

Abstract
L-type CaV1.2 channels are essential for the excitation–contraction coupling in cardiomyocytes and are hetero-oligomers of a pore-forming CaVα1C assembled with CaVβ and CaVα2δ1 subunits. A direct interaction between CaVα2δ1 and Asp-181 in the first extracellular loop of CaVα1 reproduces the native properties of the channel. A 3D model of the von Willebrand factor type A (VWA) domain of CaVα2δ1 complexed with the voltage sensor domain of CaVα1C suggests that Ser-261 and Ser-263 residues in the metal ion–dependent adhesion site (MIDAS) motif are determinant in this interaction, but this hypothesis is untested. Here, coimmunoprecipitation assays and patch-clamp experiments of single-substitution variants revealed that CaVα2δ1 Asp-259 and Ser-261 are the two most important residues in regard to protein interactions and modulation of CaV1.2 currents. In contrast, mutating the side chains of CaVα2δ1 Ser-263, Thr-331, and Asp-363 with alanine did not completely prevent channel function. Molecular dynamics simulations indicated that the carboxylate side chain of CaVα2δ1 Asp-259 coordinates the divalent cation that is further stabilized by the oxygen atoms from the hydroxyl side chain of CaVα2δ1 Ser-261 and the carboxylate group of CaVα1C Asp-181. In return, the hydrogen atoms contributed by the side chain of Ser-261 and the main chain of Ser-263 bonded the oxygen atoms of CaV1.2 Asp-181. We propose that CaVα2δ1 Asp-259 promotes Ca2+ binding necessary to produce the conformation of the VWA domain that locks CaVα2δ1 Ser-261 and Ser-263 within atomic distance of CaVα1C Asp-181. This three-way network appears to account for the CaVα2δ1-induced modulation of CaV1.2 currents.
Keywords: homology modeling, molecular dynamics, patch clamp, protein–protein interaction, calcium channel, channel activation, Western blot, protein structure, structure-function, coimmunoprecipitation, MIDAS, von Willebrand factor type A domain
Introduction
The von Willebrand factor type A (VWA)4 domain is found in more than 40,000 eukaryotic proteins (1) where it mediates protein–protein interaction (2). VWA superfamily is characterized by a “β-α-β” fold consisting of alternating sequence of six-stranded β-sheets wrapped around six α-helical segments (3–5). Binding of a divalent cation (either Mg2+, Mn2+, or Ca2+) is usually necessary for controlling its protein–ligand properties (6). Cations are coordinated by a maximum of five noncontiguous residues forming the metal ion-dependent adhesion site (MIDAS) motif characterized by the conserved primary sequence in the first loop Asp-Xaa-Ser-Xaa-Ser (DXSXS) followed by a threonine residue in the second loop and an aspartate residue in the third loop (7).
Human VWA-containing proteins include extracellular matrix proteins (e.g. collagens, cochlin, and matrilin) and integral membrane proteins (e.g. integrin and anthrax receptors) (3, 8, 9). Through the capture of a divalent ion, the MIDAS motif adopts a unique conformation that facilitates ligand association (10). Structural studies carried out in integrin receptors have shown that the nature of the ion coordinating the MIDAS motif influences the folding conformation of the protein and thus its ligand-binding properties (11, 12).
L-type CaV1.2 channels are hetero-oligomeric protein complexes consisting of a main pore-forming CaVα1C (thereafter referred to simply CaV1.2 for conciseness) assembled with CaVβ and CaVα2δ1 auxiliary subunits (13–16). Like many integrin proteins, CaVα2δ1 is glycosylated at multiple asparagine residues and is retained at the plasma membrane by a relatively short glycosylphosphatidylinositol anchor following the cleavage of its unique transmembrane domain (17, 18). Although CaV1.2 is responsible for the passage of Ca2+ through the pore, its association with CaVα2δ1 is necessary to reproduce the biophysical properties of the native channel (17, 18), in particular the channel activation at negative membrane potentials. We have recently shown that two adjacent negatively charged residues Asp-180 and Asp-181 in the S1S2 loop in the repeat I of CaV1.2 are required to recapitulate the complex characteristics of the CaVα2δ1-induced modulation of CaV1.2 currents (19). Whereas Asp-180 confers the voltage-dependent shift in channel activation, CaV1.2 Asp-181 anchors the physical interaction with CaVα2δ1, a necessary step to exert its function.
Based on the recent cryo-electron microscopy (cryo-EM) structure of the homologous CaV1.1 channel complex (CaVα1S with auxiliary subunits) (16), we have built a three-dimensional (3D) model of the VWA domain of the rat CaVα2δ1 in complex with the first voltage sensor (IS1S4) of CaV1.2 (19). Within the VWA domain of CaVα2δ1 lies a so-called “perfect” MIDAS motif with the typical Asp-Xaa-Ser-Xaa-Asp signature (20). As seen in Fig. 1A, a single divalent cation is coordinated by the multiple oxygen atoms contributed by the clockwise arrangement of Asp-259, Asp-363, Ser-263, Ser-261, and Thr-331 (rat numbering). In the model, CaVα2δ1 Ser-261 and Ser-263 are predicted to be within sufficient distance of CaV1.2 Asp-181 to form a salt bridge and anchor the proteins' interface. Herein, we aimed to identify the molecular determinants in CaVα2δ1 responsible for the protein and functional interaction with CaV1.2, using a multipronged experimental strategy combining site-directed mutagenesis, coimmunoprecipitation assays, patch-clamp experiments, and molecular dynamics (MD) simulations. We are currently reporting that mutating simultaneously the N-terminal residues Asp-259, Ser-261, and Ser-263 to the hydrophobic alanine residues within the MIDAS motif prevented the expression of CaVα2δ1 at the cell surface and consequently the increase of Ca2+ currents through CaV1.2. Single-point mutations D259A and S261A impaired the ability of CaVα2δ1 to coimmunoprecipitate with CaV1.2 and failed to up-regulate the channel activity. Using MD simulations, we propose that the side chain at position Asp-259 and the oxygen atom of the side chain of Ser-261 in CaVα2δ1 foster the folding of the MIDAS site by clasping the divalent cation within its binding site. In turn, this conformation is essential for promoting the optimal electrostatic interaction between CaV1.2 Asp-181 and the hydrogen atoms of the hydroxyl groups of CaVα2δ1 Ser-261 and Ser-263.
Figure 1.
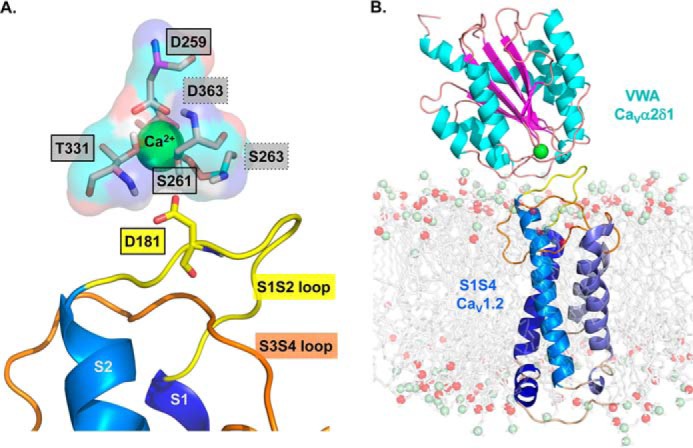
3D homology model of the S1 to S4 region from domain I of CaV1.2 in complex with the VWA domain of CaVα2δ1. A homology model of the CaV1.2–CaVα2δ1 interface was built based using the molecular coordinates of the cryo-EM structure of the skeletal muscle CaV1.1 complex (PDB code 5GJV). This model differs slightly from the 3D model of the same region published previously (19) in regard to the orientation of the IS3S4 extracellular loop. A, residues forming the protein interface are zoomed in with emphasis on the MIDAS motif. The main-chain groups of amino acids forming the MIDAS motif (Asp-259, Ser-261, Ser-263, Thr-331, and Asp-363) are shown as pale rose sticks with carboxyl groups in red, amine groups are blue, and hydrogen atoms are white. Residues Asp-259, Ser-261, and Thr-331 identified within full-lined black boxes are in the foreground, and Ser-263 and Asp-363 identified within the dot-lined boxes are in the background. MIDAS residues are facing the IS1S2 loop in CaV1.2. CaV1.2 Asp-181 is shown in stick representation. B, VWA domain of the rat CaVα2δ1 is shown in schematic representation in which α-helices appear in cyan, and β-strands are shown in pink. A single Ca2+ ion (green) is shown as being coordinated by the MIDAS residues. The region spanning from the S1 to the S4 segments in repeat I (IS1S4) of CaV1.2 is shown in schematic representation, and the transmembrane helices S1 to S4 are colored from the darker (S1) to the paler shade of blue (S4). Modeled DPPC lipids are shown in stick representation with carbon, oxygen, and phosphorus atoms being shown in gray, red, and pale green, respectively. Modeling was achieved with Modeler 9.17. The figure was produced using PyMOL (DeLano Scientific).
Results
Electrostatic interactions are mediated by MIDAS residues at the CaV1.2–CaVα2δ1 interface
We recently identified the negatively charged Asp-181 in the first extracellular loop of CaV1.2 as an essential determinant for the interaction with the extracellular CaVα2δ1 protein (19). To identify the molecular determinants of the interface, we have produced a 3D model of the IS1S4 region using the molecular coordinates of the cryo-EM structure of the homologous CaV1.1 channel complex (PDB code 5GJV) (16). The sequence identity between IS1S4 in CaV1.1 and CaV1.2 is 69.6%, whereas VWA is perfectly conserved. Nonetheless, the molecular coordinates of the IS3S4 loop were not reported in the cryo-EM structure and were reconstructed using Modeler (see details in “Experimental procedures”). The 3D model was selected using a DOPE score of −37032 (between −37,100 and −35,578) and a molpdf score of 2147 (between 2144 and 3085). MD simulations suggest that Gly-262 and/or Ser-263 in the VWA domain of CaVα2δ1 were the most likely residues to interact with Asp-181 in the first extracellular loop of CaV1.2. Along with Asp-259, Ser-261, Thr-331, and Asp-363, Ser-263 is one of the five polar residues that form a perfect MIDAS motif in the rat isoform of CaVα2δ1 (Fig. 1A). X-ray crystallography structures of integrin proteins position a single Mg2+ enclosed within MIDAS residues, although Ca2+, Zn2+, or Mn2+ could also occupy the binding site (21). Ca2+ and Mg2+ are both plausible candidates to occupy the MIDAS at the CaVα2δ1–CaV1.2 interface given that the concentration of Ca2+ and Mg2+ ions is roughly equivalent (in the millimolar range) in the extracellular medium of eukaryotic cells. We chose to perform the MD simulations in the presence of a Ca2+ ion, mostly because the ion was included in the coordinates of the published cryo-EM structure (16). The Ca2+ ion was virtually inserted in the geometric center of the motif, and its average position was adjusted by MD simulations.
MD simulations were carried out after the transmembrane helices of CaV1.2 (membrane protein) were embedded in a model dipalmitoylphosphatidylcholine (DPPC) lipid bilayer containing 128 DPPC molecules (Fig. 1B). The hydrophobic tails of the lipid molecules in the plasma membrane appear to stabilize hydrophobic residues Ala-253, Leu-254, Gly-255, and Gly-256 of the IS3S4 region within the plasma membrane. The protein–membrane system was solvated with water in the presence of 143 mm NaCl, 2 mm MgCl2, and 2 mm CaCl2. This 3D model reached a steady state after 1.1 ns equilibration with r.m.s.d. variations around 4.5 Å for simulations up to 25 ns, and this observation stood for three distinct trajectories. As seen, the negatively charged side chain of CaV1.2 Asp-181 is predicted in this model to face the hydrogen atoms from the hydroxyl groups of Ser-261 at a distances of 1.7 Å thus suggesting a key role for CaVα2δ1 Ser-261 in enabling the interaction with CaV1.2.
Multiple mutations of the MIDAS residues prevent cell-surface expression of CaVα2δ1 and physical interaction with CaV1.2
Coimmunoprecipitation assays were carried out to explore the contribution of MIDAS residues in promoting the interaction between CaV1.2 and CaVα2δ1. The importance of the polar side chains was examined after substitution with the hydrophobic alanine residue at multiple MIDAS positions with CaVα2δ1 D259A/S261A/S263A (DSS/A), D259A/S261A/S263A/T331A (DSST/A), and D259A/S261A/S263A/D363A (DSSD/A). Assays were performed with untagged CaV1.2, mCherry-CaVα2δ1-HA, and CaVβ3–c-Myc using anti-c-Myc–coated beads to bait the channel complex. For conciseness, the mCherry-CaVα2δ1-HA construct will hereafter be referred to as CaVα2δ1-HA. Under this protocol, CaVβ3–c-Myc binds to the magnetic beads and pulls down the CaV1.2–CaVα2δ1 complex (Fig. 2A). As seen in Fig. 2B, input lane, CaV1.2, CaVα2δ1-HA WT and mutants, and CaVβ3 proteins were translated at the expected molecular masses of 250, 175, and 60 kDa, respectively, after recombinant expression in HEKT cells. CaVβ3–c-Myc was captured by the beads under all experimental conditions, and CaV1.2 successfully pulled down CaVα2δ1 WT (Fig. 2C). The ablation of the negative or polar side chain in the MIDAS residues prevented the coimmunoprecipitation of any of the multiple mutants in CaVα2δ1, even after a 200-s exposure. Two-color flow-cytometry assays (17–19) were performed to determine whether disrupting MIDAS also altered the cell-surface expression of CaVα2δ1. As detailed elsewhere (17, 18), the cell-surface expression of the constructs, relative to mCherry-CaVα2δ1-HA WT, is assessed using the fluorescence intensity of an anti-hemagglutinin tag conjugated to FITC (anti-HA FITC) antibody directed against the extracellular HA tag inserted after position Arg-676 in mCherry–CaVα2δ1 (Fig. 2D). Fluorescence intensity of the anti-HA tag antibody conjugated to FITC measured in intact nonpermeabilized cells provides an estimation of the cell-surface expression of the construct, whereas the fluorescence intensity measured after cell permeabilization is used as a marker of total protein expression. As seen, multiple mutations of the MIDAS residues significantly reduced the cell-surface expression of mCherry-CaVα2δ1-HA to ≈10% for the mutants (Fig. 2E). More importantly, multiple mutations were also seen to reduce the protein stability with a sharp decrease in the relative fluorescence for FITC measured in permeabilized cells (Table 1). This observation is in agreement with previous reports showing that coexpression of CaVα2δ1 increases the total protein stability of CaVβ3-bound CaV1.2 proteins, and conversely coexpression of CaV1.2 increases the total protein stability of CaVα2δ1 in the presence of CaVβ3 (22). Altogether, these results indicate that the polar or negatively charged side chains of the MIDAS residues contribute to the stability of CaVα2δ1, its cell-surface expression, and its interaction with the pore-forming subunit of CaV1.2.
Figure 2.

Multiple mutations of MIDAS residues abolish the coimmunoprecipitation of CaV1.2 with CaVα2δ1. A, schematic demonstrating the coimmunoprecipitation assay using CaVβ3–c-Myc as the bait. B, HEKT cells were transiently transfected with pCMV-CaV1.2, pCMV-CaVβ3–c-Myc, and pmCherry-CaVα2δ1-HA WT or mutants as shown. Cell lysates were immunoprecipitated (IP) overnight with anti-c-Myc magnetic beads to capture CaVβ3, eluted in a Laemmli buffer and fractionated by SDS-PAGE using 8% gels. B, immunoblotting (IB) was carried out on total proteins (20 μg) before coimmunoprecipitation assay (input lane) to confirm that all proteins have been correctly translated. Constructs are from left to right: mCherry-CaVα2δ1-HA WT; mCherry-CaVα2δ1-HA DSS/A (D259A/S261A/S263A); mCherry-CaVα2δ1-HA DSST/A (D259A/S261A/S263A/T331A); and mCherry-CaVα2δ1-HA DSSD/A (D259A/S261A/S263A/D363A). The signal for the housekeeping protein GAPDH is shown. C, immunoblotting was carried out after eluting the protein complexes (IP–c-Myc lane) from the beads with anti-CaV1.2, anti-CaVα2δ1, and anti-CaVβ3 antibodies (from top to bottom, as indicated). All immunoblots were carried out in parallel under the same transfection and extraction conditions. The signal for CaVα2δ1 was revealed after three exposures times (1, 50, and 200 s). CaVβ3 and CaV1.2 proteins migrated, respectively, at 60 and 250 kDa. All CaVα2δ1 proteins migrated at ≈175 kDa, which is consistent with the molecular mass of the mCherry-CaVα2δ1-HA WT reported in previous studies (19). These assays were repeated three times over the course of 6 months with different cell preparations. Assays carried out with anti-HA–coated beads to pull down the channel complex with CaVα2δ1-HA as the bait (as seen in Fig. 6) yielded similar results. D, schematic illustrating the relative position of the extracellular HA epitope and the intracellular mCherry translated after the C terminus on the mCherry-CaVα2δ1-HA construct used to carry the flow cytometry assays. The construct allows for detection of intracellular and extracellular fluorescence using, respectively, a FITC-conjugated anti-HA and the constitutive fluorescence of mCherry. E, representative two-dimensional plots of mCherry versus FITC fluorescence. The cell-surface expression of the mCherry-CaVα2δ1-HA construct WT and mutants from the surface fluorescence emitted by the FITC-conjugated anti-HA as measured using a flow cytometry assay (10,000 intact cells) is shown. From top to bottom, the constructs that were tested are as follows: (a) mCherry-CaVα2δ1-HA WT; (b) mCherry-CaVα2δ1-HA DSS/A (D259A/S261A/S263A); (c) mCherry-CaVα2δ1-HA DSSD/A (D259A/S261A/S263A/D363A); and (d) mCherry-CaVα2δ1-HA DSST/A (D259A/S261A/S263A/T3331A). Left panel: relative intensity of the fluorescence signal produced by the FITC-conjugated anti-HA (x axis) and produced by the mCherry (y axis) yields an estimate of the cell surface and intracellular expression, respectively. The robust mCherry signal (y axis) confirms that the proteins were translated up to the end of the coding sequence, an observation also obtained from carrying out routine Western blotting. Middle panel: histogram of the relative fluorescence intensity for the FITC-conjugated anti-HA. Right panel: histogram of the relative fluorescence intensity for the constitutive mCherry signal. The cell-surface fluorescence for FITC, calculated as ΔMedFI, as explained under “Experimental procedures,” was significantly smaller for CaVα2δ1-HA mutants than for the WT construct (see Table 1 for numerical values). Furthermore, the mCherry signal, which reflects the total protein density, was also decreased suggesting that these mutations impaired protein stability.
Table 1.
Cell-surface expression of CaVα2δ1
Stable HEKT CaVβ3 cells were transiently transfected simultaneously with 4 μg of pCMV-CaV1.2 and 4 μg of pmCherry-CaVα2δ1 HA WT or mutants. Flow cytometry was used to determine the proportion of CaVα2δ1 proteins expressed at the cell surface as reported in the “intact cells” column, whereas the “permeabilized cells” column represent the total expression level. Fluorescence intensity was estimated using FlowJo software® as detailed elsewhere (39). The relative cell-surface expression of CaVα2δ1-HA constructs was estimated by calculating the ratio of the ΔMedFI for the FITC fluorophore over ΔMedFI values quantified for the FITC fluorophore for the WT construct (ΔMedFI mutant/ΔMedFI WT) in intact nonpermeabilized cells for the experiments carried out the same day under the same conditions. The relative total expression of CaVα2δ1 constructs was estimated by calculating the ratio of the ΔMedFI values quantified for the FITC fluorophore over ΔMedFI values quantified for the FITC fluorophore for the WT construct after cell permeabilization. These ratios are expressed herein as a percentage. The total number of experiments is provided in parentheses, with each experiment being the result of a different transfection in a separate cell dish (>10,000 cells). Statistical analysis was carried out against the ΔMedFI for FITC measured with pmCherry-CaVα2δ1-HA WT. *, p < 0.05, and **, p < 0.001.
| CaV1.2 + CaVβ3 | Relative ΔMedFI (%) FITC-conjugated anti-HA |
|
|---|---|---|
| Cell surface, intact cells | Total, permeabilized cells | |
| + mCherry-CaVα2δ1-HA WT | 92 ± 3 (12) | 97 ± 0.9 (12) |
| D259A/S261A/S263A | 10 ± 0.05 (3)** | 36 ± 0.3 (3)** |
| D259A/S261A/S263A/D363A | 6 ± 0.09 (3)** | 31 ± 0.3 (3)** |
| D259A/S261A/S263A/T331A | 11 ± 0.1 (3)** | 33 ± 0.5 (3)** |
| D259A | 55 ± 4 (3)** | 81 ± 3 (3)** |
| D259R | 20 ± 0.8 (3)** | 73 ± 1 (3)** |
| V260A | 77 ± 7 (3)* | 99 ± 1 (3) |
| V260R | 48 ± 3 (3)** | 97 ± 1 (3) |
| S261A | 36 ± 2 (3)** | 72 ± 2 (3)** |
| S261R | 39 ± 0.1 (3)** | 66 ± 1 (3)** |
| G262A | 110 ± 8 (3)* | 95 ± 4 (3) |
| G262R | 92 ± 0.5 (3) | 87 ± 2 (3)** |
| S263A | 97 ± 4 (3) | 94 ± 0.3 (3) |
| S263R | 105 ± 2 (3)* | 86 ± 0.7 (3)** |
| T331A | 133 ± 5 (3)** | 106 ± 2 (3)* |
| T331G | 103 ± 7 (3) | 98 ± 2 (3) |
| T331R | 108 ± 8 (3) | 88 ± 2 (3)** |
| D363A | 113 ± 9 (3) | 89 ± 1 (3)* |
| D363G | 92 ± 5 (3) | 85 ± 1 (3)** |
| D363R | 43 ± 3 (3)** | 66 ± 1 (3)** |
Functional modulation is intact with CaVα2δ1 V260R
Individual contributions from the side chains of MIDAS amino acids were analyzed after substitutions to the positively charged arginine (Figs. 3 and 4) and to the neutral alanine residue (Figs. 5 and 6). This two-step strategy was meant to exclude residues that would remain functional following nonconserved substitutions predicted to alter the local electrical field. MIDAS residues as well as the intervening Val-260 and Gly-262 residues were sequentially mutated. All mCherry-CaVα2δ1-HA single mutants expressed the mCherry tag and were trafficked at the plasma membrane (Table 1). Coimmunoprecipitation assays were carried out with c-Myc–coated beads to bait the channel complex. As seen in Fig. 3A, input lane, CaV1.2, CaVα2δ1-HA WT and mutants, and CaVβ3 proteins were translated at the expected molecular masses of 250, 175, and 60 kDa, respectively, after recombinant expression in HEKT cells. CaVβ3 was captured by the beads under all experimental conditions, and CaV1.2 successfully pulled down CaVα2δ1-HA WT (Fig. 3B). Not too surprisingly, the mutation to an arginine residue significantly impaired the ability for most CaVα2δ1 mutants, save for V260R, to interact with CaV1.2. Longer exposure times allowed us to detect a small interaction with CaVα2δ1-HA G262R and CaVα2δ1-HA S263R, but no interaction was ever detected with CaVα2δ1-HA D259R and CaVα2δ1-HA S261R. A separate series of experiments demonstrated that protein interaction was also somewhat conserved with CaVα2δ1-HA T331R and CaVα2δ1-HA D363R in a fashion reminiscent of CaVα2δ1-HA G262R and CaVα2δ1-HA S263R (data not shown). These results indicate that the side chain at position Val-260 is not directly involved in protein interaction, whereas the side chains at Asp-259 and Ser-261 play a critical role in this process. Examination of the 3D model concurs that the main-chain atoms of Val-260 are facing the Ca2+ ion and that side-chain substitution does not alter that preferred configuration.
Figure 3.

CaVβ3/CaV1.2 protein complex coimmunoprecipitated with CaVα2δ1 V260R. HEKT cells were transiently transfected with pCMV-CaV1.2 and pCMV-CaVβ3–c-Myc and either pmCherry-CaVα2δ1-HA WT, or pmCherry-CaVα2δ1-HA D259R, or pmCherry-CaVα2δ1-HA V260R, or pmCherry-CaVα2δ1-HA S261R, or pmCherry-CaVα2δ1-HA G262R, or pmCherry-CaVα2δ1-HA S263R. Cell lysates were immunoprecipitated (IP) overnight with anti-c-Myc magnetic beads to capture CaVβ3, eluted in a Laemmli buffer, and fractionated by SDS-PAGE using 8% gels. A, immunoblotting (IB) was carried out on total proteins (20 μg) before the immunoprecipitation assay (input lane) to confirm that constructs were expressed at the expected molecular weight. The signal for the housekeeping protein GAPDH is shown. B, immunoblotting was carried out after eluting the protein complexes from the beads with anti-CaV1.2, anti-CaVα2δ1, and anti-CaVβ3 antibody (from top to bottom, as indicated). Images for CaVα2δ1 were captured after shorter (1 s) or longer exposure times until the signal was saturated (20 s). All immunoblots were carried out in parallel under the same transfection and extraction conditions. These assays were successfully repeated three times with similar results.
Figure 4.

CaVα2δ1 V260R stimulates CaV1.2 whole-cell currents. A, representative whole-cell Ca2+ current traces recorded after recombinant expression of CaV1.2 + CaVβ3 and either pmCherry-CaVα2δ1-HA WT or mutants D259R, V260R, S261R, G262R, S263R, T331R, and D363R (from left to right). Currents were recorded in the presence of 2 mm Ca2+ from a holding potential of −100 mV. Time scale was 100 ms throughout. The current density scale is 5 pA/pF, as indicated. B, averaged current-voltage relationships. Coexpression with mCherry-CaVα2δ1-HA V260R produced whole-cell CaV1.2 currents with properties similar to currents measured in the presence of mCherry-CaVα2δ1-HA WT. Coexpression with mCherry-CaVα2δ1-HA D259R, S261R, G262R, S263R, T331R, and D363R impaired current up-regulation to different degrees. Statistical analyses were carried out with a one-way ANOVA test. 1 indicates that the mutant CaVα2δ1 proteins are significantly different from the mock vector at p < 0.01, and 2 indicates that the mutant CaVα2δ1 proteins are significantly different from mCherry-CaVα2δ1-HA WT at p < 0.05. The complete set of values is shown in Table 2. C, bar graph reporting the distribution of the free energies of activation (ΔGact) measured with each CaVα2δ1 mutant. The distribution of the ΔGact values for mCherry-CaVα2δ1-HA D259R, S261R, G262R, and D363R was right-shifted as compared with mCherry-CaVα2δ1-HA WT and overlapped with the ΔGact values measured in the presence of mock vector (pmCherry-no CaVα2δ1).
Figure 5.

CaVα2δ1 S263A and T331A stimulate CaV1.2 whole-cell currents. A, representative whole-cell Ca2+ current traces recorded after recombinant expression of CaV1.2 + CaVβ3 and either pmCherry-CaVα2δ1-HA WT, D259A, S261A, G262A, S263A, G262A/S263A, T331A, or D363A (from left to right). Currents were recorded in the presence of 2 mm Ca2+ from a holding potential of −100 mV. Time scale is 100 ms throughout. The current density scale ranged from 5 to 10 pA/pF, as indicated. B, averaged current-voltage relationships. Coexpression with mCherry-CaVα2δ1-HA S263A or T331A produced whole-cell CaV1.2 currents with properties similar to currents measured in the presence of mCherry-CaVα2δ1-HA WT. Coexpression with mCherry-CaVα2δ1-HA D259A, S261A, G262A, G262A/S263A, and D363A impaired current up-regulation to different degrees. Statistical analyses were carried out with a one-way ANOVA test. 1 indicates that the mutant CaVα2δ1 proteins are significantly different from the mock vector at p < 0.01, and 2 indicates that the mutant CaVα2δ1 proteins are significantly different from mCherry-CaVα2δ1-HA WT at p < 0.05. Complete set of values is shown in Table 2. C, bar graph reporting the distribution of the free energies of activation (ΔGact) measured with each CaVα2δ1 mutant. The distribution of the ΔGact values for mCherry-CaVα2δ1-HA D259A, S261A, G262A, and G262A/S263A was right-shifted as compared with CaVα2δ1 WT and overlapped with the ΔGact values measured in the presence of mock vector (pmCherry-no CaVα2δ1). Note that the distribution for the ΔGact obtained with mCherry-CaVα2δ1-HA S263A was very wide and may reflect different conformations of the mutant.
Figure 6.

Single alanine substitutions of Asp-259, Ser-261, and Ser-263 in CaVα2δ1 impair coimmunoprecipitation of CaV1.2 and CaV2.3 proteins. A and B, HEKT cells were transiently transfected with pCMV-CaV1.2 and pCMV-CaVβ3–c-Myc and either pmCherry-CaVα2δ1-HA WT or pmCherry-CaVα2δ1-HA D259A, V260A, S261A, G262A, S263A, T331A, or D363A. Cell lysates were immunoprecipitated (IP) overnight with anti-HA magnetic beads to capture CaVα2δ1-HA, eluted in a Laemmli buffer and fractionated by SDS-PAGE using 8% gels. A, immunoblotting (IB) was carried out on total proteins (20 μg) before the immunoprecipitation assay to confirm protein expression in all samples (input lane). The signal for the housekeeping protein GAPDH is shown below. B, immunoblotting was carried out after eluting the protein complexes from the beads with anti-CaV1.2 and anti-CaVα2δ1 antibodies (from top to bottom, as indicated). All immunoblots were carried out in parallel under the same transfection and extraction conditions. In these experiments, CaVα2δ1-HA is bound to the beads such that the interaction is probed by investigating the signal for its CaV1.2 partner. The image was obtained using the automatic mode of the ChemiDoc Touch system, which optimizes the signal. As shown, the signal for CaV1.2 was significantly reduced for mCherry-CaVα2δ1-HA D259A, S261A, and S263A. C and D, HEKT cells were transiently transfected with pcDNA3-CaV2.3 and pCMV-CaVβ3–c-Myc and either pmCherry-CaVα2δ1-HA WT or pmCherry-CaVα2δ1-HA D259A, S261A, S263A, T331A, or D363A. Cell lysates were immunoprecipitated overnight with anti-c-Myc magnetic beads to capture CaVβ3–c-Myc, eluted in a Laemmli buffer and fractionated by SDS-PAGE using 8% gels. C, immunoblotting was carried out on total proteins (20 μg) before the immunoprecipitation assay (input lane). The signal for the housekeeping protein GAPDH is shown. D, immunoblotting was carried out after eluting the protein complexes from the beads with anti-CaV2.3, anti-CaVα2δ1, and anti-CaVβ3 antibodies (from top to bottom, as indicated). In these experiments, CaVβ3/CaV2.3 is bound to the beads such that the interaction is probed by investigating the signal for CaVα2δ1. As shown, the signal for CaVα2δ1 was significantly reduced for mCherry-CaVα2δ1-HA D259A, S261A, S263A, and D363A. All immunoblots were carried out in parallel under the same transfection and extraction conditions.
To establish a correlation between physical interaction and functional modulation, the impact of these mutations on channel function was examined after their recombinant expression in the presence of CaV1.2 and CaVβ3 in HEKT cells (Fig. 4A). As published previously (17–19, 22), coexpression of CaV1.2 and CaVβ3 with the CaVα2δ1-HA WT construct increases the peak current density by 7–10-fold from −2.5 ± 0.3 pA/pF (25) (no insert in the pmCherry vector) to −18 ± 1 pA/pF (n = 359) (Fig. 4B). CaVα2δ1-HA also caused a ≈ −15-mV leftward shift in the activation potential of CaV1.2 from E0.5, act = +8 ± 2 mV (n = 25) in the absence of CaVα2δ1 to E0.5, act = −8.7 ± 0.4 mV (n = 359) in the presence of CaVα2δ1-HA. Although the negative shift was significant in our sample size, we observed some overlap between the two conditions with the corresponding free energy of activation (ΔGact) ranging from −2 to +0.2 kcal mol−1 (33% peak at −0.6 kcal mol−1) for the WT channel combination, whereas ΔGact ranged from −0.2 to +1.4 kcal mol−1 (40% peak at +0.2 kcal mol−1) in the absence of CaVα2δ1 (Fig. 4C). Up-regulation of CaV1.2 currents was severely disrupted with all arginine mutants save for CaVα2δ1-HA V260R. Peak current density measured with CaVα2δ1-HA D259R, CaVα2δ1-HA S261R, CaVα2δ1-HA G262R, CaVα2δ1-HA S263R, CaVα2δ1-HA T331R, and CaVα2δ1-HA D363R was significantly different from whole-cell currents measured with CaVα2δ1-HA WT (p < 0.05). Furthermore, modulation by CaVα2δ1-HA S261R, CaVα2δ1-HA G262R, and CaVα2δ1-HA D363R was indistinguishable from the current density obtained with the mock vector (Table 2). Altogether, these results rule out a significant role for the side chain at position 260. The addition of the long arginine side chain at position 262 could locally impair flexibility in a region that plays a role in the interaction with the IS1S2 loop of CaV1.2.
Table 2.
Biophysical properties of mCherry-CaVα2δ1-HA WT and mutants
CaV1.2 WT (or mutant in a few cases) was coexpressed with CaVβ3 and either pmCherry-mock vector or pmCherry-CaVα2δ1-HA WT or mutants using a 4:4:4 μg ratio. Biophysical parameters were measured in the presence of 2 mm Ca2+ as described elsewhere (17–19). Activation properties (E0.5, act and ΔGact) were estimated from the mean I–V relationships and fitted to a Boltzmann equation. Only the GFP-positive cells with voltage-activated currents were kept for further analysis. Null-current cells outnumbered the cells with inward currents for mCherry-CaVα2δ1-HA D259A/S261A/S263A/T331A, mCherry-CaVα2δ1-HA D259A/S261A/S263A/D363A, mCherry-CaVα2δ1-HA D259A/S261A/S263A, mCherry-CaVα2δ1-HA S261A/G262A/S263A, and mCherry-CaVα2δ1-HA G262A/ 263A. The data are shown as the mean ± S.E. of the number of cells (one cell per experiment), and the total number of experiments carried over several months is provided in parentheses. Statistical analysis was carried out against the values obtained in the presence of the mock pmCherry-N1 vector with 1 = p < 0.01 and against mCherry-CaVα2δ1-HA WT with 2 = p < 0.05.
| mCherry-CaVα2δ1-HA WT or mutants with CaV1.2 WT + CaVβ3 in HEKT cells | Electrophysiological properties in 2 mm Ca2+ |
||
|---|---|---|---|
| Peak current density | E0.5, act | ΔGact | |
| pA/pF | mV | kcal mol−1 | |
| mCherry mock vector | −2.5 ± 0.3 (25)2 | +8 ± 2 | +0.5 ± 0.1 |
| CaVα2δ1 WT | −18 ± 1 (359)1 | −8.7 ± 0.4 | −0.77 ± 0.04 |
| D259A/S261A/S263A/T331A | −1.7 ± 0.3 (8)2 | +19 ± 5 | +0.6 ± 0.2 |
| D259A/S261A/S263A/D363A | −1.6 ± 0.2 (8)2 | +24 ± 5 | +1 ± 0.2 |
| D259A/S261A/S263A | −1.9 ± 0.4 (8)2 | +24 ± 3 | +1.1 ± 0.1 |
| S261A/G262A/S263A | −1.9 ± 0.4 (7)2 | +9 ± 8 | +0.2 ± 0.2 |
| G262A/S263A | −2 ± 1 (8)2 | +12 ± 5 | +0.9 ± 0.2 |
| D259A | −3.7 ± 0.9 (14)2 | +3 ± 2 | +0.1 ± 0.1 |
| D259E | −21 ± 5 (8)1 | −11 ± 1 | −1.1 ± 0.2 |
| D259R | −5 ± 2 (8)2 | −1 ± 2 | −0.1 ± 0.2 |
| V260A | −10 ± 2 (26)1 | −5 ± 1 | −0.5 ± 0.1 |
| V260G | −14 ± 4 (8)1 | −10 ± 1 | −0.9 ± 0.1 |
| V260R | −16 ± 2 (30)1 | −7 ± 1 | −0.63 ± 0.08 |
| S261A | −5 ± 2 (19)2 | +2 ± 2 | +0.1 ± 0.1 |
| S261G | −1.5 ± 0.7 (6)2 | +15 ± 6 | +0.5 ± 0.4 |
| S261R | −3 ± 1 (7)2 | +4 ± 2 | +0.2 ± 0.1 |
| S261T | −33 ± 6 (8)1 | −1 ± 2 | −0.1 ± 0.1 |
| G262A | −8 ± 1 (42)1,2 | −3 ± 1 | −0.4 ± 0.1 |
| G262D | −2.9 ± 0.5 (8)2 | +4 ± 5 | +0.1 ± 0.2 |
| G262R | −2.2 ± 0.4 (9)2 | +6 ± 4 | +0.1 ± 0.1 |
| G262T | −6 ± 1 (13)1,2 | −1 ± 2 | −0.2 ± 0.1 |
| S263A | −13 ± 2 (26)1 | −4 ± 2 | −0.5 ± 0.1 |
| S263G | −15 ± 4 (11)1 | −9 ± 1 | −0.9 ± 0.2 |
| S263R | −6 ± 2 (8)2 | −4 ± 1 | −0.3 ± 0.1 |
| S263T | −12 ± 6 (11)1 | −8 ± 1 | −0.7 ± 0.1 |
| I330A | −14 ± 4 (8)1 | −4 ± 1 | −0.4 ± 0.1 |
| T331A | −22 ± 3 (27)1 | −12 ± 1 | −1.1 ± 0.1 |
| T331G | −30 ± 7 (9)1 | −9 ± 2 | −0.9 ± 0.2 |
| T331R | −7 ± 3 (8)2 | −5 ± 3 | −0.4 ± 0.2 |
| D332A | −8 ± 2 (8)1,2 | −4 ± 1 | −0.4 ± 0.1 |
| D363A | −8 ± 3 (18)1,2 | −2 ± 2 | −0.2 ± 0.1 |
| D363G | −13 ± 3 (9)1 | −7 ± 1 | −0.6 ± 0.1 |
| D363R | −2 ± 1 (8)2 | +2 ± 2 | +0.1 ± 0.1 |
| E366A | −38 ± 8 (11)1 | −3.4 ± 2.6 | −0.4 ± 0.2 |
| E366G | −28 ± 5 (9)1 | −2 ± 2 | −0.1 ± 0.2 |
| E366A/E367A | −13 ± 2 (14)1 | −6 ± 1 | −0.5 ± 0.1 |
| T331A + CaV1.2 S182A | −33 ± 8 (8)1 | −9 ± 2 | −1 ± 0.3 |
| T331A + CaV1.2 K257A | −22 ± 4 (9)1 | −13 ± 1 | −1.2 ± 0.1 |
| E363A + CaV1.2 K257A | −28 ± 6 (7)1 | −9 ± 3 | −1 ± 0.3 |
Functional modulation of CaV1.2 currents requires CaVα2δ1 Asp-259 and Ser-261
Mutant proteins of CaVα2δ1 were produced to investigate the molecular requirements for the protein interaction and functional modulation of CaV1.2 currents. Coexpression of CaV1.2/CaVβ3 with CaVα2δ1-HA D259A, S261A, and G262A/S263A mutants produced whole-cell currents that were not significantly different from that obtained in the presence of the mCherry mock vector (Fig. 5A), although these mutants were confirmed to form bona fide proteins that were detected at the cell surface (Table 1). In contrast, CaVα2δ1-HA G262A, S263A, T331A, and D363A stimulated whole-cell currents without any alteration in the voltage dependence of activation (Fig. 5, B and C). In fact, there was a strong association between peak current density and the negative voltage dependence of activation (ΔGact) (Fig. 5C and Table 2) such that mutants yielding large whole-cell currents activated at negative membrane potentials. We did not identify any CaVα2δ1 mutant that simply disrupted the voltage dependence of activation.
Peak current densities ranged from near-background whole-cell currents measured with the mock vector in the following sequence: CaVα2δ1 mock vector ≈ D259A ≈ G262A/S263A < S261A < G262A ≈ D363A < S263A < T331A ≈ CaVα2δ1 WT. It should be noted that conservative substitutions CaVα2δ1-HA D259E and CaVα2δ1-HA S261T produced CaV1.2 whole-cell currents that were similar to CaVα2δ1 WT (Table 2) indicating that the side chains at Asp-259 and Ser-261 contribute significantly to channel modulation.
The structural requirements at position CaVα2δ1 Gly-262 were further investigated (Table 2). A significant decrease in the channel function was also observed with CaVα2δ1-HA G262T, whereas coexpression with the negatively charged CaVα2δ1-HA G262D, predicted to project a negatively charged side chain, failed to stimulate CaV1.2 currents suggesting that a polar or a negative side chain cannot compensate for the decreased flexibility of the residue at this position.
CaVα2δ1-HA S263A, S263G, S263R, and S263T were all associated with whole-cell currents that activated in a range of negative voltages associated with CaVα2δ1-HA WT (Table 2). The peak current densities, significantly higher than those recorded with the mock vector, suggest that the side chain at this position does not contribute appreciably to the functional interaction with CaV1.2 Asp-181.
Although CaVα2δ1-HA T331R modestly stimulated whole-cell currents, CaVα2δ1-HA T331G behaved as CaVα2δ1-HA WT (Table 2). This position is not always conserved in MIDAS; hence, residues adjacent to Thr-331 were also explored with CaVα2δ1-HA I330A and D332A. Both mutant proteins stimulated whole-cell currents with average peak current densities that were not significantly smaller than whole-cell currents obtained with CaVα2δ1-HA WT with p = 0.14 (p > 0.05). In addition, mutations of the negatively charged Glu-366 and Glu-367, located 3 and 4 amino acids away from Asp-363, with CaVα2δ1-HA E366A, CaVα2δ1-HA E366G, and CaVα2δ1-HA E366A/E367A failed to reveal additional contribution from negatively charged residues outside MIDAS (Table 2).
Channel function was also characterized by combining mutations from residues in CaVα2δ1-HA and CaV1.2 that could come into atomic proximity in the virtual model of the protein interface with CaVα2δ1-HA T331A + CaV1.2 S182A, CaVα2δ1-HA T331A + CaV1.2 K257A, and CaVα2δ1-HA E363A + CaV1.2 K257A in the presence of CaVβ3. These three pairs of mutants produced depolarization-activated whole-cell currents that were not significantly different from currents produced by CaVα2δ1-HA WT + CaV1.2 WT when recorded under the same conditions (Table 2). Altogether, charged mutations of MIDAS residues CaVα2δ1 Asp-259 and CaVα2δ1 Ser-261 were seen to impact more significantly on channel function than any other position herein tested.
Mutations of MIDAS residues impair coimmunoprecipitation of CaVα2δ1 with CaV1.2 and CaV2.3
To investigate whether functional modulation requires high-affinity interaction between the two proteins at specific sites, coimmunoprecipitation assays were carried out with the single alanine mutants. CaVα2δ1-HA was used to bait the CaV1.2–CaVβ3 complex using anti-HA–coated magnetic beads (Fig. 6, A and B). As shown, CaVα2δ1-HA WT, V260A, and T331A successfully pulled down CaV1.2 and CaVβ3 (Fig. 6B). In contrast, coimmunoprecipitation of the CaV1.2–CaVβ3 complex was abolished with CaVα2δ1-HA D259A and S261A and significantly reduced, but not eliminated, with CaVα2δ1-HA S263A and D363A. It is important to stress that the pulldown assays remain a tool largely qualitative. Coimmunoprecipitation assays carried out using anti-c-Myc–coated beads to bait the CaV1.2–CaVβ3 complex yielded similar results (data not shown).
Combining the G262A with the S263A mutation in the CaVα2δ1-HA G262A/S263A mutant completely eradicated coimmunoprecipitation suggesting that local conformation plays a role in these processes (data not shown). In contrast, CaVα2δ1-HA E366A/E367A, situated just outside MIDAS, was easily pulled down by CaV1.2 (data not shown) in agreement with the patch-clamp data. The signal for the CaV1.2 protein obtained in the presence of CaVα2δ1-HA T331A remained remarkably strong confirming that the nature of the side chain at this position does not contribute directly or indirectly to protein interaction.
The importance of MIDAS residues in mediating protein interaction was further established by carrying similar experiments with the neuronal CaV2.3 channel (CaVα1E with auxiliary subunits) (23). CaVβ3–c-Myc was used to bait the CaV2.3–CaVα2δ1-HA complex using anti-c-Myc–coated magnetic beads (Fig. 6, C and D). As seen, CaVβ3 successfully pulled down CaV2.3/CaVα2δ1-HA WT and CaV2.3/CaVα2δ1-HA T331A (Fig. 6D). In contrast, coimmunoprecipitation was abolished with CaVα2δ1-HA D259A, S261A, S263A, and D363A. CaVα2δ1 Asp-259 and Ser-261 were herein shown to contribute to the interaction with CaV1.2 and CaV2.3. CaVα2δ1 Ser-263 and CaVα2δ1 Asp-363 play a more prominent role in the interaction with CaV2.3 than with CaV1.2 suggesting that the interface could be slightly different. In both channels, however, substitution of the residue CaVα2δ1-HA Thr-331 did not impair protein interaction. Hence, four MIDAS residues contribute to different degrees to protein interaction in high-voltage–activated Ca2+ channels.
Discussion
The glycosylphosphatidylinositol-anchored CaVα2δ1 protein is an intrinsic subunit of high voltage-activated Ca2+ channels of the CaV1 and CaV2 families. It is an extracellular protein that comprises several structural domains, including four CACHE domains in tandem with a single VWA domain. VWA-containing domains mediate protein–protein interaction in a wide variety of proteins located at the cell surface, including integrins, collagens and complement factor (2), and cartilage matrix proteins (24). The VWA domain of CaVα2δ1 bears high sequence identity with the domain I of integrin (15, 16, 25). This biological function is usually mediated via a conserved MIDAS motif that coordinates divalent cations at the protein–ligand interface (10). The recently published high-resolution cryo-EM structure of the homologous CaV1.1 heteromeric complex supports a close interaction between residues of the VWA domain in CaVα2δ1 and extracellular loops from repeats I, II, and III of the pore-forming CaVα1 subunit (16). It also shows that the VWA domain of CaVα2δ1 adopts a classic α/β Rossmann fold (26) and contains a “perfect” MIDAS motif formed by residues Asp-259, Ser-261, Ser-263, Thr-331, and Asp-363 (rat numbering) (16). Multiple primary sequence alignments carried out with the four isoforms of CaVα2δ (CaVα2δ1–4) in human, rabbit, and rat species, revealed that four of five MIDAS residues (Asp-259, Ser-261, Ser-263, and Asp-363) are strictly conserved with the 4th position occupied by threonine being infrequent.
N-terminal residues in MIDAS mediate interaction with CaV1.2
As we have previously shown, the interaction between CaV1.2 and CaVα2δ1 requires a negatively charged aspartate or glutamate residue at position Asp-181 in the first extracellular loop in the voltage sensor domain in repeat I of CaV1.2 (19). A single point mutation at this position is sufficient to prevent Ca2+ influx above background levels. In contrast, we are currently demonstrating that many residues in the VWA domain of CaVα2δ1 play a role in the protein interaction and functional modulation of whole-cell Ca2+ currents by CaVα2δ1. One of the most intriguing observations is the role played by CaVα2δ1 Asp-259 in mediating protein interaction with CaV1.2. MD simulations carried out with our 3D model of the WT complex (CaV1.2 IS1S4–CaVα2δ1 VWA) indicate that the minimal distance between the two α-carbons (Cα) of CaVα2δ1 Asp-259 and CaV1.2 Asp-181 is never smaller than 10.5 Å during the complete course of 25-ns simulations carried out with three trajectories. Within the limitations of simulations performed with a virtual 3D model, such distance suggests that a direct interaction between the two proteins is not very likely.
In contrast to CaVα2δ1 Asp-259, CaVα2δ1 Ser-261 is freely exposed at the cell surface and positioned to mediate the interaction with CaV1.2 Asp-181. MD simulations show that Ser-261 adopts many conformations over the 25-ns period. The oxygen atom of the side-chain hydroxyl group of Ser-261 interacts with the Ca2+ ion, and its hydrogen atom is seen to stabilize alternatively the carboxylate groups in the side chains of CaVα2δ1 Asp-259 and CaV1.2 Asp-181. The hydroxyl group is a stringent structural requirement as illustrated by the observation that CaVα2δ1-HA S261T mutant behaved essentially like CaVα2δ1-HA WT in patch-clamp experiments. Interactions are also possible between CaVα2δ1 Ser-263 and CaV1.2 Asp-181. MD simulations, however, show that the backbone of CaVα2δ1 Ser-263 undergoes a high degree of fluctuations, which may account for the observation that CaVα2δ1-HA S263A and CaVα2δ1-HA S263R somewhat modulated CaV1.2 currents.
Without being part of the signature sequence of MIDAS, glycine residues often play essential structural role in the formation of loops (27). Mutations of CaVα2δ1 Gly-262 to hydrophobic or hydrophilic residues reduced the modulation of whole-cell currents. The analysis of MD simulations suggests that the carboxylate side chain from Asp-181 interacts only occasionally with the main-chain hydrogen of CaVα2δ1 Gly-262. Nevertheless, altering simultaneously the chemical properties of the side chains at positions Gly-262 and Ser-263 disrupts the functional modulation of CaV1.2 currents.
Interaction between CaV1.2 and CaVα2δ1 was also shown to mutually improve the stability of both proteins. This interaction needs to be sufficiently strong to withstand the pulldown assays. We could not however determine the strength of this interaction using isothermal calorimetric and/or surface plasmon resonance assays since it would require purifying large quantities of CaVα2δ1, which were shown to be unstable when produced in regular bacterial systems (28).
Thr-331 plays a minor role in the interface in CaV1.2 and CaV2.3 channels
The polar residue Thr-331 is also predicted to participate in Ca2+ binding even though the fourth position in MIDAS is not strictly conserved, often being substituted by a flexible glycine residue (29). Mutations of Thr-331 to the hydrophobic residue with T331A produced only minor changes in protein–protein interaction in CaV1.2 and CaV2.3 channels and produced modest alterations, if any, in the functional modulation of CaV1.2 currents. MD simulations of CaVα2δ1 T331A showed that the mutation induced a large structural reorganization of the MIDAS motif that preserved, however, the interaction of Ser-261 with CaV1.2 Asp-181. A role for Thr-331 cannot be completely excluded because functional modulation of CaV1.2 currents was decreased with CaVα2δ1 T331R.
These results contrast with recent data published on the integrin-like regulatory subunit CLCA1. CLCA1 up-regulates TMEM16A currents through a Mg2+- and MIDAS-dependent mechanism (30) that stabilizes TMEM16A at the cell surface by preventing its internalization (31). Although the CLCA1 VWA domain also contains a five-residue MIDAS motif (Asp-312, Ser-314, Ser-316, Thr-383, and Asp-412) similar to CaVα2δ1, the molecular determinants of modulation might be slightly different between the two families of ion channels. Disruption of the MIDAS motif, in particular mutation of Ser-316 or Thr-383 to Ala (equivalent to Ser-263 and Thr-331), reduced the ability of CLCA1 VWA to modulate surface protein levels and activity of TMEM16A (32). It is suggested that the molecular determinants responsible for the modulation of channel function by MIDAS residues in integrin-like proteins might command different parameters.
Although the mutations in the N-terminal region of MIDAS disrupted protein interaction and modulation, mutations of the structurally similar Asp-363 were seen to produce a modest impact by decreasing but not abolishing interaction with the CaV1.2 protein and functional modulation of CaV1.2 currents. MD simulations suggest that this may result from the relatively high exposure of CaVα2δ1 Asp-363 to the solvent providing some additional degree of freedom to the side chain.
Divalent cation binding within MIDAS could stabilize protein–protein interaction
By analogy with integrin I proteins, cation binding within the MIDAS motif of CaVα2δ1 could play a role in strengthening the interaction with the pore-forming subunit of CaV1.2 (33). Mutations of the MIDAS motif have been shown to prevent the binding of α2 I domain of integrin with collagen ligand (33, 34). High-resolution structures of the αL I domain of integrin in complex with the domain 1 of the intracellular adhesion molecules ICAM-1 and ICAM-3 have shown that the divalent cation within the MIDAS motif is mostly stabilized by five local residues with an additional contribution from the aspartate residue of the ligand (35, 36). In the absence of the ligand, this site is filled in with a water molecule (37). To gain additional mechanistic insight, we performed 25-ns MD simulations with the minimal 3D model after virtually inserting the mutation CaVα2δ1 D259A (Fig. 7). Comparison of the minimum-energy conformations in Fig. 7, A (CaVα2δ1 WT) and B (CaVα2δ1 D259A), shows that the substitution of Asp-259 by an alanine residue is associated with a change in the conformation of the MIDAS residues around the Ca2+ ion. The hydrogen atom from the hydroxyl group contributed by the side chain from CaVα2δ1 Ser-261 switches from mostly coordinating the Ca2+ ion to forming a hydrogen bond with the carboxyl group of the side chain from CaVα2δ1 Asp-363. As a result, the Ca2+ ion is seen as being pushed away from MIDAS and away from CaV1.2 Asp-181 toward the extracellular aqueous medium. This structural rearrangement could account for the large adverse impact of CaVα2δ1 D259A on the coimmunoprecipitation and modulation of CaV1.2 currents. Hence, the binding of the divalent cation within MIDAS could be an essential component in the three-part network that stabilizes the interaction between CaVα2δ1 and CaV1.2. This proposition is congruent with structural studies showing that the transition between the open and the closed conformations of integrins is mostly driven by the interaction with a divalent ion within the MIDAS motif (38). We remain acutely aware that this working hypothesis is contingent to our virtual 3D model and the parameters we used for the MD simulations. As mentioned earlier, the 3D model of the pore-forming subunit CaVα1C was built from the atomic determinants of CaVα1S with an adequate but far from perfect identity at the primary structural level (69%). In addition, the template failed to provide electronic density in some regions, notably the extracellular IS3S4 loop that was fully reconstructed. It thus remains to be seen whether longer MD simulations carried out with a virtual 3D model of the two complete proteins (>500,000 atoms) would provide a similar picture.
Figure 7.
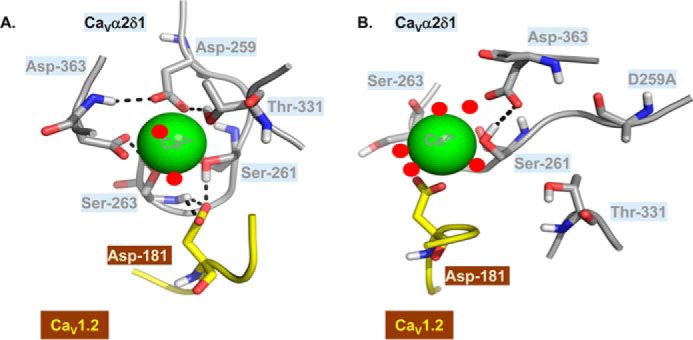
CaVα2δ1 D259A could destabilize Ca2+ binding within MIDAS. CaVα2δ1 MIDAS residues are shown in the lowest energy conformations obtained from three 25-ns MD trajectories produced with the 3D virtual models of CaVα2δ1 WT (A) and CaVα2δ1 D259A (B). Asp-181 in CaV1.2 and CaVα2δ1 MIDAS residues are shown in stick representation and are colored in yellow and gray, respectively. The Ca2+ ion is shown as a green sphere, and water molecules are visualized as red dots. Within residues, oxygen atoms are in red, nitrogen atoms are in blue, and hydrogen atoms are in white. Potential interactions between atoms are indicated by black dashed lines. The 3D model was relatively stable with little fluctuations in the backbone atoms over the 25-ns simulation. In particular, the r.m.s.d. of the Cα for the whole complex (calculated after equilibration and between each time point of the 25-ns simulation) fluctuated in average 2.52 ± 0.59 Å (mean ± S.D.) and ≈80% of the data points deviate less than 3 Å when compared with the structure shown in A. As shown, the oxygen atoms from CaV1.2 Asp-181 are within a few angstroms of the side-chain hydrogen atom of CaVα2δ1 Ser-261 and the main-chain hydrogen atom of Ser-263. Elimination of the negatively charged side chain at position 259 in CaVα2δ1 D259A (B) produced a substantial alteration in the conformation of MIDAS, whereas the hydrogen atom from the hydroxyl group of CaVα2δ1 Ser-261 switches from coordinating the Ca2+ ion and CaV1.2 Asp-181 to facing the carboxyl group in the side chain from CaVα2δ1 Asp-363. In the CaVα2δ1 D259A mutant, the Ca2+ ion is being more freely exposed to the solvent and becomes coordinated by five water molecules (B) instead of two water molecules in the WT protein complex (A). This conformational change disrupts the network of stabilizing interactions between CaVα2δ1 and CaV1.2 Asp-181. PDB files are available upon request.
Experimental procedures
Recombinant DNA techniques
The rabbit CaV1.2 (GenBankTM X15539) and the rat CaVβ3 (GenBankTM M88751) were subcloned in commercial vectors under the control of the CMV promoter as described elsewhere (23, 39). The human CaV2.3 (GenBankTM L27745) was a generous gift from Dr. Toni Schneider (40). The coding sequence (1091 residues) of the rat brain CaVα2δ1 clone (GenBankTM NM_012919) (41) was subcloned in the pmCherry-N1 vector, and the hemagglutinin (HA) epitope (YPYDVPDYA) was inserted in the extracellular domain of CaVα2 between Asp-676 and Arg-677, such that the HA epitope is accessible from the extracellular compartment, and the mCherry is translated after the C terminus. This construct enables the detection of the CaVα2δ1 proteins expressed at the cell surface as described previously (17–19, 39). Point mutations were produced with the Q5 site-directed mutagenesis kit (New England Biolabs) in the pmCherry-CaVα2δ1-HA construct (or in a few rare instances in the pCMV-CaV1.2 construct) according to the manufacturer's instructions as described elsewhere (17, 22). Briefly, substitutions were created by incorporating the desired mutation in the center of the forward primer, and the reverse primer is designed so that the 5′ ends of the two primers anneal back-to-back. Following the PCR, a kinase/ligase/DpnI enzyme mixture was added to the amplified DNA for circularization and template removal before transformation into high-efficiency DH5-α-competent E. coli (New England Biolabs). All constructs were verified by automated double-stranded sequence analysis (Genomics Platform, IRIC, Université de Montréal, Quebec, Canada). The protein expression at the correct molecular weight was confirmed by standard Western blot analysis for each construct as described below (17–19, 22).
Coimmunoprecipitation assay and Western blot analysis
HEKT cells were transiently transfected with 4 μg of pCMV-CaVβ3–c-Myc and pCMV-CaV1.2 and pmCherry-CaVα2δ1-HA WT or mutants. One series of experiments was carried out (Fig. 6, C and D) with pCMV-CaVβ3–c-Myc and pcDNA3-CaV2.3 and pmCherry-CaVα2δ1-HA WT or mutants. Two days after transfection, cells were homogenized in 20 mm NaMOPS (pH 7.4), 300 mm NaCl, and 1% digitonin supplemented with protease inhibitors (Thermo Fisher Scientific). Homogenates were sonicated, incubated for 1 h at 4 °C, and centrifuged at 16,000 × g for 30 min. The supernatant was kept, and the cell pellet was discarded. The protein concentration in the supernatant was determined using BCA protein assay (Thermo Fisher Scientific). A small fraction (20 μg) of the supernatant was kept as the “input fraction.” The remaining ≈600 μg of proteins were diluted with an equal volume of 20 mm NaMOPS (pH 7.4) and 300 mm NaCl to have a final concentration of digitonin at 0.5%. Two different protocols were used for the coimmunoprecipitation assays using either CaVα2δ1-HA or CaVβ3–c-Myc as the bait. In some cases (Fig. 6, A and B), CaVα2δ1-HA was used as the bait to immunoprecipitate the CaV1.2 complex. Appropriate control experiments carried out with the three constructs confirmed that CaVα2δ1-HA was the only protein pulled out by the anti-HA–coated beads (data not shown). The supernatant was then incubated overnight with 50 μl of anti-HA magnetic beads (Thermo Fisher Scientific) previously washed according to the manufacturer's instructions. In some other experiments (Figs. 2, B and C, 3, and 6, C and D), CaVβ3–c-Myc was used as the bait to immunoprecipitate the CaV1.2–CaVα2δ1 or the CaV2.3–CaVα2δ1 complex. In these experiments, the supernatant was incubated overnight with 50 μl of anti-c-Myc magnetic beads (Thermo Fisher Scientific) previously washed according to the manufacturer's instructions. We have performed both assays with similar success, although the signal for the pore-forming subunit CaVα1 of CaV1.2 and CaV2.3 was larger in the presence of the anti-c-Myc beads.
Beads were collected using a PureProteome magnetic rack (Millipore). Magnetic beads were washed three times with 300 μl of buffer containing 20 mm NaMOPS (pH 7.4), 300 mm NaCl, and 0.2% digitonin. The bound proteins were eluted with 20 μl of Laemmli buffer 2× at 95 °C for 5 min, electrophoresed on an 8% SDS-polyacrylamide gel, and transferred onto a nitrocellulose membrane. Western blotting was carried out with anti-CaVβ3 (Alomone, 1:10,000), anti-CaV1.2 (Alomone, 1:5000), anti-CaVα2δ1 (Alomone, Jerusalem, Israel, 1:1000), and anti-GAPDH (Sigma, 1:10,000) and then incubated with an anti-rabbit as secondary antibody (Jackson ImmunoResearch, 1:10,000). Signals were detected with the ECL chemiluminescent substrate (Thermo Fisher Scientific), and blots were visualized with the ChemiDoc Touch system (Bio-Rad).
Flow-cytometry assays
Flow-cytometry experiments were carried out to evaluate the cell-surface expression of the mCherry-CaVα2δ1-HA WT and mutants. Stably CaVβ3 cells were transiently transfected with 4 μg of pCMV-CaV1.2 WT and 4 μg of pmCherry-CaVα2δ1-HA WT or mutants. Experiments were conducted and analyzed as published before (17–19, 22) and are described in greater detail elsewhere (39). Briefly, the cell-surface expression of the mCherry-CaVα2δ1-HA WT was detected with the FITC-conjugated mouse monoclonal anti-HA epitope tag antibody at 5 μg/ml (Sigma). To determine the total quantity of both intracellular and extracellular expression of the tagged proteins, cells were fixed and permeabilized using BD Cytofix/CytopermTM fixation/permeabilization solution kit (BD Biosciences). Roughly 10,000 cells were counted using a FACSAria III® SORP flow cytometer (BD Biosciences). The control conditions were carried out in triplicate with each series of experiments: (a) untransfected CaVβ3 cells without the anti-HA FITC antibody; (b) untransfected CaVβ3 cells with the anti-HA FITC antibody to assess the level of background staining; (c) CaVβ3 cells transfected with pCMV-CaV1.2 WT and pmCherry-CaVα2δ1-HA WT, the latter serving as a quality control of transfection. Expressing mCherry-CaVα2δ1-HA WT in HEKT cells produced a 1-log increase in the FITC (x axis) and a 3-log increase in mCherry fluorescence (y axis) on two-dimensional dot plots.
Flow cytometry data were analyzed using the FlowJo software, version 10 (TreeStar, Ashland, OR), as described in Ref. 39. Relative expression of CaVα2δ1 was calculated based on Δmedian fluorescence intensity (ΔMedFI) for each fluorophore (mCherry or FITC). ΔMedFI for FITC measured in intact nonpermeabilized cells was used as a relative index of the steady-state cell-surface expression of the HA-tagged CaVα2δ1, whereas the ΔMedFI for mCherry attested that the protein was translated until the C terminus. ΔMedFI values were normalized to the maximum value measured the same day for mCherry-CaVα2δ1-HA WT expressed under the same conditions. The normalized ΔMedFI values for mCherry measured for each mutant in intact and permeabilized cells were not significantly different from one another (p > 0.1) (data not shown) suggesting that the cell permeabilization procedure did not distort significantly the relative fluorescence readout under most conditions.
Patch-clamp experiments in HEKT cells
Whole-cell patch-clamp experiments were carried out on isolated cells after transfection in HEKT cells in the presence of the peGFP vector coding for the green fluorescence protein (GFP) (0.2 μg) as a control for transfection efficiency. Only the GFP-positive cells were patched. Electrodes were filled with a solution containing (in mm): 140 CsCl; 0.6 NaGTP; 3 MgATP; 10 EGTA; 10 HEPES; titrated to pH 7.3 with NaOH with a resistance varying between 2.8 and 3.2 megohms. Cells were bathed in a modified Earle's saline solution (in mm): 135 NaCl; 20 TEACl; 2 CaCl2; 1 MgCl2; 10 HEPES, titrated to pH 7.3 with KOH. On-line data acquisition was achieved with the Axopatch 200-B amplifier (Molecular Devices, Sunnyvale, CA) connected with the PClamp software Clampex 10.5 through the Digidata 1440A acquisition system (Molecular Devices) (19). A series of 450-ms voltage pulses were applied from a holding potential of −100 mV at a frequency of 0.2 Hz, from −60 to +70 mV at 5-mV intervals. Series resistance was compensated to ∼85% after on-line capacitive transient cancellation. Unless stated otherwise, whole-cell currents were sampled at 5 kHz and filtered at 1 kHz. PClamp software Clampfit 10.4 was used for data analysis. The mid-potential of activation values (E0.5, act) was estimated from the peak I-V curves obtained for each channel composition and was reported as the mean of individual measurements ± S.E. (22). The free energy of activation was calculated using the mid-activation potential shown in Equation 1,
| (1) |
where z is the effective charge displacement during activation, and F is the Faraday constant. The r100 ratio, defined as the ratio of peak whole-cell currents remaining after a depolarizing pulse of 100 ms (I100 ms/Ipeak), was calculated for each mutant. As there was no significant change in the channel kinetics, these values were not reported herein. To assess for internal consistency, the experiments carried out with novel mutants systematically included a control experiment performed with pmCherry-CaVα2δ1-HA WT (pCMV-CaV1.2 WT + pCMV-CaVβ3 + pmCherry-CaVα2δ1-HA WT) thus explaining the larger sample size for CaVα2δ1 WT. Previous experiments confirmed that mCherry-CaVα2δ1-HA WT sustains the functional modulation of CaV1.2 currents (22). Experiments performed under the same conditions yielded peak current densities that could vary by as much as ± 45% between each series of transfections. This variation appeared to be essentially linked to minor changes in the cell density at the time of transfection. Data from all experiments performed under the same conditions were pooled, and biophysical properties are reported in Table 2. Experiments were performed at room temperature (22 °C).
3D homology modeling
The atomic coordinates of the CaVα1 protein from CaV1.1 (PDB 5GJV) were used to explore the 3D structure of CaV1.2. The primary sequence of the transmembrane segment S1 to S4 in repeat I (residues 32–179) of the rabbit CaV1.2 (GenBankTM X15539) shares 69.6% identity with the same region of the rabbit CaV1.1. The primary sequence of the VWA domain of the rat CaVα2δ1 (our construct, GenBankTM NM_012919) between amino acids 249 and 439 is perfectly conserved between both species. Amino acids 135–281 in CaV1.2 and amino acids 249–439 of the rat CaVα2δ1 were simultaneously aligned to the atomic coordinates of PDB 5GJV using the align2D algorithm in Modeler 9.17 (43). Modeler 9.17 was used to generate 100 3D models, and the models with the lowest values for the molpdf parameters and Discrete Optimized Protein Energy (DOPE) score (44) were selected. The DOPE parameter is a statistical potential used to access the energy of the protein model generated through many iterations by Modeler, which produces homology models by the satisfaction of spatial restraints.
The extracellular loop linking helix S3 and helix S4 was missing in the cryo-EM structure and was reconstructed using the “automatic loop refinement” protocol in Modeler with slow refinement level parameter. Of the more than 100 3D models generated that contain the S3-S4 loop, we selected the “best model” with the lowest DOPE score and molpdf parameters to conduct MD simulations.
Molecular dynamics simulations
MD simulations of the CaV1.2–CaVα2δ1 interface were conducted using GROMACS version 2016.3, single-precision (45). We used the 3D model (Fig. 1) as the input structure, and a Ca2+ ion was virtually inserted into the MIDAS site. The ion was initially positioned at the geometric center of the negatively polarized/charged groups of the five MIDAS residues. The side-chain oxygen position was used for the serine and threonine amino acids, and the midpoint of the two side-chain oxygen atoms was used for the two aspartic acid residues. This crude positioning was deemed sufficient because the ion was not constrained during energy minimization and could thus adopt a more favorable position.
The transmembrane regions S1 to S4 from repeat I of CaV1.2 (IS1S4) were inserted in a membrane bilayer consisting of 128 DPPC molecules. The bilayer model was also obtained from Dr. Peter Tieleman's website in accordance with the protocol developed by Berger et al. (46). The insertion in the membrane was done by aligning hydrophobic residues in the bilayer and comparing the result with the cryo-EM electron density map to optimize the protein positioning. The membrane was then packed around the proteins using the InflateGRO protocol (47).
Minimization, equilibration, and production
The GROMOS53A6 force field (48) was used for the simulations, with the parameters for the DPPC molecules obtained from Dr. Peter Tieleman's website as described above. Solvation was done using the InflateGRO protocol with temporarily enlarged van der Waals radii for carbon atoms to prevent water molecules from being inserted in the membrane (1.5 Å is the standard value, 3.75 Å was used). A saline solution containing the physiological concentrations 143 mm NaCl, 2 mm CaCl2, and 2 mm MgCl2 was used, and the protein charge was neutralized with Cl− ions. The equilibration phases and production simulations were conducted at 323 K, a value slightly above the phase transition of DPPC, which was measured to be at 315 K (42). This ensured proper fluidity as opposed to gel-like behavior under the phase-transition temperature.
Energy minimization was conducted with a target maximal force (Fmax) of no more than 100 kJ·mol−1·nm−1, a maximal number of steps of 50,000, and a step size of 0.1 Å. Fmax converged to the desired value in less than 750 steps for all the trajectories presented in this work. Equilibration was first conducted using an NVT ensemble (constant Number of particles, Volume, and Temperature) for 100 ps, followed by a second phase using an NPT ensemble (constant Number of particles, Pressure, and Temperature) for 1 ns. Production runs of 25 ns were then computed with a 2-fs time step, saving coordinates, energy, and velocities every 25 ps.
For the CaVα2δ1 WT and CaVα2δ1 D259A models of the CaV1.2–CaVα2δ1 interface, the above steps were repeated for five replicates. Because large regions of the CaV1.2 protein are missing in our 3D model, we kept for each form of the complex the three trajectories that showed the least structural divergence across the duration of the 25-ns trajectory as determined by their r.m.s.d. values compared with the initial structure (data not shown).
Statistics
Results were expressed as mean ± S.E. Tests of significance were carried out using the unpaired one-way ANOVA with the Tukey test embedded in the Origin 7.0 analysis software (OriginLab Corp., Northampton, MA). Unless stated otherwise, data were considered statistically significant at p < 0.05.
Author contributions
J. B., O. M., and L. P. data curation; J. B., O. M., B. B., M.-P. T., and L. P. formal analysis; J. B., O. M., M.-P. T., and B. B. investigation; J. B., O. M., B. B., M.-P. T., and L. P. methodology; J. B. and L. P. writing-original draft; J. B., O. M., B. B., M.-P. T., R. N., and L. P. writing-review and editing; O. M. and R. N. software; R. N. and L. P. supervision; R. N. and L. P. validation; L. P. conceptualization; L. P. funding acquisition; L. P. project administration.
Acknowledgments
We thank Dr. Rémy Sauvé for critical reading of the manuscript and the Bevan lab for their tutorial on membrane protein simulations, which can be found on line.
This work was supported in part by Operating Grant 130256 from the Canadian Institutes of Health Research and Grant G-15-0009329 from the Canadian Heart and Stroke Foundation (to L. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- VWA
- von Willebrand factor type A
- DPPC
- dipalmitoylphosphatidylcholine
- ΔGact
- free energy of activation
- MD simulation
- molecular dynamics simulation
- MIDAS
- metal ion-dependent adhesion site
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank
- ANOVA
- analysis of variance
- pF
- picofarad
- HA
- hemagglutinin
- ΔMedFI
- Δmedian fluorescence intensity.
References
- 1. Letunic I., Doerks T., and Bork P. (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 10.1093/nar/gku949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whittaker C. A., and Hynes R. O. (2002) Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 13, 3369–3387 10.1091/mbc.E02-05-0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lacy D. B., Wigelsworth D. J., Scobie H. M., Young J. A., and Collier R. J. (2004) Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 6367–6372 10.1073/pnas.0401506101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pihlajamaa T., Kajander T., Knuuti J., Horkka K., Sharma A., and Permi P. (2013) Structure of Plasmodium falciparum TRAP (thrombospondin-related anonymous protein) A domain highlights distinct features in apicomplexan von Willebrand factor A homologues. Biochem. J. 450, 469–476 10.1042/BJ20121058 [DOI] [PubMed] [Google Scholar]
- 5. Edwards Y. J., and Perkins S. J. (1995) The protein fold of the von Willebrand factor type A domain is predicted to be similar to the open twisted β-sheet flanked by α-helices found in human ras-p21. FEBS Lett. 358, 283–286 10.1016/0014-5793(94)01447-9 [DOI] [PubMed] [Google Scholar]
- 6. Craig D., Gao M., Schulten K., and Vogel V. (2004) Structural insights into how the MIDAS ion stabilizes integrin binding to an RGD peptide under force. Structure 12, 2049–2058 10.1016/j.str.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 7. Springer T. A. (2006) Complement and the multifaceted functions of VWA and integrin I domains. Structure 14, 1611–1616 10.1016/j.str.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colombatti A., and Bonaldo P. (1991) The superfamily of proteins with von Willebrand factor type A-like domains: one theme common to components of extracellular matrix, hemostasis, cellular adhesion, and defense mechanisms. Blood 77, 2305–2315 [PubMed] [Google Scholar]
- 9. Tuckwell D. (1999) Evolution of von Willebrand factor A (VWA) domains. Biochem. Soc. Trans. 27, 835–840 10.1042/bst0270835 [DOI] [PubMed] [Google Scholar]
- 10. Shimaoka M., Takagi J., and Springer T. A. (2002) Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31, 485–516 10.1146/annurev.biophys.31.101101.140922 [DOI] [PubMed] [Google Scholar]
- 11. Lee J. O., Bankston L. A., Arnaout M. A., and Liddington R. C. (1995) Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure 3, 1333–1340 10.1016/S0969-2126(01)00271-4 [DOI] [PubMed] [Google Scholar]
- 12. Valdramidou D., Humphries M. J., and Mould A. P. (2008) Distinct roles of β1 metal ion-dependent adhesion site (MIDAS), adjacent to MIDAS (ADMIDAS), and ligand-associated metal-binding site (LIMBS) cation-binding sites in ligand recognition by integrin α2β1. J. Biol. Chem. 283, 32704–32714 10.1074/jbc.M802066200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Petegem F., Clark K. A., Chatelain F. C., and Minor D. L. (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429, 671–675 10.1038/nature02588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davies A., Hendrich J., Van Minh A. T., Wratten J., Douglas L., and Dolphin A. C. (2007) Functional biology of the α(2)δ subunits of voltage-gated calcium channels. Trends Pharmacol. Sci. 28, 220–228 10.1016/j.tips.2007.03.005 [DOI] [PubMed] [Google Scholar]
- 15. Wu J., Yan Z., Li Z., Yan C., Lu S., Dong M., and Yan N. (2015) Structure of the voltage-gated calcium channel Cav1.1 complex. Science 350, aad2395 10.1126/science.aad2395 [DOI] [PubMed] [Google Scholar]
- 16. Wu J., Yan Z., Li Z., Qian X., Lu S., Dong M., Zhou Q., and Yan N. (2016) Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 537, 191–196 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- 17. Tétreault M. P., Bourdin B., Briot J., Segura E., Lesage S., Fiset C., and Parent L. (2016) Identification of glycosylation sites essential for surface expression of the CaVα2δ1 subunit and modulation of the cardiac CaV1.2 channel activity. J. Biol. Chem. 291, 4826–4843 10.1074/jbc.M115.692178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segura E., Bourdin B., Tétreault M. P., Briot J., Allen B. G., Mayer G., and Parent L. (2017) Proteolytic cleavage of the hydrophobic domain in the CaVα2δ1 subunit improves assembly and activity of cardiac CaV1.2 channels. J. Biol. Chem. 292, 11109–11124 10.1074/jbc.M117.784355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bourdin B., Briot J., Tétreault M. P., Sauvé R., and Parent L. (2017) Negatively charged residues in the first extracellular loop of the L-type CaV1.2 channel anchor the interaction with the CaVα2δ1 auxiliary subunit. J. Biol. Chem. 292, 17236–17249 10.1074/jbc.M117.806893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dolphin A. C. (2013) The α2δ subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 1828, 1541–1549 10.1016/j.bbamem.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 21. San Sebastian E., Mercero J. M., Stote R. H., Dejaegere A., Cossío F. P., and Lopez X. (2006) On the affinity regulation of the metal-ion-dependent adhesion sites in integrins. J. Am. Chem. Soc. 128, 3554–3563 10.1021/ja054142a [DOI] [PubMed] [Google Scholar]
- 22. Bourdin B., Shakeri B., Tétreault M. P., Sauvé R., Lesage S., and Parent L. (2015) Functional characterization of CaVα2δ mutations associated with sudden cardiac death. J. Biol. Chem. 290, 2854–2869 10.1074/jbc.M114.597930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shakeri B., Bourdin B., Demers-Giroux P. O., Sauvé R., and Parent L. (2012) A quartet of leucine residues in the guanylate kinase domain of CaVβ determines the plasma membrane density of the CaV2.3 channel. J. Biol. Chem. 287, 32835–32847 10.1074/jbc.M112.387233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker A. K., Mikolajek H., Werner J. M., Paulsson M., and Wagener R. (2015) Characterization of recombinantly expressed matrilin VWA domains. Protein Expr. Purif. 107, 20–28 10.1016/j.pep.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cantí C., Nieto-Rostro M., Foucault I., Heblich F., Wratten J., Richards M. W., Hendrich J., Douglas L., Page K. M., Davies A., and Dolphin A. C. (2005) The metal-ion-dependent adhesion site in the von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 102, 11230–11235 10.1073/pnas.0504183102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossmann M. G., Moras D., and Olsen K. W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature 250, 194–199 10.1038/250194a0 [DOI] [PubMed] [Google Scholar]
- 27. Kwasigroch J. M., Chomilier J., and Mornon J. P. (1996) A global taxonomy of loops in globular proteins. J. Mol. Biol. 259, 855–872 10.1006/jmbi.1996.0363 [DOI] [PubMed] [Google Scholar]
- 28. Briot J., D'Avanzo N., Sygusch J., and Parent L. (2016) Three-dimensional architecture of the L-type calcium channel: structural insights into the CaVα2δ1 auxiliary protein. Biochem. Mol. Biol. J. 2, 10.21767/2471-8084.100025 [DOI] [Google Scholar]
- 29. Yan B. X., and Sun Y. Q. (1997) Glycine residues provide flexibility for enzyme active sites. J. Biol. Chem. 272, 3190–3194 10.1074/jbc.272.6.3190 [DOI] [PubMed] [Google Scholar]
- 30. Patel A. C., Brett T. J., and Holtzman M. J. (2009) The role of CLCA proteins in inflammatory airway disease. Annu. Rev. Physiol. 71, 425–449 10.1146/annurev.physiol.010908.163253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sala-Rabanal M., Yurtsever Z., Nichols C. G., and Brett T. J. (2015) Secreted CLCA1 modulates TMEM16A to activate Ca2+-dependent chloride currents in human cells. eLife 4, 05875 10.7554/eLife.05875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sala-Rabanal M., Yurtsever Z., Berry K. N., Nichols C. G., and Brett T. J. (2017) Modulation of TMEM16A channel activity by the von Willebrand factor type A (VWA) domain of the calcium-activated chloride channel regulator 1 (CLCA1). J. Biol. Chem. 292, 9164–9174 10.1074/jbc.M117.788232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamata T., Liddington R. C., and Takada Y. (1999) Interaction between collagen and the α(2)I-domain of integrin α(2)β(1). Critical role of conserved residues in the metal ion-dependent adhesion site (MIDAS) region. J. Biol. Chem. 274, 32108–32111 10.1074/jbc.274.45.32108 [DOI] [PubMed] [Google Scholar]
- 34. Bergelson J. M., and Hemler M. E. (1995) Integrin-ligand binding. Do integrins use a ‘MIDAS touch’ to grasp an Asp? Curr. Biol. 5, 615–617 10.1016/S0960-9822(95)00124-2 [DOI] [PubMed] [Google Scholar]
- 35. Luo B. H., Carman C. V., and Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 10.1146/annurev.immunol.25.022106.141618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimaoka M., Xiao T., Liu J. H., Yang Y., Dong Y., Jun C. D., McCormack A., Zhang R., Joachimiak A., Takagi J., Wang J. H., and Springer T. A. (2003) Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 112, 99–111 10.1016/S0092-8674(02)01257-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mahalingam B., Ajroud K., Alonso J. L., Anand S., Adair B. D., Horenstein A. L., Malavasi F., Xiong J. P., and Arnaout M. A. (2011) Stable coordination of the inhibitory Ca2+ ion at the metal ion-dependent adhesion site in integrin CD11b/CD18 by an antibody-derived ligand aspartate: implications for integrin regulation and structure-based drug design. J. Immunol. 187, 6393–6401 10.4049/jimmunol.1102394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang K., and Chen J. (2012) The regulation of integrin function by divalent cations. Cell Adh. Migr. 6, 20–29 10.4161/cam.18702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bourdin B., Segura E., Tetreault M. P., Lesage S., and Parent L. (2016) Determination of the relative cell surface and total expression of recombinant ion channels using flow cytometry. J. Vis. Exp. 10.3791/54732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pereverzev A., Leroy J., Krieger A., Malécot C. O., Hescheler J., Pfitzer G., Klöckner U., and Schneider T. (2002) Alternate splicing in the cytosolic II-III loop and the carboxy terminus of human E-type voltage-gated Ca2+ channels: electrophysiological characterization of isoforms. Mol. Cell. Neurosci. 21, 352–365 10.1006/mcne.2002.1179 [DOI] [PubMed] [Google Scholar]
- 41. Williams M. E., Feldman D. H., McCue A. F., Brenner R., Velicelebi G., Ellis S. B., and Harpold M. M. (1992) Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron 8, 71–84 10.1016/0896-6273(92)90109-Q [DOI] [PubMed] [Google Scholar]
- 42. Nagle J. F. (1993) Area/lipid of bilayers from NMR. Biophys. J. 64, 1476–1481 10.1016/S0006-3495(93)81514-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Webb B., and Sali A. (2017) Protein structure modeling with MODELLER. Methods Mol. Biol. 1654, 39–54 10.1007/978-1-4939-7231-9_4 [DOI] [PubMed] [Google Scholar]
- 44. Shen M. Y., and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 10.1110/ps.062416606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mark James Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 [Google Scholar]
- 46. Berger O., Edholm O., and Jähnig F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013 10.1016/S0006-3495(97)78845-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidt T. H., and Kandt C. (2012) LAMBADA and InflateGRO2: efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J. Chem. Inf. Model. 52, 2657–2669 10.1021/ci3000453 [DOI] [PubMed] [Google Scholar]
- 48. Oostenbrink C., Villa A., Mark A. E., and van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 10.1002/jcc.20090 [DOI] [PubMed] [Google Scholar]