

Abstract
The key regulatory enzymes of glycogenolysis are phosphorylase kinase, a hetero-oligomer with four different types of subunits, and glycogen phosphorylase, a homodimer. Both enzymes are activated by phosphorylation and small ligands, and both enzymes have distinct isoforms that are predominantly expressed in muscle, liver, or brain; however, whole-transcriptome high-throughput sequencing analyses show that in brain both of these enzymes are likely composed of subunit isoforms representing all three tissues. This Minireview examines the regulatory properties of the isoforms of these two enzymes expressed in the three tissues, focusing on their potential regulatory similarities and differences. Additionally, the activity, structure, and regulation of the remaining enzyme necessary for glycogenolysis, glycogen-debranching enzyme, are also reviewed.
Keywords: brain, energy metabolism, glycogen, neurobiology, phosphorylase
Introduction
As discussed throughout this Minireview series, glycogenolysis, especially in astrocytes, is central to a variety of biological processes carried out in brain. Thus, the regulation of glycogenolysis is critical for understanding control of these processes. There are but three enzymes devoted solely to glycogenolysis: phosphorylase kinase (PhK),2 which activates glycogen phosphorylase (GP); GP, which phosphorolyzes α-1,4 glycosidic bonds to form glucose-1-P; and glycogen-debranching enzyme (GDE), which is both a glucosyltransferase and a glucosidase that hydrolyzes α-1,6-glycosidic branch point bonds to release glucose. The first two enzymes constitute an unusually selective pair, in that PhK is the only known protein kinase to activate GP, which in turn is the only generally acknowledged physiological substrate for PhK. As will be discussed, however, it is possible that glycogen synthase (GS) may also be a biological substrate for PhK. Besides being activated by phosphorylation, PhK and GP respond to small-molecule signals to regulate glycogenolysis, whereas GDE is directly involved in regulating glycogen synthesis.
The structure, activity, and regulation of PhK
Almost everything known about PhK has come from 6 decades of study on the enzyme from rabbit fast-twitch skeletal muscle, which fortunately has turned out to be a relatively homogeneous isoform. This form of the enzyme will be referred to as mPhK, to distinguish it from the liver (lPhK) and brain (bPhK) forms, which at first approximation seem to share similar subunit compositions and regulatory features as mPhK, albeit with distinguishing differences in their regulation (1). The mPhK is a hexadecameric oligomer composed of four copies each of four distinct subunits, (αβγδ)4, that tightly associate in a 1.3-MDa complex. The α, β, and δ subunits are regulatory and exert fine control over the activity of the catalytic γ subunit (1). Within the complex each type of subunit directly interacts with the three remaining types, consistent with extensive quaternary interactions (2). Moreover, cryo-EM reconstructions and subunit localizations show the three regulatory subunits to be arrayed around the catalytic subunit (Fig. 1) (3–7), and their interactions with it are, at least in part, through the γ subunit's C-terminal regulatory domain (γCRD) (8). Adding the masses of the γCRD (12.7 kDa) and the three regulatory subunits (α, 138 kDa; β, 125 kDa; δ, 16.7 kDa) indicates that 90% of mPhK's mass is involved in its regulation. It is assumed that the regulation of the bPhK will be equally complex if not more complex than mPhK, given that there are a greater variety of subunit isoforms in the brain.
Figure 1.
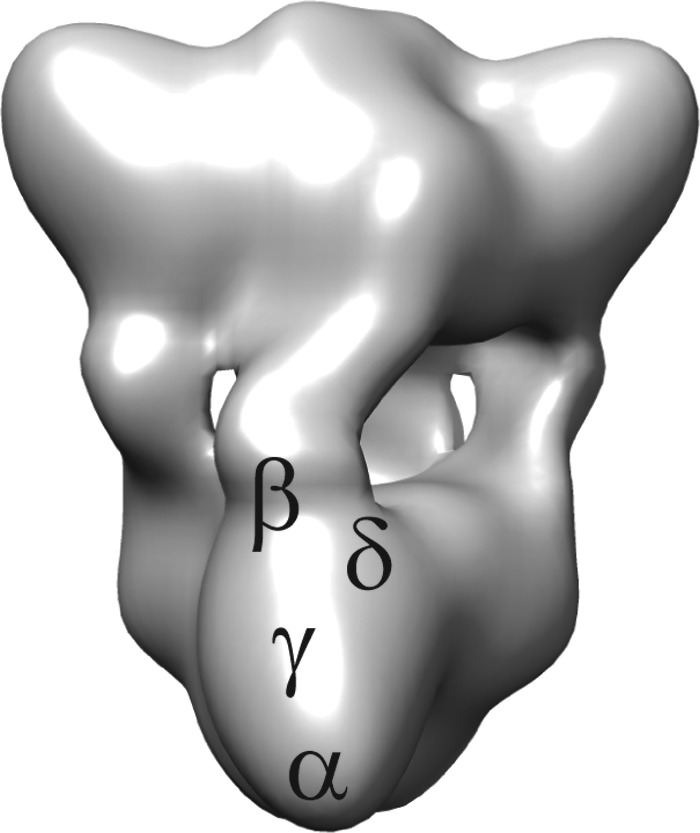
PhK structure and subunit locations. The cryo-EM reconstruction of the (αβγδ)4 mPhK complex (6) shows the relative locations of the four subunits within a single αβγδ protomer determined by immunoelectron microscopy or nanogold derivatization (3–5). The bridges between the octameric lobes are composed of β subunits (94). The lobe tips, which contain a C-terminal region of the α subunit (4), are separated by 176 Å (intralobal) and 213 Å (interlobal) (6).
There are two primary activators of mPhK, one being Ca2+ (9) and the other phosphorylation (10), with that catalyzed by cAMP-dependent protein kinase (PKA) as part of the fight or flight response being the most thoroughly characterized (1). The mPhK is a calmodulin (CaM)-dependent protein kinase; however, in this regard it is atypical of that kinase family because of four features. The first is that CaM, its integral δ subunit (11), does not associate and dissociate from the complex depending on the concentration of free Ca2+. The second is that CaM (δ) binds to the γCRD in a novel mode using a salt-bridge involving its third EF-hand (12). The third is that mPhK is activated by and binds only three Ca2+ ions per δ subunit (13), consistent with its third EF-hand being involved in the binding of δ to γ. The fourth difference between mPhK and other CaM-dependent kinases is that Ca2+ remains an obligatory cofactor even after the enzyme has been activated via phosphorylation (14), although the sensitivity to Ca2+ does change (9, 15, 16). It should be remembered, however, that Ca2+ and phosphorylation are co-dependent activators, in that neither alone causes significant activation, at least at neutral pH (15). This last point is important, because activation schemes showing the glycogenolysis cascade often imply that Ca2+ alone can significantly activate PhK, which is not true for mPhK, at least at neutral pH values or below. As for other PhK isoforms, including brain, their overall Ca2+ dependence will require similarly thorough analyses. At alkaline pH values, which activate mPhK by eliminating its need for phosphorylation, its requirement for Ca2+ ions for expression of that activity remains (15).
It might also be noted that PhK directly binds to glycogen, which stimulates its protein kinase activity (17). Moreover, acarbose, which is a transition-state analogue inhibitor of α-glucosidases, binds tightly to PhK and stimulates its catalytic activity, presumably through glycogen-binding site(s) on its α and/or β subunit(s) (18).
Besides phosphorylating GP, mPhK from rabbit has also been shown by numerous groups to phosphorylate rabbit muscle GS in vitro (19–22). The characteristics of this PhK-dependent phosphorylation completely mirror those with GP as substrate, including Ca2+ dependence. Moreover, the kinetic parameters for both substrates are quite similar: equivalent Km values, with Vmax twice as high for GP (21). As is the case with GP, the GS phosphorylation site is near its N terminus (Ser-7), which occurs within a sequence similar to the GP phosphorylation site (21). Most importantly, this phosphorylation of GS also leads to its inactivation (19, 20, 22, 23). It is not known whether PhK also phosphorylates GS in vivo, although they both bind glycogen (19) and thus may be proximal. The difficulty in establishing whether GS is a physiological substrate for PhK is that there are five other protein kinases that phosphorylate Ser-7 of GS (19).
In contrast to mPhK, very little is known about the structure, activity, and regulation of bPhK. The PhK activity in brain lysates is ∼7% that of muscle, normalized by protein concentration (24). It should be noted, however, that these lysates would represent glial, neuronal, endothelial, and smooth muscle cells. The mass of bPhK does not appear to have been estimated; however, it most likely has the same quaternary structure as mPhK and lPhK, because all four subunits are expressed in brain (25). Moreover, both Ca2+ and PKA have been shown to activate bPhK in lysates, further suggesting a similar subunit composition and regulation of the PhK from various tissues, although there were significant qualitative differences in the activation profiles (24). It should be noted, however, and is detailed in subsequent sections, transcripts of the muscle isoforms of the α, β, and γ subunits are all found in brain, but the brain additionally has transcripts corresponding to the γ subunit isoform associated with liver and a β subunit isoform that predominates in brain.
Regarding these isoforms of PhK, multiple genes and alternative splicing give rise to a wide variety of potential complexes containing different subunits. There are two genes for the α subunit on the X-chromosome: PHKA1 and PHKA2 (26, 27). The A1 gene encodes the α subunit found in mPhK (28); however, alternative RNA splicing removes an internal segment of 59 amino acids to form α′ (29), which predominates in PhK from cardiac and slow-twitch muscle. The A2 gene encodes the predominant α subunit in lPhK (30). There is but a single gene for the β subunit on chromosome 16 (27), but its RNA can undergo two different types of alternative splicing, resulting in β subunits with different N termini and two different mutually exclusive internal segments of 28 residues (29). Of the four possible β permutations, one predominates in brain (βB) (29), and it is significantly different from that of muscle (βM) (29). There are two genes for the γ subunit: PHKG1 on chromosome 7 (31) and PHKG2 on chromosome 16 (32). As for the δ subunit, there are three different genes for CaM on chromosomes 2, 14, and 19 (33); however, all three genes encode the identical protein, and of course, CaM has multiple targets. Because of all the different potential combinations of subunits in the hexadecameric complex, only mPhK, which is nearly all (α1βMγ1δ)4, and to a much lesser extent heart PhK (α′1βMγ1δ)4 have been characterized as purified, mostly homogeneous isoforms (1). Liver PhK has been purified and studied (34, 35); however, those enzyme preparations likely contained multiple isoforms. No purified bPhK with any identified isoform has ever been characterized; however, in this regard, it should be noted that an expression system that would allow this is now available, as hexadecameric mPhK that mimics the enzyme purified from skeletal muscle has now been expressed (36).
The relative amounts and types of RNA characterized by whole-transcriptome high-throughput sequencing of skeletal muscle, liver, and brain tissues is shown in Table 1. Comparing hippocampus and astrocytes, one sees similar expression of the different isoforms, albeit in somewhat different ratios. It should be noted, however, that the hippocampal data are from humans, and the astrocytic data are from mice (25). Considering the α subunit in astrocytic PhK, α1 predominates over α2. No α′1 was observed, and it is not known whether α′2 is produced in any tissue. The γ subunit exhibits a similar ratio of the muscle to liver forms as observed for α in astrocytes, with γ1 predominating. Of the three genes for the δ subunit (CaM), only one is significantly transcribed in astrocytes. The greatest difference between bPhK and PhK from other tissues occurs in the β subunit. The βB isoform predominantly contains the mutually exclusive internal exon found in βL (29); however, its N terminus is distinct from the other β isoforms in being seven residues shorter and having altered phosphorylatable regulatory sequences (29), as described in detail below. Thus, the predominant makeup of astrocytic PhK is (α1βBγ1δ)4. It must be noted, however, that the RNA for α2, βM, and γ2 isoforms is also present and could give rise to a large variety of PhK complexes, likely having different regulatory, or even catalytic, properties. Moreover, that complexity would be even further increased if all subunit isoforms could freely associate within a given hexadecameric complex, e.g. different α isoforms within the same complex, different β isoforms, etc. The selectivity of association of subunit isoforms within an individual PhK complex is simply not known.
Table 1.
Isoforms of PhK Subunits and PhKaa

a Expression in human skeletal muscle, liver, hippocampus, and mouse astrocytes was estimated from RNA-sequence data sets, with FPKM shown in the separate heat maps for PHK and CALM/GP by the central double line (52). PhK subunit splice variants were evaluated using previously reported values from rabbit (29).
b Deletion of 59 amino acids (AA's) (654–712) through alternative splicing (29).
c Residues 779–806 are unique due to mutually exclusive exons (29).
d Residues 779–806 are the same in the β subunits of all non-muscle tissues (29).
e N.D. means not determined.
f Data are similar to βL, but with a unique N terminus (29).
g All three CALM genes encode the identical protein (33).
One of most salient differences between the brain and muscle PhKs concerns their potential regulation by phosphorylation. In the case of mPhK, its activation by PKA occurs mainly through phosphorylation of Ser-26 (37) of the β subunit (Fig. 2); but once this residue is phosphorylated, additional activation occurs through phosphorylation of multiple seryl residues in the C-terminal region of the α subunit (38). Phosphorylation of mPhK is associated with an increased self-association of the β subunits within the (αβγδ)4 complex (8, 39) and a concomitantly large increase in the kcat of its γ subunit (40). From results with zero-length oxidative cross-linking and a phosphomimetic peptide of the N terminus of βM (8), a structural model was recently proposed to explain the phospho-activation of mPhK (41). The model posits that the nonactivated conformer of PhK is stabilized by interactions between the γ subunit and the nonphosphorylated N terminus of β and that phosphorylation of β weakens this inhibitory interaction, leading to the activation of γ. The βB isoform has a very different primary structure at its N terminus than at βM (Fig. 2), which makes it highly probable that bPhK is regulated by phosphorylation differently from mPhK. The difference could be potentially manifested in three different ways: autophosphorylation, phosphorylation by PKA, and phosphorylation by Pro-directed protein kinases. The first two potential differences were explicitly noted by Harmann et al. (29) in their groundbreaking study on the isoform diversity of PhK's α and β subunits created by alternative RNA splicing. They noted that Ser-11 of βM, which is an autophosphorylation and autoactivation site in vitro (8, 42), is eliminated in βB (Fig. 2), although it is not known whether mPhK autophosphorylation occurs in vivo. They also noted that the primary structure immediately surrounding Ser-26 in βM, the activation site during phosphorylation by PKA (37), is conserved in βB as Ser-19 and surrounding residues (Fig. 2); however, N-terminal to that, only one of the six basic residues present in βM is retained in βB, which would undoubtedly negatively affect phosphorylation by the basophilic PKA. The third and most intriguing difference between βM and βB in their N-terminal regions is the introduction of three additional seryl residues in the latter (five total versus two), with two of the residues in βB occurring as Ser-Pro pairs (Fig. 2). These pairs could be targets for members of the large superfamily of Pro-directed protein kinases, e.g. GSK3, JNKs, MAPKs, ERKs, etc. No Pro-directed protein kinase has been previously shown to phosphorylate PhK from any tissue. The location of these Ser-Pro pairs at the N terminus of βB makes it highly likely that their phosphorylation would lead to activation of that PhK, and thus glycogenolysis. Thus, conditions or receptor agonists acting independently of cAMP could stimulate brain glycogenolysis.
Figure 2.

N-terminal domains of βM and βB. Serines in red represent known PKA phosphorylation sites for βM (1, 37) and the putative PKA site for βB. The green residues in the βB sequence show the potential Pro-directed protein kinase phosphorylation sites.
To fully understand the regulation of bPhK by phosphorylation and Ca2+ ions, it will be necessary to co-express defined isoforms of the relevant subunits found in brain and then to purify and characterize the resultant complexes using the brain isoform of GP as substrate. All studies to date examining bPhK activity in lysates or slices have utilized only the muscle isoform of GP as substrate; as discussed in the below, there are significant differences in these two GP isoforms.
Intracellular degradation of glycogen
Release of glucosyl monomers from glycogen occurs through the concerted actions of GP and GDE, which produce glucose 1-phosphate and glucose, respectively, through phosphorolysis and hydrolysis of 1,4 and 1,6 glycosidic bonds in glycogen. Regulatory mechanisms governing the activity of GP are for the most part well characterized through extensive studies of rabbit mGP (43), whereas the regulation of GDE activity is less well understood.
The structure, activity, and regulation of GP
GP (1,4-α-d-glucan:orthophosphate α-d-glycosyltransferase, EC 2.4.1.1) is a member of the clan 35 family of retaining glycosyltransferases (GT), which in the case of GP transfer sugar moieties (glucose) from donor (glycogen) to phosphate acceptor molecules (44). Each GP monomer (97.4 kDa) contains a pyridoxal phosphate prosthetic group that is attached through a Schiff base with a Lys in its active site (45). The phosphate group of pyridoxal phosphate protonates Pi in the GP-active site, which in turn protonates the hemiacetal oxygen of the α-1,4 glycosidic bond, resulting in its cleavage and the formation of a glucosyl carbocation that bonds with phosphate to form glucose 1-phosphate. Biologically, GP exists as a homodimer of subunits associated in a nonactive conformation, in which both subunits' catalytic sites are partially buried (46). To achieve full activation, GP undergoes tiered conformational changes in response to phosphorylation of Ser-14 by PhK and/or the binding of the allosteric activator AMP, promoting active conformations in which moderate changes in tertiary structure lead to large changes in quaternary structure, rotating the subunits ∼10° with respect to each other to expose fully solvent-accessible active sites (46–48).
As is the case with PhK, there are three structurally related mammalian isoforms of GP, named for the tissues in which they predominate: muscle (mGP), liver (lGP), and brain (bGP). Each of the isoforms is encoded by a unique gene, and the three genes are located on different chromosomes (49–51). In addition to liver, lGP is expressed, at least at low levels, ubiquitously in other tissues, including brain (52). In contrast, mGP is expressed to a great extent only in muscle, with low levels of expression observed in brain, primarily in astrocytes (52–54). For lGP and mGP, RNA levels mirror protein expression in most tissues (52). The bGP isoform is expressed predominantly in fetal tissues, with its expression levels attenuating in most tissues to different extents after birth, giving rise to tissue-specific ratios of GP isoform expression (55). In brain, bGP is expressed in both astrocytes and neurons (53).
The activity of GP is tightly controlled by activating (AMP) and inhibiting (glucose, glucose-6-P, and purine nucleosides) allosteric effectors (56). As revealed by X-ray crystallographic analyses of mGP–ligand complexes, each allosteric ligand binds to a distinct region of the GP dimer, except glucose-6-P and AMP, whose binding sites partially overlap (57). It has also been suggested that glycogen itself may act as an allosteric activator through binding at a glycogen storage site distinct from the active site (46). GP is also activated, as described above, by upstream signaling through phosphorylation of Ser-14 by PhK in response to hormonal stimuli and increased intracellular Ca2+ ions. Nonphosphorylated GP is termed GPb, and phosphorylated GP is termed GPa.
There are significant differences in the allosteric activation of the three GP isoforms in response to AMP and phosphorylation. Crystal structures of all GPs show that the phosphorylation and AMP-binding sites are proximal (12 Å) and close to the subunit interface in the dimeric complex (58). Activation of GP through these two sites is thought to involve similar conformational changes (47, 48, 58–60), involving specific rearrangements in tertiary and quaternary structure, confined primarily to a region comprising residues 1–120, termed the activation subdomain (61). Phosphorylation results in nearly full activation of mGP and lGP; however, bGP is less potently activated by phosphorylation (61–64) and also requires the binding of AMP to achieve the extent of activation observed with mGPa and lGPa (63). The three isoforms also respond differently to AMP, with lGP showing little to no activation by the nucleotide (61). In contrast, bGPb is sensitive to AMP activation and has a greater affinity for the nucleotide than does mGPb; however, it binds AMP noncooperatively (61, 65), whereas the binding of AMP by mGPb is highly cooperative (61). Additionally, AMP significantly lowers the Km value of bGP for glycogen, which otherwise would be much greater than that for the muscle isoform (62).
Although likely not as problematic as for PhK, the simultaneous presence of multiple isoforms of GP in brain complicates understanding the overall regulation of the enzyme. In astrocytes, where the bulk of glycogenolysis occurs, both the mGP and bGP isoforms are expressed (65); however, there is also evidence for the expression of lGP in cultured astrocytes (53), and low levels of RNA for this isoform have been detected in brain by whole-transcriptome shotgun sequencing (52). Thus, there is the possibility that all three isoforms exist in brain, allowing for isoform-dependent variations in response to activation by AMP and other effectors, as well as to phosphorylation by PhK (65). As discussed above, upstream signaling by phosphorylation is further complicated by the presence of the βB subunit of PhK in astrocytes. Moreover, AMP signaling may also be complicated by the susceptibility of bGP to reactive oxygen species (ROS) (66). Two Cys residues (318 and 326) are conserved within all brain isoforms of GP in higher vertebrates and occupy positions in the adenine loop that is part of the AMP-binding site (60). Oxidation of bGP with H2O2 has been shown to reduce AMP-induced activation by 75% and activation by phosphorylation by 20%. Both effects were reversible, in line with several redox proteomics studies (67–69) that suggested potential regulation of bGP by ROS (66). Because only bGP is expressed in neurons (53, 54), its exposure to ROS could potentially limit its activation to predominantly extracellular signaling pathways through phosphorylation, rather than in response to neuronal intracellular AMP.
Another potential allosteric activator of bGP is Rac1, although this particular activation mechanism has yet to be examined with the brain isoform. Rac1 (77), a member of the Rho family of small GTPases, does however activate mGP (70). Moreover (78–83), muscle contraction has been shown to increase activation of Rac1, leading in turn to activation of the protein kinase PAK1, a Rac1 target, and to increased glucose uptake through actin cytoskeletal remodeling and GLUT4 translocation (71). PAK1 is activated by the binding of Rac1 to its auto-inhibitory domain, which shares homology with residues 191–270 of mGP, a region required for the binding and activation of mGP by active Rac1 (70). The Rac1-binding region of mGP is near the dimer interface, proximal to the tower helices, which undergo large conformational changes upon activation (46). These results suggest that glycogenolysis may be stimulated during muscle contraction through multiple activators that target GP, including Rac1.
Because Rac1 is expressed widely and to a great extent in brain (52) and because bGP essentially conserves the entire Rac1-binding site found in mGP (70), it is possible that (73–76) brain glycogenolysis may also be mediated by Rac1 similar to its effect in muscle. In a recent report, Fernandez et al. (72) demonstrated that glucose uptake in forebrain through GLUT1 translocation is synergistically stimulated by insulin and insulin-like growth factor-1 (IGF1) through mitogen-activated protein kinase/protein kinase D activation of Rac1 in astrocytes, which have been reported to express insulin, IGF1, and insulin receptors (73, 74). In response to sensory stimulation, blockade of IGF1 receptors in the somato-sensory cortex was shown to diminish neuronal activity and glucose uptake by astrocytes (72). Correspondingly, both Rac1 and mGP levels were diminished in post-mortem samples of the dorsolateral prefrontal cortex of patients with schizophrenia and in astroglia-enriched cultures from rodent models of schizophrenia, with samples from both exhibiting changes in energy-regulating pathways (75–77). Together these studies provide evidence for altered regulation of the glycogenolytic pathway in chronic schizophrenia, leading Pinacho et al. (78) to hypothesize the potential down-regulation of Rac1 and mGP as a mechanism for diminishing the transfer of astrocytic energy stores to neurons (77, 79, 92). Rac1 activation through Wnt5a signaling in brain influences synaptic plasticity and memory formation (79), and through its binding and activation of GP it may potentially couple actin dynamics and glycogenolysis, both linked to memory consolidation (80). Furthermore, as a modulator of actin dynamics (81), Rac1 also plays an important role in neuronal migration and synaptic plasticity (79, 82, 83).
The activity, structure, and regulation of GDE
In the breakdown of glycogen, GDE expresses both GT and glycosidase (GC) activities following the reactions of GP. Phosphorolysis by GP of α-1,4 glycosidic bonds in the glycogen polymer halts at four glucose units from an α-1,6 branch point, with those remaining four glucosyl residues targeted by GDE. First, three glucose units are cleaved from the branch and transferred as a trisaccharide to the reducing end of a nearby outer chain by the GT activity, and then the α-1,6 branch point glucose is hydrolyzed by the GC activity. Together, these two activities of GDE produce a linear polyglucose outer chain that is accessible to further phosphorolysis by GP.
Until recently, little was known structurally about eukaryotic GDEs, including whether GT and GC catalysis was carried by a single or separate catalytic sites; however, the first crystal structure of GDE from Candida glabrata (cgGDE), which shares 38% sequence similarity with human GDE, unequivocally demonstrated the presence of N- and C-terminal catalytic domains that possess, respectively, 4-α-d-glucotransferase (GT: EC 2.4.1.25) and amylo-α-1,6-glucosidase (GC: EC 3.2.1.33) activities (84). The N-terminal GT domain contains three subdomains and is structurally similar to glycosyl hydrolase (GH) 13 family members (44), which contain conserved catalytic Glu and Asp residues. Structural analyses of the GT site with oligosaccharide model substrates demonstrated that it is highly selective for glycogen branches with four glucose units, i.e. the end products of glycogen digestion by GP (85). Mutation of either of the predicted GT site catalytic residues abolished the enzyme's glucotransferase activity without affecting its glucosidase activity. The C-terminal catalytic domain is a structural homolog of the GH15 family, which contains funnel-shaped active sites with two carboxyl-containing catalytic residues (86). Using the methodology described above, the C-terminal GC domain was shown to be selective for single branch oligosaccharides mimicking the α-1,6 branches of glucose that are resistant to phosphorolysis by GP. Mutation of either of the catalytic residues in the GH site abolished glycosidase activity, but not GDE's transferase activity. In addition to the catalytic sites that bind glycogen, an intervening domain contains a glycogen contact region that is conserved among GDEs from other organisms (84).
Kinetic analyses of native and mixed catalytically dead CT and GC combinations of cgGDE suggest a mechanism whereby the GT hydrolysis product must first dissociate from the GDE before the enzyme binds to an α-1,6 branch for hydrolysis by its GC domain (84). Besides being substrates, the binding of oligosaccharides by GDE has been reported to stimulate its GT activity (87, 88).
Tissue-specific regulation of GDE and the extent and number of its interactions with other cellular proteins remain poorly understood. GDE levels may be influenced by fluctuations in glycogen stores through signals that, in part, alter its association with glycogen. Cheng et al. (89) demonstrated that conditions possibly leading to down-regulation of cAMP/PKA signaling and dissociation of GDE from glycogen influence the translocation of GDE from the cytosol to the nucleus to form GDE–malin complexes, ultimately leading to a decrease in GDE levels. Because malin is a ubiquitin ligase (90), its association with GDE suggests a potential model for regulating GDE levels by ubiquitination (89). Moreover Liu et al. (91) later showed that degradation of lafora bodies requires an assembly of proteins that include malin, GS, and bGP, plus the most common splice variant of the GDE gene (91), suggesting additional potential binding partners for GDE in polyglucan degradation pathways in brain and other tissues.
GDE may target other proteins that associate with glycogen, e.g. it reportedly binds the β subunit of AMP-activated kinase (AMPK), resulting in the stimulation of AMPK activity (92). Phosphorylation of GS by AMPK reportedly inhibits GS activity in concert with GSK (93), potentially down-regulating glycogen synthesis.
Summary
Continuing work on the regulation of glycogenolysis indicates that it is more complex than previously appreciated, especially in brain. In part, this is because of the probable wide variety of PhK and GP isoforms in brain, representing those enzymes from all tissues. Furthermore, the specific brain isoforms of both enzymes are regulated differently from their isoforms of other tissues, e.g. the unique N-terminal regulatory phosphorylation region of bPhK's βB subunit and the oxidation of bGP. To better understand the allosteric regulation of glycogenolysis in brain, the actual amounts of the different subunit isoforms of PhK and GP present in the different regions of brain must be determined, as well as their association, followed by determination of their resultant activities under controlled conditions. It would also be necessary, of course, to estimate the relative contributions to overall activation by the various allosteric activators acting on the two enzymes (phosphorylation, Ca2+, AMP, Rac-1, oxidation, etc.). The rapidly accessible energy source supplied by glycogenolyis in supporting numerous brain functions requires few enzymes, but they are under complex control.
This is the second article in the Thematic Minireview Series “Brain glycogen metabolism.” The authors declare that they have no conflicts of interest with the contents of this article.
- PhK
- phosphorylase kinase
- GS
- glycogen synthase
- GDE
- glycogen-debranching enzyme
- GP
- glycogen phosphorylase
- PTG
- protein targeting to glycogen
- AMPK
- AMP-activated kinase
- γCRD
- γ subunit's C-terminal regulatory domain
- ROS
- reactive oxygen species
- mPhK
- muscle PhK
- lPhK
- liver PhK
- bPhK
- bPhK
- GT
- 4-α-d-glucotransferase
- GC
- glycosidase
- CaM
- calmodulin
- GH
- glycosyl hydrolase.
References
- 1. Brushia R. J., and Walsh D. A. (1999) Phosphorylase kinase: the complexity of its regulation is reflected in the complexity of its structure. Front. Biosci. 4, D618–D641 10.2741/Brushia [DOI] [PubMed] [Google Scholar]
- 2. Lane L. A., Nadeau O. W., Carlson G. M., and Robinson C. V. (2012) Mass spectrometry reveals differences in stability and subunit interactions between activated and nonactivated conformers of the (αβγδ)4 phosphorylase kinase complex. Mol. Cell. Proteomics 11, 1768–1776 10.1074/mcp.M112.021394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Traxler K. W., Norcum M. T., Hainfeld J. F., and Carlson G. M. (2001) Direct visualization of the calmodulin subunit of phosphorylase kinase via electron microscopy following subunit exchange. J. Struct. Biol. 135, 231–238 10.1006/jsbi.2001.4411 [DOI] [PubMed] [Google Scholar]
- 4. Wilkinson D. A., Marion T. N., Tillman D. M., Norcum M. T., Hainfeld J. F., Seyer J. M., and Carlson G. M. (1994) An epitope proximal to the carboxyl terminus of the α-subunit is located near the lobe tips of the phosphorylase kinase hexadecamer. J. Mol. Biol. 235, 974–982 10.1006/jmbi.1994.1051 [DOI] [PubMed] [Google Scholar]
- 5. Wilkinson D. A., Norcum M. T., Fizgerald T. J., Marion T. N., Tillman D. M., and Carlson G. M. (1997) Proximal regions of the catalytic γ and regulatory β subunits on the interior lobe face of phosphorylase kinase are structurally coupled to each other and with enzyme activation. J. Mol. Biol. 265, 319–329 10.1006/jmbi.1996.0739 [DOI] [PubMed] [Google Scholar]
- 6. Nadeau O. W., Gogol E. P., and Carlson G. M. (2005) Cryoelectron microscopy reveals new features in the three-dimensional structure of phosphorylase kinase. Protein Sci. 14, 914–920 10.1110/ps.041123905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vénien-Bryan C., Jonic S., Skamnaki V., Brown N., Bischler N., Oikonomakos N. G., Boisset N., and Johnson L. N. (2009) The structure of phosphorylase kinase holoenzyme at 9.9 angstroms resolution and location of the catalytic subunit and the substrate glycogen phosphorylase. Structure 17, 117–127 10.1016/j.str.2008.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nadeau O. W., Anderson D. W., Yang Q., Artigues A., Paschall J. E., Wyckoff G. J., McClintock J. L., and Carlson G. M. (2007) Evidence for the location of the allosteric activation switch in the multisubunit phosphorylase kinase complex from mass spectrometric identification of chemically crosslinked peptides. J. Mol. Biol. 365, 1429–1445 10.1016/j.jmb.2006.10.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brostrom C. O., Hunkeler F. L., and Krebs E. G. (1971) The regulation of skeletal muscle phosphorylase kinase by Ca2+. J. Biol. Chem. 246, 1961–1967 [PubMed] [Google Scholar]
- 10. Krebs E. G., and Fischer E. H. (1956) The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 20, 150–157 10.1016/0006-3002(56)90273-6 [DOI] [PubMed] [Google Scholar]
- 11. Cohen P., Burchell A., Foulkes J. G., Cohen P. T., Vanaman T. C., and Nairn C. (1978) Identification of the Ca2+-dependent modulator protein as the fourth subunit of rabbit skeletal muscle phosphorylase kinase. FEBS Lett. 92, 287–293 10.1016/0014-5793(78)80772-8 [DOI] [PubMed] [Google Scholar]
- 12. Jeyasingham M. D., Artigues A., Nadeau O. W., and Carlson G. M. (2008) Structural evidence for co-evolution of the regulation of contraction and energy production in skeletal muscle. J. Mol. Biol. 377, 623–629 10.1016/j.jmb.2007.12.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burger D., Cox J. A., Fischer E. H., and Stein E. A. (1982) The activation of rabbit skeletal muscle phosphorylase kinase requires the binding of 3 Ca2+ per δ subunit. Biochem. Biophys. Res. Commun. 105, 632–638 10.1016/0006-291X(82)91481-4 [DOI] [PubMed] [Google Scholar]
- 14. Picton C., Shenolikar S., Grand R., and Cohen P. (1983) Calmodulin as an integral subunit of phosphorylase kinase from rabbit skeletal muscle. Methods Enzymol. 102, 219–227 10.1016/S0076-6879(83)02023-6 [DOI] [PubMed] [Google Scholar]
- 15. Cohen P. (1980) The role of calcium ions, calmodulin and troponin in the regulation of phosphorylase kinase from rabbit skeletal muscle. Eur. J. Biochem. 111, 563–574 10.1111/j.1432-1033.1980.tb04972.x [DOI] [PubMed] [Google Scholar]
- 16. Ozawa E. (1973) Activation of phosphorylase kinase from brain by small amounts of calcium ion. J. Neurochem. 20, 1487–1488 10.1111/j.1471-4159.1973.tb00263.x [DOI] [PubMed] [Google Scholar]
- 17. DeLange R. J., Kemp R. G., Riley W. D., Cooper R. A., and Krebs E. G. (1968) Activation of skeletal muscle phosphorylase kinase by adenosine triphosphate and adenosine 3′,5′-monophosphate. J. Biol. Chem. 243, 2200–2208 [PubMed] [Google Scholar]
- 18. Nadeau O. W., Liu W., Boulatnikov I. G., Sage J. M., Peters J. L., and Carlson G. M. (2010) The glucoamylase inhibitor acarbose is a direct activator of phosphorylase kinase. Biochemistry 49, 6505–6507 10.1021/bi101006j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roach P. J. (1990) Control of glycogen synthase by hierarchal protein phosphorylation. FASEB J. 4, 2961–2968 10.1096/fasebj.4.12.2168324 [DOI] [PubMed] [Google Scholar]
- 20. Roach P. J., DePaoli-Roach A. A., and Larner J. (1978) Ca2+-stimulated phosphorylation of muscle glycogen synthase by phosphorylase b kinase. J. Cyclic Nucleotide Res. 4, 245–257 [PubMed] [Google Scholar]
- 21. Soderling T. R., Sheorain V. S., and Ericsson L. H. (1979) Phosphorylation of glycogen synthase by phosphorylase kinase: stoichiometry, specificity and site of phosphorylation. FEBS Lett. 106, 181–184 10.1016/0014-5793(79)80723-1 [DOI] [PubMed] [Google Scholar]
- 22. Vila J., Salavert A., Itarte E., and Guinovart J. J. (1982) Phosphorylation of glycogen synthase by cyclic AMP-independent glycogen synthases kinase-1 (GSK-1) comparative study with cyclic AMP-dependent protein kinase and phosphorylase kinase. Arch. Biochem. Biophys. 218, 1–7 10.1016/0003-9861(82)90313-7 [DOI] [PubMed] [Google Scholar]
- 23. Soderling T. R., Srivastava A. K., Bass M. A., and Khatra B. S. (1979) Phosphorylation and inactivation of glycogen synthase by phosphorylase kinase. Proc. Natl. Acad. Sci. U.S.A. 76, 2536–2540 10.1073/pnas.76.6.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taira T., Kii R., Sakai K., Tabuchi H., Takimoto S., Nakamura S., Takahashi J., Hashimoto E., Yamamura H., and Nishizuka Y. (1982) Comparison of glycogen phosphorylase kinases of various rat tissues. J. Biochem. 91, 883–888 10.1093/oxfordjournals.jbchem.a133776 [DOI] [PubMed] [Google Scholar]
- 25. Petryszak R., Keays M., Tang Y. A., Fonseca N. A., Barrera E., Burdett T., Füllgrabe A., Fuentes A. M., Jupp S., Koskinen S., Mannion O., Huerta L., Megy K., Snow C., Williams E., et al. (2016) Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44, D746–D752 10.1093/nar/gkv1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davidson J. J., Ozçelik T., Hamacher C., Willems P. J., Francke U., and Kilimann M. W. (1992) cDNA cloning of a liver isoform of the phosphorylase kinase α subunit and mapping of the gene to Xp22.2-p22.1, the region of human X-linked liver glycogenosis. Proc. Natl. Acad. Sci. U.S.A. 89, 2096–2100 10.1073/pnas.89.6.2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Francke U., Darras B. T., Zander N. F., and Kilimann M. W. (1989) Assignment of human genes for phosphorylase kinase subunits α (PHKA) to Xq12-q13 and β (PHKB) to 16q12-q13. Am. J. Hum. Genet. 45, 276–282 [PMC free article] [PubMed] [Google Scholar]
- 28. Zander N. F., Meyer H. E., Hoffmann-Posorske E., Crabb J. W., Heilmeyer L. M. Jr, and Kilimann M. W. (1988) cDNA cloning and complete primary structure of skeletal muscle phosphorylase kinase (α subunit). Proc. Natl. Acad. Sci. U.S.A. 85, 2929–2933 10.1073/pnas.85.9.2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harmann B., Zander N. F., and Kilimann M. W. (1991) Isoform diversity of phosphorylase kinase α and β subunits generated by alternative RNA splicing. J. Biol. Chem. 266, 15631–15637 [PubMed] [Google Scholar]
- 30. Hirono H., Hayasaka K., Sato W., Takahashi T., and Takada G. (1995) Isolation of cDNA encoding the human liver phosphorylase kinase α subunit (PHKA2) and identification of a missense mutation of the PHKA2 gene in a family with liver phosphorylase kinase deficiency. Biochem. Mol. Biol. Int. 36, 505–511 [PubMed] [Google Scholar]
- 31. Jones T. A., da Cruz e Silva E. F., Spurr N. K., Sheer D., and Cohen P. T. (1990) Localisation of the gene encoding the catalytic γ subunit of phosphorylase kinase to human chromosome bands 7p12-q21. Biochim. Biophys. Acta 1048, 24–29 10.1016/0167-4781(90)90017-V [DOI] [PubMed] [Google Scholar]
- 32. Burwinkel B., Shiomi S., Al Zaben A., and Kilimann M. W. (1998) Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum. Mol. Genet. 7, 149–154 10.1093/hmg/7.1.149 [DOI] [PubMed] [Google Scholar]
- 33. Berchtold M. W., Egli R., Rhyner J. A., Hameister H., and Strehler E. E. (1993) Localization of the human bona fide calmodulin genes CALM1, CALM2, and CALM3 to chromosomes 14q24-q31, 2p21.1-p21.3, and 19q13.2-q13.3. Genomics 16, 461–465 10.1006/geno.1993.1211 [DOI] [PubMed] [Google Scholar]
- 34. Chrisman T. D., Jordan J. E., and Exton J. H. (1982) Purification of rat liver phosphorylase kinase. J. Biol. Chem. 257, 10798–10804 [PubMed] [Google Scholar]
- 35. Sakai K., Matsumura S., Okimura Y., Yamamura H., and Nishizuka Y. (1979) Liver glycogen phosphorylase kinase. Partial purification and characterization. J. Biol. Chem. 254, 6631–6637 [PubMed] [Google Scholar]
- 36. Boulatnikov I. G., Peters J. L., Nadeau O. W., Sage J. M., Daniels P. J., Kumar P., Walsh D. A., and Carlson G. M. (2009) Expressed phosphorylase b kinase and its αγδ subcomplex as regulatory models for the rabbit skeletal muscle holoenzyme. Biochemistry 48, 10183–10191 10.1021/bi901429y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeaman S. J., and Cohen P. (1975) The hormonal control of activity of skeletal muscle phosphorylase kinase. Phosphorylation of the enzyme at two sites in vivo in response to adrenalin. Eur. J. Biochem. 51, 93–104 10.1111/j.1432-1033.1975.tb03910.x [DOI] [PubMed] [Google Scholar]
- 38. Ramachandran C., Goris J., Waelkens E., Merlevede W., and Walsh D. A. (1987) The interrelationship between cAMP-dependent α and β subunit phosphorylation in the regulation of phosphorylase kinase activity. Studies using subunit specific phosphatases. J. Biol. Chem. 262, 3210–3218 [PubMed] [Google Scholar]
- 39. Fitzgerald T. J., and Carlson G. M. (1984) Activated states of phosphorylase kinase as detected by the chemical cross-linker 1,5-difluoro-2,4-dinitrobenzene. J. Biol. Chem. 259, 3266–3274 [PubMed] [Google Scholar]
- 40. Newsholme P., and Walsh D. A. (1992) A kinetic re-interpretation of the regulation of rabbit skeletal-muscle phosphorylase kinase activity by Ca2+ and phosphorylation. Biochem. J. 283, 845–848 10.1042/bj2830845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thompson J. A., Nadeau O. W., and Carlson G. M. (2015) A model for activation of the hexadecameric phosphorylase kinase complex deduced from zero-length oxidative crosslinking. Protein Sci. 24, 1956–1963 10.1002/pro.2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kilimann M. W., Zander N. F., Kuhn C. C., Crabb J. W., Meyer H. E., and Heilmeyer L. M. Jr. (1988) The α and β subunits of phosphorylase kinase are homologous: cDNA cloning and primary structure of the β subunit. Proc. Natl. Acad. Sci. U.S.A. 85, 9381–9385 10.1073/pnas.85.24.9381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buchbinder J. L., Rath V. L., and Fletterick R. J. (2001) Structural relationships among regulated and unregulated phosphorylases. Annu. Rev. Biophys. Biomol. Struct. 30, 191–209 10.1146/annurev.biophys.30.1.191 [DOI] [PubMed] [Google Scholar]
- 44. Henrissat B., and Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 10.1016/S0959-440X(97)80072-3 [DOI] [PubMed] [Google Scholar]
- 45. Oikonomakos N. G., Johnson L. N., Acharya K. R., Stuart D. I., Barford D., Hajdu J., Varvill K. M., Melpidou A. E., Papageorgiou T., Graves D. J., and et al. (1987) Pyridoxal phosphate site in glycogen phosphorylase b: structure in native enzyme and in three derivatives with modified cofactors. Biochemistry 26, 8381–8389 10.1021/bi00399a053 [DOI] [PubMed] [Google Scholar]
- 46. Barford D., and Johnson L. N. (1989) The allosteric transition of glycogen phosphorylase. Nature 340, 609–616 10.1038/340609a0 [DOI] [PubMed] [Google Scholar]
- 47. Sprang S. R., Acharya K. R., Goldsmith E. J., Stuart D. I., Varvill K., Fletterick R. J., Madsen N. B., and Johnson L. N. (1988) Structural changes in glycogen phosphorylase induced by phosphorylation. Nature 336, 215–221 10.1038/336215a0 [DOI] [PubMed] [Google Scholar]
- 48. Sprang S. R., Withers S. G., Goldsmith E. J., Fletterick R. J., and Madsen N. B. (1991) Structural basis for the activation of glycogen phosphorylase b by adenosine monophosphate. Science 254, 1367–1371 10.1126/science.1962195 [DOI] [PubMed] [Google Scholar]
- 49. Lebo R. V., Gorin F., Fletterick R. J., Kao F. T., Cheung M. C., Bruce B. D., and Kan Y. W. (1984) High-resolution chromosome sorting and DNA spot-blot analysis assign McArdle's syndrome to chromosome 11. Science 225, 57–59 10.1126/science.6587566 [DOI] [PubMed] [Google Scholar]
- 50. Newgard C. B., Fletterick R. J., Anderson L. A., and Lebo R. V. (1987) The polymorphic locus for glycogen storage disease VI (liver glycogen phosphorylase) maps to chromosome 14. Am. J. Hum. Genet. 40, 351–364 [PMC free article] [PubMed] [Google Scholar]
- 51. Newgard C. B., Littman D. R., van Genderen C., Smith M., and Fletterick R. J. (1988) Human brain glycogen phosphorylase. Cloning, sequence analysis, chromosomal mapping, tissue expression, and comparison with the human liver and muscle isozymes. J. Biol. Chem. 263, 3850–3857 [PubMed] [Google Scholar]
- 52. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 53. Pfeiffer-Guglielmi B., Bröer S., Bröer A., and Hamprecht B. (2000) Isozyme pattern of glycogen phosphorylase in the rat nervous system and rat astroglia-rich primary cultures: electrophoretic and polymerase chain reaction studies. Neurochem. Res. 25, 1485–1491 10.1023/A:1007676109206 [DOI] [PubMed] [Google Scholar]
- 54. Pfeiffer-Guglielmi B., Fleckenstein B., Jung G., and Hamprecht B. (2003) Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J. Neurochem. 85, 73–81 10.1046/j.1471-4159.2003.01644.x [DOI] [PubMed] [Google Scholar]
- 55. Sato K., Satoh K., Sato T., Imai F., and Morris H. P. (1976) Isozyme patterns of glycogen phosphorylase in rat tissues and transplantable hepatomas. Cancer Res. 36, 487–495 [PubMed] [Google Scholar]
- 56. Griffiths J. R., Dwek R. A., and Radda G. K. (1976) Heterotropic interactions of ligands with phosphorylase b. Eur. J. Biochem. 61, 243–251 10.1111/j.1432-1033.1976.tb10017.x [DOI] [PubMed] [Google Scholar]
- 57. Johnson L. N., Snape P., Martin J. L., Acharya K. R., Barford D., and Oikonomakos N. G. (1993) Crystallographic binding studies on the allosteric inhibitor glucose-6-phosphate to T state glycogen phosphorylase b. J. Mol. Biol. 232, 253–267 10.1006/jmbi.1993.1380 [DOI] [PubMed] [Google Scholar]
- 58. Barford D., Hu S. H., and Johnson L. N. (1991) Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J. Mol. Biol. 218, 233–260 10.1016/0022-2836(91)90887-C [DOI] [PubMed] [Google Scholar]
- 59. Barford D., and Johnson L. N. (1992) The molecular mechanism for the tetrameric association of glycogen phosphorylase promoted by protein phosphorylation. Protein Sci. 1, 472–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sprang S., Goldsmith E., and Fletterick R. (1987) Structure of the nucleotide activation switch in glycogen phosphorylase a. Science 237, 1012–1019 10.1126/science.3616621 [DOI] [PubMed] [Google Scholar]
- 61. Crerar M. M., Karlsson O., Fletterick R. J., and Hwang P. K. (1995) Chimeric muscle and brain glycogen phosphorylases define protein domains governing isozyme-specific responses to allosteric activation. J. Biol. Chem. 270, 13748–13756 10.1074/jbc.270.23.13748 [DOI] [PubMed] [Google Scholar]
- 62. Lowry O. H., Schulz D. W., and Passonneau J. V. (1967) The kinetics of glycogen phosphorylases from brain and muscle. J. Biol. Chem. 242, 271–280 [PubMed] [Google Scholar]
- 63. Mathieu C., Li de la Sierra-Gallay I., Duval R., Xu X., Cocaign A., Léger T., Woffendin G., Camadro J. M., Etchebest C., Haouz A., Dupret J. M., and Rodrigues-Lima F. (2016) Insights into brain glycogen metabolism: The structure of human brain glycogen phosphorylase. J. Biol. Chem. 291, 18072–18083 10.1074/jbc.M116.738898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mathieu C., Dupret J. M., and Rodrigues Lima F. (2017) The structure of brain glycogen phosphorylase–from allosteric regulation mechanisms to clinical perspectives. FEBS J. 284, 546–554 10.1111/febs.13937 [DOI] [PubMed] [Google Scholar]
- 65. Müller M. S., Pedersen S. E., Walls A. B., Waagepetersen H. S., and Bak L. K. (2015) Isoform-selective regulation of glycogen phosphorylase by energy deprivation and phosphorylation in astrocytes. Glia 63, 154–162 10.1002/glia.22741 [DOI] [PubMed] [Google Scholar]
- 66. Mathieu C., Duval R., Cocaign A., Petit E., Bui L. C., Haddad I., Vinh J., Etchebest C., Dupret J. M., and Rodrigues-Lima F. (2016) An isozyme-specific redox switch in human brain glycogen phosphorylase modulates its allosteric activation by AMP. J. Biol. Chem. 291, 23842–23853 10.1074/jbc.M116.757062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang C., Weerapana E., Blewett M. M., and Cravatt B. F. (2014) A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79–85 10.1038/nmeth.2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., and Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 10.1038/nature09472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang J., Gupta V., Tallman K. A., Porter N. A., Carroll K. S., and Liebler D. C. (2015) Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 10, 1022–1037 10.1038/nprot.2015.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arrizabalaga O., Lacerda H. M., Zubiaga A. M., and Zugaza J. L. (2012) Rac1 protein regulates glycogen phosphorylase activation and controls interleukin (IL)-2-dependent T cell proliferation. J. Biol. Chem. 287, 11878–11890 10.1074/jbc.M111.297804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sylow L., Jensen T. E., Kleinert M., Mouatt J. R., Maarbjerg S. J., Jeppesen J., Prats C., Chiu T. T., Boguslavsky S., Klip A., Schjerling P., and Richter E. A. (2013) Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes 62, 1139–1151 10.2337/db12-0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fernandez A. M., Hernandez-Garzón E., Perez-Domper P., Perez-Alvarez A., Mederos S., Matsui T., Santi A., Trueba-Saiz A., Garcia-Guerra L., Pose-Utrilla J., Fielitz J., Olson E. N., Fernandez de la Rosa R., García-Garcia L., Pozo M. A., et al. (2017) Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 66, 64–74 10.2337/db16-0861 [DOI] [PubMed] [Google Scholar]
- 73. Albrecht J., Wróblewska B., and Mossakowski M. J. (1982) The binding of insulin to cerebral capillaries and astrocytes of the rat. Neurochem. Res. 7, 489–494 10.1007/BF00965500 [DOI] [PubMed] [Google Scholar]
- 74. Baron-Van Evercooren A., Olichon-Berthe C., Kowalski A., Visciano G., and Van Obberghen E. (1991) Expression of IGF-I and insulin receptor genes in the rat central nervous system: a developmental, regional, and cellular analysis. J. Neurosci. Res. 28, 244–253 10.1002/jnr.490280212 [DOI] [PubMed] [Google Scholar]
- 75. Lewis D. A., and Moghaddam B. (2006) Cognitive dysfunction in schizophrenia: convergence of γ-aminobutyric acid and glutamate alterations. Arch. Neurol. 63, 1372–1376 10.1001/archneur.63.10.1372 [DOI] [PubMed] [Google Scholar]
- 76. Moghaddam B., and Javitt D. (2012) From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37, 4–15 10.1038/npp.2011.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Molina V., Solera S., Sanz J., Sarramea F., Luque R., Rodríguez R., Jiménez-Arriero M. A., and Palomo T. (2009) Association between cerebral metabolic and structural abnormalities and cognitive performance in schizophrenia. Psychiatry Res. 173, 88–93 10.1016/j.pscychresns.2008.09.009 [DOI] [PubMed] [Google Scholar]
- 78. Pinacho R., Vila E., Prades R., Tarragó T., Castro E., Ferrer I., and Ramos B. (2016) The glial phosphorylase of glycogen isoform is reduced in the dorsolateral prefrontal cortex in chronic schizophrenia. Schizophr. Res. 177, 37–43 10.1016/j.schres.2016.04.024 [DOI] [PubMed] [Google Scholar]
- 79. Chen C. M., Orefice L. L., Chiu S. L., LeGates T. A., Hattar S., Huganir R. L., Zhao H., Xu B., and Kuruvilla R. (2017) Wnt5a is essential for hippocampal dendritic maintenance and spatial learning and memory in adult mice. Proc. Natl. Acad. Sci. U.S.A. 114, E619–E628 10.1073/pnas.1615792114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gibbs M. E. (2015) Role of glycogenolysis in memory and learning: regulation by noradrenaline, serotonin and ATP. Front. Integr. Neurosci. 9, 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sarowar T., and Grabrucker A. M. (2016) Actin-dependent alterations of dendritic spine morphology in shankopathies. Neural Plast. 2016, 8051861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tashiro A., Minden A., and Yuste R. (2000) Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb. Cortex 10, 927–938 10.1093/cercor/10.10.927 [DOI] [PubMed] [Google Scholar]
- 83. Zamboni V., Armentano M., Sarò G., Ciraolo E., Ghigo A., Germena G., Umbach A., Valnegri P., Passafaro M., Carabelli V., Gavello D., Bianchi V., D'Adamo P., de Curtis I., El-Assawi N., et al. (2016) Disruption of ArhGAP15 results in hyperactive Rac1, affects the architecture and function of hippocampal inhibitory neurons and causes cognitive deficits. Sci. Rep. 6, 34877 10.1038/srep34877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhai L., Feng L., Xia L., Yin H., and Xiang S. (2016) Crystal structure of glycogen debranching enzyme and insights into its catalysis and disease-causing mutations. Nat. Commun. 7, 11229 10.1038/ncomms11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Adeva-Andany M. M., González-Lucán M., Donapetry-García C., Fernández-Fernández C., and Ameneiros-Rodríguez E. (2016) Glycogen metabolism in humans. BBA Clin. 5, 85–100 10.1016/j.bbacli.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mizuno M., Tonozuka T., Suzuki S., Uotsu-Tomita R., Kamitori S., Nishikawa A., and Sakano Y. (2004) Structural insights into substrate specificity and function of glucodextranase. J. Biol. Chem. 279, 10575–10583 10.1074/jbc.M310771200 [DOI] [PubMed] [Google Scholar]
- 87. Watanabe Y., Makino Y., and Omichi K. (2006) Activation of 4-α-glucanotransferase activity of porcine liver glycogen debranching enzyme with cyclodextrins. J. Biochem. 140, 135–140 10.1093/jb/mvj129 [DOI] [PubMed] [Google Scholar]
- 88. Yamamoto E., Watanabe Y., Makino Y., and Omichi K. (2009) Inspection of the activator binding site for 4-α-glucanotransferase in porcine liver glycogen debranching enzyme with fluorogenic dextrins. J. Biochem. 145, 585–590 10.1093/jb/mvp012 [DOI] [PubMed] [Google Scholar]
- 89. Cheng A., Zhang M., Gentry M. S., Worby C. A., Dixon J. E., and Saltiel A. R. (2007) A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease. Genes Dev. 21, 2399–2409 10.1101/gad.1553207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roach P. J. (2015) Glycogen phosphorylation and Lafora disease. Mol. Aspects Med. 46, 78–84 10.1016/j.mam.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu Y., Zeng L., Ma K., Baba O., Zheng P., Liu Y., and Wang Y. (2014) Laforin-malin complex degrades polyglucosan bodies in concert with glycogen debranching enzyme and brain isoform glycogen phosphorylase. Mol. Neurobiol. 49, 645–657 10.1007/s12035-013-8546-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sakoda H., Fujishiro M., Fujio J., Shojima N., Ogihara T., Kushiyama A., Fukushima Y., Anai M., Ono H., Kikuchi M., Horike N., Viana A. Y., Uchijima Y., Kurihara H., and Asano T. (2005) Glycogen debranching enzyme association with β-subunit regulates AMP-activated protein kinase activity. Am. J. Physiol. Endocrinol. Metab. 289, E474–E481 10.1152/ajpendo.00003.2005 [DOI] [PubMed] [Google Scholar]
- 93. Halse R., Fryer L. G., McCormack J. G., Carling D., and Yeaman S. J. (2003) Regulation of glycogen synthase by glucose and glycogen: a possible role for AMP-activated protein kinase. Diabetes 52, 9–15 10.2337/diabetes.52.1.9 [DOI] [PubMed] [Google Scholar]
- 94. Nadeau O. W., Lane L. A., Xu D., Sage J., Priddy T. S., Artigues A., Villar M. T., Yang Q., Robinson C. V., Zhang Y., and Carlson G. M. (2012) Structure and location of the regulatory β subunits in the (αβγδ)4 phosphorylase kinase complex. J. Biol. Chem. 287, 36651–36661 10.1074/jbc.M112.412874 [DOI] [PMC free article] [PubMed] [Google Scholar]