205 nm (73).
Surface plasmon resonance
SPR was performed on a Biacore T200 with a buffer comprising 20 mm HEPES and 150 mm NaCl, pH 7.5, as both the immobilization and running buffer. The active and reference flow cells of a Biacore T200 CM5 series S sensor chip (GE Healthcare) were activated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and N-hydroxysuccinimide at 25 °C according to the manufacturer's instructions. BRD3-ET (100 μg ml−1) in 10 mm sodium acetate, pH 4, was then injected for 15 min at 10 μl min−1, and the flow cells then blocked with an injection of 1 m ethanolamine. Following protein immobilization, the system was cooled to 4 °C. Duplicate samples of BRG1, INO80B, NSD3, or CHD4 peptides (0.02–56 μm in running buffer) were injected at a flow rate of 20 μl min−1 over immobilized BRD3-ET. Equilibrium dissociation constants (Kd) were calculated by nonlinear least-squares fitting to a simple 1:1 Langmuir binding isotherm, as implemented in the Biacore T200 Evaluation software. Each affinity was measured at least twice on different days, and the quoted uncertainty is an estimate based on the spread of measured affinities as well as estimates of uncertainty in protein concentrations.
Structure determination by NMR spectroscopy
15N/13C-Labeled BRD3-ET samples (500 μm) were prepared in either 20 mm sodium phosphate, pH 6.5, 50 mm NaCl, 1 mm DTT (CHD4), or 20 mm MES (pH 6.5), 50 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine (TCEP) and then titrated with unlabeled CHD4 (1.1 molar equivalents) or BRG1 (2 molar equivalents) peptide. For acquisition of HCCH-TOCSY and 13C NOESY-HSQC spectra, the protein–peptide complex samples were lyophilized and reconstituted in an equal volume of D2O. For all other NMR experiments, 5–10% (v/v) D2O was added directly to the samples. 2,2-Dimethyl-2-silapentane-5-sulfonic acid was added as a chemical shift reference to all samples to a final concentration of 150–300 μm. Spectra were acquired at 298 K on Bruker Avance III 600 or 800 MHz NMR spectrometers, each fitted with a cryogenic TCI probe head and using standard pulse sequences from the Bruker library. The spectra were processed using TOPSPIN3 (Bruker, Karlsruhe, Germany), and NMRFAM-SPARKY (74, 75) was used for spectral analysis.
Backbone assignments of peptide-bound BRD3-ET were made using HNCACB, CBCA(CO)NH, HNCA, HNCO, and HN(CA)CO spectra. Side-chain resonances were assigned using HBHA(CO)NH, H(C)(CO)NH-TOCSY, CC(CO)NH-TOCSY, and HCCH-TOCSY spectra, and aromatic spin systems were assigned using 2D 1H,1H-NOESY spectra (150 ms mixing time). Resonances of the BRD3-bound peptide were assigned using 2D 15N/13C (F2,F1)-filtered 1H,1H TOCSY and 15N/13C (F2,F1)-filtered 1H, 1H NOESY spectra (150 ms mixing time). Intramolecular protein NOEs were identified in 15N- and 13C-edited NOESY-HSQC spectra (150 ms mixing time), and intermolecular NOEs were identified in 15N/13C (F1)-filtered NOESY-HSQC spectra (100 ms mixing time). φ/ψ torsion angle restraints were predicted from chemical shifts using TALOS+ (76).
CYANA 3.0 (77–79) was used for automated NOE assignment and initial structure calculations. 200–1000 starting conformers were generated and subjected to simulated annealing with 20,000 torsion angle dynamics steps. Further molecular dynamics under the Crystallographic and NMR System (CNS) force field was performed on the lowest energy conformer from the CYANA calculation, using the final set of NOEs from the CYANA calculations, and the 100 (CHD4 complex) or 500 (BRG1 complex) lowest-energy structures from this process were refined with a 7-Å shell of water molecules according to the RECOORD protocol (80). Structural statistics of the ensemble of 20 lowest-energy structures at the end of water refinement were analyzed using the Protein Validation Software Suite web server (81). The structures of the BRD3-ET–CHD4 (PDB code 6BGG) and BRD3-ET–BRG1 (PDB code 6BGH) complexes have been deposited in the PDB.
Chemical shift perturbation experiments
15N HSQC spectra of 200 μm 15N-labeled BRD3-ET, BRD3-L, and CHD4-Ne were acquired, and unlabeled peptide or protein was titrated into the sample in increments of 0.25 m eq (up to 2 m eq); 15N HSQC spectra were acquired after each addition. The 15N HSQC spectrum of BRD3-ET was assigned at the completion of each titration from HNCACB and CBCA(CO)NH spectra. The 15N HSQC spectrum of CHD4-Ne (residues 265–310) was partially assigned using 2D 1H,1H NOESY/TOCSY spectra and a 15N-separated NOESY (200-ms mixing time). Chemical shift perturbation values were calculated as a weighted average of changes in 1H and 15N chemical shift between free and peptide-saturated BRD3-ET, using Equation 1 (82),
| (1) |
where ΔδHN is the chemical shift change in the proton dimension, and ΔδN is the chemical shift change in the nitrogen dimension.
Author contributions
D. C. W., T. N. S., A. E. C., C. K., and J. P. M. conceptualization; D. C. W. and T. N. S. data curation; D. C. W., T. N. S., A. E. C., C. K., L. E. W.-W., A. P. G. S., A. H. K., R. G., J. N. C., W. M. P., B. L., C. R. V., G. B., and J. P. M. formal analysis; D. C. W., A. P. G. S., J. K. L., A. H. K., R. G., B. L., C. R. V., G. B., and J. P. M. supervision; D. C. W., B. L., C. R. V., G. B., and J. P. M. funding acquisition; D. C. W., T. N. S., A. E. C., C. K., L. E. W.-W., A. P. G. S., J. K. L., A. H. K., R. G., J. N. C., W. M. P., B. L., and G. B. investigation; D. C. W., A. E. C., C. K., A. P. G. S., J. K. L., and J. P. M. methodology; D. C. W., T. N. S., and J. P. M. writing-original draft; D. C. W. and J. P. M. project administration; D. C. W., W. M. P., and J. P. M. writing-review and editing.
Supplementary Material
Acknowledgment
We acknowledge the use of the Bosch Molecular Biology Facility at the University of Sydney for providing access to SPR infrastructure.
This work was supported by Australian National Health and Medical Research Council Grants APP1063301 and APP1058916 (to J. P. M.) and National Institutes of Health Grant R01DK054937 (to G. A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2, Tables S1–S5, and PDB validation report.
The atomic coordinates and structure factors (codes 6BGG and 6BGH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The NMR chemical shift data of BRD3 ET domain bound to CHD4 can be accessed through the Biological Magnetic Resonance Bank (BMRB) under accession number 30367. The NMR chemical shift data for BRD3 ET domain bound to BRG1 can be accessed through the BMRB under accession number 30368.
A. Kuntimaddi, B. I. Leach, J. H. Bushweller, unpublished data.
- BET
- bromodomain and extra-terminal domain
- ET
- extra-terminal
- PMSF
- phenylmethylsulfonyl fluoride
- AHD
- ANC1 homology domain
- PDB
- Protein Data Bank
- SPR
- surface plasmon resonance
- IPTG
- isopropyl β-d-thiogalactopyranoside
- HSQC
- heteronuclear single quantum coherence
- MLV-IN
- murine leukemia virus integrase
- KSHV–LANA
- Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen.
References
- 1. Lin Y.-J., Umehara T., Inoue M., Saito K., Kigawa T., Jang M.-K., Ozato K., Yokoyama S., Padmanabhan B., and Güntert P. (2008) Solution structure of the extraterminal domain of the bromodomain-containing protein BRD4. Protein Sci. 17, 2174–2179 10.1110/ps.037580.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dey A., Chitsaz F., Abbasi A., Misteli T., and Ozato K. (2003) The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U.S.A. 100, 8758–8763 10.1073/pnas.1433065100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.-P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T., Gingras A.-C., Arrowsmith C. H., and Knapp S. (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 10.1016/j.cell.2012.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. LeRoy G., Rickards B., and Flint S. J. (2008) The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 30, 51–60 10.1016/j.molcel.2008.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheung K. L., Zhang F., Jaganathan A., Sharma R., Zhang Q., Konuma T., Shen T., Lee J.-Y., Ren C., Chen C.-H., Lu G., Olson M. R., Zhang W., Kaplan M. H., Littman D. R., et al. (2017) Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol. Cell 65, 1068–1080 10.1016/j.molcel.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gamsjaeger R., Webb S. R., Lamonica J. M., Billin A., Blobel G. A., and Mackay J. P. (2011) Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell. Biol. 31, 2632–2640 10.1128/MCB.05413-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang B., Yang X.-D., Zhou M.-M., Ozato K., and Chen L.-F. (2009) Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 29, 1375–1387 10.1128/MCB.01365-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi J., Wang Y., Zeng L., Wu Y., Deng J., Zhang Q., Lin Y., Li J., Kang T., Tao M., Rusinova E., Zhang G., Wang C., Zhu H., Yao J., et al. (2014) Disrupting the interaction of BRD4 with diacetylated twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25, 210–225 10.1016/j.ccr.2014.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsume M., Kimura-Yoshida C., Mochida K., Shibukawa Y., Amazaki S., Wada Y., Hiramatsu R., Shimokawa K., and Matsuo I. (2012) Brd2 is required for cell cycle exit and neuronal differentiation through the E2F1 pathway in mouse neuroepithelial cells. Biochem. Biophys. Res. Commun. 425, 762–768 10.1016/j.bbrc.2012.07.149 [DOI] [PubMed] [Google Scholar]
- 10. Wang F., Liu H., Blanton W. P., Belkina A., Lebrasseur N. K., and Denis G. V. (2009) Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J. 425, 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dey A., Ellenberg J., Farina A., Coleman A. E., Maruyama T., Sciortino S., Lippincott-Schwartz J., and Ozato K. (2000) A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell. Biol. 20, 6537–6549 10.1128/MCB.20.17.6537-6549.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houzelstein D., Bullock S. L., Lynch D. E., Grigorieva E. F., Wilson V. A., and Beddington R. S. (2002) Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 22, 3794–3802 10.1128/MCB.22.11.3794-3802.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gyuris A., Donovan D. J., Seymour K. A., Lovasco L. A., Smilowitz N. R., Halperin A. L., Klysik J. E., and Freiman R. N. (2009) The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim. Biophys. Acta 1789, 413–421 10.1016/j.bbagrm.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shang E., Wang X., Wen D., Greenberg D. A., and Wolgemuth D. J. (2009) The double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev. Dyn. 238, 908–917 10.1002/dvdy.21911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamonica J. M., Deng W., Kadauke S., Campbell A. E., Gamsjaeger R., Wang H., Cheng Y., Billin A. N., Hardison R. C., Mackay J. P., and Blobel G. A. (2011) Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. U.S.A. 108, E159–E168 10.1073/pnas.1102140108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stonestrom A. J., Hsu S. C., Jahn K. S., Huang P., Keller C. A., Giardine B. M., Kadauke S., Campbell A. E., Evans P., Hardison R. C., and Blobel G. A. (2015) Functions of BET proteins in erythroid gene expression. Blood 125, 2825–2834 10.1182/blood-2014-10-607309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsu S. C., Gilgenast T. G., Bartman C. R., Edwards C. R., Stonestrom A. J., Huang P., Emerson D. J., Evans P., Werner M. T., Keller C. A., Giardine B., Hardison R. C., Raj A., Phillips-Cremins J. E., and Blobel G. A. (2017) The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 66, 102–116 10.1016/j.molcel.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McBride A. A., and Jang M. K. (2013) Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 5, 1374–1394 10.3390/v5061374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu S.-Y., Lee A.-Y., Hou S. Y., Kemper J. K., Erdjument-Bromage H., Tempst P., and Chiang C.-M. (2006) Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20, 2383–2396 10.1101/gad.1448206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. You J., Croyle J. L., Nishimura A., Ozato K., and Howley P. M. (2004) Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117, 349–360 10.1016/S0092-8674(04)00402-7 [DOI] [PubMed] [Google Scholar]
- 21. Dawson M. A., Prinjha R. K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.-I., Robson S. C., Chung C.-W., Hopf C., Savitski M. M., Huthmacher C., Gudgin E., Lugo D., Beinke S., Chapman T. D., et al. (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533 10.1038/nature10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mertz J. A., Conery A. R., Bryant B. M., Sandy P., Balasubramanian S., Mele D. A., Bergeron L., and Sims R. J. 3rd. (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A. 108, 16669–16674 10.1073/pnas.1108190108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. French C. A. (2012) Pathogenesis of NUT midline carcinoma. Annu. Rev. Pathol. 7, 247–265 10.1146/annurev-pathol-011811-132438 [DOI] [PubMed] [Google Scholar]
- 24. Zuber J., Shi J., Wang E., Rappaport A. R., Herrmann H., Sison E. A., Magoon D., Qi J., Blatt K., Wunderlich M., Taylor M. J., Johns C., Chicas A., Mulloy J. C., Kogan S. C., et al. (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 10.1038/nature10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. French C. A. (2010) Demystified molecular pathology of NUT midline carcinomas. J. Clin. Pathol. 63, 492–496 10.1136/jcp.2007.052902 [DOI] [PubMed] [Google Scholar]
- 26. Brand M., Measures A. R., Wilson B. G., Cortopassi W. A., Alexander R., Höss M., Hewings D. S., Rooney T. P., Paton R. S., and Conway S. J. (2015) Small molecule inhibitors of bromodomain–acetyl-lysine interactions. ACS Chem. Biol. 10, 22–39 10.1021/cb500996u [DOI] [PubMed] [Google Scholar]
- 27. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., et al. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Z., Wang P., Chen H., Wold E. A., Tian B., Brasier A. R., and Zhou J. (2017) Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 60, 4533–4558 10.1021/acs.jmedchem.6b01761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Viejo-Borbolla A., Ottinger M., Brüning E., Bürger A., König R., Kati E., Sheldon J. A., and Schulz T. F. (2005) Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79, 13618–13629 10.1128/JVI.79.21.13618-13629.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hellert J., Weidner-Glunde M., Krausze J., Richter U., Adler H., Fedorov R., Pietrek M., Rückert J., Ritter C., Schulz T. F., and Lührs T. (2013) A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog. 9, e1003640 10.1371/journal.ppat.1003640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ottinger M., Christalla T., Nathan K., Brinkmann M. M., Viejo-Borbolla A., and Schulz T. F. (2006) Kaposi's Sarcoma-Associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80, 10772–10786 10.1128/JVI.00804-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Q., Zeng L., Shen C., Ju Y., Konuma T., Zhao C., Vakoc C. R., and Zhou M.-M. (2016) Structural mechanism of transcriptional regulator NSD3 recognition by the ET domain of BRD4. Structure 24, 1201–1208 10.1016/j.str.2016.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crowe B. L., Larue R. C., Yuan C., Hess S., Kvaratskhelia M., and Foster M. P. (2016) Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. U.S.A. 113, 2086–2091 10.1073/pnas.1516813113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Narlikar G. J., Sundaramoorthy R., and Owen-Hughes T. (2013) Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 10.1016/j.cell.2013.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nitarska J., Smith J. G., Sherlock W. T., Hillege M. M., Nott A., Barshop W. D., Vashisht A. A., Wohlschlegel J. A., Mitter R., and Riccio A. (2016) A functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep. 17, 1683–1698 10.1016/j.celrep.2016.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Torchy M. P., Hamiche A., and Klaholz B. P. (2015) Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 72, 2491–2507 10.1007/s00018-015-1880-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang L., Nogales E., and Ciferri C. (2010) Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 102, 122–128 10.1016/j.pbiomolbio.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hodges C., Kirkland J. G., and Crabtree G. R. (2016) The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb. Perspect. Med. 6, a026930 10.1101/cshperspect.a026930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conaway R. C., and Conaway J. W. (2009) The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem. Sci. 34, 71–77 10.1016/j.tibs.2008.10.010 [DOI] [PubMed] [Google Scholar]
- 40. Gerhold C. B., and Gasser S. M. (2014) INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol. 24, 619–631 10.1016/j.tcb.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 41. Gregory G. D., Miccio A., Bersenev A., Wang Y., Hong W., Zhang Z., Poncz M., Tong W., and Blobel G. A. (2010) FOG1 requires NuRD to promote hematopoiesis and maintain lineage fidelity within the megakaryocytic-erythroid compartment. Blood 115, 2156–2166 10.1182/blood-2009-10-251280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hong W., Nakazawa M., Chen Y. Y., Kori R., Vakoc C. R., Rakowski C., and Blobel G. A. (2005) FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J. 24, 2367–2378 10.1038/sj.emboj.7600703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miccio A., Wang Y., Hong W., Gregory G. D., Wang H., Yu X., Choi J. K., Shelat S., Tong W., Poncz M., and Blobel G. A. (2010) NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J. 29, 442–456 10.1038/emboj.2009.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanno T., Kanno Y., Siegel R. M., Jang M. K., Lenardo M. J., and Ozato K. (2004) Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 13, 33–43 10.1016/S1097-2765(03)00482-9 [DOI] [PubMed] [Google Scholar]
- 45. Fox A. H., Liew C., Holmes M., Kowalski K., Mackay J., and Crossley M. (1999) Transcriptional cofactors of the FOG family interact with GATA proteins by means of multiple zinc fingers. EMBO J. 18, 2812–2822 10.1093/emboj/18.10.2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rahman S., Sowa M. E., Ottinger M., Smith J. A., Shi Y., Harper J. W., and Howley P. M. (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 31, 2641–2652 10.1128/MCB.01341-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yokoyama A., Lin M., Naresh A., Kitabayashi I., and Cleary M. L. (2010) A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17, 198–212 10.1016/j.ccr.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silva A. P., Ryan D. P., Galanty Y., Low J. K., Vandevenne M., Jackson S. P., and Mackay J. P. (2016) The N-terminal region of chromodomain helicase DNA-binding protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) box-like-domain that can bind poly(ADP-ribose). J. Biol. Chem. 291, 924–938 10.1074/jbc.M115.683227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 41, W349–W357 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aiyer S., Swapna G. V., Malani N., Aramini J. M., Schneider W. M., Plumb M. R., Ghanem M., Larue R. C., Sharma A., Studamire B., Kvaratskhelia M., Bushman F. D., Montelione G. T., and Roth M. J. (2014) Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 42, 5917–5928 10.1093/nar/gku175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kowalski K., Liew C. K., Matthews J. M., Gell D. A., Crossley M., and Mackay J. P. (2002) Characterization of the conserved interaction between GATA and FOG family proteins. J. Biol. Chem. 277, 35720–35729 10.1074/jbc.M204663200 [DOI] [PubMed] [Google Scholar]
- 52. Obenauer J. C., Cantley L. C., and Yaffe M. B. (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 10.1093/nar/gkg584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin J., Cai Y., Yao T., Gottschalk A. J., Florens L., Swanson S. K., Gutiérrez J. L., Coleman M. K., Workman J. L., Mushegian A., Washburn M. P., Conaway R. C., and Conaway J. W. (2005) A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. J. Biol. Chem. 280, 41207–41212 10.1074/jbc.M509128200 [DOI] [PubMed] [Google Scholar]
- 54. Denis G. V., McComb M. E., Faller D. V., Sinha A., Romesser P. B., and Costello C. E. (2006) Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 5, 502–511 10.1021/pr050430u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matangkasombut O., Buratowski R. M., Swilling N. W., and Buratowski S. (2000) Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 14, 951–962 [PMC free article] [PubMed] [Google Scholar]
- 56. Roberts T. C., Etxaniz U., Dall'Agnese A., Wu S.-Y., Chiang C.-M., Brennan P. E., Wood M. J. A., and Puri P. L. (2017) BRD3 and BRD4 BET bromodomain proteins differentially regulate skeletal myogenesis. Sci. Rep. 7, 6153 10.1038/s41598-017-06483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang W., Aubert A., Gomez de Segura J. M., Karuppasamy M., Basu S., Murthy A. S., Diamante A., Drury T. A., Balmer J., Cramard J., Watson A. A., Lando D., Lee S. F., Palayret M., Kloet S. L., et al. (2016) The nucleosome remodeling and deacetylase complex NuRD is built from preformed catalytically active sub-modules. J. Mol. Biol. 428, 2931–2942 10.1016/j.jmb.2016.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu S.-Y., Lee A. Y., Lai H.-T., Zhang H., and Chiang C.-M. (2013) Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 49, 843–857 10.1016/j.molcel.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuntimaddi A., Achille N. J., Thorpe J., Lokken A. A., Singh R., Hemenway C. S., Adli M., Zeleznik-Le N. J., and Bushweller J. H. (2015) Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 Di- and Tri-methylation on target genes and transformation potential. Cell Rep. 11, 808–820 10.1016/j.celrep.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meyer C., Hofmann J., Burmeister T., Gröger D., Park T. S., Emerenciano M., Pombo de Oliveira M., Renneville A., Villarese P., Macintyre E., Cavé H., Clappier E., Mass-Malo K., Zuna J., Trka J., et al. (2013) The MLL recombinome of acute leukemias in 2013. Leukemia 27, 2165–2176 10.1038/leu.2013.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leach B. I., Kuntimaddi A., Schmidt C. R., Cierpicki T., Johnson S. A., and Bushweller J. H. (2013) Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 21, 176–183 10.1016/j.str.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin C., Smith E. R., Takahashi H., Lai K. C., Martin-Brown S., Florens L., Washburn M. P., Conaway J. W., Conaway R. C., and Shilatifard A. (2010) AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 37, 429–437 10.1016/j.molcel.2010.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li Y., Sabari B. R., Panchenko T., Wen H., Zhao D., Guan H., Wan L., Huang H., Tang Z., Zhao Y., Roeder R. G., Shi X., Allis C. D., and Li H. (2016) Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol. Cell 62, 181–193 10.1016/j.molcel.2016.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li Y., Wen H., Xi Y., Tanaka K., Wang H., Peng D., Ren Y., Jin Q., Dent S. Y. R., Li W., Li H., and Shi X. (2014) AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571 10.1016/j.cell.2014.09.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Delmore Jake E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., et al. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Erb M. A., Scott T. G., Li B. E., Xie H., Paulk J., Seo H. S., Souza A., Roberts J. M., Dastjerdi S., Buckley D. L., Sanjana N. E., Shalem O., Nabet B., Zeid R., Offei-Addo N. K., et al. (2017) Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274 10.1038/nature21688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shen C., Ipsaro Jonathan J., Shi J., Milazzo J. P., Wang E., Roe J.-S., Suzuki Y., Pappin D. J., Joshua-Tor L., and Vakoc C. R. (2015) NSD3–short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol. Cell 60, 847–859 10.1016/j.molcel.2015.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Finn R. D., Clements J., and Eddy S. R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lejon S., Thong S. Y., Murthy A., AlQarni S., Murzina N. V., Blobel G. A., Laue E. D., and Mackay J. P. (2011) Insights into association of the NuRD complex with FOG-1 from the crystal structure of an RbAp48·FOG-1 complex. J. Biol. Chem. 286, 1196–1203 10.1074/jbc.M110.195842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Loyola A., and Almouzni G. (2004) Histone chaperones, a supporting role in the limelight. Biochim. Biophys. Acta 1677, 3–11 10.1016/j.bbaexp.2003.09.012 [DOI] [PubMed] [Google Scholar]
- 72. Lovén J., Hoke H. A., Lin C. Y., Lau A., Orlando D. A., Vakoc C. R., Bradner J. E., Lee T. I., and Young R. A. (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 10.1016/j.cell.2013.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Anthis N. J., and Clore G. M. (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 22, 851–858 10.1002/pro.2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee W., Tonelli M., and Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Goddard T. D., and Kneller D. G. (2006) SPARKY 3. University of California, San Francisco, CA [Google Scholar]
- 76. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program Dyana11. J. Mol. Biol. 273, 283–298 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 78. Güntert P. (2004) in Protein NMR Techniques (Downing A. K., ed) pp. 353–378, Humana Press, Totowa, NJ [Google Scholar]
- 79. Güntert P., and Buchner L. (2015) Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471 10.1007/s10858-015-9924-9 [DOI] [PubMed] [Google Scholar]
- 80. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., and Bonvin A. M. (2005) RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672 10.1002/prot.20408 [DOI] [PubMed] [Google Scholar]
- 81. Bhattacharya A., Tejero R., and Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 82. Ayed A., Mulder F. A., Yi G.-S., Lu Y., Kay L. E., and Arrowsmith C. H. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Biol. 8, 756–760 10.1038/nsb0901-756 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.