

Abstract
NADH (NAD+) and its reduced form NADH serve as cofactors for a variety of oxidoreductases that participate in many metabolic pathways. NAD+ also is used as substrate by ADP-ribosyl transferases and by sirtuins. NAD+ biosynthesis is one of the most fundamental biochemical pathways in nature, and the ubiquitous NAD+ synthetase (NadE) catalyzes the final step in this biosynthetic route. Two different classes of NadE have been described to date: dimeric single-domain ammonium-dependent NadENH3 and octameric glutamine-dependent NadEGln, and the presence of multiple NadE isoforms is relatively common in prokaryotes. Here, we identified a novel dimeric group of NadEGln in bacteria. Substrate preferences and structural analyses suggested that dimeric NadEGln enzymes may constitute evolutionary intermediates between dimeric NadENH3 and octameric NadEGln. The characterization of additional NadE isoforms in the diazotrophic bacterium Azospirillum brasilense along with the determination of intracellular glutamine levels in response to an ammonium shock led us to propose a model in which these different NadE isoforms became active accordingly to the availability of nitrogen. These data may explain the selective pressures that support the coexistence of multiple isoforms of NadE in some prokaryotes.
Keywords: NAD biosynthesis, nitrogen metabolism, glutamine, glutamine synthase, ammonia, ammonia assimilation, NadE, nitrogenase
Introduction
NADH (NAD+) and its reduced form NADH serve as cofactors for a variety of oxidoreductases that participate in a diverse range of metabolic pathways. More recently, NAD+ has also been implicated in signal transduction mechanisms. Studies in mammals showed that a conserved Nudix domain NAD+ sensor protein regulates DNA repair mechanisms, thereby relating NAD+ levels to cancer and aging (1). NAD+ is also used as substrate for signaling pathways by mono- and poly-ADP-ribosyl-transferases and by sirtuins (2–4). Furthermore, manipulation of the NAD+/NADH levels in microorganisms can help to develop or improve a plethora of biotechnological processes.
NAD+ biosynthesis is a fundamental biochemical process in all cells, it can occur de novo or through salvage pathways. The last reaction step in de novo pathway and in some of the NAD+ salvage pathways is the amidation of nicotinic acid adenine dinucleotide (NaAD)2 to form NAD+, a reaction catalyzed by the ubiquitous NAD+ synthetase (NadE) (5). In the first reaction step, NadE uses NaAD and ATP as substrates, forms a NaAD-AMP intermediate (thereby activating the carboxyl group of nicotinamide) and releases PPi. In the second reaction step, NH3 acts as a nucleophile to attack NaAD-AMP releasing NAD+ and AMP as final products (6).
Two different classes of NadE are found in nature. Single domain ammonium-dependent NadENH3 (EC 6.3.1.5) is present in Bacteria and Archaea and uses external ammonium as the N donor. These enzymes are homodimers and have been extensively characterized biochemically and structurally (7–9). The second class is the glutamine-dependent NadEGln (EC 6.3.5.1) which is present in Eukarya, Bacteria, and Archaea and is able to use l-glutamine as N donor because of the presence of an extra N-terminal glutaminase domain (CN hydrolase domain) (9). Just a few NadEGln have been characterized in prokaryotes (10–12).
The best studied NadEGln is from Mycobacterium tuberculosis (MtNadEGln), this enzyme is arranged as a homooctamer where the N-terminal glutaminase domain from one subunit connects with the C-terminal synthetase domain from another subunit. The ammonia released from glutamine hydrolysis is directed to the synthetase domain through a 40 Å intersubunit ammonia tunnel (13). The MtNadEGln exhibits strong synergism between the two catalytic domains such that efficient glutamine hydrolysis only occurs in the presence of NaAD and ATP and depends of the formation of the NaAD-AMP intermediate. Conversely, the presence of glutamine significantly decreases the Km for NaAD and ATP.
Extensive genomic context analysis revealed that a few prokaryotic single-domain NadE (presumably NadENH3 as judged by domain organization) are actually clustered with a separate gene encoding a glutaminase and, at least in the case of Thermus thermophilus, these domains can interact to form a holoenzyme that is able to utilize glutamine to direct NAD+ formation in vitro (9). Based on these studies, an evolutionary scenario has been postulated where a single-domain NadENH3 was present in last universal common ancestor (LUCA), and the two-domain NadEGln evolved via recruitment and fusion of the N-terminal glutaminase domain (9).
Genome analysis revealed that some prokaryotes can encode up to three different NadE (9). Two different genes encoding two-domain NadEGln have been identified in the diazotrophic endophytic β-Proteobacterium Herbaspirillum seropedicae. In a previous study we presented the biochemical characterization of HsNadE1Gln, which resembled the properties of the archetypical NadEGln from M. tuberculosis; it is also arranged as a homooctamer and preferentially uses glutamine and N donor (12).
Here we describe the biochemical characterization of the second NadEGln from H. seropedicae (named NadE2Gln). Surprisingly, we observed that HsNadE2Gln forms a lower oligomeric structure (probably a homodimer) and can use both ammonium and glutamine as N donor.
From now on we will use the term NadENH3 to describe single-domain homodimeric enzymes. NadEGln will be used to describe enzymes carrying the additional N-terminal glutaminase domain with numbers 1 and 2, indicating quaternary structures arranged as octamers or dimers, respectively.
Oligomerization studies of the two NadEGln detected in the diazotrophic α-Proteobacterium Azospirillum brasilense also showed that AbNadE1Gln forms an octamer, whereas AbNadE2Gln is a dimer. Furthermore, PDB searches identified two structures of dimeric NadE2Gln: one in the β-Proteobacterium Burkholderia thailandensis (PDB code 4F4H) and the other in the δ-Proteobacterium Acinetobacter baumannii (PDB code 5KHA). Phylogenetic analysis showed that all these dimeric NadE2Gln form a separate group distinct from the octameric NadE1Gln. Structural comparisons between dimeric A. baumannii and octameric MtNadE1Gln show that the dimeric and octameric proteins possess similar domain interaction geometry between the glutaminase and synthetase domains achieved through an interprotein domain swapping in the octameric enzymes. In addition to the substrate-binding sites, a conserved intersubunit ammonia tunnel could be detected. We speculate that dimeric NadE2Gln are evolutionary intermediates between single-domain dimeric NadENH3 and two-domain octameric NadE1Gln.
Results
Characterization of H. seropedicae NadE2Gln (HsNadE2)
A previous report indicated the presence of two putative glutamine-depended NAD+ synthetase encoded by the genome of H. seropedicae. One of these enzymes, namely HsNadE1Gln, has been characterized previously, and its oligomerization and biochemical properties resemble the characteristics of MtNadE1Gln (12). The second glutamine-dependent NadE from H. seropedicae (Uniprot D81X05), namely HsNadEGln, has the two domains typically present in other types of NadEGln according to Pfam: an N-terminal glutaminase domain (CN hydrolase, amino acids 3–252) and a C-terminal NAD synthase domain (amino acids 281–542). Sequence alignments with other characterized NadEGln showed that HsNadE2Gln carries the conserved glutamine catalytic triad EKC in the N-terminal region and the conserved residues involved in NaAD and ATP binding in the C-terminal region (Fig. 1).
Figure 1.

Sequence alignment of different NadEGln. The sequences of M. tuberculosis, H. seropedicae NadE1Gln and NadE2Gln, A. brasilense NadE1Gln and NadE2Gln, and B. thailandensis were aligned using Clustal W. The threshold for shading was set as 60%. The conserved catalytic triad EKC present in the glutaminase domain is indicated in red. Conserved residues involved in NaAD and ATP binding are indicated in yellow and orange, respectively. Residues forming the ammonia tunnel constriction are indicated in blue. The interdomain loop in MtNadE1Gln is underlined.
The gene encoding HsNadE2Gln was cloned into pET28a, and the protein was expressed as a N-terminal His6 tag fusion protein using Escherichia coli BL21 (λDE3) as host. HsNadE2Gln was purified by Ni2+-affinity chromatography. Analytical gel filtration was performed, and HsNadE2Gln eluted as a single homogeneous peak with the elution volume corresponding to 85 kDa (Figs. S1 and S2). Because the calculated mass for each monomer is 61 kDa, the gel-filtration analysis supports that HsNadE2 is arranged in a different oligomeric state, contrasting with the octameric MtNadE1Glnand HsNadE1Gln.
The activity of HsNadE2Gln was determined by coupling NAD+ formation with the activity of alcohol dehydrogenase and following the absorbance of NADH at 340 nm. The substrates NaAD and ATP were kept at saturating levels, and the experiments were performed with increasing concentrations of ammonium or glutamine to determine kinetic parameters for these two substrates. For both ammonium and glutamine the initial velocity versus substrate concentration exhibited typical Michaelis–Menten hyperbolic response (Fig. S2). The determined Km for ammonium and glutamine were in the same order of magnitude, being 240 and 130 μm, respectively (Table 1). This is in contrast MtNadE1Gln and HsNadE1Gln, which have a Km for glutamine at least 1 order of magnitude lower that for ammonium (Table 1). The Km values of HsNadE2Gln are more related to the values obtained for a NadEGln characterized in the deep-branching Bacteria Thermotoga maritima (Table 1). Note that no information about the oligomerization state is available for TmNadEGln. Comparison of the kcat/Km values, which are indicative of substrate preference, showed that HsNadE2Gln and TmNadEGln have no significant preference N donor substrates. On the other hand, HsNadE1Gln and MtNadE1Gln showed a clear preference to use glutamine as N donor (Table 1).
Table 1.
Kinetic properties in different Gln-dependent NadE
| Organism | Substrate | Km | kcat | kcat/Km | Preference | Reference |
|---|---|---|---|---|---|---|
| mm | s−1 | s−1/mm | ||||
| M. tuberculosis | NH4+ | 20 ± 2 | 3.0 ± 0.1 | 0.15 ± 0.02 | 2.8 | Ref. 13 |
| MtNadE1 | Gln | 1.3 ± 0.1 | 0.55 ± 0.01 | 0.42 ± 0.03 | Gln > NH4+ | |
| T. maritima | NH4+ | 0.33 ± 0.1 | 0.24 ± 0.04 | 0.7 ± 0.3 | 0.9 | Ref. 11 |
| Gln | 0.42 ± 0.05 | 0.29 ± 0.01 | 0.68 ± 0.08 | NH4+ > Gln | ||
| T. thermophilus | NH4+ | 0.03 ± 0.01 | 0.09 ± 0.03 | 3 | 50 | Ref. 9 |
| Gln | 0.73 ± 0.19 | 0.04 ± 0.01 | 0.06 | NH4+ > Gln | ||
| HsNadE1 | NH4+ | 5.7 ± 0.61 | 0.05 ± 0.001 | 0.009 ± 0.0001 | 9 | Ref. 12 |
| Gln | 0.37 ± 0.08 | 0.03 ± 0.02 | 0.08 ± 0.02 | Gln > NH4+ | ||
| HsNadE2 | NH4+ | 0.24 ± 0.07 | 0.07 ± 0.01 | 0.29 ± 0.08 | 2.4 | This work |
| Gln | 0.13 ± 0.02 | 0.09 ± 0.01 | 0.69 ± 0.05 | Gln > NH4+ | ||
| AbNadE1 | NH4+ | 10.01 ± 0.84 | 0.23 ± 0.01 | 0.02 ± 0.001 | 140 | This work |
| Gln | 0.08 ± 0.005 | 0.23 ± 0.003 | 2.8 ± 0.23 | Gln > NH4+ | ||
| AbNadE2 | NH4+ | 1.85 ± 0.13 | 0.06 ± 0.004 | 0.03 ± 0.001 | 1.6 | This work |
| Gln | 0.82 ± 0.08 | 0.04 ± 0.001 | 0.05 ± 0.006 | Gln > NH4+ |
The preference for the utilization of ammonium or glutamine was compared between HsNadE1Gln and HsNadE2Gln using LC/MS. For this analysis, unlabeled glutamine is mixed with 15NH4Cl, and the ratio of 15N NAD+ and 14N NAD+ (NH4+/Gln) formed is determined using LC coupled to high resolution MS. The results confirmed that HsNadE1Gln has a stronger preference for glutamine than HsNadE2Gln (Table 2). When both ammonium and glutamine were available at equimolar concentration (2 mm), HsNadE2Gln was able to incorporated 11.9-fold more ammonium in relation to glutamine than HsNadE1Gln (Table 2).
Table 2.
Ammonium versus glutamine competition in H. seropedicae NadE1 and NadE2
| Substrate concentrations | HsNadE1 NH4+/Gln | HsNadE2 NH4+/Gln | HsNadE2/HsNadE1 ratio |
|---|---|---|---|
| 2 mm glutamine and 0.2 mm NH4Cl | 0.05 | 0.34 | 6.8 |
| 0.2 mm glutamine and 2 mm NH4Cl | 0.18 | 5.26 | 29.2 |
| 2 mm glutamine and 2 mm NH4Cl | 0.15 | 1.78 | 11.9 |
Characterization of the three NadE from A. brasilense
An inspection on the genome of the diazotrophic plant associative α-Proteobacterium A. brasilense Sp245 revealed the presence of three NadE-like genes: first a single-domain NadENH3 type (Uniprot G8ATC0), namely AbNadENH3, and the other two belonging to the NadEGln type, namely AbNadE1Gln (Uniprot G8AIW8) and AbNadE2Gln (Uniprot G8ATT3). The genes encoding these proteins were cloned and expressed as recombinant proteins in E. coli BL21 (λDE3). The proteins were purified to homogeneity as judged by SDS-PAGE (Fig. S3).
Analytical gel filtration revealed that all three NadE eluted as a homogeneous symmetric peak with elution volumes corresponding to 600, 103, and 72 kDa for AbNadE1Gln, AbNadE2Gln, and AbNadENH3, respectively (Fig. 2 and Fig. S4). The determined molecular weight fits with dimer oligomerization for AbNadE2Gln and AbNadENH3 (calculated masses are 120 kDa for dimer AbNadE2 and 74 kDa for AbNadE3). On the other hand, AbNadE1Gln is arranged as an octamer (calculated mass for octamer is 605 kDa); hence AbNadE1Gln resembles the oligomeric organization found in HsNadE1Gln and MtNadE1Gln(all octameric).
Figure 2.
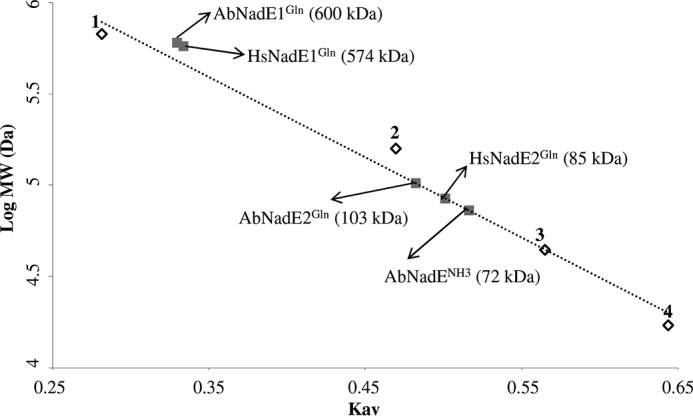
Gel filtration analysis of the different NadE. Gel filtration was performed on a Superose 6 column (GE Healthcare), which was calibrated with a range of molecular mass standards (Bio-Rad): point 1, thyroglobulin; point 2, bovine γ-globulin; point 3, chicken ovalbumin; and point 4, equine myoglobin. The arrows indicate the estimated molecular weight of each NadE protein.
The AbNadENH3 belongs to the NadENH3 type and showed a dimeric organization. As expected, AbNadENH3 was not able to use glutamine as N donor for NAD+ production (data not shown). On the other hand, the identification of a dimeric NadEGln also in A. brasilense supports the presence of a novel subgroup of NadEGln, which is different from the archetypical octameric NadEGln described in M. tuberculosis and eukaryotes such as Saccharomyces cerevisiae.
The kinetic characterization of the two A. brasilense NadEGln revealed the same trend as observed for the H. seropedicae enzymes. The dimeric AbNadE2Gln showed a Km for ammonium and glutamine in the same order of magnitude being 1,850 and 820 μm, respectively (Table 1). On the other hand, the octameric AbNadE1Gln had a Km for glutamine more than 2 orders of magnitude lower that for ammonium (Table 1). The kcat/Km values clearly show that AbNadE1Gln has a strong preference for glutamine as N donor, whereas AbNadE2Gln can use both substrates with nearly similar preference (Table 1).
Intracellular glutamine levels in A. brasilense
It is well-established that the intracellular glutamine levels in Proteobacteria correlates with the availability of ammonium in the culture media (14, 15). Particularly, in some nitrogen fixing organisms, such as A. brasilense and H. seropedicae, nitrogen fixation only occurs when the intracellular glutamine levels are low (16, 17).
We used untargeted LC/MS to determine the intracellular levels of glutamine in A. brasilense. Under nitrogen-fixing conditions, the glutamine levels ranged between 74 and 79 μm (Fig. 3A). After the addition of 200 μm of NH4Cl to the cells, the intracellular levels of glutamine augmented to 830 μm in just 30 s and remained at similar levels after 1 min (Fig. 3A). 2 min after the ammonium shock, the ammonium added is consumed by the cellular metabolism, and the intracellular glutamine levels declined to 200 μm, decreasing even further after 20 min (Fig. 3A). These fluctuations in intracellular glutamine were well-synchronized with the covalent modification of the nitrogenase component NifH and glutamine synthetase GS (Fig. 3B). Both NifH and GS are known to suffer reversible covalent modification that inactivates both enzymes in response to an ammonium shock (18, 19).
Figure 3.
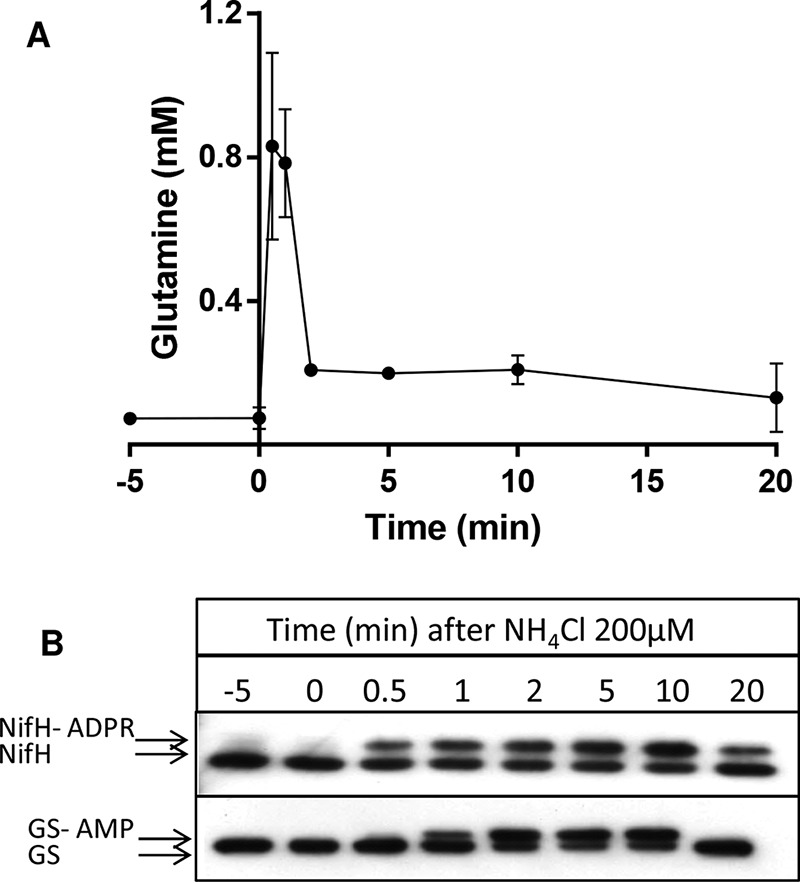
Intracellular levels of glutamine and NifH and GS modification after an ammonium shock in A. brasilense. A, average intracellular glutamine levels from triplicate experiments ± S.D. B, Western blotting analysis showing of the reversible post-translational modification of NifH and GS. The upper bands represent the ADP-ribosylated NifH (NifH-ADPR) or adenylylated GS (GS-AMP).
Under nitrogen-fixing conditions, the intracellular levels of glutamine were nearly identical to the AbNadE1Gln Km for glutamine (∼80 μm) and ∼10-fold below the AbNadE2Gln Km for glutamine (Table 1 and Fig. 3A). These data suggest that AbNadE1Gln is much more active than AbNadE2Gln under nitrogen-fixing conditions. In contrast, upon an ammonium shock, the intracellular levels of glutamine reached the AbNadE2Gln Km for glutamine (∼800 μm). Hence, upon an ammonium shock, AbNadE2Gln should become more active, whereas AbNadE1Gln should be operating at Vmax (Fig. S2)
The LC/MS metabolome data were submitted to XCMS software (20) to identify other potential metabolites that fluctuate in response to the ammonium shock. However, only glutamine showed statistical significant fluctuations (p < 0.05). Even though other metabolites such as NADH, NAD+, ATP, ADP, AMP, and 2-oxoglutarate could be detected in the analysis (as suggested by comparison of retention times and m/z to authenticated standards), their low signal and/or high variation among biological replicates did not allow their quantification.
Phylogeny analysis of dimeric NadEGln
The presence of other representatives of dimeric NadE2Gln in nature is supported by at least two structures deposited in the PDB. The NadE2Gln from the β-Proteobacterium B. thailandensis (PDB code 4F4H) and from the δ-Proteobacterium A. baumannii (PDB code 5KHA). A Neighbor-joining phylogenetic tree was constructed using the alignment of NadEGln sequences derived from organisms belonging to all domains of life (Fig. 4). The sequences were separated into three distinct groups; 1) the eukaryotic NadE1Gln; 2) a group containing the octameric NadE1Gln from M. tuberculosis, H. seropedicae and A. brasilense; and 3) a more diverse group comprising all the NadE2Gln described as dimeric. This last group includes the sequences of deep branching Bacteria (Thermotoga and Aquifex) and Archaea (Fig. 4).
Figure 4.

Phylogenetic analysis of selected NadEGln. The sequence of NadEGln was retrieved from NCBI and aligned using Clustal W. The phylogentic three was constructed by Neighbor-joining using MEGA 7. All positions containing gaps and missing data were eliminated from the data set. Bootstrap values were adjusted to 1000 replicates. Three groups were observed: 1) eukaryotic representatives; 2) NadE1-like octameric NadEGln; and 3) NadE2-like dimeric NadEGln. The relevant proteins with experimentally determined quaternary structure are indicated by arrows. A. baumannii, WP_065718975.1; Akkermansia muciniphila ATCC BAA-835, ACD04457.1; Aquifex aeolicus, NP_213654.1; Arabidopsis thaliana, NP_175906.1; Ardenticatena maritima, KPL88222.1; A. brasilense, AIB10872.1; A. brasilense, AIB14429.1; Azotobacter vinelandii DJ, YP_002798395.1; B. thailandensis, AOJ56104.1; Chloroflexus aurantiacus, ABY34602.1; Clostridium cellulolyticum H10, YP_002505537.1; Danio rerio, NP_001092723.1; Dehalococcoides mccartyi 195, YP_181837.1; Drosophila melanogaster, NP_572913.1; Fervidobacterium nodosum Rt17-B1, YP_001410277.1; Gluconacetobacter diazotrophicus PA1 5, YP_001601200.1; Haloferax mediterranei, AHZ23047.1; H. seropedicae, AKN65438.1; H. seropedicae, AKN67808.1; Homo sapiens, NP_060631.2; Methanoregula formicica, AGB01500.1; Methanosaeta thermophila PT, YP_843157.1; Methylobacterium extorquens CM4, YP_002423109.1; Mus musculus, NP_084497.1; Mycobacterium bovis AF2122/97, NP_856111.1; Ralstonia eutropha H16, YP_725265.1; S. cerevisiae S288C, NP_011941.1; Schizosaccharomyces pombe 972h-, NP_587771.1; Theionarchaea archaeon, KYK35595.1; Thermosipho africanus TCF52B, YP_002335497.1; T. maritima, AKE27162.1; Thermotoga neapolitana DSM 4359, YP_002534863.1; and M. tuberculosis, AMP30329.1.
Structural analysis of dimeric NadE2Gln
Given the availability of a 1.7 Å resolution structure of dimeric NadE2Gln from B. thailandensis (PDB code 4F4H) obtained by the Burkholderia structome project (21), we used this protein to analyze the structural features of the subgroup of dimeric NadE2Gln. Structural alignments with other available NadEGln structures revealed that B. thailandensis is highly related to the dimeric NadE2Gln from A. baumannii (data not shown) as anticipated by their degree of phylogenetic relationship (Fig. 4), hence supporting that B. thailandensis NadE2Gln may serve as a prototype for this group of enzymes. Alignments with NAD synthetase NadENH3 from Bacillus subtilis (22) revealed structural conservation at the NAD synthetase domain structures (Fig. S5).
Comparison between the dimeric B. thailandensis NadE2 and the octameric M. tuberculosis NadE1 (13) revealed that the individual domains are structurally very similar with conserved residues involved in substrate binding and domain folding (Fig. 1). However, they display a rearrangement of the relative domain positions within the protomers (Fig. 5). In B. thailandensis NadE2, the glutaminase and synthetase domains within the same polypeptide make extensive contacts and are located in the same plane (Fig. 5, A–C). On the other hand, in the case of M. tuberculosis NadE1, the glutaminase domain of one polypeptide extends to make contact with the NaAD sythetase domain of the next polypeptide in the octamer through a flexible loop comprising resides 313–331 (MtNadE1Gln numbering) (Fig. 5, A–C). This particular segment is shorter in the dimeric subgroup of proteins (Fig. 1). This flexible loop facilitates the extensive intersubunit contacts in the octameric structure of M. tuberculosis. The superimposed picture of the two structures (Fig. 5A) shows how the glutaminase domain of M. tuberculosis translates by ∼40 Å compared with the same domain in B. thailandensis and makes most of the surface contact with the next chain, which suggests that the glutaminase and the NaAD sythetase function of the domain likely occur within domains of two different polypeptides. This arrangement indicates that the relative orientations between the N and G domains remain the same though through a domain-swap interaction in the octameric case (Fig. 5D). This is illustrated by the high structural similarity of the dimeric protein to the octameric when allowing for structural superpositions over different monomers. In this case the dimeric (PDB code 4F4H) structure can be superposed on the octameric (PDB code 3DLA) structure with an RMSD of 1.85 Å over 741 aligned residues (i.e. superposing the boxed domains in Fig. 5D).
Figure 5.
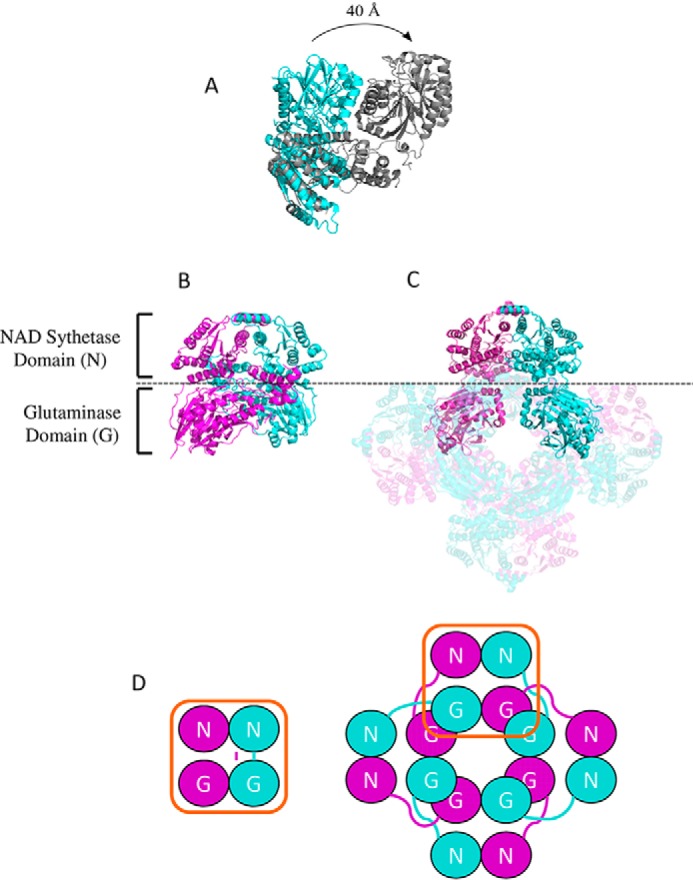
Comparison between the structures of dimeric and octameric NadEGln. A, superimposition of the single chain of NadE2Gln from B. thailandensis (cyan) and NadE1Gln from M. tuberculosis (gray). The PDB structures 4FH4 and 3DLA were superimposed using COOT and visualized using PyMOL. Structures are colored by chain. B and C, comparison between the organization of dimeric NadEGln from B. thailandensis (B) and octameric NadEGln from M. tuberculosis (C). Only one dimer of the octameric M. tuberculosis NadE is shown without transparency for comparison with B. thailandensis. D, schematic organization of the NAD synthetase domain N and the glutaminase domain G in the dimeric and in the octameric conformation of NadEGln. Note that the relative orientations between the N and G domains remain the same though through a domain-swap interaction in the octameric case. The PDB structures 4FH4 and 3DLA were visualized using PyMOL.
Surface representation of B. thailandensis NadE2Gln showed that the conserved residues forming the glutaminase catalytic triad and the intersubunit NaAD–ATP bind sites are located in solvent-exposed pores (Fig. 6, A and B). A phosphate is present in the NaAD binding site of B. thailandensis NadE (not shown) and makes similar contacts as observed for the NaAD phosphates in the M. tuberculosis NadE structure (residues Asn-471 and Lys-635, Mt numbering) (23), hence suggesting similar substrate binding modes between octameric and dimeric NadEGln enzymes.
Figure 6.

Structure of the dimeric NadE2Gln from B. thailandensis. A and B, surface (A) and cartoon (B) representations; the monomers are colored green and cyan. The glutaminase catalytic triade residues are in red sticks. Residues involved in NaAD and ATP binding are shown as yellow and orange sticks, respectively. The dashed line indicates the separation of the NAD synthetase domain on the top and the glutaminase domain on the bottom. C and D, the ammonia tunnel connecting the glutaminase and NaAD synthetase domain in different protomers (C) and within the same protomer (D). The detailed structure of the intersubunit ammonia tunnel of the dimeric NadE2Gln from B. thailandensis is shown. E, residues forming the intersubunit ammonia tunnel from different promoters are indicated as green and cyan sticks, respectively. F, conserved residues forming the major constriction within the ammonia tunnel are show in cyan, and the glutaminase catalytic residues are in red. The figures were generated using PyMOL and the PDB entry 4F4H. The ammonia tunnel was identified by CAVER.
Given that the NaAD- and glutamine-binding sites are separated 40–50 Å in space (within the same protomer or between the two different promoters), we hypothesized that B. thailandensis NadE2Gln should have an internal tunnel to translocate ammonia between the two catalytic domains as observed in MtNadE1Gln. We used CAVER to search for internal tunnels within the B. thailandensis NadE2Gln structure. This analysis identified two continuous tunnels that are potential routes for ammonia translocation. A 57.6 Å tunnel connects the glutaminase site from one promoter to the synthetase site in the other promoter (Fig. 6C). Another 58.3 Å tunnel connects the two catalytic domains within the same protomer (Fig. 6D). The interprotomer tunnel is U-shaped resembling the ammonia tunnel identified in the M. tuberculosis NadE, whereas the intrasubunit tunnel is Z-shaped (Fig. 6). Considering the protein symmetry, there are actually four possible pathways for ammonia translocation (G1 to S1, G1 to S2, G2 to S1, and G2 to S2); all these routes are interconnected by a major empty space located within the interprotomer/interdomain interface (Fig. 6).
A major bottleneck constriction (0.94 Å) in the ammonia tunnel is formed just after the glutaminase catalytic triad (Fig. 6, E and F). Given that the ammonia van der Waals radius is ∼1.6 Å (24), it is likely that BtNadE2Gln must go through conformational changes to allow ammonia transfer between the two catalytic domains. The bottleneck constriction is mostly hydrophobic and formed by residues that are conserved in both octameric and dimeric forms of NadEGln (Figs. 1 and 6). The hydrophobic nature of the constriction suggests that ammonia rather than ionic ammonium may be the translocated species. A hydrophobic ammonia bottleneck or gate is also present in other ammonia-translocating proteins such as AmtB and GlmS (24, 25). In AmtB, conserved amino acid residues help to deprotonate ammonium before translacation (25), an important step to drive the equilibrium toward NH3 formation under physiological pH considering the NH4+ pKa of 9.2. More studies will be necessary to determine the nature of the translocated species in NadE2Gln.
Discussion
In a previous study we reported the characterization of a NadE1Gln from H. seropedicae, HsNadE1Gln. We found that HsNadE1Gln oligomerization and biochemical properties resemble the characteristics reported for the intensively studied MtNadE1Gln and the S. cerevisiae NadE1Gln. Here we report the characterization of a second type of NadE2Gln encoded by the genome of H. seropedicae, HsNadE2Gln. Surprisingly, we found that HsNadE2Gln is arranged in a completely novel oligomeric state (Fig. 2). Even though the experimentally determined molecular mass for HsNadE2Gln was 85 kDa (which is between a monomer and dimer), it is more likely that HsNadE2Gln is arranged as a dimer based on the phylogenetic relationship with other dimeric NadE2Gln synthetases (Fig. 4).
Additional studies in A. brasilense revealed that this organism also encodes a dimeric form of NadE2Gln, AbNadE2Gln (Fig. 2). Further inspection in the PDB revealed the presence of dimeric NadE2Gln in A. baumannii and B. thailandensis. These data suggest the presence of a novel group of dimeric NadE2Gln. Phylogenetic analysis of NadEGln sequences from various organisms with diverse lifestyles support the presence two distinct groups of NadEGln in prokaryotes (Fig. 4). It is tempting to speculate that the group containing MtNadE1Gln, AbNadE1Gln, and HsNadE1Gln (all octameric), is likely to be formed mostly by octameric types of NadE1Gln, whereas the group containing HsNadE2Gln, AbNadE2Gln, A. baumannii NadE2Gln, and B. thailandensis NadE2Gln (all dimeric) should be mostly composed of dimeric forms of NadE2Gln.
Structural analysis of B. thailandensis dimeric NadE2Gln showed that this enzyme shares some features with octameric MtNadE1Gln: the residues involved in substrate binding and catalysis and the residues forming a long intersubunit tunnel that are likely to carry ammonia from the glutaminase to the synthetase domain (Figs. 1 and 6). However, although BtNadE2Gln has an intensive intrachain glutaminase-synthetase domain contact, such contacts are virtually absent in the MtNadE1Gln enzyme (Fig. 5). The octameric NadE1Gln-like forms presumably evolved to make much more intersubunit contacts, a property that may explain the strong domain synergism detected in MtNadE1Gln (13) and the little efficiency of NadE1Gln-like in using ammonium as N source (Tables 1 and 2).
The sequences of deep branching Bacteria (Thermotoga and Aquifex) and Archaea cluster in the NadE2Gln-like dimeric group (Fig. 4). A previous phylogentic analysis suggested that a single-domain NadENH3 was present in last universal common ancestor and the two-domain NadEGln evolved via recruitment and fusion of the N-terminal glutaminase domain (9). If this assumption is correct, we speculate that the dimeric NadE2Gln could be an intermediate evolutionary link between the single-domain dimeric NadENH3 and two domain octameric NadE1Gln. Indeed, the enzymatic characteristics of the NadE2Gln enzymes (Tables 1 and 2) show intermediate properties between dimeric NadENH3 and octameric NadE1Gln (i.e. NadE2Gln can use both ammonium and glutamine with similar efficiencies). The biogeochemical history of earth support that although ammonium was readily available to the ancient life forms (that could support NadENH3 activity), ammonium is very limited in most habitats nowadays, and this could explain the need to recruit a glutaminase domain to support NadE function during evolution.
More than one copy of NadE has been detected in the genome of a range of prokaryotes (see Table S3 in Ref. 9). One intriguing question remaining to be answered is why some organisms encode multiple isoforms of NadE. Different expression in response to nitrogen availability would be one possible explanation; however, at least in the case of A. brasilense, proteomic analysis revealed that AbNadE1Gln, AbNadE2Gln, and AbNadENH3 levels did not change in response to ammonium availability.3 Hence, the different kinetic properties and/or substrate preferences may constitute the only selective pressure to maintain three NadE isoforms within the A. brasilense genome. Our data allowed us to propose a model for the role of these isoforms in A. brasilense. Under nitrogen-fixing conditions, the intracellular levels of glutamine are low (∼80 μm), and NadE1Gln operates at Vmax/2 and thereby prompts responses to small fluctuations in glutamine availability within this range accordingly to the NadE1 Michaelis–Menten curve (Fig. 7, A and D). Upon an ammonium shock of NH4+ 200 μm, the intracellular glutamine raises to ∼800 μm, NadE1 operates at Vmax, whereas NadE2Gln operates at Vmax/2 and thereby prompts responses to small fluctuations in glutamine availability within this range (Fig. 7, B and D). Hence, the presence of two glutamine-dependent NadEs would allow better adjustment of NAD+ production over a broad range of glutamine concentrations (Fig. 7).
Figure 7.
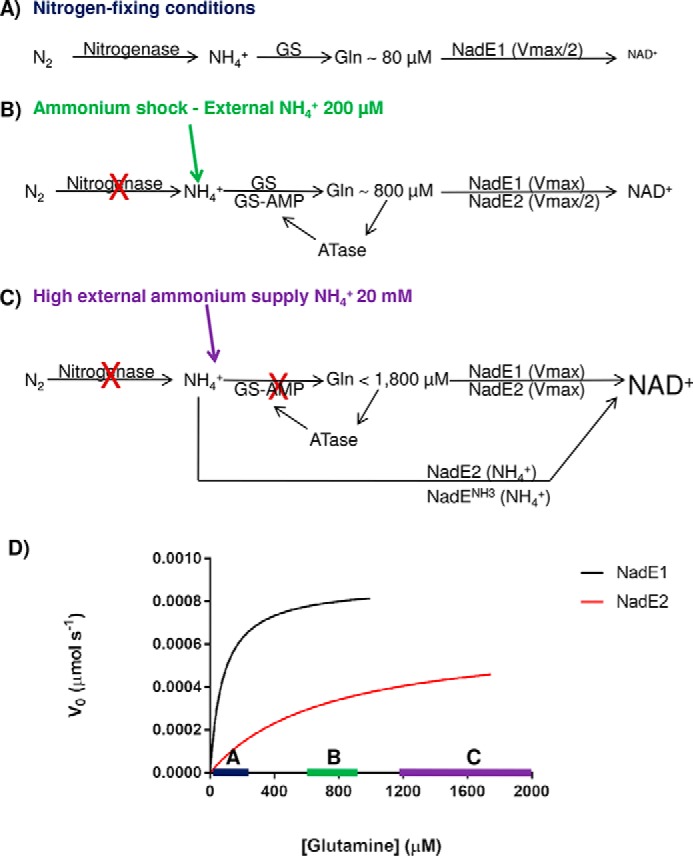
Model for the role of the three NadE in A. brasilense. A, under nitrogen-fixing conditions nitrogenase feeds ammonium into GS, and intracellular levels of glutamine are low (∼80 μm). NadE1Gln operates at Vmax/2 and promptly responds to small fluctuations in glutamine availability within this range (see the blue bar with Michaelis–Menten curves in D). NadE2Gln and NadENH3 activities are negligible leading to slow production of NAD+. B, upon an ammonium shock of NH4+ 200 μm, the intracellular glutamine rises to ∼800 μm, leading to total inhibition of nitrogenase by ADP-ribosylation and nearly full inhibition of GS by adenylylation. NadE1Gln operates at Vmax, whereas NadE2Gln operates at Vmax/2 and promptly responds to small fluctuations in glutamine availability within this range (see the green bar with Michaelis–Menten curves in D). NAD+ production is elevated. C, when external levels of ammonium are high, the high intracellular glutamine will lead to NadE1Gln and NadE2Gln to operate at Vmax using glutamine as substrate. Furthermore, because GS is fully inactive due adenylylation, free intracellular ammonium could be directly assimilated into NAD+ by NadE2Gln and presumably also by NadENH3 bypassing GS activity. The concert action of all three isoforms of NadE is likely to further enhance NAD+ production. D, velocities of the different A. brasilense NadE isoforms accordingly to the intracellular glutamine levels.
We did not measure the intracellular glutamine concentrations under a high ammonium regime; however, in E. coli concentrations of 1.9–5 mm under high ammonium regime were detected (26–28). When A. brasilense receives an ammonium shock of 20 mm NH4Cl, GS is fully adenylylated and inactive after 2 min.4 If we assume that intracellular ammonium levels could be similar to external levels because of ammonia diffusion through the cytoplasmic membrane (29) and lack of active GS under 20 mm NH4Cl, then NadE2Gln (and presumably also NadENH3) could bypass GS and directly assimilate ammonium into NAD+ (Fig. 7C).
The presence of NadE isoforms that are able to bypass GS under high ammonium regimes would be energetic advantageous giving that GS activity may consume up to 15% of the cellular ATP (30). Energy saving may be particularly important in dynamic and competitive niches such as soil and rhizosphere, the habitats were A. brasilense is found.
Experimental procedures
Plasmid construction
Isolation of plasmid DNA, gel electrophoresis, bacterial transformation, and cloning were performed using standard protocols. T4 DNA ligase and the restriction enzymes NdeI and BamHI were purchase from Fermentas and were used following the manufacturers' instructions. The genes nadE2 from H. seropedicae SMR1, nadE1, nadE2, and nadE3 from A. brasilense FP2 were amplified by PCR using DNA from boiled culture as template and high-fidelity polymerase Pfu (Fermentas). PCR fragments and expression vectors pET28a, pET29a, and pTEV5 were digested with NdeI and BamHI and ligated. The primers used in PCR are listed on Table S1. DNA sequencing was performed using dye-labeled terminators (BigDye® ABI PRISM) in an automated DNA sequencer ABI 3500 from Applied Biosystems. Recombinant plasmids obtained in this work were: pASnade2 (Express H. seropedicae NadE2Gln with a N-terminal His6 tag in pET28a), pASnade3 (Express A. brasilense NadENH3 with a N-terminal His6 tag in pTEV5), pLHnade2 (Express native A. brasilense NadE2Gln in pET29a), and pASnade1 (Express A. brasilense NadE1 with a N-terminal His6 tag in pTEV5).
Protein purification
NadE proteins were overexpressed in freshly transformed E. coli BL21 (λDE3) and purified adapting previous protocols (12). Typically, the cells were grown in LB medium to an A600 nm of 0.7 at 37 °C. The incubation temperature was changed to 16 °C, and protein expression was induced by adding 0.5 mm isopropyl β-d-thiogalactopyranoside. The cells were harvested after 12 h of shaking at 200 rpm. For His-tagged protein purification (AbNadE1Gln, AbNadENH3, HsNadE1Gln, and HsNadE2Gln), the cell pellets from 0.8 liter of culture were resuspended in 40 ml of buffer A: 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 20% (v/v) glycerol. The cells were lysed by sonication and centrifuged. The soluble fraction used for His-NadE purification using Ni2+ affinity chromatography on a Hi-trap chelating column (GE Healthcare); the proteins were eluted using a imidazole gradient. Protein concentration was measured at 595 nm by Bradford reaction assay (Bio-Rad).
When indicated, TEV protease was used to remove the N-terminal His6 tag fusion as follows: purified AbNadE1Gln and AbNadENH3 proteins were mixed with His-TEV protease at a 9:1 ratio in 50 mm Tris-HCl, pH 8, 100 mm NaCl, 1 mm DTT. The mixture was incubated at 8 °C for 20 h. His-TEV and uncleaved NadE protein were removed using a Ni2+ Hi-trap chelating column (GE Healthcare). Cleavage was verified by SDS-PAGE electrophoresis. The final preparation has four additional residues (Gly–Ala–Ser–His) at the N terminus of the TEV protease cleavage site.
The A. brasilense NadE2Gln was purified without tags. The plasmid pLHnadE2 was used to express untagged NadE2Gln in E. coli BL21 (λDE3) as described above. The cell pellet was resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 20% (v/v) glycerol) and sonicated. The soluble fraction was loaded onto a Q-Sepharose column, and proteins were eluted with 0.1–1 m linear gradient of NaCl. Fractions containing NadE were pooled and loaded onto a gel filtration HiLoad Superdex 200 16/60 column (GE Healthcare).
Gel filtration and MW determination
Analytical size-exclusion chromatography was performed in a 24-ml Superose 6 column (GE Healthcare) using 50 mm Tris-HCl, pH 8, and 100 mm NaCl as buffer. The column was calibrated with the molecular weight standards: thyroglobulin, bovine γ-globulin, chicken ovalbumin, and equine myoglobin (Bio-Rad). The log of molecular weight was plotted against the Kav (partition coefficient of each standard) to build a linear calibration curve. The molecular weight of each protein was determined by comparing the Kav against the calibration curve.
NadE kinetic analysis
The NadE activity was monitored by coupling the NAD+ production to its reduction to NADH using alcohol dehydrogenase from yeast (Sigma) and monitoring NADH formation at 340 nm as described previously (12). The assays were carried out at 30 °C in 62 mm Tris-HCl, pH 8.5, 20 mm KCl, 20 mm MgCl2, 1.6% (v/v) ethanol, 10 mm DTT, 6 units of yeast alcohol dehydrogenase (Sigma) and 50 nm of NadE (monomer concentration). To determine the kinetic constants, the substrates ATP and NaAD were kept at saturating concentrations (10 and 1 mm, respectively), whereas glutamine or ammonium (NH4Cl) varied from 2.5 to 10,000 μm and from 50 to 100,000 μm, respectively. The assays were performed in triplicate, and the initial velocity data were fitted to the Michaelis–Menten equation using GraphPad Prism software package.
LC/MS analysis ammonium versus glutamine substrate competition
Competition assays were performed combining different amounts of glutamine and 15N-labeled NH4Cl (Sigma). Reactions containing the two substrates were performed in 20 mm Tris-HCl, pH 8, 2 mm ATP, 2 mm NaAD, 5 mm MgCl2, and 1 mm DTT. The reactions were started by adding 50 nm of NadE (monomer concentration) and incubated at 30 °C for 5 min. Acetic acid to 10% (v/v) was added to quench the reactions. The samples were centrifuged at 20,000 × g for 5 min at 4 °C, and 10 μl of the supernatant was used for LC/MS analysis as described (31). Metabolites were separated using a UFLC Prominence (Shimadzu) in a C18 column of 2.6 μm and 50 × 2.1 mm (Phenomenex) kept at 40 °C. The mobile phases were composed of 15 mm acetic acid and 10 mm tributylamine (solvent A) or methanol (solvent B). Samples were placed into vials and kept in an auto-sampler at 4 °C; 10 μl of each sample was injected at 0.2 ml min−1 in duplicate. Metabolites were eluted from the column using a linear gradient (0–100% of solvent B) in 30 min. The detection of the metabolites was performed by coupling the LC with a MicroTOF-QII (Bruker Daltonics), equipped with an electrospray ionization source. The retention time of NAD+ was confirmed using a standard solution.
The 15N incorporation was determined by the formula: intensity peak m/z 663 − (0.227 × intensity peak m/z 662). This value represents the increase in the m/z 663.1 signal because of 15N ammonium incorporation subtracted from the theoretical 22.7% isotopic contribution of 1-[13C-14N]NAD+). The value obtained (which corresponds only to [12C-15N]NAD+) was divided by the signal of [14N]glutamine incorporation at 662 m/z to give the 15N NH4+/[14N]Gln incorporation ratio.
Intracellular levels of glutamine and protein post-translational modification analysis
A. brasilense FP2 was cultured in 110 ml of NfbHP medium containing 5 mm glutamate as nitrogen source, and nitrogenase activity was determined by the acetylene reduction method using GC exactly as described previously (18). The A. brasilense cells were subjected to an ammonium shock (NH4Cl 200 μm) while being kept in a rotatory shaker incubator at 120 rpm and 30 °C. The samples were collected at −5, 0, 0.5, 1, 2, 5, 10, and 20 min after ammonium addition. To analyze the effectiveness of the ammonium shock, the post-translational modification of the GS and NifH proteins were monitored by Western blotting exactly as described previously (18).
For the extraction of metabolites, 2-ml samples were rapidly collected and quickly cooled in liquid nitrogen. The samples were centrifuged for 2 min at 20,000 × g at 4 °C, and the cell pellets were resuspended in 60 μl of ice-cold solvent (40:40:20 acetonitrile:methanol:water and 0.1 m formic acid) and kept for 20 min at −20 °C as described (26). The extracts were centrifuged at 20,000 × g at 4 °C for 3 min, the supernatant was transferred to a new tube, and the pellet was subjected to another round of metabolite extraction. The supernatant from the two extractions were combined and neutralized with 4 μl of ammonium hydroxide. The samples were centrifuged (20,000 × g, 20 min, 4 °C), and supernatant was subject to LC/MS analysis.
Separation and detection of metabolites was performed using a LC/MS equipment as described for NadE glutamine versus ammonium completion assays (see above). The high resolution MicroTOF-QII (Bruker Daltonics) was set to an acquisition interval of 50–1000 m/z and sampling rate of 1 s operating on negative MS mode. The capillary voltage was maintained at 3,500 V, and end-plate offset was −500 V. The nebulizer gas flow was 2.0 bar, and the dry gas was kept at 6.0 liters min−1 at 180 °C. All samples were subject to technical replicate runs. The equipment was calibrated with a standard curve of glutamine (R2 > 0.98) before each analysis. All samples were analyzed within the linear range of the calibration curve. The data were processed using QuantiAnalysis (version 4.0 SP1 Bruker). The metabolites were extracted using calculated m/z with an error window of ± 0.01 m/z, and retentions times were confirmed by comparison with standards.
To calculate intracellular glutamine concentrations, the number of viable cells in each experiment was counted by serial dilution plating. Metabolite intracellular concentration was calculated based on the number of cells and the estimated intracellular volume of A. brasilense of 2.35 μm3 (1-μm diameter × 3-μm length).
Author contributions
A. R. S. S., E. C. M. G., F. O. P., E. M. S., M. H., and L. F. H. conceptualization; A. R. S. S., E. C. M. G., V. R. M., R. D., M. H., and L. F. H. data curation; A. R. S. S., E. C. M. G., R. D., and M. H. formal analysis; A. R. S. S., E. C. M. G., and L. F. H. investigation; A. R. S. S., E. C. M. G., V. R. M., and L. F. H. methodology; E. C. M. G., V. R. M., and L. F. H. supervision; F. O. P., E. M. S., and L. F. H. funding acquisition; F. O. P., E. M. S., and L. F. H. project administration; R. D. writing-review and editing; M. H. and L. F. H. writing-original draft; L. F. H. software.
Supplementary Material
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Araucária, Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES), and CNPq-Instituto Nacional de Ciência e Tecnologia (INCT). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S5.
C. Kukolj and Souza EM, personal communication.
L. F. Huergo, unpublished observations.
- NaAD
- nicotinic acid adenine dinucleotide
- PDB
- Protein Data Bank.
References
- 1. Li J., Bonkowski M. S., Moniot S., Zhang D., Hubbard B. P., Ling A. J., Rajman L. A., Qin B., Lou Z., Gorbunova V., Aravind L., Steegborn C., and Sinclair D. A. (2017) A conserved NAD+ binding pocket that regulates protein–protein interactions during aging. Science 355, 1312–1317 10.1126/science.aad8242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moure V. R., Costa F. F., Cruz L. M., Pedrosa F. O., Souza E. M., Li X. D., Winkler F., and Huergo L. F. (2015) Regulation of nitrogenase by reversible mono-ADP-ribosylation. Curr. Top. Microbiol. Immunol. 384, 89–106 [DOI] [PubMed] [Google Scholar]
- 3. Aravind L., Zhang D., de Souza R. F., Anand S., and Iyer L. M. (2015) The natural history of ADP-ribosyltransferases and the ADP-ribosylation system. Curr. Top. Microbiol. Immunol. 384, 3–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bheda P., Jing H., Wolberger C., and Lin H. (2016) The substrate specificity of sirtuins. Annu. Rev. Biochem. 85, 405–429 10.1146/annurev-biochem-060815-014537 [DOI] [PubMed] [Google Scholar]
- 5. Gazzaniga F., Stebbins R., Chang S. Z., McPeek M. A., and Brenner C. (2009) Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 73, 529–541 10.1128/MMBR.00042-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brenner C. (2002) Catalysis in the nitrilase superfamily. Curr. Opin. Struct. Biol. 12, 775–782 10.1016/S0959-440X(02)00387-1 [DOI] [PubMed] [Google Scholar]
- 7. Rizzi M., Nessi C., Mattevi A., Coda A., Bolognesi M., and Galizzi A. (1996) Crystal structure of NH3-dependent NAD+ synthetase from Bacillus subtilis. EMBO J. 15, 5125–5134 [PMC free article] [PubMed] [Google Scholar]
- 8. Jauch R., Humm A., Huber R., and Wahl M. C. (2005) Structures of Escherichia coli NAD synthetase with substrates and products reveal mechanistic rearrangements. J. Biol. Chem. 280, 15131–15140 10.1074/jbc.M413195200 [DOI] [PubMed] [Google Scholar]
- 9. de Ingeniis J., Kazanov M. D., Shatalin K., Gelfand M. S., Osterman A. L., and Sorci L. (2012) Glutamine versus ammonia utilization in the NAD synthetase family. PLoS One 7, e39115 10.1371/journal.pone.0039115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bellinzoni M., De Rossi E., Branzoni M., Milano A., Peverali F. A., Rizzi M., and Riccardi G. (2002) Heterologous expression, purification, and enzymatic activity of Mycobacterium tuberculosis NAD+ synthetase. Protein. Expr. Purif. 25, 547–557 10.1016/S1046-5928(02)00041-4 [DOI] [PubMed] [Google Scholar]
- 11. Resto M., Yaffe J., and Gerratana B. (2009) An ancestral glutamine-dependent NAD+ synthetase revealed by poor kinetic synergism. Biochim. Biophys. Acta 1794, 1648–1653 10.1016/j.bbapap.2009.07.014 [DOI] [PubMed] [Google Scholar]
- 12. Laskoski K., Santos A. R., Bonatto A. C., Pedrosa F. O., Souza E. M., and Huergo L. F. (2016) In vitro characterization of the NAD+ synthetase NadE1 from Herbaspirillum seropedicae. Arch. Microbiol. 198, 307–313 10.1007/s00203-016-1190-z [DOI] [PubMed] [Google Scholar]
- 13. LaRonde-LeBlanc N., Resto M., and Gerratana B. (2009) Regulation of active site coupling in glutamine-dependent NAD+ synthetase. Nat. Struct. Mol. Biol. 16, 421–429 10.1038/nsmb.1567 [DOI] [PubMed] [Google Scholar]
- 14. van Heeswijk W. C., Westerhoff H. V., and Boogerd F. C. (2013) Nitrogen assimilation in Escherichia coli: putting molecular data into a systems perspective. Microbiol. Mol. Biol. Rev. 77, 628–695 10.1128/MMBR.00025-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huergo L. F., and Dixon R. (2015) The emergence of 2-oxoglutarate as a master regulator metabolite. Microbiol. Mol. Biol. Rev. 79, 419–435 10.1128/MMBR.00038-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huergo L. F., Pedrosa F. O., Muller-Santos M., Chubatsu L. S., Monteiro R. A., Merrick M., and Souza E. M. (2012) PII signal transduction proteins: pivotal players in post-translational control of nitrogenase activity. Microbiology 158, 176–190 10.1099/mic.0.049783-0 [DOI] [PubMed] [Google Scholar]
- 17. Dixon R., and Kahn D. (2004) Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631 10.1038/nrmicro954 [DOI] [PubMed] [Google Scholar]
- 18. Huergo L. F., Souza E. M., Araujo M. S., Pedrosa F. O., Chubatsu L. S., Steffens M. B., and Merrick M. (2006) ADP-ribosylation of dinitrogenase reductase in Azospirillum brasilense is regulated by AmtB-dependent membrane sequestration of DraG. Mol. Microbiol. 59, 326–337 10.1111/j.1365-2958.2005.04944.x [DOI] [PubMed] [Google Scholar]
- 19. Huergo L. F., Chandra G., and Merrick M. (2013) P(II) signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol. Rev. 37, 251–283 10.1111/j.1574-6976.2012.00351.x [DOI] [PubMed] [Google Scholar]
- 20. Tautenhahn R., Patti G. J., Rinehart D., and Siuzdak G. (2012) XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem. 84, 5035–5039 10.1021/ac300698c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baugh L., Gallagher L. A., Patrapuvich R., Clifton M. C., Gardberg A. S., Edwards T. E., Armour B., Begley D. W., Dieterich S. H., Dranow D. M., Abendroth J., Fairman J. W., Fox D. 3rd, Staker B. L., Phan I., et al. (2013) Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS One 8, e53851 10.1371/journal.pone.0053851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Symersky J., Devedjiev Y., Moore K., Brouillette C., and DeLucas L. (2002) NH3-dependent NAD+ synthetase from Bacillus subtilis at 1A resolution. Acta Crystallogr. D Biol. Crystallogr. 58, 1138–1146 10.1107/S0907444902006698 [DOI] [PubMed] [Google Scholar]
- 23. Chuenchor W., Doukov T. I., Resto M., Chang A., and Gerratana B. (2012) Regulation of the inter subunit ammonia tunnel in Mycobacterium tuberculosis glutamine-dependent NAD+ synthetase. Biochem. J. 443, 417–426 10.1042/BJ20112210 [DOI] [PubMed] [Google Scholar]
- 24. Floquet N., Mouilleron S., Daher R., Maigret B., Badet B., and Badet-Denisot M. A. (2007) Ammonia channeling in bacterial glucosamine-6-phosphate synthase (Glms): molecular dynamics simulations and kinetic studies of protein mutants. FEBS Lett. 581, 2981–2987 10.1016/j.febslet.2007.05.068 [DOI] [PubMed] [Google Scholar]
- 25. Javelle A., Lupo D., Ripoche P., Fulford T., Merrick M., and Winkler F. K. (2008) Substrate binding, deprotonation, and, selectivity at the periplasmic entrance of the Escherichia coli ammonia channel AmtB. Proc. Natl. Acad. Sci. U.S.A. 105, 5040–5045 10.1073/pnas.0711742105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., and Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 10.1038/nchembio.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan J., Doucette C. D., Fowler W. U., Feng X. J., Piazza M., Rabitz H. A., Wingreen N. S., and Rabinowitz J. D. (2009) Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 5, 302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okano H., Hwa T., Lenz P., and Yan D. (2010) Reversible adenylylation of glutamine synthetase is dynamically counterbalanced during steady-state growth of Escherichia coli. J. Mol. Biol. 404, 522–536 10.1016/j.jmb.2010.09.046 [DOI] [PubMed] [Google Scholar]
- 29. Kleiner D. (1985) Bacterial ammonium transport. FEMS Microbiol. Lett. 32, 87–100 10.1111/j.1574-6968.1985.tb01185.x [DOI] [Google Scholar]
- 30. Reitzer L. (2003) Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 57, 155–176 10.1146/annurev.micro.57.030502.090820 [DOI] [PubMed] [Google Scholar]
- 31. Rodrigues T. E., Gerhardt E. C., Oliveira M. A., Chubatsu L. S., Pedrosa F. O., Souza E. M., Souza G. A., Müller-Santos M., and Huergo L. F. (2014) Search for novel targets of the PII signal transduction protein in Bacteria identifies the BCCP component of acetyl-CoA carboxylase as a PII binding partner. Mol. Microbiol. 91, 751–761 10.1111/mmi.12493 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.