

Abstract
The twin-arginine translocation (Tat) pathway transports folded proteins across bacterial membranes. Tat precursor proteins possess a conserved twin-arginine (RR) motif in their signal peptides that is involved in the binding of the proteins to the membrane-associated TatBC receptor complex. In addition, the hydrophobic region in the Tat signal peptides also contributes to TatBC binding, but whether regions beyond the signal-peptide cleavage site are involved in this process is unknown. Here, we analyzed the contribution of the early mature protein part of the Escherichia coli trimethylamine N-oxide reductase (TorA) to productive TatBC receptor binding. We identified substitutions in the 30 amino acids immediately following the TorA signal peptide (30aa-region) that restored export of a transport-defective TorA[KQ]-30aa-MalE precursor, in which the RR residues had been replaced by a lysine–glutamine pair. Some of these substitutions increased the hydrophobicity of the N-terminal part of the 30aa-region and thereby likely enhanced hydrophobic substrate–receptor interactions within the hydrophobic TatBC substrate-binding cavity. Another class of substitutions increased the positive net charge of the region's C-terminal part, presumably leading to strengthened electrostatic interactions between the mature substrate part and the cytoplasmic TatBC regions. Furthermore, we identified substitutions in the C-terminal domains of TatB following the transmembrane segment that restored transport of various transport-defective TorA–MalE derivatives. Some of these substitutions most likely affected the orientation or conformation of the flexible, carboxy-proximal helices of TatB. Therefore, we propose that a tight accommodation of the folded mature region by TatB contributes to productive binding of Tat substrates to TatBC.
Keywords: Escherichia coli (E. coli), membrane transport, protein targeting, protein translocation, protein export, early mature region, protein folding, secretion pathway, substrate receptor, twin arginine translocation
Introduction
The twin-arginine translocation (Tat)3 system operates in parallel with the well-studied general secretion (Sec) pathway (1) and serves the unique and remarkable role of transporting fully folded, cofactor-containing or even large oligomeric proteins across the cytoplasmic membrane of bacteria, archaea, and the thylakoid membrane of plant chloroplasts (2–9).
Tat-specific precursor proteins are targeted to the Tat machinery by N-terminal signal peptides that have a tripartite structure with a basic n-region at the N terminus bearing a characteristic (S/T)RRXFLK consensus motif, a central stretch consisting of hydrophobic residues (the so-called h-region), and a polar C-terminal c-region that contains the signal peptidase recognition site (10). The twin-arginine (RR) pair in the Tat consensus motif, which gave rise to the pathway's name, is of crucial importance for productive recognition and binding of Tat signal peptides by the Tat translocase (11–17).
The Tat system of Escherichia coli is composed of the three major membrane-integral components TatA, TatB, and TatC along with the minor component TatE, which is functionally equivalent to TatA but is expressed at a 50–200-fold lower level and is not essential for Tat transport (18, 19). TatC, the most conserved Tat component, is a polytopic membrane protein that encompasses six predicted transmembrane helices with the N and C termini of the protein being exposed to the cytoplasmic face of the membrane. TatA, TatB, and TatE each contain a single N-terminal transmembrane domain and a cytoplasmically located amphipathic helix that is followed by a less structured C terminus. TatB and TatC form higher-order 1:1 complexes that interact with twin-arginine signal peptides and act as the receptor for Tat substrates (13, 20–23). Some studies provide evidence that Tat precursors are first inserted into the membrane bilayer via their signal peptides prior to their specific interaction with the TatBC receptor complex (24–26). Site-specific cross-linking revealed that the cytosolic N terminus and the first cytosolic loop of TatC are directly involved in the initial recognition and binding of the conserved Tat consensus motif present in Tat signal peptides (13, 15, 17, 22, 27, 28). Subsequently, the signal peptide insertase activity of TatC promotes a deep, hairpin loop-like insertion of the signal peptide and most likely also the early mature protein region into a membrane-embedded, enclosed binding cavity formed by both subunits of the substrate receptor (29), a process that was commonly thought to strictly depend on an intact RR motif (13). However, recent studies showed also that Tat signal peptides lacking the highly conserved arginine residues in the Tat consensus motif can be threaded deep into the TatBC-binding cavity, if compensatory mutations in the signal peptides were present, which enhance the substrate–receptor interactions at a non-twin-arginine position and restore productive recognition and/or binding of the otherwise export-defective Tat precursor protein by the TatBC receptor complex (30, 31). In the so-called stage of advanced binding deep within the TatBC-binding cavity, the signal peptide is primarily located in close proximity to TatB which, in addition to the Tat consensus motif, also contacts the h-region of the signal peptide as well as the folded mature domain, suggesting a cage-like structure of the TatB monomers transiently accommodating the folded Tat substrate before its translocation (13, 32). Substrate binding to TatBC subsequently triggers the protonmotive force-dependent recruitment of TatA oligomers (33), which are proposed to mediate the passage of the folded protein through the membrane probably either by forming size-fitting translocation channels (34, 35) or, due to fact that the TatA transmembrane domain is too short to fully span the lipid bilayer, by destabilizing/weakening the membrane (36, 37).
In previous studies, we have used suppression genetics to identify regions within the TatBC receptor complex that interact with Tat signal peptides, and a number of suppressor mutations within TatB and/or TatC have been characterized that restored export of Tat precursor proteins bearing defective signal peptides (23, 38). Now, we have attempted to determine such functionally relevant regions within the Tat precursor protein that are directly involved in TatBC substrate receptor binding and significantly contribute to its overall binding affinity to the TatBC-binding site.
Various site-directed mutagenesis studies have shown that the two adjacent arginine residues within the Tat consensus motif are indisputably the most important determinants for productive recognition and binding of Tat signal peptides by the Tat translocase, because the replacement of one or both arginines usually severely affects membrane translocation of the respective precursor protein (11–17). Recently, we have elucidated the role of the hydrophobic core of Tat signal peptides in productive TatBC receptor binding and isolated mutations in the h-region of the Tat-specific trimethylamine N-oxide reductase signal peptide, TorA, which restored Tat-dependent transport of a TorA[KQ]-30aa-MalE Tat precursor protein, harboring an inactivating KQ mutation of the RR motif, by enhancing the hydrophobic substrate–receptor interactions within the TatBC-binding cavity at the stage of advanced binding. These results clearly showed that the h-region of Tat signal peptides significantly supports binding of Tat precursor proteins to the TatBC substrate receptor and thus is a second major binding determinant of Tat precursors to the TatBC receptor complex besides the RR motif (30).
Now, we have addressed the question of whether the early mature protein part is also directly involved in TatBC receptor binding by undertaking a similar genetic screen to isolate intragenic mutations within the first 30 amino acids of the early mature region of TorA that can likewise restore export of the otherwise transport-defective TorA[KQ]-30aa-MalE precursor protein. The successful identification of such mutations confirms that the protein region immediately following the signal peptide is not only a passive passenger domain, but also contributes to the productive recognition and/or binding of the Tat precursor protein by the Tat translocase. Because the deep insertion mode of the signal peptide places the cytoplasmically-exposed early mature region in juxtaposition with the cytosolic domains of TatB, we further asked whether these regions of the TatBC receptor complex might represent the corresponding interaction site for the (early) mature protein region of Tat substrates. In fact, we found mutations distributed over the cytosolic domains of TatB that restored export of the TorA[KQ]-30aa-MalE precursor protein, providing the first genetic evidence that interactions between the flexible cytosolic helices of TatB and the mature protein region might further support productive binding of Tat substrates to the TatBC receptor complex.
Results
Amino acid substitutions within the early mature protein region can restore Tat-dependent transport of an export-defective precursor protein that lacks the crucial twin arginine residues in its signal peptide
As described earlier, the Tat-specific fusion protein TorA-MalE allows an easy in situ detection of Tat-dependent protein translocation on indicative media (23, 38). The plasmid encoded TorA-MalE reporter protein consists of the signal peptide of the Tat substrate trimethylamine N-oxide reductase (TorA) and the first eight amino acid residues of the mature TorA protein fused to the mature protein part of the normally Sec-dependent maltose-binding protein (MalE) via a linker region consisting of the three amino acids Glu, Phe, and Asp. Because the presence of MalE in the periplasm is strictly required for maltose uptake (39), the Tat-specific export of TorA-MalE into the periplasm is directly linked with the ability of cells to utilize maltose and thus to grow on maltose minimal medium (MMM) as well as to form red colonies on MacConkey maltose (MCM) agar plates (23, 38, 40, 41). In this study, we chose a derivative of the TorA-MalE reporter protein, TorA-30aa-MalE that, in addition to the TorA signal peptide, also contains the first 30 amino acid residues of the mature part of TorA (named 30aa-region) fused to the mature portion of MalE.
We recently demonstrated that a set of mutations in the h-region of the TorA signal peptide can suppress the export defect of a transport-defective TorA[KQ]-30aa-MalE precursor protein, in which the crucial RR residues had been replaced with a lysine–glutamine pair, by enhancing the hydrophobic interactions between the TorA[KQ] signal peptide and the TatBC-binding site deep within the membrane and thus restoring productive binding required for successful translocation (30). To assess the contribution of the early mature protein region of Tat substrates to TatBC receptor binding, we now performed a similar genetic screen for suppressor mutations specifically located within the 30aa-region of the TorA[KQ]-30aa-MalE precursor.
Using pTorA[KQ]-30aa-MalE as template, the entire 30aa-region was mutagenized by error-prone (ep)-PCR. The resulting ep-PCR fragments were used to replace the corresponding gene fragment encoding the unaltered 30aa-region in the low-copy vector pBBR1MCS-2 carrying the torA[KQ]-30aa-MalE fusion gene, thereby creating a pool of pTorA[KQ]-30aa-MalE plasmid variants harboring randomly mutagenized 30aa-regions. Alternatively, 20 different amino acid positions in the 30aa-region were randomly chosen and subjected to saturation mutagenesis (Fig. 1). Here, a set of degenerate oligonucleotides was used as primers to individually replace each of the selected amino acid residues with all 20 naturally occurring amino acids (encoded by the codon set NNN), thereby generating a library of TorA[KQ]-30aa-MalE reporter variants harboring single amino acid substitutions at the pre-determined target positions in the 30aa-region.
Figure 1.

Generation of a mutant library of TorA[KQ]-30aa-MalE by site-directed mutagenesis for the identification of suppressing mutations in the 30aa-region. The amino acid sequence of TorA[KQ]-30aa-MalE encompassing the entire TorA signal peptide (n-region, h-region, and c-region), the first 30 amino acids of the mature TorA protein (30aa-region), and the first five residues of the mature MalE protein is shown. The Tat consensus motif is underlined (dashed line); the position of the twin arginines is marked by arrows. The KQ mutation of the RR motif is highlighted in black. A total of 20 residues in the 30aa-region (Ala-40, Gln-41, Ala-42, Ala-43, Thr-44, Asp-45, Ile-48, Ser-49, Lys-50, Glu-51, Gly-52, Thr-55, Gly-56, Ser-57, His-58, Gly-60, Arg-63, Thr-65, Lys-67, and Asp-68; highlighted in black) has been separately subjected to saturation mutagenesis (*) to generate a library of TorA[KQ]-30aa-MalE reporter variants harboring single amino acid substitutions at the predetermined positions in the 30aa-region.
These two constructed mutant plasmid libraries were used for transformation of GSJ101 (pHSG-TatABCE) expressing plasmid-borne tat genes. For the selection of intragenic suppressors of the TorA[KQ]-30aa-MalE export defect, the resulting transformants were plated onto MMM agar plates, which were subsequently incubated for up to 6 days until a formation of single colonies could be observed. A total of 280 colonies were randomly chosen, tested for reproducible growth on MMM, and further analyzed with respect to the DNA sequence of the mutated torA[KQ]-30aa-MalE fusion genes.
Two different types of mutated TorA[KQ]-30aa-MalE reporter variants harboring the single amino acid substitution K50L and K50I, respectively, in the 30aa-region could be isolated using the mutant library generated by ep-PCR. A strikingly higher diversity among the isolated suppressor mutant reporters was obtained utilizing site-saturation mutagenesis for the creation of a pool of mutated pTorA[KQ]-30aa-MalE plasmid variants. Here, DNA sequencing revealed 48 different types of mutated TorA[KQ]-30aa-MalE precursor proteins carrying single amino acid alterations in the 30aa-region (also including the mutations K50I and K50L that had been isolated before from the epPCR-generated mutant library), each of which was sufficient to allow significant export of the otherwise transport-incompetent reporter into the periplasm reflected by the regained ability of the respective strains to grow with maltose as the sole carbon and energy source. Surprisingly, six more suppressors were isolated, which harbored double mutations predominantly located at the positions Thr-55, Thr-65, and/or Asp-68 in the C-terminal half of their 30aa-region. Because these TorA[KQ]-30aa-MalE reporter variants have been selected by using the mutant library generated by one-codon site-directed mutagenesis for transformation of E. coli GSJ101 (pHSG-TatABCE), the respective second mutations must have occurred spontaneously in these cases.
First, we tested whether the isolated TorA[KQ]-30aa-MalE variants (subsequently designated iKQ-X for intragenic KQ suppressor mutant reporter with X being the respective amino acid alteration) were still strictly exported via the Tat pathway by analyzing their in situ phenotypes on indicative media in the absence of a functional Tat translocase in the ΔmalEΔtatABCE deletion mutant GSJ101. In all cases, no growth on MMM and the formation of pale colonies on MCM agar plates was observed, excluding the possibility that export of the TorA[KQ]-30aa-MalE suppressor mutant reporters had occurred via the Sec pathway (Table 1 and Table S1).
Table 1.
Phenotype of E. coli GSJ101 expressing various TorA[KQ]-30aa-MalE reporter variants in the presence or absence of a functional Tat translocase on maltose minimal medium (MMM) and MacConkey maltose (MCM) agar plates
E. coli strains were streaked on MacConkey agar plates containing 1% maltose (MCM) or minimal medium agar plates containing 0.4% maltose (MMM) as the sole carbon and energy source and incubated for up to 48 h at 37 °C. −, no growth; +, slow growth; ++, moderate growth; +++, good growth. Substitutions chosen for further analysis are indicated in bold and underlined.
| Suppressor category | Location of mutation(s) in 30aa-region | Reporter variant X | Color of colonies of E. coli GSJ101 (X, TatABCE) on MCM, incubation time, 24 h | Growth of E. coli GSJ101 (X, TatABCE) on MMM |
Color of colonies of E. coli GSJ101 (X) on MCM, incubation time, 24 h | Growth of E. coli GSJ101 (X) on MMM, incubation time, up to 48 h | |
|---|---|---|---|---|---|---|---|
| Incubation time | |||||||
| 24 h | 48 h | ||||||
| I | N-terminal half | iKQ-A43L/D45E | Pale | + | +++ | Pale | − |
| iKQ-T44I | Pale | + | +++ | Pale | − | ||
| iKQ-K50I | Light red | + (+) | +++ | Pale | − | ||
| iKQ-K50L | Light red | + (+) | +++ | Pale | − | ||
| iKQ-K50V | Light red | + (+) | +++ | Pale | − | ||
| iKQ-K50M | Light red | + (+) | +++ | Pale | − | ||
| iKQ-K50F | Light red | + (+) | +++ | Pale | − | ||
| iKQ-K50C | Light red | + (+) | +++ | Pale | − | ||
| iKQ-E51I | Pale | + | +++ | Pale | − | ||
| iKQ-E51W | Pale | + | +++ | Pale | − | ||
| C-terminal half | iKQ-T55R/T65R | Light red | + (+) | +++ | Pale | − | |
| iKQ-T55S/D68R | Pale | + | +++ | Pale | − | ||
| iKQ-S57L | Pale | (+) | ++ | Pale | − | ||
| iKQ-H58I | Pale | + | +++ | Pale | − | ||
| iKQ-H58F | Pale | + | +++ | Pale | − | ||
| iKQ-H58W | Pale | (+) | ++ | Pale | − | ||
| iKQ-G60F | Pale | (+) | ++ | Pale | − | ||
| iKQ-T65R | Pale | + | +++ | Pale | − | ||
| iKQ-D68R | Pale | + | +++ | Pale | − | ||
| II | N-terminal half | iKQ-A43F | Pale | − | + | Pale | − |
| iKQ-A43F | Pale | − | + | Pale | − | ||
| iKQ-D45L | Pale | − | ++ | Pale | − | ||
| C-terminal half | iKQ-T55R/T65L | Pale | − | ++ | Pale | − | |
| iKQ-T55A/D68G | Pale | − | ++ | Pale | − | ||
| iKQ-G56R | Pale | − | + | Pale | − | ||
| iKQ-G56W | Pale | − | + | Pale | − | ||
| iKQ-S57R | Pale | − | + | Pale | − | ||
| iKQ-S57W | Pale | − | + | Pale | − | ||
| iKQ-H58Y | Pale | − | + | Pale | − | ||
| iKQ-H58R | Pale | − | + | Pale | − | ||
| iKQ-H58L | Pale | − | + | Pale | − | ||
| Controls | TorA-30aa-MalE | Red | +++ | +++ | Pale | − | |
| TorA[KQ]-30aa-MalE | pale | − | − | Pale | − | ||
Second, the mutated TorA[KQ]-30aa-MalE reporter variants were further analyzed with respect to the suppressing activities of the corresponding mutations by analyzing the Tat-dependent export of MalE into the periplasm of E. coli GSJ101 indirectly by MMM and MCM plate assays. As shown in Table 1 and Table S1, GSJ101 strains coexpressing the tat genes and the various mutated reporter proteins showed diverse in situ phenotypes in the plate assays because they exhibited different growth rates with maltose as the sole carbon and energy source. The suppressing activities of the respective 30aa-region-located mutations seemed to be generally quite low because no formation of red colonies on MCM of the corresponding strains could be observed in contrast to the positive control GSJ101 (pHSG-TatABCE) containing an unaltered TorA-30aa-MalE precursor protein. Nevertheless, in all cases, significant growth on MMM was observed in contrast to the negative control GSJ101 (pHSG-TatABCE) expressing the export-defective TorA[KQ]-30aa-MalE reporter with an unaltered 30aa-region, showing that the amino acid alterations present in the 30aa-region allow a weak suppression of the transport defect.
Depending on the particular relative suppressing activities of the introduced amino acid substitutions, the isolated TorA[KQ]-30aa-MalE reporter variants can be roughly divided into three groups. The first suppressor category consisting of 19 reporters contains single or double amino acid alterations in the 30aa-region that permit a comparatively effective Tat-dependent transport of the normally export-defective precursor protein, resulting in significant growth of the respective strains on MMM within 24 h of incubation (Table 1, class I). Somewhat weaker suppressor phenotypes were promoted by the second category of mutant reporter proteins. In these cases, the incubation period had to be extended up to 48 h before any significant growth of the strains GSJ101 (pHSG-TatABCE) coexpressing the respective TorA[KQ]-30aa-MalE reporter variants with maltose as the sole carbon and energy source could be observed (Table 1, class II). The third group of mutated TorA[KQ]-30aa-MalE precursors harbored amino acid alterations that exhibited the lowest suppressing activities allowing for visible growth of the corresponding cells on MMM after at least 48 h of incubation (Table S1, class III). Because only very low levels of MalE export were promoted by the third class of suppressing mutations, this group of suppressor mutant reporters was not further considered in the following analyses.
The isolated amino acid substitutions were distributed over 15 different positions in the 30aa-region. Interestingly, mutations that replaced hydrophilic amino acids with hydrophobic residues were in most cases located in the N-terminal half, whereas the presence of amino acid substitutions that introduced basic amino acids and thus increased the positive net charge of the early mature protein part was found to be restricted to the C-terminal half of the 30aa-region. These observations indicate that these two types of suppressing mutations might operate in a different manner. From all isolated suppressing mutations, amino acid substitutions occupying position Lys-50 in the N-terminal half of the 30aa-region, which replaced a basic lysine residue with more hydrophobic amino acids (e.g. Leu, Ile, or Val), showed the highest suppressing activities, resulting in the formation of light red colonies on MCM and the fastest growth on MMM of the corresponding strains. A similar suppression phenotype was promoted by the spontaneous double mutation T55R/T65R that introduced two positively charged arginine residues into the C-terminal half of the 30aa-region. These findings clearly show that mutations in the 30aa-region can compensate to a certain degree for the loss of the crucial RR residues and restore export of the TorA[KQ]-30aa-MalE reporter either by increasing the hydrophobicity of the early mature protein part immediately following the signal peptide or by increasing the positive net charge of the more distal part of the 30aa-region. The isolation of these two different types of suppressing mutations indicates that the early mature protein region of TorA[KQ]-30aa-MalE could interact with different parts of the TatBC receptor complex. Considering the presumed deep hairpin loop-like insertion of the signal peptide and a certain number of amino acid residues of the early mature protein region into the TatBC-binding cavity, the newly introduced hydrophobic residues in the membrane-embedded portion of the 30aa-region might interact with the hydrophobic TatBC-binding cavity, whereas the positively charged amino acids in the cytoplasmically-exposed part of this region could strengthen the interactions between the precursor and the nonhydrophobic domains of the TatBC receptor complex located in the cytosol.
Hence, the present results provide the first evidence for a direct interaction of the early mature region with the Tat translocase and support the hypothesis that, besides the Tat signal peptide, this protein region is in fact also directly involved in the productive binding of the Tat substrate to the TatBC receptor complex.
Combinations of amino acid substitutions in the early mature protein region further increase Tat-dependent export of the TorA[KQ]-30aa-MalE precursor protein
As shown before, the presence of a single mutation in the 30aa-region was sufficient to allow low but nevertheless significant export of a TorA[KQ]-30aa-MalE precursor protein reflected by the ability of the corresponding cells to grow with maltose as the sole carbon and energy source. Next, we addressed the question whether combinations of the identified 30aa-region-located mutations would further enhance export of a TorA[KQ]-30aa-MalE precursor. For the following analysis, we chose the single amino acid substitutions T44I, D45L, K50L, T65R, and D68R, respectively, as well as the double mutation T55R/T65R, which differed in their position within the 30aa-region, the particular suppressing activity, and/or their mode of action (Table 1, chosen substitutions are indicated in bold and underlined; Fig. 2A, lines 3–8). For the construction of the six double or triple-mutated reporter proteins iKQ-T44I/K50L, iKQ-K50L/T65R, iKQ-T44I/T65R, iKQ-D45L/D68R, iKQ-K50L/D68R, and iKQ-K50L/T55R/T65R, the corresponding single or double mutations were introduced into the pTorA[KQ]-30aa-MalE vector by site-directed mutagenesis, and the resulting pTorA[KQ]-30aa-MalE plasmid variants (Fig. 2A, lines 9–14) were subsequently transformed into GSJ101 coexpressing the tatABCE genes. The export of the various TorA[KQ]-30aa-MalE reporter proteins was analyzed either indirectly by in situ MMM/MCM plate assays or directly by determining the amount of exported MalE in the periplasm in cell fractionation experiments (Fig. 2B; Fig. S1). Here, EDTA-lysozyme spheroplasting was used to yield a combined cytosolic/membrane (C/M) fraction and a periplasmic fraction (P) that were subsequently separated by SDS-PAGE followed by Western blotting using MalE-specific antibodies. The relative export efficiency (reflected by the amount of mature-sized MalE present in the periplasm) of the unaltered TorA-30aa-MalE reporter protein, to which all further relative translocation efficiencies described in this work will be related, was set to 100%.
Figure 2.

Mutations in the 30aa-region cooperate with each other in the suppression of the export defect of a TorA[KQ]-30aa-MalE precursor protein. A, amino acid sequences of TorA-30aa-MalE reporter variants encompassing the Tat consensus motif (dashed line) in the TorA signal peptide (residues 1–39), the entire 30aa-region (residues 40–69), and the first five amino acid residues of the mature MalE protein (residues 70–439). Positions of the particular mutations in the 30aa-region are highlighted in red and underlined. The KQ mutation of the RR motif is highlighted in black. B, export of the various TorA[KQ]-30aa-MalE reporter variants has been analyzed by MMM/MCM plate assays in the presence (Tat+) or absence (Tat−) of the Tat translocase in E. coli GSJ101 and on the protein level. The phenotypes of the respective strains on MMM (−, no growth; +, slow growth; ++, moderate growth; +++, good growth) and MCM (pale, light red, pink, or red color of colonies) after 24 h of incubation are shown in the boxes at the bottom of the figure. Cells were fractionated into a periplasmic fraction and a combined cytosol/membrane fraction by EDTA-lysozyme spheroplasting. The samples of the fractions corresponding to an identical amount of cells were subjected to SDS-PAGE and immunoblotting using anti-MalE antibodies. Subsequently, relative export efficiencies were calculated by determining the amount of exported MalE protein in the periplasmic fraction of strains GSJ101 coexpressing the tatABCE genes and TorA-30aa-MalE or one of the mutated TorA[KQ]-30aa-MalE reporter variants in at least three different independent experiments via quantification of the chemiluminescence signals. The signals were recorded by a CCD camera and subsequently analyzed by the program AIDA 4.50 (Raytest). The average values are indicated by horizontal marker lines and standard deviations by error bars. The relative export efficiency of the positive control GSJ101 (pTorA-30aa-MalE and pHSG-TatABCE) was set to 100% (wt; lane 1). The other samples correspond to GSJ101 coexpressing the tatABCE genes and the export-defective TorA[KQ]-30aa-MalE reporter (KQ; lane 2) or one of the TorA[KQ]-30aa-MalE reporter variants containing the single, double, or triple mutations in the 30aa-region indicated below lanes 3–14. wt, WT.
As described above, only low levels of MalE export were promoted by the mutations T44I, D45L, K50L, T65R, D68R, and T55R/T65R, respectively, allowing for slow growth on MMM and the formation of pale or, in case of K50L, light red colonies on MCM of the corresponding strains (Fig. 2B, lanes 3–8; Fig. S1, sectors 3–8). Quantification of the chemiluminescence signals further confirmed that the export efficiencies were generally low compared with the positive control (100%), with values between 0.8% observed for the weakest suppressor mutant reporter iKQ-D45L and 3.2% for iKQ-K50L (Fig. 2B, lanes 3–8). A substantial increase in the relative translocation efficiency could be observed when the mutations T44I, D45L, and K50L, which increased the hydrophobicity of the N-terminal half of the 30aa-region, were combined with amino acid substitutions that introduced basic residues into the C-terminal half of this early mature protein part (i.e. T55R/T65R, T65R, and D68R). The resulting double or triple-mutated reporter proteins iKQ-T44I/T65R, iKQ-D45L/D68R, iKQ-K50L/T65R, iKQ-K50L/D68R, and iKQ-K50L/T55R/T65R promoted a clearly stronger suppression phenotype in the in situ plate assays reflected by faster growth on MMM and the formation of pink or red colonies on MCM of the corresponding strains compared with the suppressing activities conferred by the respective single or double mutations (Fig. 2B, lanes 10–14; Fig. S1, sectors 10–14). Importantly, the newly constructed TorA[KQ]-30aa-MalE reporter variants were strictly Tat-specific because no export was detectable in the in situ plate assays in the absence of a functional Tat translocase (Fig. 2B, lowest panel, lanes 10–14). In full agreement with the in situ phenotypes, a clear increase of MalE export into the periplasm conferred by the combined mutations was also observed on a protein level. As shown in Fig. 2B, the respective single mutations were found to synergistically contribute to the suppression of the TorA[KQ]-30aa-MalE export defect because the export efficiencies observed for the double or triple-mutated reporter variants iKQ-T44I/T65R (4.5%), iKQ-D45L/D68R (3.9%), iKQ-K50L/T65R (5.8%), iKQ-K50L/D68R (5.7%), and iKQ-K50L/T55R/T65R (5.9%) were higher than the sum of the translocation efficiencies determined for the corresponding single or double-mutated reporters iKQ-T44I (1.8%), iKQ-D45L (0.8%), iKQ-K50L (3.2%), iKQ-T65R (1.3%), iKQ-D68R (1.6%), and iKQ-T55R/T65R (2.1%) (Fig. 2B, compare lanes 10–14 with 3–8).
In comparison, a combination of the two mutations T44I and K50L, both of which exerted their effect by increasing the overall hydrophobicity of the N-terminal half of the 30aa-region, had rather an additive effect on the export of TorA[KQ]-30aa-MalE. The translocation efficiency of the resulting double-mutated reporter variant iKQ-T44I/K50L (4.1%) was even somewhat lower than the value that would result from a summation of the export efficiencies of the corresponding single mutant proteins iKQ-T44I (1.8%) and iKQ-K50L (3.2%) (Fig. 2B, compare lane 9 with lanes 3 and 5).
Obviously, the overall effect of combinations of the various suppressing mutations on TorA[KQ]-30aa-MalE export depends on their nature and their particular localization in the 30aa-region (i.e. either in the N- or C-terminal half of the 30aa-region). Nevertheless, because in all cases the transport efficiency of TorA[KQ]-30aa-MalE could be significantly increased, the identified mutations in the early mature protein region evidently cooperate with each other in restoring export of the otherwise transport-defective TorA[KQ]-30aa-MalE precursor protein. This finding therefore strongly suggests that the entire early mature protein region contributes to the binding of the normally export-defective Tat precursor to the membrane-embedded and/or cytosolically-localized parts of the TatBC receptor complex.
Amino acid substitutions in the hydrophobic (h-) region of the TorA signal peptide and in the early mature protein act together in restoring export of the TorA[KQ]-30aa-MalE precursor protein
Next, we analyzed whether the export of the normally export-defective TorA[KQ]-30aa-MalE precursor protein can be further improved by combining the 30aa-region-located mutations with the previously reported suppressor mutations located in the h-region of the TorA[KQ] signal peptide (30). To test this, we combined the amino acid substitutions A16V, T22A, G25W, G28W, and G25W/G28W, which were distributed all over the hydrophobic core and exhibited various suppressing activities in the in vivo transport assays, with the 30aa-region-located mutations K50L, H58I, T65R, and D68R, respectively, giving rise to the reporter variants iKQ-A16V/H58I, iKQ-T22A/T65R, iKQ-T22A/D68R, iKQ-G28W/H58I, iKQ-G25W/K50L, iKQ-G28W/K50L, and iKQ-G25W/G28W/K50L (Fig. 3A, lines 12–18). The export of the newly constructed TorA[KQ]-30aa-MalE variants was then analyzed on indicator plates and directly by cell-fractionation experiments (Fig. 3B and Fig. S2). As shown in Fig. 3B (lower panel), no growth on MMM and, consequently, no formation of colored colonies on MCM could be observed for E. coli GSJ101 (ΔtatABCE) expressing the various reporters in the absence of a functional Tat translocase, confirming that the export of the newly constructed TorA[KQ]-30aa-MalE reporter proteins occurred exclusively via the Tat pathway.
Figure 3.

Mutations in the h-region of the TorA signal peptide and the 30aa-region cooperate with each other in the suppression of the export defect of a TorA[KQ]-30aa-MalE precursor protein. A, amino acid sequences of TorA-30aa-MalE reporter variants encompassing the Tat consensus motif (dashed line) and the entire h-region of the TorA signal peptide (residues 1–39), the entire 30aa-region (residues 40–69), and the first five amino acid residues of the mature MalE protein (residues 70–439). Positions of the particular mutations in the h-region of the TorA signal peptide (or, in case of the mutation A16V, at the boundary between the n-region with the Tat consensus motif and the h-region) and/or the 30aa-region are highlighted in red and underlined. The KQ mutation of the RR motif is highlighted in black. B, export of the various TorA[KQ]-30aa-MalE reporter variants has been analyzed by MMM/MCM plate assays in the presence (Tat+) or absence (Tat−) of the Tat translocase in E. coli GSJ101 and on the protein level. The phenotypes of the respective strains on MMM (−, no growth; +, slow growth; ++, moderate growth; +++, good growth) and MCM (pale, light red, pink, or red color of colonies) after 24 h of incubation are shown in the boxes at the bottom of the figure. Cells were fractionated into a periplasmic fraction and a combined cytosol/membrane fraction by EDTA-lysozyme spheroplasting. The samples of the fractions corresponding to an identical amount of cells were subjected to SDS-PAGE and immunoblotting using anti-MalE antibodies. Subsequently, relative export efficiencies were calculated by determining the amount of exported MalE protein in the periplasmic fraction of strains GSJ101 coexpressing the tatABCE genes and TorA-30aa-MalE or one of the mutated TorA[KQ]-30aa-MalE reporter variants in at least three different independent experiments via quantification of the chemiluminescence signals. The signals were recorded by a CCD camera and subsequently analyzed by the program AIDA 4.50 (Raytest). The average values are indicated by horizontal marker lines and standard deviations by error bars. The relative export efficiency of the positive control GSJ101 (pTorA-30aa-MalE, pHSG-TatABCE) was set to 100% (wt; lane 1). The other samples correspond to GSJ101 coexpressing the tatABCE genes and the export-defective TorA[KQ]-30aa-MalE reporter (KQ; lane 2) or one of the TorA[KQ]-30aa-MalE reporter variants containing the single, double, or triple mutations in the h-region of the TorA signal peptide and/or the 30aa-region indicated below the lanes 3–18. wt, WT.
As already described by Ulfig et al. (30), the double mutation G25W/G28W located in the C-terminal part of the h-region of the TorA[KQ] signal peptide efficiently restored the export of the otherwise translocation-incompetent TorA[KQ]-30aa-MalE precursor protein into the periplasm (21.7%), allowing fast growth on MMM and the formation of red colonies on MCM of the respective strain (Fig. 3B, lane 7; Fig. S2, sector 7). No significant difference in the in situ phenotype could be observed for GSJ101 coexpressing the tatABCE genes and the triple-mutated reporter iKQ-G25W/G28W/K50L harboring the additional 30aa-region-located mutation K50L (Fig. 3B, compare lanes 18 and 7; Fig. S2, compare sectors 18 and 7). However, compared with iKQ-G25W/G28W, a slight increase in the translocation efficiency could be detected for iKQ-G25W/G28W/K50L (24.9%) strongly suggesting that, in this case, the mutations in the h- and 30aa-region contribute additively to the suppression of the export defect of the TorA[KQ]-30aa-MalE precursor protein.
The cooperative action of both types of suppressor mutations in promoting export of the otherwise transport-incompetent reporter becomes even more evident when the 30aa-region-located mutation K50L was combined with the single amino acid substitution G25W or G28W in the h-region, both of which did not possess sufficient suppressing activities to allow for the formation of red colonies on MCM. As shown in Fig. 3B and Fig. S2, GSJ101 strains expressing the resulting double-mutated reporter proteins iKQ-G25W/K50L or iKQ-G28W/K50L showed faster growth on MMM and the formation of pink or, in case of iKQ-G25W/K50L, even red colonies on MCM compared with cells harboring the corresponding single-mutated reporter variants iKQ-G25W, iKQ-G28W, or iKQ-K50L (Fig. 3B, compare lanes 16 and 17 with 5, 6, and 8; Fig. S2, compare sectors 16 and 17 with 5, 6, and 8). In total agreement with the in situ phenotypes, a significant increase of MalE export into the periplasm conferred by the combined mutations was also observed on a protein level. The translocation efficiencies of the reporter proteins iKQ-G25W/K50L and iKQ-G28W/K50L (6.4 and 5.1%, respectively) corresponded approximately to the sum of the export efficiencies observed for the respective single mutant reporter variants, directly demonstrating the additive effects of the individual suppressor mutations on the export of the TorA[KQ]-30aa-MalE precursor protein (Fig. 3B, compare lanes 16 and 17 with 5, 6, and 8).
To test whether these observed effects of combinations of the h-region-located mutations with the amino acid alterations in the 30aa-region depend on their particular position in the hydrophobic core, the amino acid substitutions A16V and G28W located in the extreme N and C termini of the h-region, respectively, were combined with the mutation H58I residing in the center of the 30aa-region giving rise to the double-mutated reporter variants iKQ-A16V/H58I and iKQ-G28W/H58I (Fig. 3A, lines 12 and 15). Also in these cases, the export of the single mutant reporters iKQ-A16V and iKQ-G28W could be significantly increased by the additional mutation in the early mature region. Both double-mutated reporter proteins promoted a stronger suppression phenotype in the in situ plate assays reflected by faster growth on MMM and the formation of pink colonies on MCM of the corresponding strains compared with the suppressing activities conferred by the respective single mutations A16V, G28W, and H58I (i.e. slow growth on MMM; pale colonies on MCM) (Fig. 3B, compare lanes 12 and 15 with 3, 6, and 9; Fig. S2, compare sectors 12 and 15 with 3, 6, and 9). The translocation efficiencies of the reporter proteins iKQ-A16V/H58I and iKQ-G28W/H58I (3.8 and 4.1%, respectively) corresponded approximately to somewhat more than the sum of the export efficiencies observed for the respective single mutant reporter variants. These results clearly show that the isolated suppressing mutations in the h-region of the TorA[KQ] signal peptide and the 30aa-region, when combined, act at least additively in restoring export of the otherwise transport-incompetent TorA[KQ]-30aa-MalE precursor protein, irrespective of their particular position within the hydrophobic core of the TorA signal peptide and the early mature region, respectively.
All mutations being combined so far suppressed the export defect of the TorA[KQ]-30aa-MalE precursor protein by increasing the overall hydrophobicity of the TorA signal peptide and the N-terminal half of the 30aa-region, respectively. Next, we analyzed the export of the reporter variants iKQ-T22A/T65R and iKQ-T22A/D68R, in which the h-region-located suppressing mutation T22A was combined with either T65R or D68R positioned in the C-terminal part of the 30aa-region (Fig. 3A, lines 13 and 14). Unlike the other suppressing mutations, these two 30aa-region-localized amino acid substitutions restored the export of TorA[KQ]-30aa-MalE by introducing an additional positively charged arginine residue into the respective part of the early mature protein region. However, also in case of these combinations, a cooperative action of the respective single mutations could be detected in the plate assay as well as on the protein level. Because of the significantly higher translocation efficiency of iKQ-T22A/T65R (3.1%) and iKQ-T22A/D68R (4.4%), E. coli strain GSJ101 (pHSG-TatABCE) expressing these double-mutated reporter proteins exhibited faster growth rates with maltose as the sole carbon and energy source and formed light red or pink colonies on MCM compared with the strains that harbored the corresponding single mutant reporters iKQ-T22A (1.0%), iKQ-T65R (1.3%), and iKQ-D68R (1.6%) (i.e. slow growth on MMM; pale colonies on MCM) (Fig. 3B, compare lanes 13 and 14 with 4, 10, and 11; Fig. S2, compare sectors 13 and 14 with 4, 10, and 11).
Taken together, these combined results clearly demonstrate that suppressing mutations in the h-region of the signal peptide and the early mature protein part cooperate with each other in the suppression of the export defect of TorA[KQ]-30aa-MalE and support the hypothesis that the newly identified mutations in the 30aa-region, similarly to the h-region-located amino acid alterations, increase the overall binding affinity of the Tat substrate to the TatBC receptor complex, providing further evidence for a direct involvement of the early mature protein region in the binding process to the Tat translocase.
Combinations of amino acid substitutions in the early mature protein region and in the TatBC receptor complex strongly increase export of the TorA[KQ]-30aa-MalE precursor protein
Finally, we asked whether the transport of a TorA[KQ]-30aa-MalE precursor protein can also be further improved by combining the various 30aa-region-located amino acid substitutions with the previously identified suppressor mutation L9F residing in the cytoplasmically localized N terminus of the TatC component of the TatBC receptor complex (23, 38). To test this, we analyzed the export of eight TorA[KQ]-30aa-MalE reporter variants harboring single (i.e. T44I, D45L, K50L, T65R, or D68R) or double mutations (i.e. T44I/K50L, T44I/T65R, or D45L/D68R) in their early mature protein regions by the mutated TatABC[L9F]E translocase on indicator plates and directly by cell-fractionation experiments (Fig. 4 and Fig. S3).
Figure 4.

Combinations of mutations in the 30aa-region and the mutation L9F in TatC synergistically suppress the export-defect of a TorA[KQ]-30aa-MalE precursor protein. A–D, subcellular localization of TorA-30aa-MalE-derived polypeptides. Cells were fractionated into a periplasmic (P) fraction and a combined cytosol/membrane (C/M) fraction by EDTA-lysozyme spheroplasting. The samples of the fractions corresponding to an identical amount of cells were subjected to SDS-PAGE and immunoblotting using anti-MalE antibodies. The positive controls were E. coli GSJ101 containing plasmids pTorA-30aa-MalE and pHSG-TatABCE (lane 1) or pHSG-TatABC[L9F]E (lane 2). The other samples correspond to GSJ101 coexpressing the WT tatABCE or mutated tatABC[L9F]E genes and the export-defective TorA[KQ]-30aa-MalE reporter (lanes 3 and 4, respectively) or one of the TorA[KQ]-30aa-MalE reporter variants containing the single mutations or the double mutations in the 30aa-region as indicated above the lanes (lanes 5–20). wt, WT; p, WT or mutated TorA-30aa-MalE precursor in the C/M fraction; m, mature form of MalE in the P fraction; asterisk, WT or mutated TorA-30aa-MalE-derived degradation products in the C/M fraction. Positions of molecular weight markers are indicated on the left margin. The phenotypes of the respective strains on MMM (−, no growth; +, slow growth; ++, moderate growth; +++, good growth) and MCM (pale, light red (light r.), pink, red) agar plates after 24 h of incubation are shown in the boxes at the bottom of the figure. E, relative export efficiencies of the analyzed TorA-30aa-MalE reporter variants in strains expressing the WT or mutant TatABC[L9F]E translocase. The amount of exported MalE protein in the P fraction of strains GSJ101 coexpressing the WT tatABCE or mutated tatABC[L9F]E genes and TorA-30aa-MalE, TorA[KQ]-30aa-MalE, or one of the mutated TorA[KQ]-30aa-MalE reporter variants was determined in at least three different independent experiments via quantification of the chemiluminescence signals. The signals were recorded by a CCD camera and subsequently analyzed by the program AIDA 4.50 (Raytest). The average values are indicated by horizontal marker lines; standard deviations by error bars. The relative export efficiency of the positive control GSJ101 (pTorA-30aa-MalE, pHSG-TatABCE) was set to 100%. The lane numbers in E correspond to the lane numbers in A–D.
As described previously (23, 30), the mutation L9F in TatC was sufficient to partially restore export of the otherwise translocation-defective TorA[KQ]-30aa-MalE precursor protein into the periplasm (8.8%), allowing moderate growth on MMM and the formation of pink colonies on MCM of the respective strain (Fig. 4, lane 4; Fig. S3, sector 4). The presence of the additional mutations in the 30aa-region, however, resulted in a substantial increase in the translocation efficiency of the TorA[KQ]-30aa-MalE precursor protein by the mutant TatABC[L9F]E translocase. Although comparatively low translocation efficiencies of the TorA[KQ]-30aa-MalE reporter variants harboring the various 30aa-region-located suppressing mutations were promoted by the WT Tat-translocase resulting in moderate growth on MMM and the formation of pink colonies on MCM at best, all strains carrying the mutated tatABC[L9F]E genes instead of the WT tat genes exhibited a uniform phenotype on the indicator plates characterized by efficient growth on MMM and red-colored colonies on MCM (Fig. 4, A–D, compare even-numbered with odd-numbered lanes 5–20; Fig. S3, compare even-numbered with odd-numbered sectors 5–20). Quantification of the exported MalE amounts in the periplasm of these strains further showed that the respective 30aa-region-located mutations and the amino acid substitution L9F in TatC act even synergistically in the suppression of the export defect of TorA[KQ]-30aa-MalE (Fig. 4E). As shown in Fig. 4, A–D, various MalE-derived polypeptides corresponding to the unprocessed precursor and its cytosolic degradation products can be detected in the C/M fractions of GSJ101 expressing either the unaltered TorA-30aa-MalE reporter (positive control; lanes 1 and 2), the export-defective TorA[KQ]-30aa-MalE (negative control; lanes 3 and 4), or the various TorA[KQ]-30aa-MalE reporter variants harboring 30aa-region-located suppressor mutations (lanes 5–20) in the presence of the WT or mutated TatABC[L9F]E translocase. Except for the unaltered TorA-30aa-MalE protein, which was efficiently exported by both translocases, visible amounts of mature-sized MalE were only observed in the P fractions of GSJ101 possessing the mutant TatABC[L9F]E translocase, confirming that the exported amounts of the various 30aa-region-coupled suppressor mutant reporters by the WT Tat translocase are indeed very low and not visible to the naked eye in the Western blot analysis of the respective periplasmic fractions. Strikingly, in the case of two TorA[KQ]-30aa-MalE reporter variants that harbored the amino acid substitution K50L in their 30aa-region (i.e. iKQ-K50L and iKQ-T44I/K50L), two protein bands of similar signal intensity could be identified in the P fraction of the respective strains corresponding to the MalE protein lacking its TorA[KQ] signal peptide and a second, smaller-sized processing product (Fig. 4D, lanes 10 and 20). Possibly, the mutation K50L introduces a recognition and cleavage site for a periplasmic protease that causes an N-terminal truncation of some mature MalE proteins. In these cases, both chemiluminescence signals were quantified and summarized to obtain the actual export efficiency of these TorA[KQ]-30aa-MalE reporter variants by the mutant TatABC[L9F]E translocase.
Quantification of the chemiluminescence signals in the P fractions of all analyzed strains further confirmed that the translocation efficiencies of the various TorA[KQ]-30aa-MalE reporter variants with single or double mutations in the 30aa-region promoted by the WT Tat translocase were much lower compared with the ones observed for the TatABC[L9F]E translocase, with values between 0.8% determined for the weakest suppressor mutant reporter iKQ-D45L and 4.5% for the double-mutated protein iKQ-T44I/T65R (Fig. 4E, odd lanes 5–19). As shown in Fig. 4E, even lanes 6–20, except for D68R, the respective amino acid substitutions in the 30aa-region and the TatC mutation L9F were found to synergistically contribute to the suppression of the TorA[KQ]-30aa-MalE export defect because the export efficiencies of the 30aa-region-coupled suppressor mutant reporters by the mutant TatABC[L9F]E translocase, with values between 14.9% (iKQ-T65R) and 45.0% (iKQ-T44I/K50L), were strikingly higher than the sum of the translocation efficiencies conferred by the TatC mutation L9F (8.8%) and the respective 30aa-region-located mutations (0.8%–4.5%). In case of iKQ-D68R, however, the additional mutation in the TatBC receptor complex had a rather additive effect resulting in a comparatively small increase of the export from 1.6 to 10.2% (Fig. 4E, lanes 13 and 14). In comparison, the strongest increase in translocation efficiency (from 4.1 to 45.0%) could be observed for the reporter variant iKQ-T44I/K50L characterized by the highest overall hydrophobicity of the N-terminal part of the 30aa-region (Fig. 4E, lanes 19 and 20).
Evidently, combinations of the amino acid substitution L9F in TatC with the suppressing mutations in the early mature protein region, which introduce hydrophobic residues into the N-terminal half of the 30aa-region and thus likely exert their effect by enhancing the hydrophobic substrate–receptor interactions within the TatBC-binding cavity, have a stronger positive effect on the transport efficiency of the normally export-defective TorA[KQ]-30aa-MalE reporter than combinations of L9F with mutations located in the C-terminal part of the 30aa-region, which likely act rather differently, probably by improving the interaction of the Tat substrate with a polar, cytoplasmically-localized domain of the substrate receptor (i.e. T65R and D68R).
Amino acid substitutions in the cytosolic domains of TatB restore export of various transport-defective Tat precursor proteins
As mentioned above, some of the isolated suppressing mutations increased the positive net charge of the C-terminal part of the 30aa-region of the respective TorA[KQ]-30aa-MalE mutant reporters by replacing neutral or negatively charged amino acids with one or two basic arginine residues (i.e. T65R, D68R, and T55R/T65R). Although the N-terminal half of the 30aa-region is likely embedded within the TatBC-binding pocket due to the deep hairpin loop-like insertion of the TorA signal peptide and some amino acid residues of the early mature protein part at the stage of advanced binding, the more distal C-terminal part of this early mature protein region might be located outside this cavity in the cytoplasm. Because in vitro cross-linking studies revealed that the folded mature protein domain of Tat substrates is encapsulated by the cytoplasmically-localized domains of TatB monomers prior to translocation (32), we speculated that the additional positively charged residues in the 30aa-region of TorA[KQ]-30aa-MalE might enhance the electrostatic interactions between oppositely charged amino acid side chains residing at the interface between the surrounded (early) mature part of the Tat substrate and the circumjacent cytoplasmic regions of TatB.
The predicted structure of E. coli TatB shows an elongated “L-shape” conformation consisting of four well-defined α-helices as follows: a TMH (residues Ser-7–Leu-19) α1; an APH (residues Val-27–Gln-47) α2; and two highly hydrophilic and flexible helices α3 (Leu-56–Leu-71) and α4 (Glu-77–Tyr-96) (42). To investigate whether the C-terminal domains of TatB following the transmembrane segment contribute to productive substrate binding, we performed a genetic screen for suppressing mutations located in these regions that were able to restore transport of the TorA[KQ]-30aa-MalE precursor protein. For this purpose, the respective tatB gene fragment within the tatABCE operon on plasmid pHSG-TatABCE was randomly mutagenized by ep-PCR. The resulting pool of pHSG-TatABCE plasmid variants was then used for transformation of GSJ101 (pTorA[KQ]-30aa-MalE) expressing the transport-defective TorA[KQ]-30aa-MalE reporter variant. For the selection of TatB-coupled suppressors of the TorA[KQ]-30aa-MalE export defect, the transformants were plated onto MMM agar plates that were subsequently incubated for 4 days. Twenty five clones that appeared on the selection plates were randomly chosen, tested for reproducible growth on MMM, and further analyzed with respect to the DNA sequence of the mutated tatB gene.
The identified amino acid substitutions in TatB appeared to cluster within the APH, including I36N that occurred in two clones and R37H that appeared in five clones, whereas P26L, K30I, and A38E were each found once (Table 2; Fig. 5, green dots). It should be mentioned that the amino acid substitutions K30I and I36N have been already identified as suppressors of inactivating mutations in the AmiA or SufI signal peptide in an independent recent study by Huang et al. (43). We also identified mutations that were located in the more distal C-terminal domains of TatB encompassing helices α3 and α4 and the respective intermediate regions. Substitutions T51A and T51I, isolated two and four times, respectively, resided in the linker region between the APH and helix α3, whereas the adjacent mutations T75M and P76L, each found four times, were located within the intermediate sequence connecting helices α3 and α4. The last identified substitution A89V appeared only in one of the isolated clones and mapped to the carboxy-proximal helix α4.
Table 2.
Export of various TorA-MalE derivatives by the isolated Tat translocases harboring suppressing mutations in the cytosolic domains of TatB
The export of the various TorA-MalE derivatives by the unaltered Tat translocase and the mutated TatAB[X]CE (with X being the respective amino acid alteration in TatB) translocases was analyzed by a MMM plate assay in E. coli GSJ101. Phenotypes of the respective strains on MMM after 24 and 48 h of incubation: −, no growth; +, slow growth; ++, moderate growth; +++, good growth.
| Suppressing mutation X in TatB | Phenotype of E. coli GSJ101 on MMM co-expressing TatAB[X]CE and the reporter variant: |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TorA-30aa-MalE |
TorA[KQ]-30aa-MalE |
TorA[KQ]-MalE |
TorA[QQ]-MalE |
TorA[KQ]-30aa(NapA)-MalE |
iKQ-K50L |
iKQ-T55R, T65R |
iKQ-D68R |
|||||||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| None | +++ | +++ | − | − | − | − | − | − | − | − | + (+) | +++ | + (+) | +++ | + | ++ (+) |
| P26L | +++ | +++ | − | ++ | − | + | − | − | + | ++ | ++ | +++ | ++ (+) | +++ | + (+) | +++ |
| K30I | +++ | +++ | (+) | ++ (+) | − | + | − | − | (+) | ++ | ++ (+) | +++ | ++ | +++ | ++ (+) | +++ |
| I36N | +++ | +++ | (+) | ++ (+) | (+) | ++ (+) | − | − | + | +++ | ++ | +++ | ++ | +++ | ++ | +++ |
| R37H | +++ | +++ | − | ++ (+) | − | + (+) | − | − | + | +++ | ++ | +++ | ++ (+) | +++ | + (+) | +++ |
| A38E | +++ | +++ | − | ++ (+) | − | ++ (+) | − | − | + | +++ | ++ | +++ | ++ (+) | +++ | ++ (+) | +++ |
| T51A | +++ | +++ | − | + (+) | − | + | − | − | + | ++ | + (+) | +++ | + (+) | +++ | + (+) | +++ |
| T51I | +++ | +++ | − | ++ | − | ++ | − | − | + | +++ | + (+) | +++ | ++ (+) | +++ | ++ (+) | +++ |
| T75M | +++ | +++ | − | ++ | − | + | − | − | + | ++ | ++ | +++ | ++ | +++ | ++ | +++ |
| P76L | +++ | +++ | − | ++ | − | ++ | − | − | + | +++ | ++ | +++ | ++ | +++ | ++ | +++ |
| A89V | +++ | +++ | − | + | − | + | − | − | (+) | ++ | ++ | +++ | ++ | +++ | + (+) | +++ |
| A | B | C | D | E | F | G | H | |||||||||
Figure 5.
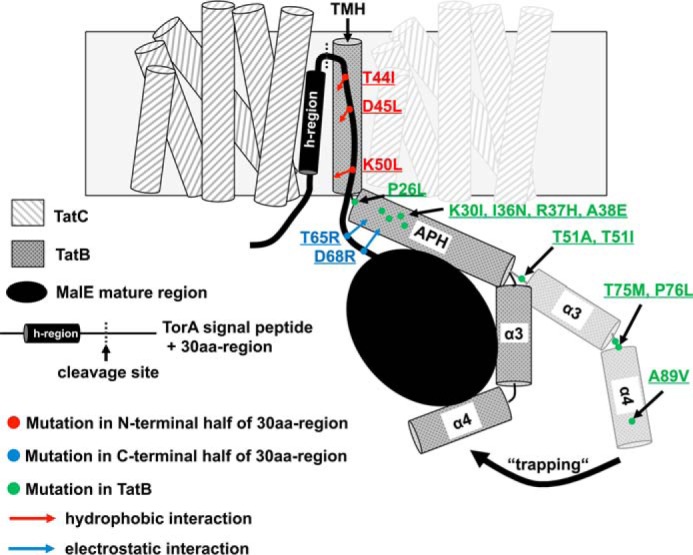
Model of possible TatBC–precursor interactions. The framed light gray bar represents the lipid bilayer. For clarity, only one TatB monomer (checkered cylinder) and two adjacent TatC monomers (diagonally hatched cylinders) of the multimeric TatBC receptor complex are shown. Each TatC monomer is depicted by six transmembrane helices. The TatB monomer consists of a membrane-embedded transmembrane helix (TMH), a cytosolic amphipathic helix (APH), and flexible C-terminal domains (helices α3 and α4) encapsulating the folded Tat substrate. The membrane-embedded TorA signal peptide with the h-region forming an α-helix (black cylinder) and the following 30aa-region of the TorA-30aa-MalE precursor protein is represented by a black line; the folded MalE mature region is represented by a black ellipse. The positions of selected suppressor mutations in the membrane-embedded N-terminal half of the 30aa-region are indicated by red dots, substitutions in the cytoplasmically-located C-terminal half of the 30aa-region by blue dots, and the TatB-located suppressing mutations of the TorA[KQ]-30aa-MalE export defect by green dots. The indicated positions of the newly introduced hydrophobic (red arrows) or electrostatic (blue arrows) precursor–TatBC interactions by the 30aa-region-located mutations are speculative. The effects of the more C-terminal TatB mutations (T51A, T51I, T75M, P76L, and A89V) on the orientation/conformation of the flexible cytosolic domains are likewise hypothetical. A possible scenario could be that, upon insertion of the signal peptide and the partially unfolded N-terminal part of the early mature protein into the hydrophobic TatBC-binding cavity, these substitutions alter the conformation or orientation of the C-terminal domains of TatB such that they allow them to wrap more tightly around the folded part of the non-native substrate MalE, thereby promoting a stronger fixation (“trapping”) of the otherwise too weakly bound Tat precursor to the Tat translocase.
First, the amounts of the proteins TatA, TatB, and TatC in the membrane fraction of the corresponding E. coli strains were analyzed by Western blotting using specific antibodies (Fig. S4). In all cases, the amounts of the Tat proteins present in the membrane fractions of the cells expressing the mutant Tat translocases were equal or slightly lower compared with the amounts of the respective Tat proteins in the membrane fraction of the cells expressing a WT Tat translocase. This finding excludes that the observed suppression of the export defect of TorA[KQ]-30aa-MalE by the various mutations in TatB is due to an increased expression level of the Tat proteins in the strains possessing the mutant Tat translocases and indicates that suppression is indeed caused by the corresponding amino acid substitutions in TatB.
Second, we tested the ability of the identified TatB-located suppressing mutations to also support export of other transport-defective derivatives of the TorA-30aa-MalE reporter protein (Table 2; Fig. S5, A–D). To determine whether the observed suppression was, at least in some cases, specific for the early mature protein region of TorA, we generated another construct where a different 30aa-region comprising the first 30 amino acids of the mature part of the nitrate reductase NapA, which is also an authentic Tat substrate in E. coli, was fused in between the TorA signal peptide and the mature region of MalE (TorA[KQ]-30aa(NapA)-MalE) (Fig. S5, B and E). Furthermore, we additionally chose the reporter variants TorA[KQ]-MalE and TorA[QQ]-MalE in which the respective mutated signal peptide and the first eight amino acids of mature TorA were fused to the mature part of MalE via a linker region consisting of the three amino acids Glu, Phe, and Asp (Fig. S5, C, D, and F). Compared with TorA[KQ]-MalE, TorA[QQ]-MalE harbors a far less conservative QQ substitution of the crucial twin arginine residues in the TorA signal peptide, likewise resulting in a complete defect in transport by the WT Tat translocase. Finally, to test whether the TatB substitutions can cooperate with the previously identified mutations in the 30aa-region in the suppression of the TorA[KQ]-30aa-MalE export defect, we also analyzed the export of the 30aa-region-coupled suppressor mutant reporters iKQ-K50L, iKQ-T55R/T65R, and iKQ-D68R by the various mutant Tat translocases.
As shown in Table 2, the export of the unaltered TorA-30aa-MalE precursor protein was not significantly affected by the various mutations in the TatB component of the Tat translocase (column A). As noted above, each of the selected mutant Tat translocases allowed for the transport of the normally export-defective TorA[KQ]-30aa-MalE precursor, albeit with various efficiencies, as judged by the regained ability of the respective strains to significantly grow with maltose as the sole carbon and energy source after up to 48 h of incubation (Table 2, column B). These in situ suppressor phenotypes were generally stronger or comparable with those observed with GSJ101 coexpressing the various Tat translocases together with the reporter variant TorA[KQ]-MalE, demonstrating that, in many cases, the presence of a shorter part (i.e. only the first 8 amino acid residues) of the early mature region of TorA clearly decreased the export efficiency by the mutant Tat translocases (Table 2, compare columns B and C). Because the first 8 amino acids of the mature TorA protein part are most likely sequestered within the intramembrane TatBC-binding groove and all isolated substitutions in the cytosolic TatB domains still significantly rescued growth on MMM also in the absence of the cytoplasmically-located portion of the early mature TorA region (i.e. C-terminal half of the full-length 30aa-region) in the TorA[KQ]-MalE precursor, these suppressing mutations obviously do not act by specifically introducing new or enhancing already existing binding contacts between the early mature region of TorA and the respective cytosolic domain of TatB. Strikingly, all TatB substitutions promoted even more efficient export of TorA[KQ]-30aa(NapA)-MalE, harboring a completely different 30aa-region compared with the TorA[KQ]-30aa-MalE precursor protein (against which they were originally isolated), as reflected by generally faster growth rates on MMM of the respective strains (Table 2, compare columns B and E). The latter finding therefore further supports the assumption that the observed suppression by the TatB mutations is in fact not specific for the early mature protein region of TorA and indicates that, compared with the early mature region of TorA, the first 30 amino acid residues of the mature NapA protein might generally promote higher-affinity binding of the Tat substrate to the TatBC receptor complex, being responsible for the significantly increased export efficiency by the various Tat translocases. Finally, none of the isolated TatB-located suppressing mutations was able to suppress the QQ substitution in the TorA signal peptide variant of the TorA[QQ]-MalE precursor protein, suggesting that the respective suppressing activities were not sufficient to compensate for this nonconservative exchange of the crucial arginine residues in the Tat motif of the TorA signal peptide (Table 2, column D).
Interestingly, in nearly all cases, the identified TatB suppressor mutations further improved export of the TorA[KQ]-30aa-MalE reporter variants harboring the amino acid substitutions K50L, T55R/T65R, or D68R, respectively, in their 30aa-region (Table 2, columns F–H). Accordingly, the respective strains GSJ101 coexpressing these reporter variants and the various mutant Tat translocases exhibited significantly faster growth rates on MMM already after 24 h of incubation compared with GSJ101 (pHSG-TatABCE), which possessed a WT Tat translocase. Because none of the TatB-located suppressing mutations was, by itself, sufficient to rescue detectable growth on MMM within the first 24 h (Table 2, column B), we conclude that the mutations in the 30aa-region and TatB tightly cooperate with each other in restoring export of the TorA[KQ]-30aa-MalE precursor protein. Although we do not have direct experimental evidence for that, we consider it likely that all these mutations facilitate productive binding of this nonnative substrate to the TatBC receptor complex either directly by strengthening the substrate–receptor interactions or, in case of TatB substitutions, possibly also indirectly by somehow altering the conformation of the TatBC substrate receptor now being capable of exporting the otherwise too weakly bound precursor into the periplasm.
Although no specific binding contacts between the early mature region of Tat substrates and the cytosolically-localized domains of TatB could be identified so far, we found that even suppressing mutations located in the highly flexible carboxy-proximal regions of TatB encompassing helices α3 and α4 and the respective intermediate regions can significantly restore export of an otherwise transport-defective Tat precursor. A possible scenario could be that these mutations act by promoting an optimized adaptation of the flexible hydrophilic C-terminal helices to the folded nonnative Tat substrate MalE, providing a first indication that interactions between these cytosolic domains of TatB and the mature region of a Tat precursor generally contribute to productive substrate binding.
Discussion
In this study, we addressed the role of the early mature part of Tat substrates in E. coli TatBC receptor binding by using an in vivo genetic approach. To examine whether the protein region immediately following the signal peptide contributes to the productive interaction of a Tat precursor with the substrate receptor, we aimed to identify mutations within the first 30 residues of the mature TorA protein (so-called 30aa-region) which restore export of a TorA[KQ]-30aa-MalE Tat precursor, in which the crucial RR residues have been substituted by a lysine–glutamine pair. Recently, we identified such mutations in the hydrophobic region of the TorA signal peptide that significantly and, when combined, even synergistically suppressed the export defect of the TorA[KQ]-30aa-MalE precursor protein by increasing the overall hydrophobicity of this central hydrophobic stretch. Assuming that the binding affinity of the TorA[KQ]-30aa-MalE precursor was too low to establish a productive interaction with the TatBC receptor complex resulting in effective transport, we showed that upon insertion into the TatBC-binding cavity, the newly introduced hydrophobic residues in the h-region restored productive binding of the otherwise transport-incompetent Tat precursor by enhancing the hydrophobic interactions between the signal peptide and residues encompassing the hydrophobic signal peptide-binding site on the TatBC substrate receptor (30).
In this work, we identified 54 different single or double amino acid substitutions in the 30aa-region, each of which was able to significantly restore Tat-dependent export of a TorA[KQ]-30aa-MalE precursor protein, providing the first indirect evidence for interaction of the early mature protein region with the Tat translocase. However, the observed export efficiencies were considerably lower than those conferred by the previously identified mutations in the h-region of the TorA signal peptide (30), suggesting that binding of the early mature protein region to the TatBC substrate receptor occurs with a significantly lower affinity. In line with these results, Alami et al. (13) reported contacts between TatB and an amino acid position more than 20 residues beyond the signal peptide cleavage site. Whereas interaction of the signal peptide with TatB was independent of the amount of TatB, the formation of cross-links of TatB with the mature part of a Tat substrate required a high TatB expression level to become detectable (13).
Interestingly, a common trend in the distribution of the 30aa-region-located substitutions could be observed. Mutations that replaced hydrophilic amino acids with hydrophobic residues were predominantly located in the N-terminal half of the 30aa-region immediately following the signal peptide, whereas substitutions that introduced positively charged amino acids preferentially occurred within the distal C-terminal part of this early mature region. It is therefore tempting to speculate that both types of suppressing mutations act in a different manner.
Numerous biochemical studies revealed that, once the Tat signal peptide has been recognized by the TatBC receptor complex, it remains N-terminally anchored to the surface of the Tat translocase and is threaded deep into the TatBC-binding cavity resulting in the formation of a hairpin loop topology consisting of the signal peptide and, depending on the depth of insertion that might actually differ for different precursor proteins, a certain number of amino acid residues beyond the signal peptide cleavage sites that are derived from the early mature protein region (16, 29). Furthermore, the first 10 amino acids of the mature TorA protein, which are thus likely sequestered within the TatBC-binding groove, were exclusively found in close vicinity of TatB (17), suggesting a juxtaposition of the early mature region with the transmembrane domain of TatB. One might therefore speculate that at least some part of the early mature region provides a potential interaction site for residues encompassing the membrane-embedded hydrophobic TatBC substrate-binding site. Such a precursor topology as shown in Fig. 5 prompted us to hypothesize that, similar to the previously reported mutations in the h-region of the TorA signal peptide (30), the newly introduced hydrophobic residues in the N-terminal part of the early mature TorA region might enhance the hydrophobic substrate–receptor interactions within the intramembrane TatBC-binding groove and thus, if correct, then sufficiently increase the overall binding affinity required for productive binding of the otherwise export-defective TorA[KQ]-30aa-MalE precursor protein (Fig. 5, red arrows). In support of this hypothetical working model for the mode of action of these amino acid substitutions, we showed that a combination of the two single suppressing mutations T44I and K50L, leading to a further increase of the overall hydrophobicity of this protein region (as reflected by a substantially higher GRAVY index, see Table S2), clearly improves export of the TorA[KQ]-30aa-MalE reporter and, in addition, that the amino acid substitutions in the early mature region tightly cooperate both with the previously identified suppressing amino acid substitutions in the h-region (30) and with the extragenic suppressor mutation L9F located in the cytosolically exposed N terminus of TatC (23, 38).
A different mode of action might be speculated for the amino acid substitutions located in the more distal part of the 30aa-region that seem to exert their suppressing activities by increasing the regions' positive net charge (Table S2, rows 6–8). Because only few residues of the mature region are thought to be sequestered within the TatBC-binding cavity upon signal peptide insertion, the vast majority of binding of the Tat substrate to the TatBC receptor likely occurs outside this groove in the cytoplasm (Fig. 5). Along this line, Maurer et al. (32) reported multiple contact sites both on the predicted transmembrane and amphipathic helices of TatB and on the major part of a Tat precursors' surface, suggesting a cage-like structure of the cytosolic TatB domains transiently accommodating the folded Tat substrate prior to its translocation. Although these cross-linking studies indicated an association of TatB with the mature part of the Tat precursor protein, they did not assess whether these interactions actually contribute to productive substrate binding by the TatBC receptor complex. Our present work might thus provide a first although indirect indication that electrostatic interactions between the early mature region and the hydrophilic cytoplasmic domains of TatB indeed support precursors binding to the Tat translocase (Fig. 5, blue arrows).
For the Sec pathway, Chatzi et al. (44) recently showed that the mature domains of unfolded translocation-competent Sec substrates harbor essential hydrophobic targeting signals that interact with SecA prior to secretion. First experimental evidence that such targeting signals might also exist in Tat precursors is provided by a study by Huang et al. (43) who identified mutations in TatB that allowed Tat-dependent export of a signal peptide-less substrate. Because proper membrane targeting was not impaired by the lack of the Tat signal peptide, at least some targeting information must be located in the mature region of the Tat substrate. Because of the fully folded conformation of targeting-competent Tat precursors, such Tat targeting signals beyond the signal peptide might rather consist of surface-exposed hydrophilic residues than of hydrophobic amino acids being predominantly hidden in the core of the folded substrate. The observed suppression of the TorA[KQ]-30aa-MalE export defect by the second type of 30aa-region-located mutations could therefore also be explained by the speculative but plausible scenario where the newly introduced positively charged residues in the mature TorA domain further optimize such a hydrophilic targeting sequence being directly involved in productive recognition and binding of the Tat substrate by the Tat translocase. Consistent with the finding that folded Tat substrates are entirely encapsulated by TatB monomers prior to their translocation (32), the respective receptor-binding site could be realized by hydrophilic stretches residing in the cytosolic domains of TatB, provided that these regions actually contribute to the recognition/binding of Tat substrates by the TatBC receptor complex. To address this issue, we performed a genetic screen for mutations specifically located in the C-terminal domains following the TMH of TatB, which can suppress the export defect of a TorA[KQ]-30aa-MalE precursor protein. Indeed, several amino acid substitutions could be identified, which allowed for low but nevertheless detectable export into the periplasm in the absence of the RR residues. Mutations present in half of the isolated clones mapped to the predicted APH of TatB, and the remaining amino acid substitutions were distributed over the more distal C-terminal helices and the respective linker regions (Fig. 5, green dots). In full agreement with the results of several truncation analyses (45, 46), no suppressing mutations were identified in the extreme C terminus of TatB beyond residue 101, which was found not to be essential for TatB function. Strikingly, suppression of the TorA[KQ]-30aa-MalE export defect by the various TatB substitutions was not drastically impaired by a severe truncation of the 30aa-region. Furthermore, even the replacement of the 30aa-region of TorA by a completely unrelated 30aa-region of another Tat substrate, NapA, did not abolish the suppressing activities of the mutated TatB proteins, clearly indicating that none of the isolated TatB mutations restored export of the corresponding export-defective precursor proteins by strengthening specific interactions between the respective early mature protein regions and TatB, but that rather must have acted in a different way. Interestingly, all TatB substitutions showed an even stronger suppressing activity toward the export-defective Tat substrate when the 30aa-region from NapA was present. Although the underlying reason for this observation is unknown, one might speculate that, compared with the early mature region of TorA, the first 30 amino acid residues of the mature NapA protein might intrinsically possess a higher affinity to the TatBC receptor complex. If so, then this might generally imply that the contribution of the early mature part of Tat substrates to TatBC receptor binding significantly differs between different Tat precursor proteins.
The strongest suppressor phenotypes were promoted by the amino acid substitutions K30I and I36N in the APH of TatB. Interestingly, exactly the same mutations were previously found to also suppress the export defect of AmiA and SufI Tat precursor proteins carrying inactivating substitutions in their signal peptides (43), demonstrating that these mutations evidently act in a rather indirect and substrate-unspecific manner. Recent studies showed that the TMHs of TatB and TatA share a common binding site on TatC, which is occupied by TatB in the absence of substrate binding. Signal peptide binding to the TatBC complex is suggested to trigger movement of the TMH of TatB from this binding site allowing TatA to bind, oligomerize, and induce formation of an active TatABC translocase (43, 47). Because the orientation between the TMH and APH of TatB was found to be rather rigid (42), substitutions in the APH might indeed lead to slight conformational alterations of the TatBC-binding cavity, thereby reducing the affinity of the TatB TMH for the respective binding site on TatC, as was previously proposed for the TatB mutations K30I and I36N, which restored some export of otherwise export-defective AmiA and SufI Tat precursor variants (43). As a consequence, weak residual binding of the mutated signal peptide was found to be sufficient to trigger these essential conformational changes in the TatBC substrate receptor leading to effective transport of the normally export-defective Tat precursor across the membrane.
Although such an indirect and substrate-unspecific effect on the conformation of the hydrophobic TatBC substrate-binding pocket might likewise be caused by the other mutations found in the APH and the preceding linker region, we, however, regard it less likely for the remaining mutations located in the more distal C-terminal regions of TatB. Studies on TatB structure revealed that, in contrast to the TMH and APH of TatB, the two C-terminal helices α3 and α4 are highly flexible and thus can change their relative orientations by flexible movements at the respective connecting regions for an optimal adaptation to various sizes and shapes of Tat substrates (42). It is therefore difficult to imagine that mutations in these flexible parts of TatB could have such a long range conformational effect that is transmitted to and alters positioning of the TMH of TatB with respect to the TatA/TatB-binding site on TatC. We would rather speculate that, upon insertion of the signal peptide and the partially unfolded N-terminal part of the early mature protein into the TatBC-binding cavity, these substitutions alter the conformation of the C-terminal domains of TatB allowing them to wrap more tightly around the folded part of the nonnative substrate MalE, thereby promoting a stronger fixation of the otherwise too weakly bound Tat precursor to the Tat translocase, as indicated in the model depicted in Fig. 5. Because significant MalE export was restored by these mutations, these results provide a first genetic evidence that association of the cytosolically-localized domains of TatB with folded Tat substrates indeed contributes to the productive interaction of Tat precursors with the TatBC receptor complex.
Taken together, the results of our genetic approach strongly support the view that productive recognition and binding of Tat substrates by the Tat translocase does not exclusively depend on the Tat signal peptide but also involves a more extended area encompassing at least the early mature protein region. Thus, the early mature part is not only a passenger domain playing a passive role in the binding process but also seems to contribute to the productive binding of the Tat substrate to the TatBC receptor complex. Likewise, domains of the receptor complex located outside the hydrophobic TatBC-binding cavity were found to be involved in substrate binding, providing further evidence that the TatBC receptor complex forms a large binding pocket that accommodates the entire Tat substrate.
Experimental procedures
Bacterial strains, plasmids, and culture conditions
E. coli strains and plasmids used in this study are listed in Table 3. Cells were grown at 37 °C in Luria Bertani medium (48), minimal medium (49) supplemented with 0.4% (w/v) maltose (MMM), or MacConkey agar base medium (Difco) supplemented with 1% (w/v) maltose (MCM). If required, isopropyl β-d-thiogalactopyranoside was used at a 0.1 mm concentration. Antibiotics were supplemented at the following concentrations: kanamycin, 50 mg/liter; chloramphenicol, 25 mg/liter.
Table 3.
Bacterial strains and plasmids used in this study
| Relevant propertiesa | Source | |
|---|---|---|
| E. coli strains | ||
| DH5α | supE44, ΔlacU169 (φ80 lacZΔM15) hsdR17 recA1 endA1 hsdR gyrA relA thi | 51 |
| GSJ100 | MC4100 X P1.MM129>TcR ΔmalE444 zjb729::Tn10 | 40 |
| GSJ101 | DADE X P1.MM129>TcR ΔmalE444 zjb729::Tn10 | 40 |
| Plasmids | ||
| pHSG575 | pSC101 replicon, lacZα+; CmR | 52 |
| pHSG-TatABCE | pHSG575 derivative; carrying the tatABCE genes of E. coli | 41 |
| pHSG-TatABC[L9F]E | pHSG-TatABCE harboring L9F mutation in TatC | 23 |
| pHSG-TatAB[P26L]CE | pHSG-TatABCE harboring P26L mutation in TatB | This study |
| pHSG-TatAB[K30I]CE | pHSG-TatABCE harboring K30I mutation in TatB | This study |
| pHSG-TatAB[I36N]CE | pHSG-TatABCE harboring I36N mutation in TatB | This study |
| pHSG-TatAB[R37H]CE | pHSG-TatABCE harboring R37H mutation in TatB | This study |
| pHSG-TatAB[A38E]CE | pHSG-TatABCE harboring A38E mutation in TatB | This study |
| pHSG-TatAB[T51A]CE | pHSG-TatABCE harboring T51A mutation in TatB | This study |
| pHSG-TatAB[T51I]CE | pHSG-TatABCE harboring T51I mutation in TatB | This study |
| pHSG-TatAB[T75M]CE | pHSG-TatABCE harboring T75 M mutation in TatB | This study |
| pHSG-TatAB[P76L]CE | pHSG-TatABCE harboring P76L mutation in TatB | This study |
| pHSG-TatAB[A89V]CE | pHSG-TatABCE harboring A89V mutation in TatB | This study |
| pTorA-MalE | pBBR1MCS-2 carrying the torA-malE fusion gene, KmR | 40 |
| pTorA-30aa-MalE | pBBR1MCS-2 carrying the torA-30aa-malE fusion gene with linker region encoding the first 30 aa of mature TorA, KmR | 30 |
| pNapA-30aa(NapA)-MalE | pBBR1MCS-2 carrying the napA-30aa-malE fusion gene with linker region encoding the first 30 aa of mature NapA, KmR | This study |
| pTorA-30aa(NapA)-MalE | pBBR1MCS-2 carrying the torA-30aa(napA)-malE fusion gene with linker region encoding the first 30 aa of mature NapA, KmR | This study |
| pTorA[KQ]-30aa(NapA)-MalE | pTorA-30aa(NapA)-MalE (R11K and R12Q) | This study |
| pTorA[KQ]-MalE | pTorA-MalE (R11K and R12Q) | 23 |
| pTorA[QQ]-MalE | pTorA-MalE (R11Q and R12Q) | This study |
| pTorA[KQ]-30aa-MalE | pTorA-30aa-MalE (R11K and R12Q) | 30 |
| pTorA[KQ,A16V]-30aa-MalE | pTorA-30aa-MalE (R11K, R12Q, A16V) | 30 |
| pTorA[KQ,T22A]-30aa-MalE | pTorA-30aa-MalE (R11K, R12Q, T22A) | 30 |
| pTorA[KQ,G25W]-30aa- | pTorA-30aa-MalE (R11K, R12Q, G25W) | 30 |
| pTorA[KQ,G28W]-30aa-MalE | pTorA-30aa-MalE (R11K, R12Q, G28W) | 30 |
| pTorA[KQ,G25W/G28W]-30aa-MalE | pTorA-30aa-MalE (R11K, R12Q, G25W, G28W) | 30 |
| pTorA[KQ]-30aa[T44I]-MalE | pTorA-30aa-MalE (R11K, R12Q, T44I) | This study |
| pTorA[KQ]-30aa[D45L]-MalE | pTorA-30aa-MalE (R11K, R12Q, D45L) | This study |
| pTorA[KQ]-30aa[H58I]-MalE | pTorA-30aa-MalE (R11K, R12Q, H58I) | This study |
| pTorA[KQ]-30aa[K50L]-MalE | pTorA-30aa-MalE (R11K, R12Q, K50L) | This study |
| pTorA[KQ]-30aa[T55R/T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, T55R, T65R) | This study |
| pTorA[KQ]-30a[T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, T65R) | This study |
| pTorA[KQ]-30aa[D68R]-MalE | pTorA-30aa-MalE (R11K, R12Q, D68R) | This study |
| pTorA[KQ]-30aa[T44I/K50L]-MalE | pTorA-30aa-MalE (R11K, R12Q, T44I, K50L) | This study |
| pTorA[KQ]-30aa[T44I/T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, T44I, T65R) | This study |
| pTorA[KQ]-30aa[K50L/T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, K50L, T65R) | This study |
| pTorA[KQ]-30aa[D45L/D68R]-MalE | pTorA-30aa-MalE (R11K, R12Q, D45L, D68R) | This study |
| pTorA[KQ]-30aa[K50L/D68R]-MalE | pTorA-30aa-MalE (R11K, R12Q, K50L, D68R) | This study |
| pTorA[KQ]-30aa[K50L/T55R/T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, K50L, T55R, T65R) | This study |
| pTorA[KQ,A16V]-30aa[H58I]-MalE | pTorA-30aa-MalE (R11K, R12Q, A16V, H58I) | This study |
| pTorA[KQ,T22A]-30aa[T65R]-MalE | pTorA-30aa-MalE (R11K, R12Q, T22A, T65R) | This study |
| pTorA[KQ,T22A]-30aa[D68R]-MalE | pTorA-30aa-MalE (R11K, R12Q, T22A, D68R) | This study |
| pTorA[KQ,G28W]-30aa[H58I]-MalE | pTorA-30aa-MalE (R11K, R12Q, G28W, H58I) | This study |
| pTorA[KQ,G25W]-30aa[K50L]-MalE | pTorA-30aa-MalE (R11K, R12Q, G25W, K50L) | This study |
| pTorA[KQ,G28W]-30aa[K50L]-MalE | pTorA-30aa-MalE (R11K, R12Q, G28W, K50L) | This study |
| pTorA[KQ,G25W/G28W]-30aa[K50L]-MalE | pTorA-30aa-MalE (R11K, R12Q, G25W, G28W, K50L) | This study |
a KmR is kanamycin resistance; CmR is chloramphenicol resistance.
DNA manipulations
All DNA manipulations followed standard procedures (50). Oligonucleotides used in this study are listed in Table S3. The correctness of all newly constructed plasmids was verified by DNA sequencing. Plasmids pTorA[KQ]-30aa[T44I/K50L]-MalE, pTorA[KQ]-30aa[K50L/T65R]-MalE, pTorA[KQ]-30aa[K50L/D68R]-MalE, and pTorA[KQ]-30aa[K50L/T55R/T65R]-MalE harboring double or triple mutations in the 30aa-region were constructed by introducing the amino acid substitution K50L into pTorA[KQ]-30aa[X]-MalE with X being the single mutation T44I, T65R, D68R, or the double mutation T55R/T65R, respectively, using the QuikChange®II XL site-directed mutagenesis kit (Agilent) and the primers 30aa_K50L_qc_fw and 30aa_K50L_qc_rev, according to the manufacturer's instructions. The combinations of the 30aa-region-located mutations T44I and T65R or D45L and D68R were constructed by the same procedure with pTorA[KQ]-30aa[T65R]-MalE and pTorA[KQ]-30aa[D68R]-MalE, respectively, as templates and primer pairs 30aa_T44I_qc_fw/rev and 30aa_D45L_qc_fw/rev, respectively, resulting in plasmids pTorA[KQ]-30aa[T44I/T65R]-MalE and pTorA[KQ]-30aa[D45L/D68R]-MalE.
Likewise, the combination of the h-region-located mutations A16V and G28W, respectively, with the amino acid substitution H58I in the 30aa-region, resulting in plasmids pTorA[KQ,A16V]-30aa[H58I]-MalE and pTorA[KQ,G28W]-30aa[H58I]-MalE, was done using the QuikChange® II XL site-directed mutagenesis kit (Agilent) with pTorA[KQ,A16V]-30aa-MalE and pTorA[KQ,G28W]-30aa-MalE, respectively, as templates and primers 30aa_H58I_qc_fw and 30aa_H58I_qc_rev.
For the construction of pTorA[KQ,T22A]-30aa[T65R]-MalE and pTorA[KQ,T22A]-30aa[D68R]-MalE, the h-region-located amino acid substitution T22A was introduced using the QuikChange® II XL site-directed mutagenesis kit (Agilent) with pTorA[KQ]-30aa[T65R]-MalE and pTorA[KQ]-30aa[D68R]-MalE, respectively, as templates and the primers spTorA[KQ]_T22A_qc_fw and spTorA[KQ]_T22A_qc_rev. Plasmids pTorA[KQ,G25W]-30aa[K50L]-MalE, pTorA[KQ,G28W]-30aa[K50L]-MalE, and pTorA[KQ,G25W/G28W]-30aa[K50L]-MalE were constructed using the same method with pTorA[KQ,G25W]-30aa-MalE, pTorA[KQ,G28W]-30aa-MalE, and pTorA[KQ,G25W/G28W]-30aa-MalE, respectively, as templates and primers 30aa_K50L_qc_fw and 30aa_K50L_qc_rev.
Plasmid pTorA[QQ]-MalE was constructed by introducing the double mutation R11Q/R12Q into pTorA-MalE using the QuikChange® II XL site-directed mutagenesis kit (Agilent) and the primers RR-QQfor and RR-QQrev, according to the manufacturer's instructions.
Plasmid pNapA-30aa(NapA)-MalE was constructed by crossover PCR. Two DNA fragments were amplified using primer pairs Cross_NapA-NapA30-MalE1_fw and Cross_NapA-NapA30-MalE2_rev or Cross_NapA-NapA30-MalE3_fw and Cross_NapA-NapA30-MalE4_rev, respectively, and chromosomal napA gene of E. coli or pTorA-30aa-MalE plasmid as template, respectively. Subsequently, both fragments were fused and amplified using Cross_NapA-NapA30-MalE1_fw and Cross_NapA-NapA30-MalE4_rev as primers. The resulting PCR fragment was digested with BamHI and EcoRI and ligated into BamHI/EcoRI-digested linearized pTorA-30aa-MalE plasmid. Plasmids pTorA-30aa(NapA)-MalE and pTorA[KQ]-30aa(NapA)-MalE were constructed by the same procedure. For each plasmid, two DNA fragments were amplified using primer pairs Cross_TorA-NapA30-MalE1_fw and Cross_TorA-NapA30-MalE2_rev or Cross_TorA-NapA30-MalE3_fw and Cross_TorA-NapA30-MalE4_rev, respectively, and plasmid pTorA-30aa-MalE or pTorA[KQ]-30aa-MalE and pNapA-30aa(NapA)-MalE as template, respectively. These two fragment pairs were fused and amplified using Cross_TorA-NapA30-MalE1_fw and Cross_TorA-NapA30-MalE4_rev as primers. The resulting PCR fragments were digested with BamHI and EcoRI and ligated into BamHI/EcoRI digested linearized pTorA-30aa-MalE plasmid.
Construction of libraries of mutant TorA[KQ]-30aa-MalE variants and the isolation of 30aa-region-located suppressor mutations
To screen for intragenic suppressors of the inactivating KQ mutation in the RR motif of the TorA signal peptide, two separate mutagenesis libraries were constructed by ep-PCR and site-saturation mutagenesis, respectively, that contained amino acid substitutions at random or selected positions within the early mature protein region of the normally export-defective TorA[KQ]-30aa-MalE reporter protein.
For the construction of the first mutant library, the gene region encoding the entire 30aa-region of the TorA[KQ]-30aa-MalE reporter protein consisting of the first 30 amino acids of the mature TorA protein was mutagenized via ep-PCR using the GeneMorph® II random mutagenesis kit (Agilent) according to the manufacturer's instructions. Plasmid pTorA[KQ]-30aa-MalE was used as template and intramut_torA30aaMalE2_fw and intramut_torA30aaMalE_rev as primers that contained HpaI and Kpn2I restriction sites, respectively. The ep-PCR conditions were adjusted such that 1–9 mutations were introduced per kilobase of template DNA. After completion of the PCR, the amplified fragments of 229 bp in size encompassing the c-region of the TorA signal peptide, the 30aa-region, and 38 amino acids of the mature MalE protein were cut with HpaI and Kpn2I and ligated into HpaI/Kpn2I-digested linearized pTorA[KQ]-30aa-MalE. After transformation of the ligation products into E. coli DH5α, about 3000 clones were obtained from which a pool of mutagenized pTorA[KQ]-30aa-MalE plasmids was isolated.
For the construction of the second mutant library, a total of 20 different amino acid positions within the 30aa-region (Ala-40, Gln-41, Ala-42, Ala-43, Thr-44, Asp-45, Ile-48, Ser-49, Lys-50, Glu-51, Gly-52, Thr-55, Gly-56, Ser-57, His-58, Gly-60, Arg-63, Thr-65, Lys-67, and Asp-68) were randomly chosen for site-directed one-codon saturation mutagenesis using the QuikChange® II XL site-directed mutagenesis kit (Agilent) according to the manufacturer's instructions with pTorA[KQ]-30aa-MalE as template and the degenerate primers 30aa_qc_X_fw and 30aa_qc_X_rev, with X being the respective mutagenized amino acid position. The resulting pTorA[KQ]-30aa-MalE variants harboring single amino acid substitutions at the selected positions within the 30aa-region were subsequently transformed into E. coli DH5α. For each mutagenized amino acid position, about 2000 clones were randomly chosen from which a pool of pTorA[KQ]-30aa-MalE plasmid variants was isolated.
Small aliquots of both mutant library pools were transformed separately into GSJ101 (pHSG-TatABCE) by using a standard heat-shock method. The transformed cells were plated on solid minimal medium containing 0.4% (w/v) maltose and incubated at 37 °C for up to 6 days. Some of the single mutant colonies that appeared on the selection plates were randomly picked and re-streaked on the same medium, and those isolates that showed reproducible growth after 48 h of incubation at the latest were chosen for further characterization. From these isolates, plasmids pHSG-TatABCE and mutagenized pTorA[KQ]-30aa-MalE were isolated and separated, and pTorA[KQ]-30aa-MalE was subsequently used for DNA sequence analysis and further functional characterizations.
Construction of a library of mutant Tat translocases and the isolation of suppressing mutations in the cytoplasmically-located domains of TatB
For the isolation of suppressing mutations of the TorA[KQ]-30aa-MalE export defect located in the cytoplasmically-located domains of TatB, the entire APH as well as the following C-terminal domains were randomly mutagenized by ep-PCR using the GeneMorph II EZClone Domain mutagenesis kit (Agilent) with pHSG-TatABCE as template and TatB_epAPH_fw and TatB_epAPH_rev as primers, according to the manufacturer's instructions. The ep-PCR conditions were adjusted such that 1–4 mutations were introduced per kb of template DNA. The resulting mutagenized pHSG-TatABCE plasmids were subsequently transformed into E. coli DH5α. About 2400 colonies were randomly picked from which a pool of pHSG-TatABCE plasmid variants was isolated. Finally, a small aliqout of the constructed mutant library was used for transformation of GSJ101 (pTorA[KQ]-30aa-MalE) expressing the export-defective TorA[KQ]-30aa-MalE reporter variant by using a standard heat-shock method. The transformed cells were plated on solid minimal medium containing 0.4% (w/v) maltose and incubated at 37 °C for 4 days. Some of the single mutant colonies that appeared on the selection plates were randomly picked and re-streaked on the same medium, and those isolates that showed reproducible growth after 48 h of incubation at the latest were chosen for further characterization. From these isolates, mutagenized pHSG-TatABCE and pTorA[KQ]-30aa-MalE plasmid were isolated and separated, and pHSG-TatABCE was subsequently used for DNA sequence analysis and further functional characterizations.
Cell fractionation studies
Fractionation of cells into a fraction containing the C/M and a P was done using an EDTA-lysozyme spheroplasting method as described by Kreutzenbeck et al. (23). Samples of the cell fractions corresponding to an equal amount of cells were subjected to SDS-PAGE and Western blotting using MalE-specific antibodies (23). As a control for the quality of the fractionation experiments, the subcellular localization of the cytoplasmic membrane-associated protein SecA was analyzed in parallel using SecA-specific antibodies (30). Western blotting using anti-MalE or anti-SecA antibodies was performed by using the ECL Western blotting detection kit (GE Healthcare) according to the manufacturer's instructions. The chemiluminescent protein bands were recorded and quantified using the Fujifilm LAS-3000 Mini CCD camera and image-analyzing system together with the software AIDA 4.50 (Raytest).
Miscellaneous procedures
Preparation of membranes, SDS-PAGE, and Western blotting using anti-TatA, anti-TatB, and anti-TatC antibodies were performed as described previously (23).
Author contributions
A. U. and R. F. conceptualization; A. U. investigation; A. U. writing-original draft; R. F. supervision; R. F. project administration; R. F. writing-review and editing; A. U. conceived and coordinated the study and designed the experiments. A. U. performed all experiments and wrote the manuscript; analyzed the experiments; approved the final version of the manuscript. R. F. conceived and coordinated the study and designed the experiments; analyzed the experiments; revised and optimized the manuscript; approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Astrid Bida and Doris Dohmen-Olma for excellent technical assistance, Anna Katharina Heide for the help with plasmid construction, and Michael Bott for ongoing support.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5, Tables S1–S3 and supporting Ref. 1.
- Tat
- twin-arginine translocation
- aa
- amino acid
- RR
- twin-arginine
- C/M
- cytosolic/membrane
- ep
- error-prone
- TMH
- transmembrane helix
- APH
- amphipathic helix
- P
- periplasmic fraction
- MMM
- maltose minimal medium
- MCM
- MacConkey maltose.
References
- 1. Denks K., Vogt A., Sachelaru I., Petriman N. A., Kudva R., and Koch H. G. (2014) The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 31, 58–84 10.3109/09687688.2014.907455 [DOI] [PubMed] [Google Scholar]
- 2. Robinson C., Matos C. F., Beck D., Ren C., Lawrence J., Vasisht N., and Mendel S. (2011) Transport and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria. Biochim. Biophys. Acta 1808, 876–884 10.1016/j.bbamem.2010.11.023 [DOI] [PubMed] [Google Scholar]
- 3. Hou B., and Brüser T. (2011) The Tat-dependent protein translocation pathway. Biomol. Concepts 2, 507–523 [DOI] [PubMed] [Google Scholar]
- 4. Fröbel J., Rose P., and Müller M. (2012) Twin-arginine-dependent translocation of folded proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1029–1046 10.1098/rstb.2011.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palmer T., and Berks B. C. (2012) The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10, 483–496 10.1038/nrmicro2814 [DOI] [PubMed] [Google Scholar]
- 6. Goosens V. J., Monteferrante C. G., and van Dijl J. M. (2014) The Tat system of Gram-positive bacteria. Biochim. Biophys. Acta 1843, 1698–1706 10.1016/j.bbamcr.2013.10.008 [DOI] [PubMed] [Google Scholar]
- 7. Berks B. C. (2015) The twin-arginine protein translocation pathway. Annu. Rev. Biochem. 84, 843–864 10.1146/annurev-biochem-060614-034251 [DOI] [PubMed] [Google Scholar]
- 8. Cline K. (2015) Mechanistic aspects of folded protein transport by the twin arginine translocase (Tat). J. Biol. Chem. 290, 16530–16538 10.1074/jbc.R114.626820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Celedon J. M., and Cline K. (2013) Intra-plastid protein trafficking: how plant cells adapted prokaryotic mechanisms to the eukaryotic condition. Biochim. Biophys. Acta 1833, 341–351 10.1016/j.bbamcr.2012.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berks B. C. (1996) A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22, 393–404 10.1046/j.1365-2958.1996.00114.x [DOI] [PubMed] [Google Scholar]
- 11. Stanley N. R., Palmer T., and Berks B. C. (2000) The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275, 11591–11596 10.1074/jbc.275.16.11591 [DOI] [PubMed] [Google Scholar]
- 12. Buchanan G., Sargent F., Berks B. C., and Palmer T. (2001) A genetic screen for suppressors of Escherichia coli Tat signal peptide mutations establishes a critical role for the second arginine within the twin-arginine motif. Arch. Microbiol. 177, 107–112 10.1007/s00203-001-0366-2 [DOI] [PubMed] [Google Scholar]
- 13. Alami M., Lüke I., Deitermann S., Eisner G., Koch H. G., Brunner J., and Müller M. (2003) Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell 12, 937–946 10.1016/S1097-2765(03)00398-8 [DOI] [PubMed] [Google Scholar]
- 14. Strauch E. M., and Georgiou G. (2007) Escherichia coli tatC mutations that suppress defective twin-arginine transporter signal peptides. J. Mol. Biol. 374, 283–291 10.1016/j.jmb.2007.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panahandeh S., Maurer C., Moser M., DeLisa M. P., and Müller M. (2008) Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. J. Biol. Chem. 283, 33267–33275 10.1074/jbc.M804225200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fröbel J., Rose P., Lausberg F., Blümmel A. S., Freudl R., and Müller M. (2012) Transmembrane insertion of twin-arginine signal peptides is driven by TatC and regulated by TatB. Nat. Commun. 3, 1311 10.1038/ncomms2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blümmel A. S., Haag L. A., Eimer E., Müller M., and Fröbel J. (2015) Initial assembly steps of a translocase for folded proteins. Nat. Commun. 6, 7234 10.1038/ncomms8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jack R. L., Sargent F., Berks B. C., Sawers G., and Palmer T. (2001) Constitutive expression of Escherichia coli tat genes indicates an important role for the twin-arginine translocase during aerobic and anaerobic growth. J. Bacteriol. 183, 1801–1804 10.1128/JB.183.5.1801-1804.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eimer E., Fröbel J., Blümmel A. S., and Müller M. (2015) TatE as a regular constituent of bacterial twin-arginine protein translocases. J. Biol. Chem. 290, 29281–29289 10.1074/jbc.M115.696005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cline K., and Mori H. (2001) Thylakoid ΔpH-dependent precursor proteins bind to a cpTatC-Hcf106 complex before Tha4-dependent transport. J. Cell Biol. 154, 719–729 10.1083/jcb.200105149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDevitt C. A., Buchanan G., Sargent F., Palmer T., and Berks B. C. (2006) Subunit composition and in vivo substrate-binding characteristics of Escherichia coli Tat protein complexes expressed at native levels. FEBS J. 273, 5656–5668 10.1111/j.1742-4658.2006.05554.x [DOI] [PubMed] [Google Scholar]
- 22. Gérard F., and Cline K. (2006) Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J. Biol. Chem. 281, 6130–6135 10.1074/jbc.M512733200 [DOI] [PubMed] [Google Scholar]
- 23. Kreutzenbeck P., Kröger C., Lausberg F., Blaudeck N., Sprenger G. A., and Freudl R. (2007) Escherichia coli twin arginine (Tat) mutant translocases possessing relaxed signal peptide recognition specificities. J. Biol. Chem. 282, 7903–7911 10.1074/jbc.M610126200 [DOI] [PubMed] [Google Scholar]
- 24. Hou B., Frielingsdorf S., and Klösgen R. B. (2006) Unassisted membrane insertion as the initial step in ΔpH/Tat-dependent protein transport. J. Mol. Biol. 355, 957–967 10.1016/j.jmb.2005.11.029 [DOI] [PubMed] [Google Scholar]
- 25. Shanmugham A., Wong Fong Sang H. W., Bollen Y. J., and Lill H. (2006) Membrane binding of twin arginine preproteins as an early step in translocation. Biochemistry 45, 2243–2249 10.1021/bi052188a [DOI] [PubMed] [Google Scholar]
- 26. Bageshwar U. K., Whitaker N., Liang F. C., and Musser S. M. (2009) Interconvertibility of lipid- and translocon-bound forms of the bacterial Tat precursor pre-SufI. Mol. Microbiol. 74, 209–226 10.1111/j.1365-2958.2009.06862.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holzapfel E., Eisner G., Alami M., Barrett C. M., Buchanan G., Lüke I., Betton J. M., Robinson C., Palmer T., Moser M., and Müller M. (2007) The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry 46, 2892–2898 10.1021/bi062205b [DOI] [PubMed] [Google Scholar]
- 28. Zoufaly S., Fröbel J., Rose P., Flecken T., Maurer C., Moser M., and Müller M. (2012) Mapping precursor-binding site on TatC subunit of twin arginine-specific protein translocase by site-specific photo cross-linking. J. Biol. Chem. 287, 13430–13441 10.1074/jbc.M112.343798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fincher V., McCaffery M., and Cline K. (1998) Evidence for a loop mechanism of protein transport by the thylakoid ΔpH pathway. FEBS Lett. 423, 66–70 10.1016/S0014-5793(98)00066-0 [DOI] [PubMed] [Google Scholar]
- 30. Ulfig A., Fröbel J., Lausberg F., Blümmel A. S., Heide A. K., Müller M., and Freudl R. (2017) The h-region of twin arginine signal peptides supports productive binding of bacterial Tat precursor proteins to the TatBC receptor complex. J. Biol. Chem. 292, 10865–10882 10.1074/jbc.M117.788950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang Q., and Palmer T. (2017) Signal peptide hydrophobicity modulates interaction with the twin-arginine translocase. mBio 8, e00909–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maurer C., Panahandeh S., Jungkamp A. C., Moser M., and Müller M. (2010) TatB functions as an oligomeric binding site for folded Tat precursor proteins. Mol. Biol. Cell 21, 4151–4161 10.1091/mbc.E10-07-0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mori H., and Cline K. (2002) A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid ΔpH/Tat translocase. J. Cell Biol. 157, 205–210 10.1083/jcb.200202048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gohlke U., Pullan L., McDevitt C. A., Porcelli I., de Leeuw E., Palmer T., Saibil H. R., and Berks B. C. (2005) The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc. Natl. Acad. Sci. U.S.A. 102, 10482–10486 10.1073/pnas.0503558102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dabney-Smith C., Mori H., and Cline K. (2006) Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J. Biol. Chem. 281, 5476–5483 10.1074/jbc.M512453200 [DOI] [PubMed] [Google Scholar]
- 36. Brüser T., and Sanders C. (2003) An alternative model of the twin arginine translocation system. Microbiol. Res. 158, 7–17 10.1078/0944-5013-00176 [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez F., Rouse S. L., Tait C. E., Harmer J., De Riso A., Timmel C. R., Sansom M. S., Berks B. C., and Schnell J. R. (2013) Structural model for the protein-translocating element of the twin-arginine transport system. Proc. Natl. Acad. Sci. U.S.A. 110, E1092–E1101 10.1073/pnas.1219486110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lausberg F., Fleckenstein S., Kreutzenbeck P., Fröbel J., Rose P., Müller M., and Freudl R. (2012) Genetic evidence for a tight cooperation of TatB and TatC during productive recognition of twin-arginine (Tat) signal peptides in Escherichia coli. PLoS ONE 7, e39867 10.1371/journal.pone.0039867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hengge R., and Boos W. (1983) Maltose and lactose transport in Escherichia coli. Examples of two different types of concentrative transport systems. Biochim. Biophys. Acta 737, 443–478 10.1016/0304-4157(83)90009-6 [DOI] [PubMed] [Google Scholar]
- 40. Blaudeck N., Kreutzenbeck P., Freudl R., and Sprenger G. A. (2003) Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J. Bacteriol. 185, 2811–2819 10.1128/JB.185.9.2811-2819.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blaudeck N., Kreutzenbeck P., Müller M., Sprenger G. A., and Freudl R. (2005) Isolation and characterization of bifunctional Escherichia coli TatA mutant proteins that allow efficient Tat-dependent protein translocation in the absence of TatB. J. Biol. Chem. 280, 3426–3432 10.1074/jbc.M411210200 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Y., Wang L., Hu Y., and Jin C. (2014) Solution structure of the TatB component of the twin-arginine translocation system. Biochim. Biophys. Acta 1838, 1881–1888 10.1016/j.bbamem.2014.03.015 [DOI] [PubMed] [Google Scholar]
- 43. Huang Q., Alcock F., Kneuper H., Deme J. C., Rollauer S. E., Lea S. M., Berks B. C., and Palmer T. (2017) A signal sequence suppressor mutant that stabilizes an assembled state of the twin arginine translocase. Proc. Natl. Acad. Sci. U.S.A. 114, E1958–E1967 10.1073/pnas.1615056114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chatzi K. E., Sardis M. F., Tsirigotaki A., Koukaki M., Šoštarić N., Konijnenberg A., Sobott F., Kalodimos C. G., Karamanou S., and Economou A. (2017) Preprotein mature domains contain translocase targeting signals that are essential for secretion. J. Cell Biol. 216, 1357–1369 10.1083/jcb.201609022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee P. A., Buchanan G., Stanley N. R., Berks B. C., and Palmer T. (2002) Truncation analysis of TatA and TatB defines the minimal functional units required for protein translocation. J. Bacteriol. 184, 5871–5879 10.1128/JB.184.21.5871-5879.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee P. A., Orriss G. L., Buchanan G., Greene N. P., Bond P. J., Punginelli C., Jack R. L., Sansom M. S., Berks B. C., and Palmer T. (2006) Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J. Biol. Chem. 281, 34072–34085 10.1074/jbc.M607295200 [DOI] [PubMed] [Google Scholar]
- 47. Alcock F., Stansfeld P. J., Basit H., Habersetzer J., Baker M. A., Palmer T., Wallace M. I., and Berks B. C. (2016) Assembling the Tat protein translocase. eLife 5, e20718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller J. H. (1972) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 49. Tanaka S., Lerner S. A., and Lin E. C. (1967) Replacement of a phosphoenolpyruvate-dependent phosphotransferase by a nicotinamide adenine dinucleotide-linked dehydrogenase for the utilization of mannitol. J. Bacteriol. 93, 642–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 51. Hanahan D. (1983) Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 10.1016/S0022-2836(83)80284-8 [DOI] [PubMed] [Google Scholar]
- 52. Takeshita S., Sato M., Toba M., Masahashi W., and Hashimoto-Gotoh T. (1987) High-copy-number and low-copy-number plasmid vectors for lacZ α-complementation and chloramphenicol- or kanamycin-resistance selection. Gene 61, 63–74 10.1016/0378-1119(87)90365-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.