

Abstract
Genetic factors are known to significantly contribute to the etiology of psychiatric diseases such as attention deficit hyperactivity disorder (ADHD) and autism spectrum and bipolar disorders, but the underlying molecular processes remain largely elusive. The dopamine transporter (DAT) has received continuous attention as a potential risk factor for psychiatric disease, as it is critical for dopamine homeostasis and serves as principal target for ADHD medications. Constrain metrics for the DAT-encoding gene, solute carrier family 6 member 3 (SLC6A3), indicate that missense mutations are under strong negative selection, pointing to pathophysiological outcomes when DAT function is compromised. Here, we systematically characterized six rare genetic variants of DAT (I312F, T356M, D421N, A559V, E602G, and R615C) identified in patients with neuropsychiatric disorders. We evaluated dopamine uptake and ligand interactions, along with ion coordination and electrophysiological properties, to elucidate functional phenotypes, and applied Zn2+ exposure and a substituted cysteine–accessibility approach to identify shared structural changes. Three variants (I312F, T356M, and D421N) exhibited impaired dopamine uptake associated with changes in ligand binding, ion coordination, and distinct conformational disturbances. Remarkably, we found that all three variants displayed gain-of-function electrophysiological phenotypes. I312F mediated an increased uncoupled anion conductance previously suggested to modulate neuronal excitability. T356M and D421N both mediated a cocaine-sensitive leakage of cations, which for T356M was potentiated by Zn2+, concurrent with partial functional rescue. Collectively, our findings support that gain of disruptive functions due to missense mutations in SLC6A3 may be key to understanding how dopaminergic dyshomeostasis arises in heterozygous carriers.
Keywords: dopamine transporter, neurotransmitter transport, monoamine transporter, genetic polymorphism, autism, attention-deficit hyperactivity disorder, missense mutation, molecular genetics, psychiatric disease, rare variants
Introduction
A substantial proportion of the disease etiology of common psychiatric disorders, such as attention deficit hyperactivity disorder (ADHD),3 autism spectrum disorders (ASD), bipolar disorder, and schizophrenia, is attributed to genetic components (1). The underlying neurobiological mechanisms are not clear, but dopamine disturbances are believed to constitute a central component (2–7). The allelic spectrum of these dopamine-related disorders is rapidly expanding and includes both common variants and rare structural or exonic mutations, including de novo variants (1, 8–12). Moreover, an interesting overlap in the genetic architecture of psychiatric disorders has been observed, and this pleiotrophy is seen for both common and rare variants (13–18). Despite progress, much of the genetic component remains unaccounted for, and we also have the challenge ahead of translating most of the comprehensive genetic information into an understanding of underlying biological processes and subsequently into clinically applicable knowledge. Rare variants may provide a unique handle for obtaining new disease insights as they are expected to have stronger effect sizes than typical common variants, which may facilitate the identification of cellular phenotypes (76).
The dopamine transporter (DAT), encoded by SLC6A3, mediates dopamine reuptake through a Na+-dependent active transport mechanism and thereby exerts spatiotemporal regulation of extracellular dopamine and ensures sufficient intracellular dopamine stores (19, 20). DAT has been the focus of many genetic studies of psychiatric disorders, not only because of its essential role in dopaminergic neurotransmission, but also because DAT serves as an important target for prescriptive medicines, such as methylphenidate and amphetamines that are used in treatment of ADHD symptoms (21). Several studies have found associations between common genetic variants of SLC6A3 and increased risk of psychiatric disease, but deduction of their biological implications is largely elusive (22–24). During recent years, a number of rare exonic DAT variants have been identified in patients suffering from diverse neuropsychiatric and/or neurodevelopmental disorders (25–31). The negative health consequences of genetic changes in DAT are strongly supported by the constrain metrics reported in the comprehensive EXaC database, where DAT is classified as “loss-of-function-intolerant” and is reported to be missense constrained (32), indicating that mutations affecting aspects of DAT function are under considerable negative selection.
Six rare coding variants of DAT, identified in patients with psychiatric diagnoses are as follows: I312F, D421N, T356M, A559V, E602G, and R615C (25–29, 31). Despite being individually rare, the study of genetic variants with potentially large effect sizes could provide a strong tool for elucidating disease-relevant phenotypes that may pertain to more general disease etiologies (33). Indeed, many of the coding variants have been reported to have altered function or regulation (25, 28, 29, 31, 34, 35), but a systematic comparison of the coding DAT variants associated with psychiatric disease is lacking. Because obligate carriers of loss-of-function mutations do not appear to be predisposed to psychiatric disease (36, 37), key questions are whether and how coding variants in psychiatric patients may impose pathophysiological disturbances, and whether the variants share structural or functional changes. Here, our findings reveal novel aberrant phenotypes for several rare, disease-associated coding DAT variants and identify changes in transporter ion conductances as a potential common mechanism through which functional variants may cause dopaminergic dyshomeostasis.
Results
Functional impact of rare coding DAT variants correlates with allele frequency and conservation
To achieve detailed comparative knowledge about structural and functional changes in DAT, imposed by disease-associated mutations, we systematically analyzed the six coding variants, I312F, T356M, D421N, A559V, E602G, and R615C, identified in patients with diverse psychiatric disorders (25–29). I312F and T356M are located near the substrate and ion-binding sites in transmembrane domains (TM) 6 and 7, respectively. D421N is positioned in TM8 directly within the second sodium-binding site, whereas A559V is found near the beginning of TM12. Finally, E602G and R615C localize to the intracellular C-terminal tail of DAT (Fig. 1A). The variants' frequencies from reference databases are listed in Fig. 1B, along with index patient's diagnosis, sequence conservation of the affected residues, and the mutation's predicted functional impact from PolyPhen-2 scores (38). A559V and E602G were the first disease-associated coding DAT variants to be identified (26). Both variants are among the most frequently occurring missense variants in SLC6A3 (32) and are predicted by their PolyPhen-2 scores (38) to have low probability of damaging effects (Fig. 1B). In contrast, the I312F, T356M, D421N, and R615C variants are either extremely rare (T356M and R615C) or singletons (I312F and D421N), and they all affect conserved residues with predicted high probability of damaging outcome on DAT function (Fig. 1B).
Figure 1.

Occurrence, cross-species conservation, and functional assessment of disease-associated coding DAT variants. A, schematic representation of DAT illustrating the position of six coding DAT variants, identified in patients with psychiatric disease. B, for each coding variant the table lists the following: index patient's diagnosis; cross-species sequence conservation of the affected residues; the variant's reported frequencies in reference databases; and the mutations predicted probability of having damaging impact on DAT function from PolyPhen-2 scores. C, direct functional evaluation of [3H]dopamine saturation uptake with indicated dopamine concentrations into transiently transfected COS-7 cells. Dopamine uptake curves are presented as average curves (mean ± S.E.) of three experiments, each performed in triplicate and normalized to the fitted Vmax of DAT WT. BP, bipolar disorder; EOP, early-onset parkinsonism; MAF, minor allele frequency.
To compare the variants' PolyPhen-2 scores with direct functional measures, we performed [3H]dopamine uptake and [3H]CFT-binding experiments on transiently transfected COS-7 cells. Three of the six variants (I312F, T356M, and D421N) displayed functional impairments (Fig. 1C and Table 1). Both I312F and T356M showed reduced uptake capacity (∼50 and ∼40% of DAT WT, respectively), consistent with previous findings (25, 29). These reductions in uptake capacity were accompanied by significant but small reductions in apparent dopamine affinities (Km = 2.0 ± 0.2 μm for I312F and 2.2 ± 0.6 μm for T356M versus 0.84 ± 0.1 μm for DAT WT, see Fig. 1C and Table 1). D421N displayed, as reported previously (25), a markedly disruptive phenotype, evident as a linear uptake curve that could not be reliably fitted by Michaelis-Menten kinetics (Fig. 1C, and Table 1). The uptake impairments of T356M and D421N were associated with pronounced (∼50-fold) affinity losses for the high-affinity cocaine analogue, CFT, whereas the variants' maximal binding capacities were significantly higher than for DAT WT. Thus, the compromised functionality of these variants does not appear to be explained by a reduced number of binding sites (Table 1). Consistent with our previous findings (25), we did not observe any changes in [3H]CFT binding for the I312F variant, implying that the I312F mutation impairs substrate translocation rather than substrate binding. The A559V and E602G variants showed no differences in uptake kinetic parameters compared with DAT WT, as also reported elsewhere (34, 39). The uptake kinetics of R615C were also similar to DAT WT, even though the PolyPhen-2 score predicted damaging effects of the mutation. In line with the unaltered dopamine uptake by A559V, E602G, and R615C, these variants also had [3H]CFT-binding properties that were similar to those of DAT WT, suggesting that both ligand binding and substrate translocation is preserved for these variants (Fig. 1 and Table 1).
Table 1.
Kinetic parameters of [3H]dopamine uptake and [3H]CFT competition binding
Vmax and Km values from [3H]dopamine uptake experiments were obtained by fitting data to Michaelis-Menten kinetics. CFT Ki values were calculated from pIC50 values, determined by nonlinear regression analysis of [3H]CFT/CFT competition binding curves. Mean Ki values with S.E. intervals were calculated from the mean pKi ± S.E. I312F, T356M, and D421N show significant impairments in uptake kinetic parameters. T356M and D421N also have dramatically lower CFT binding affinity and significant increases in Bmax. A559V, E602G, and R615C display uptake and binding properties similar to DAT WT. Data are mean ± S.E., n = 3, and all experiments were performed in triplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA with Dunnett's multiple comparisons test.
| Km dopamine, mean ± S.E. | Vmax dopamine, mean ± S.E. (% WT) | KiCFT, mean (nm) (S.E. interval) | Bmax CFT mean ± S.E. (% of WT) | |
|---|---|---|---|---|
| μm | ||||
| WT | 0.84 ± 0.1 | 100 ± 14 | 7.80 (6.3, 9.3) | 100 ± 15 |
| I312F | 2.0 ± 0.2* | 50.5 ± 6.0* | 5.53 (5.16, 5.90) | 86.0 ± 7.1 |
| T356M | 2.2 ± 0.6* | 41.85 ± 5.8** | 298 (230, 390)*** | 197 ± 11*** |
| D421N | >10 × WT | Not fitted | 338 (267, 426)*** | 154 ± 16*** |
| A559V | 0.80 ± 0.09 | 104 ± 11 | 8.54 (7.6, 9.6) | 106 ± 11 |
| E602G | 0.93 ± 0.05 | 99.3 ± 13 | 6.92 (6.2, 7.7) | 82.1 ± 3.7 |
| R615C | 0.75 ± 0.02 | 102 ± 16 | 7.29 (6.2, 8.5) | 89.7 ± 11 |
Differential binding of psychostimulants to DAT-coding variants
ADHD, and ADHD symptoms occurring comorbidly with other psychiatric diseases, are often effectively treated with psychostimulant drugs, such as methylphenidate (e.g. Ritalin) or amphetamines (e.g. Aderall). We evaluated the ability of these drugs, alongside the classical DAT blocker cocaine, to inhibit dopamine uptake in transiently transfected COS-7 cells, expressing DAT WT or a disease-associated variant. Interestingly, the IC50 values of methylphenidate and cocaine, both of which act as blockers of DAT, were significantly smaller for I312F than for DAT WT (∼3- and ∼2.5-fold lower for methylphenidate and cocaine, respectively. See Fig. 2). By contrast amphetamine, serving as a DAT substrate, had a 5-fold lower apparent affinity for I312F than for DAT WT (Fig. 2 and Table 2). Note that we also observed a similar loss in the apparent affinity of dopamine to I312F (Table 1). For T356M, all three ligands showed significantly lower inhibitory potency than for DAT WT, but the apparent affinity loss was particularly pronounced for blockers (IC50 values for T356M were ∼7-, ∼40-, and ∼150-fold higher than for DAT WT for amphetamine, cocaine, and methylphenidate, respectively, Fig. 2 and Table 2). We did not observe any differences in ligand IC50 values for A559V, E602G, and R615C, and we could not obtain reliable IC50 values for D421N, because of a very low uptake signal.
Figure 2.
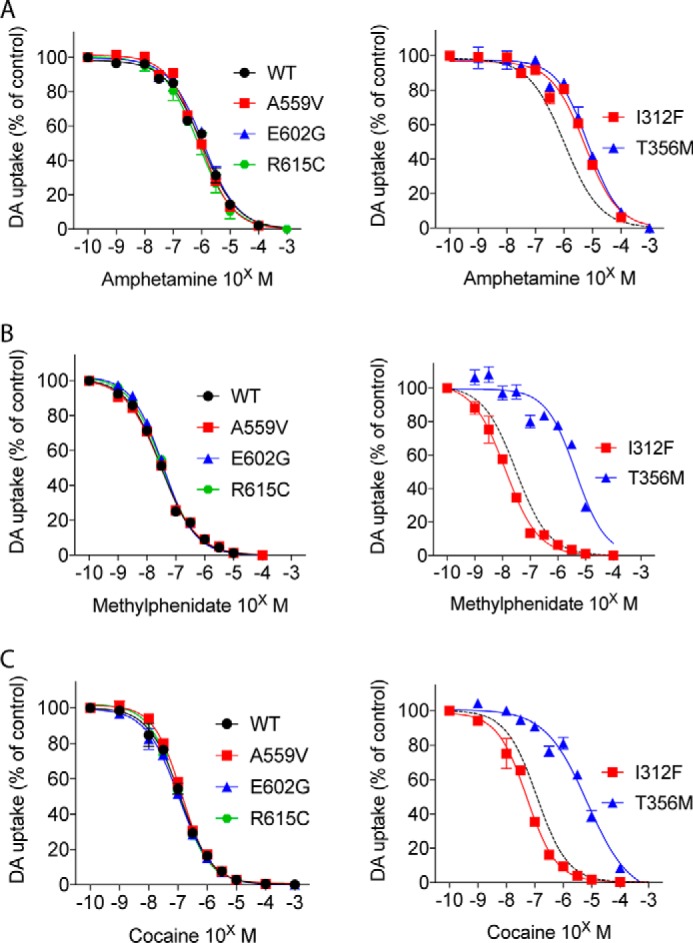
Functionally impaired DAT variants display alterations in psychostimulant interactions. Competition [3H]dopamine uptake curves with indicated concentrations of amphetamine (A), methylphenidate (B), or cocaine (C), carried out on transiently transfected COS-7 cells expressing DAT WT or a coding DAT variant. Curves are presented as means ± S.E. of three experiments, each normalized to control without inhibitor present. I312F shows decreased apparent amphetamine affinity but significantly increased apparent affinity for methylphenidate and cocaine (see Table 2). T356M displays a loss of apparent affinity for all three DAT ligands, though particularly pronounced for methylphenidate and cocaine. The dopamine uptake mediated by D421N was too low to allow meaningful data fitting.
Table 2.
IC50 values for amphetamine, methylphenidate, and cocaine
pIC50 values were derived from competition [3H]dopamine uptake curves from transiently transfected COS-7 cells expressing DAT WT or a coding DAT variant by nonlinear regression analysis. Meaningful fitting of data from D421N was not possible because of very low specific signals. Analyses for statistical differences were performed on the pIC50 values, and mean pIC50 ± S.E. were used to calculate the IC50 values and S.E. interval. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. One-way ANOVA of pIC50 values with Dunnett's multiple comparisons test. ND means not determined.
| Amphetamine IC50 (μm) (S.E. interval) | Methylphenidate IC50 (nm) (S.E. interval) | Cocaine IC50(nm) (S.E. interval) | |
|---|---|---|---|
| WT | 1.03 (0.98, 1.1) | 29.0 (26, 33) | 122 (110, 140) |
| I312F | 5.26 (5.0, 5.5)**** | 11.3 (8.6, 14)** | 51.0 (48, 54)*** |
| T356M | 6.73 (6.1, 7.4)**** | 4420 (3700, 5300)**** | 5350 (4400, 6400)**** |
| D421N | ND | ND | ND |
| A559V | 0.86 (0.85, 0.87) | 29.2 (26, 33) | 139 (130, 150) |
| E602G | 1.16 (1.0, 1.3) | 35.6 (35, 37) | 108 (98, 120) |
| R615C | 0.77 (0.60, 1.0) | 34.0 (29, 40) | 110 (100, 120) |
Ion binding is perturbed in functionally impaired DAT variants
Efficient reuptake of dopamine via DAT depends on co-transport of sodium and chloride with a predicted stoichiometry of 1DA:2Na+:1Cl− (19, 40). We evaluated the ion dependence of dopamine transport for the transporter variants by carrying out dopamine uptake measurements under increasing Cl− or Na+ concentrations, using sodium gluconate and choline chloride for equimolar ion substitutions (Fig. 3 and Table 3). The functionally impaired variants, i.e. I312F, T356M, and D421N, all showed distinct changes in ion-binding properties. I312F displayed a 4-fold reduction in apparent sodium affinity (136 ± 44 mm versus 29.1 ± 1.6 mm for DAT WT), accompanied by an even larger loss of apparent chloride affinity (Kd = 221 ± 38 mm versus 31.5 ± 1.6 mm for DAT WT) (Fig. 3 and Table 3). The T356M variant demonstrated impaired sodium binding (Kd = 145 ± 64 mm versus 29.1 ± 1.6 mm for DAT WT), whereas the apparent chloride affinity was not significantly different from that of DAT WT (Fig. 3 and Table 3). Finally, D421N showed not only disrupted sodium binding, as reported previously (25), but also produced an almost linear chloride dependence curve, suggesting that the chloride coordination is equally compromised at physiological sodium concentrations. The A559V, E602G, and R615C variants, which localize to regions outside the substrate and ion-binding sites, obtained ion affinity estimates that were practically indistinguishable from WT (Fig. 3 and Table 3).
Figure 3.

Ion binding is compromised in functionally impaired DAT variants. Sodium dependence (A) and chloride dependence (B) of [3H]dopamine uptake are shown. Choline chloride and sodium gluconate were used for equimolar cation and anion substitution, respectively. I312F, T356M, and D421N all show distinct changes in ion coordination. The derived Kd values for Na+ and Cl− are listed in Table 3. All curves are normalized and presented as mean ± S.E. from three (all variants) or six (WT) experiments, each performed in triplicate.
Table 3.
Kd values for sodium and chloride
Kd values were derived by nonlinear regression analysis of Na+ and Cl− dose–response curves, obtained by measuring [3H]dopamine uptake into transiently transfected COS-7 cells during equimolar titration of 200 mm NaCl against choline chloride and sodium gluconate (final Na+ and Cl− concentrations of 200, 130, 80, 40, 20, 10, 5, and 0 mm). *, p < 0.05; ***, p < 0.001. One-way ANOVA with Dunnett's multiple comparisons test (n = 3 for variants and n = 6 for DAT WT, with triplicate determinations). Kd values for D421N could not be reliably fitted, because of the linear nature of the ion dependency curves.
| Na+ Kd (mm), mean ± S.E. | Cl− Kd (mm), mean ± S.E. | |
|---|---|---|
| WT | 29.1 ± 1.2 | 31.5 ± 1.6 |
| I312F | 136 ± 44* | 221 ± 38*** |
| T356M | 145 ± 64* | 71.6 ± 16 |
| D421N | >WT | >WT |
| A559V | 25.7 ± 0.56 | 30.2 ± 5.8 |
| E602G | 31.5 ± 0.97 | 27.8 ± 2.3 |
| R615C | 31.2 ± 0.41 | 32.6 ± 3.7 |
Functionally impaired variants display “gain-of-function” electrophysiological properties
The ion-binding sites in DAT have been shown to be intimately related to transporter conductances (41). We applied two-electrode voltage-clamp measurements in Xenopus oocytes to elucidate possible changes in the conductance properties of the three loss-of-function variants: I312F, T356M, and D421N, and we compared them with conductances in the DAT WT and the A559V variant, which does not show reduced dopamine uptake but has been reported to mediate an anomalous constitutive dopamine efflux (34, 35). DAT is known to mediate three ion conductances: a stoichiometric current arising from the excess transport of positive changes upon dopamine translocation, a substrate-activated uncoupled anion conductance, and a cation leak current that is blocked by DAT ligands (40, 42, 43). We first evaluated dopamine-induced currents, defined here as INaCl, DA − INaCl, at different membrane potentials. As expected (40, 42), DAT WT mediated an inwardly rectifying current at negative membrane potentials, and a similar current was observed for both I312F and A559V (Fig. 4). T356M and seemingly also D421N, however, elicited a striking outward current at negative potentials when perfused with dopamine (Fig. 4). A likely explanation is that dopamine blocks a tonic inward leak conductance, thereby eliciting an apparent outward current in these mutants, although the alternative explanation that dopamine elicits an outward leak cannot be excluded.
Figure 4.

Functionally impaired DAT variants display gain-of-function electrophysiological properties. A, dopamine-induced currents through indicated DAT variants. I/V recordings were made in NaCl, and in NaSCN to isolate the uncoupled anion conductance. For I312F, Cl− substitution to SCN generates a greater left-shift in reversal potential and a larger outward current at positive potentials, compared with DAT WT, which is consistent with an enhanced uncoupled anion conductance. T356M and seemingly also D421N conduct an aberrant dopamine-associated outward current at negative potentials, which is markedly enhanced in NaSCN for D421N. B, leak conductances were evaluated by recording cocaine-sensitive currents (Icontrol − Icocaine) in NaCl and LiCl. Both T356M and D421N show enhanced leak conductances in NaCl. For D421N, the leak conductance in NaCl is similar in size and reversal potential to the leak current in LiCl. In contrast, Na+ substitution for Li+ completely abolishes the leak current through T356M, in marked contrast to DAT WT. All curves presented in A and B are mean ± S.E. from 4 to 11 experiments.
To dissect out potential changes in the substrate-activated uncoupled anion current, we substituted extracellular chloride with the more permeable anion, thiocyanate (SCN), and again recorded dopamine-induced currents (42). As expected, chloride substitution with SCN generated a negative shift in current through DAT WT, leading to a reversal potential around −40 mV (42). The observed reversal potential upon SCN substitution was strikingly more negative for I312F than for DAT WT (approximately −60 mV for I312F versus approximately −40 mV for WT), and the I312F variant also mediated a larger outward ion flux at positive membrane potentials (Fig. 4A). Together, these findings suggest that I312F possesses increased anion permeability (42). For T356M, SCN shifted the dopamine-activated aberrant outward current toward more hyperpolarized potentials, resulting in an associated change in reversal potential from approximately −20 to −60 mV and a small inward current at potentials above −60 mV. In contrast, substitution with SCN led to a remarkable increase in the dopamine-associated outward current through D421N at negative membrane potentials, without parallel changes in reversal potential (Fig. 4A). Of note, we did not observe differences in dopamine-induced currents through A559V compared with DAT WT, neither in NaCl nor NaSCN.
To uncover putative changes in leak conductances, we assessed cocaine-sensitive currents (Icontrol − Icocaine). The steady-state leak current through DAT WT was barely detectable in NaCl, as reported elsewhere (40, 44), but increased dramatically upon sodium substitution to the more permeable lithium ion (Fig. 4B). The I312F and A559V variants did not differ from DAT WT in their leak conductance properties (Fig. 4B). T356M, however, displayed an apparently larger, but still small, cocaine-sensitive leak current at hyperpolarizing potentials in NaCl. Surprisingly, this leak current through T356M was not increased when sodium was replaced by lithium (Fig. 4B). As reported recently (25), D421N also showed dramatic changes in its leak conductance, evident as a large leak current in NaCl, which was similar to the leak current in LiCl both regarding size and reversal potential (Fig. 4B). Collectively, we find that the functionally impaired DAT variants display distinct and substantial alterations in their ion-binding properties and transporter-mediated currents.
Functionally impaired coding variants show altered regulation by Zn2+ and distinct conformational changes
Changes in substrate- and ion-binding properties are likely to associate with alterations in the conformational equilibrium, because efficient dopamine translocation rely on ordered binding and release of substrate and ions to induce conformational changes. The extracellular face of DAT has been shown to contain a Zn2+-binding site, through which Zn2+ and other transition metals such as Cu2+ and Ni2+ can exert allosteric modulation of DAT function (45–47). Regulation of DAT by Zn2+ has been proposed to involve a Zn2+-dependent bias toward the outward-facing conformation of DAT, and Zn2+ responses have therefore been applied to elucidate conformational changes (46, 48–51). Moreover, Zn2+ modulation of DAT function may serve physiological functions, as Zn2+ is packaged into synaptic vesicles by Zn2+ transporters and released as a cotransmitter upon synaptic transmission (52). Accordingly, we investigated the Zn2+-dependent regulation of dopamine uptake by the disease-associated DAT variants, in the presence of increasing Zn2+ concentrations. The variants without compromised dopamine uptake or ion binding i.e. A559V, G602E, and R615C, showed, as expected, a Zn2+-dependent inhibition of uptake, similar to that obtained for DAT WT (Fig. 5A). In marked contrast, the transport function of T356M was potentiated by Zn2+, in agreement with recent findings (53), whereas I312F demonstrated an interesting increase in Zn2+ sensitivity (Fig. 5A). We also observed a tendency toward increased Zn2+ sensitivity for D421N, but the low uptake by D421N only provided a very small specific signal, and the Zn2+ response did not reach a significant difference from DAT WT (Fig. 5A) (25, 54).
Figure 5.
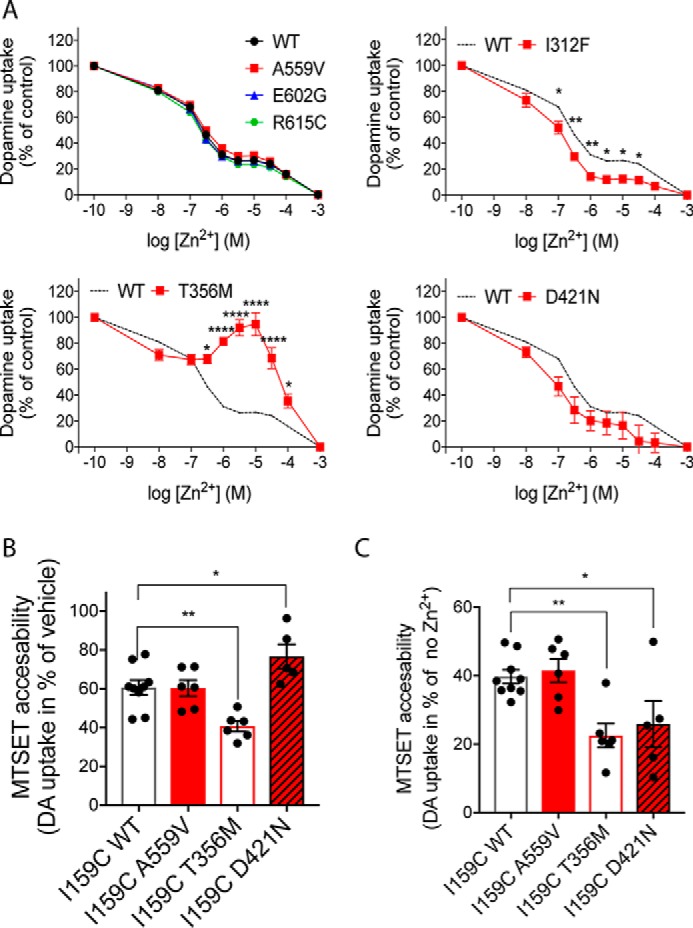
Conformational changes imposed by disease-associated variants. Conformational changes in disease-associated DAT variants were evaluated in transiently transfected COS-7 cells by assessing Zn2+ responses and MTSET accessibility. A, [3H]dopamine (DA) uptake was performed for 10 min (room temperature) in the presence of indicated Zn2+ concentrations. Data are presented as mean ± S.E. from three experiments, each performed in triplicates and normalized to uptake in the absence of Zn2+. *, p < 0.05; **, p < 0.01; ****, p < 0.0001, multiple t test with Holm-Sidak correction for multiple comparisons. B, MTSET accessibility assessed by treating COS-7 cells, expressing the indicated cysteine-engineered constructs, with vehicle or 0.5 mm MTSET prior to performing [3H]dopamine uptake. MTSET accessibility is reported as the MTSET-induced inhibition of uptake (in % of vehicle). E2C–I159C/T356M is more sensitive to MTSET than E2C–I159C–DAT WT, whereas E2C–I159C/D421N is protected from MTSET-induced inactivation. We could not obtain specific dopamine uptake from E2C–I159C/I312F even in absence of MTSET. C, Zn2+-imposed conformational changes in the variants were evaluated from MTSET-dependent inactivation of [3H]dopamine uptake following a 5-min treatment with 10 μm Zn2+ versus vehicle. Zn2+ increased MTSET accessibility for both E2C–I159C–DAT WT and coding variants, but the Zn2+-dependent increase in MTSET accessibility was significantly larger for E2C–I159C/T356M and E2C–I159C/D421N than for E2C–I159C–DAT WT. *, p < 0.05; **, p < 0.01 (n = 5–9, one-way ANOVA with Dunnett's post hoc test).
To obtain more insight into putative conformational alterations within I312F, T356M, and D421N, we undertook a substituted cysteine accessibility approach, where the membrane-impermeable and cysteine-reactive compound, MTSET, is used to probe the accessibility to cysteine-engineered constructs (48, 55). We also evaluated the A559V variant that has been associated with anomalous dopamine efflux, but otherwise shows transport properties similar to DAT WT. We first introduced I312F, T356M, D421N, and A559V (34), into a MTSET-insensitive background construct (DAT-E2C) (48), in which two endogenous cysteines has been removed (C90A and C306A). We confirmed that the variants did not alter the MTSET insensitivity of DAT-E2C (Fig. S1). Next, we introduced a cysteine at position 159, which allows MTSET to react with 159C when DAT adopts the outward conformation and thereby inhibits dopamine uptake (48). Evaluation of basal MTSET accessibility showed that E2C–I159C/T356M was significantly more sensitive to MTSET than E2C–I159C–DAT WT (residual dopamine uptake was 61 ± 4% for E2C–I159C–DAT WT versus 41 ± 3% for E2C–I159C/T356M, p < 0.01, Fig. 5B), suggesting that the conformational equilibrium of E2C–I159C/T356M is shifted toward the outward-facing state, making it more accessible for MTSET. In contrast, the E2C–I159C/D421N mutant showed an attenuated MTSET response (77 ± 6% residual uptake versus ∼61 ± 4% for E2C–I159C–DAT WT p < 0.01, Fig. 5B), consistent with a stabilized inward-facing conformation. Despite extensive efforts, we did not obtain specific dopamine uptake from E2C–I159C/I312F even in the absence of MTSET, indicating a synergistic disruptive effect of the I312F mutation in combination with I159C. The A559V mutant showed no changes in MTSET sensitivity compared with DAT WT, in agreement with the unaltered Zn2+ response.
To directly assess the structural effects that Zn2+ imposes on the different disease-associated variants, we evaluated MTSET accessibility following treatment with 10 μm Zn2+. As expected, Zn2+ increased the MTSET-dependent inhibition of DAT WT and A559V by ∼60%, compared with vehicle (Fig. 5C), consistent with a conformational shift in favor of the outward conformational state (48). Remarkably, Zn2+ produced even larger MTSET-dependent inhibition of dopamine uptake for both T356M (∼80%) and D421N (∼75%) even though these variants display opposite conformational preferences in the absence of Zn2+.
The molecular mechanisms underlying Zn2+-dependent rescue of T356M function are highly interesting and could provide important insight into molecular mechanisms of allosteric activators of DAT, which has been suggested to hold therapeutic potential (47, 53). To further elucidate the Zn2+-imposed changes in T356M function, we performed electrophysiological recordings on Xenopus oocytes expressing T356M or DAT WT in the presence or absence of Zn2+. Trace recordings of voltage-clamped oocytes showed that dopamine indeed elicits an aberrant outward current through T356M (Fig. 6A), as also indicated from the I-V curve of coupled currents (Fig. 4). Interestingly, subsequent perfusion with 10 μm Zn2+ established a large inward current through T356M, whereas DAT WT responded with a reduction in the inward current established by dopamine, conceivably reflecting substrate washout. This Zn2+-activated current through T356M was also observed without prior dopamine stimulation (Fig. 6A). Saturating concentrations of cocaine (100 μm) returned the steady-state current to baseline levels for DAT WT, while we observed a relative outward current for T356M that was similar in size to that observed for dopamine, supporting that both cocaine and dopamine inhibit an inward leak at negative potentials (see also Fig. 4).
Figure 6.
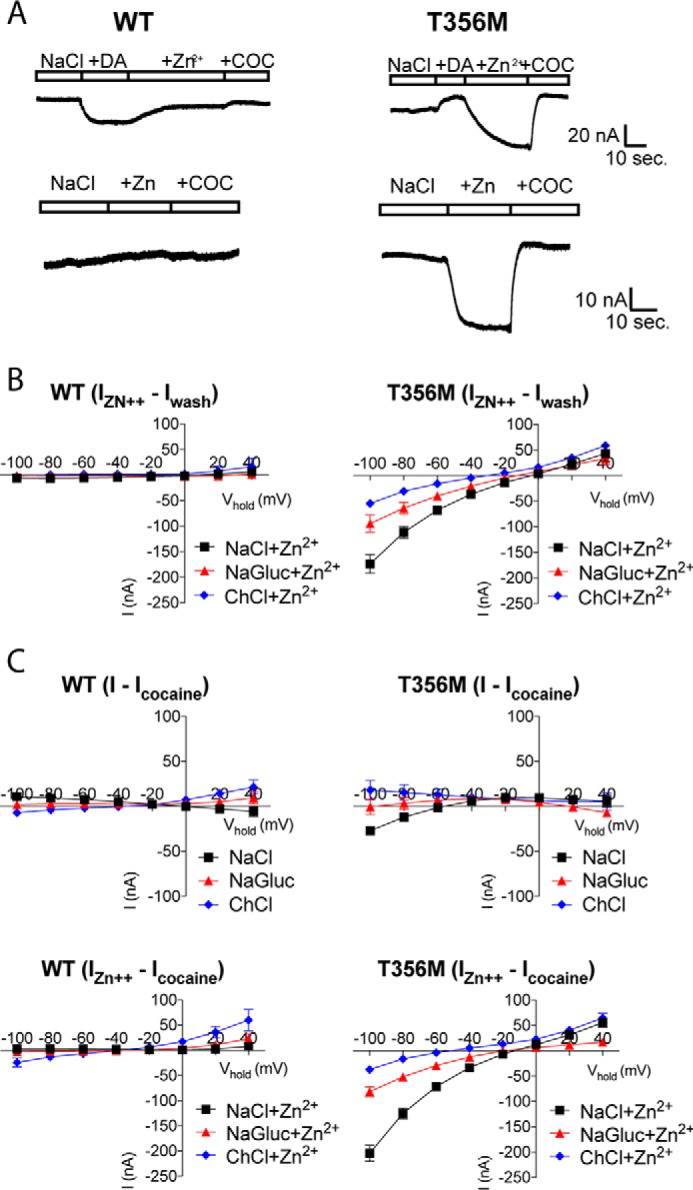
Zn2+ establishes a cation leak current through DAT T356M. A, trace recordings of voltage-clamped oocytes (at −60 mV), expressing DAT WT or T356M, following gravity perfusion with vehicle, 100 μm dopamine (DA), 10 μm Zn2+, and 100 μm cocaine (COC) (top panel) or with vehicle, 10 μm Zn2+, and 100 μm cocaine (bottom panel). Unlike DAT WT, T356M mediates an outward current in response to dopamine and cocaine, whereas Zn2+ generates a large inward current through T356M, which is observed both with and without prior stimulation with dopamine. Data are presented as representative traces. B, I-V recordings assessing the ion dependence of Zn2+-induced currents (IZn2+ − Iwash) through T356M. ChCl generates a clear left shift in reversal potential of the Zn2+-activated current through T356M, whereas sodium gluconate changes the size of the current, indicative of a chloride-modulated cation current. Data are presented as average curves (mean ± S.E.) of 4–8 experiments. C, I-V recordings assessing the ion dependence of cocaine-sensitive currents (I − Icocaine) through DAT WT and T356M, in the absence (top panels) and presence (bottom panels) of Zn2+. The Zn2+-induced currents (IZn2+ − Icocaine) through T356M are sensitive to cocaine, and ion substitutions confirm that the current is carried, at least in part, by sodium and modulated by chloride. Data are presented as average curves (mean ± S.E.) of 4–8 experiments.
We next explored the nature of the Zn2+-induced current (IZn2+ − Icontrol) through T356M by analysis of current-voltage relations with ion substitutions. The I-V plots revealed that in the case of T356M, Zn2+ induced an inward current at negative potentials with a reversal potential around −5 mV (Fig. 6B). Replacing sodium with choline generated a clear left shift in reversal potential of this Zn2+-activated current (reversal potential = ∼−30 mV), whereas chloride substitution for gluconate changed the size of the current with only a minor left shift in the reversal potential to approximately −10 mV (Fig. 6B). These data suggest that the Zn2+-dependent current through T356M is carried, at least in part, by sodium and that it can be modulated by chloride.
To ensure that all Zn2+-mediated currents were carried by DAT, we investigated the nature of the putative T356M leak current in the absence and presence of Zn2+ by analyzing cocaine subtracted I-V plots (IZn2+ − Icocaine). Although we did see an inward leak for T356M at negative potentials (<60 mV) in the absence of Zn2+, it was too small to reliably assess the consequence of ion substitution (Fig. 6C). In the presence of Zn2+, however, we observed a large current that was similar to that seen for IZn2+ − Icontrol with a reversal potential of approximately −10 mV. Importantly, ion substitutions revealed the same pattern with a left shift in reversal potential upon sodium substitution and reduced current without a prominent change in reversal potential upon chloride substitution (Fig. 6).
Discussion
Genetic variations that affect DAT function hold great potential to change dopaminergic homeostasis by disturbing the termination of dopamine signaling and the synthesis-independent supply of dopamine for vesicular storage (56, 57). Consistently, SLC6A3, encoding DAT, has been a long-standing candidate gene in psychiatric diseases, such as bipolar disorder, schizophrenia, and ADHD (22, 58–60), but the reported effect sizes of associated common variants are small, as for most other genes, and this challenges deduction of potential disease-relevant biological processes. Rare variants have been found to account for a significant component of the genetic architecture of several psychiatric disorders, and the association signal for both common and rare variants appears to be enriched in mutation-intolerant genes (1, 61–64). Indeed, DAT is classified as both loss-of-function-intolerant and missense-constrained (32). Here, we approached an understanding of DAT dysfunction in psychiatric disease from a rare variant perspective by performing a systematic characterization of six DAT missense variants, identified in patients suffering from psychiatric diseases (26–29).
We generally observed that PolyPhen-2 scores (38) correctly predicted functional deficits in dopamine uptake and that the variants' functional phenotypes correlated, at least to some extent, with their minor allele frequency. Accordingly, the extremely rare variants, I312F, T356M, and D421N, also had severe impairments in uptake kinetic parameters, whereas the most frequently occurring variants, A559V and E602E, mediated dopamine uptake comparable with DAT WT. The R615C variant, however, although extremely rare and predicted to have a high probability of damaging effects, did not display functional impairments in our hands. Of notice, the original characterization of R615C reported a significant reduction in dopamine uptake capacity in a stably transfected flip-in HEK-293 cell line (28). This difference in expression system may explain the discrepancy between the observed functional impact of R615C, as C-terminal mutations in DAT have been reported to be highly dependent on the model system (65).
The detrimental effects of mutations in DAT, however, may not arise only from impairments in dopamine uptake, but potentially also through gain of disruptive functions. For instance, the A559V variant, which was first identified in a patient with bipolar disorder and subsequently in both patients with ADHD and ASD (26, 27, 31), has been found to exhibit so-called anomalous dopamine efflux (ADE), i.e. a constitutive leak of cytoplasmic dopamine via DAT (34). Remarkably, the T356M and D421N variants also mediate ADE (25, 29), supporting that this phenomenon may have pathological implications. Of notice, neither I312F nor R615C mediate ADE (25, 28), and although we are not aware of any studies assessing ADE via E602G, one might expect that this variant shows WT-like properties as well, based on its localization in the C-terminal tail, where the R615C variant is also located. Thus, ADE is not a shared feature for all the disease-associated variants, but nevertheless, it is a remarkable gain-of-function phenotype that apparently can arise from very different mutations. Importantly, A559V knockin mice were recently found to display both behavioral and functional alteration, supporting a significant impact of ADE in vivo (66). Therefore, it is also highly interesting that the R615C variant has been found to undergo dysregulated trafficking with enhanced constitutive internalization and altered microdomain distribution (28, 67). This finding further supports that the potential pathological impact of altered DAT function is not necessarily recapitulated in direct assessment of dopamine uptake and that there might be facets of DAT function that require a native context to uncover. Our data suggest that changes in DAT ion conductances could be an underappreciated molecular mechanism through which a functionally impaired rare variant can perturb dopamine homeostasis. Additionally, our findings suggest that the structural and functional changes are unique to each transporter variant, as further discussed below.
The I312F variant was identified in a patient with ADHD and early-onset parkinsonism, who also carried the D421N mutation (25). We found that I312F has ∼50% residual uptake capacity without changes in Bmax, confirming previous data (25). Interestingly, we also found that I312F displays lower apparent affinity for substrates (dopamine and AMPH), whereas methylphenidate and cocaine, which are blockers rather than substrates, had significantly higher apparent affinity for I312F, compared with DAT WT. These data hint that the I312F mutation may compromise the catalytic activity by favoring the outward open state, which concomitantly might facilitate the interaction with inhibitors (21, 68–70). This is supported by the observation that I312F also exhibited increased sensitivity to Zn2+, which stabilizes the outward conformation of DAT as well (21). Unfortunately, direct assessment of MTSET accessibility to a cysteine-engineered I312F construct could not be conducted, as introducing the MTSET-reactive cysteine (I159C) abolished all transport activity in the I312F E2C background construct. Assessment of the ion dependence of dopamine uptake and characterization of electrophysiological properties for I312F furthermore uncovered an ∼7-fold reduction in apparent chloride affinity and a remarkably larger substrate-induced current in NaSCN, consistent with an enhanced uncoupled anion conductance. Importantly, the uncoupled anion current through DAT WT has been suggested previously to regulate tonic activity of dopaminergic neurons (42). It is therefore intriguing to speculate that the I312F mutation not only compromises dopamine homeostasis through impaired catalytic efficiency, but also mediates a “disruptive function” through a larger uncoupled anion conductance that potentially influences the neuronal excitability.
The D421N variant, localized in the Na2 site, was identified together with the I312F variant and presumably arose from a de novo mutation (25). Our characterization of D421N showed a dramatic reduction in dopamine uptake capacity and a marked loss in apparent affinity for both substrates and inhibitors of DAT, consistent with previous findings (25). Moreover, D421N did not only exhibit impairments in sodium binding (25), but also a marked loss in apparent chloride affinity at physiological sodium concentrations, supporting a functional coupling between the Na2 site and the chloride-binding site (41). Of notice, sodium and chloride have been found to mutually increase the binding affinity of one another in the serotonin transporter (71). Hence, it is possible that this loss of apparent chloride affinity is a secondary consequence of the apparent sodium affinity loss, which becomes obscured, as the experiment cannot be carried out under saturating sodium concentrations. As observed for I312F, the marked changes in ion coordination by D421N were associated with altered electrophysiological properties. D421N mediated a large constitutive leak current in NaCl, which was similar in size and reversal potential to the leak current recorded in LiCl. Ion substitution experiments have found that this leak current is carried in part by sodium and modulated by chloride (25, 41). Our I/V recordings furthermore showed a small outward current at negative potentials in the presence of dopamine, as opposed to the inward current recorded for DAT WT. Because dopamine is known to block the constitutive cation leak current in DAT WT (40), this aberrant outward current through D421N likely reflects that the cation leak current is relatively larger than the substrate-induced inward current. The alternative possibility is that dopamine elicits an outward conductance that is distinct from the leak current and carried by K+ and/or Cl−. However, a large contribution from these ions to the current observed at hyperpolarized potentials seems unlikely.
Interestingly, the dopamine-evoked outward current through D421N became markedly larger when chloride was substituted for the more permeable thiocyanate, presumably reflecting that thiocyanate increases the cation leak current through D421N (41). The dramatic functional changes that we observed for D421N were accompanied by reduced MTSET accessibility, consistent with a conformational shift toward the inward-facing state. This aligns with the general understanding that weakening of the Na2 site promotes the inward-facing transporter conformation (54, 72). Even though Zn2+ effectively promoted a shift in conformational equilibrium toward the outward-facing state, we did not observe a Zn2+-dependent activation of D421N, as reported for some inward-facing mutants (49). Collectively, the present data support that the D421N mutation compromises both sodium- and chloride-binding properties, which likely underlie a conformational change toward an inward-facing conformation, as well as impaired ligand-binding properties. Moreover, the transporter gains a large dissipating cation leak current that could be envisioned to influence the excitability of dopaminergic neurons, as suggested for the uncoupled anion conductance (42). Aside from the functional impairments assessed in this study, D421N also mediates ADE (25), which likely imposes further dopaminergic dyshomeostasis and provides further support for the importance of gain-of-function phenotypes.
The T356M variant, in TM7, was identified as the first DAT de novo mutation in a patient suffering from autism spectrum disorder and was shown to demonstrate impaired dopamine uptake and cocaine-sensitive ADE (29, 73). We also observed a reduction in uptake capacity for T356M of ∼60%, along with lower (2–5-fold) apparent affinities for substrates, i.e. dopamine and AMPH. Interestingly, methylphenidate and cocaine had EC50 values that were ∼150-fold, respectively, ∼40-fold higher for T356M than for DAT WT, suggesting that Thr-356 might be particularly important for inhibitor binding. Our assessment of ion coordination furthermore uncovered that compromised sodium, but not chloride, binding may be underlying the functional impairment of T356M. Although T356M had around 40% residual uptake capacity, current-voltage recordings of dopamine-induced conductances identified a net outward current at negative membrane potentials, rather than the expected inward current seen for DAT WT. As for D421N, this outward current might reflect inhibition of a constitutive leak current by dopamine. I/V recordings in the presence of cocaine indeed showed a larger leak current through T356M. Voltage-clamped trace recordings furthermore uncovered that both dopamine and cocaine promoted a net outward current through T356M that was similar in size, supporting that the dopamine-associated outward current is not distinct from the cocaine-sensitive leak current, as discussed previously for D421N. Remarkably, sodium substitution for lithium did not enhance the leak current through T356M, as seen for DAT WT. Instead, the leak current through T356M disappeared in LiCl, suggesting that the cation leak permeation pathway is markedly different from that of both DAT WT and of the disease-associated mutant D421N.
Our assessment of structural changes in T356M showed, in agreement with recent findings (53), that Zn2+ partly rescues uptake function of T356M. A similar Zn2+-dependent potentiation has been demonstrated for inward-facing DAT mutants and has been suggested to reflect a partial restoration of the conformational equilibrium by promoting an outward-facing conformation (48, 49). However, our MTSET accessibility assessment showed enhanced MTSET-dependent inactivation of T356M, suggesting that T356M is more prone to adopt an outward-facing conformation, as also proposed for the equivalent mutation in the prokaryotic transporter, LeuT (29). We also found that Zn2+ increased MTSET accessibility significantly more for T356M than for DAT WT, suggesting that a larger faction of T356M adopts the outward-facing state in the presence of Zn2+. Promotion of the outward-facing state does not seem to define the potentiating effect of Zn2+ per se, because Zn2+ elicits inhibitory effects on both DAT WT and D421N even though the MTSET data show that they too are stabilized in the outward conformation by Zn2+. Interestingly, the affinity for dopamine has been reported to be inversely related to the affinity for Zn2+ (46, 47). It is possible that the loss of apparent dopamine affinity for T356M and D421N explains the increased ability of Zn2+ to stabilize their outward conformation. The unique properties of T356M in the presence of Zn2+ were further substantiated by electrophysiological recordings showing a major Zn2+-induced inward current that, at least in part, is carried by sodium and may represent a potentiation of the cocaine-sensitive leak seen in the absence of Zn2+. Notably, this presumed cation conductance is strikingly different from the previously reported Zn2+-induced anion current seen upon mutation of the intracellular gate residue, Tyr-335 (43), further underlining the distinct phenotypic properties of the disease-associated T356M mutation.
Anomalous efflux of dopamine is a remarkable gain-of-function phenotype shared by T356M, D421N, and A559V. ADE was first described for A559V, and it was suggested to be an underlying mechanism through which DAT can impact the risk of psychiatric disease (34). An interesting question is whether the ADE phenotype is supported by distinct common structural changes. Our characterization of MTSET availability and Zn2+-induced conformational changes, however, suggests that this is not the case. By contrast, we found that the conformational equilibrium of T356M is shifted toward an outward-facing state, whereas D421N is inward-facing, and we did not find evidence supporting structural changes for A559V. It is interesting, however, that both T356M and D421N display impaired sodium binding and mediate a cation leak current in NaCl, and it is tempting to speculate that these common changes in electrophysiological properties may be directly linked to ADE in these functionally impaired variants.
Translation of genetic information into clinically applicable knowledge depends in large part on the well-established genotype–phenotype associations (74). One source of phenotypic variation is the distinctive effect of different mutations in the same gene that may have diverse functional outcomes, depending on the nature and severity of the mutation. Our characterization shows that the functionally impaired DAT variants all exhibited unique changes in ligand-binding properties, ion dependence, and conformational equilibrium, and identifies aberrant ion conductances as a potential underlying mechanism through which functionally impaired DAT variants could exert disruptive functions.
Experimental procedures
Molecular biology and cell culturing
The QuickChange site-directed mutagenesis kit (Stratagene) was used to introduce the mutations (I312F, T356M, D421N, A559V, E602G, and R615C) in a pRC/CMV hDAT background. All constructs were verified by DNA sequencing. COS-7 were grown in a 10% CO2 humidified incubator at 37 °C in Dulbecco's modified Eagle's medium, supplemented with 2 mm glutamine, 10% fetal bovine serum, and 1% penicillin/streptomycin. Transient transfections were performed with Lipofectamine 2000 (Invitrogen), using reagent/DNA ratio of 3:1. Assays were conducted 48 h after transfection.
PolyPhen-2 scores
The probability that the respective amino acid substitutions impose damaging effects on DAT function was evaluated using the polymorphism phenotyping version 2 software available online (38).
[3H]Dopamine uptake
Saturation dopamine uptake experiments were carried out as described previously (25). COS-7 cells were transiently transfected and seeded into polyornithine-coated 24-well plates. Approximately 48 h after transfection, cells were washed in uptake buffer (25 mm HEPES, 120 mm NaCl, 5 mm KCl, 1.2 mm CaCl2, and 1.2 mm MgSO4 supplemented with 5 mm d-glucose, 1 mm ascorbic acid, and 1 μm catechol-O-methyltransferase inhibitor Ro 41-0960, pH 7.4) and allowed to equilibrate at room temperature for 10 min. Dopamine uptake was initiated by adding a serial dilution row, containing a mix of 2,5,6-[3H]dopamine (PerkinElmer Life Sciences) and unlabeled dopamine (Sigma) with final dopamine concentrations of 6.4, 3.2, 1.6, 0.8, 0.4, 0.2, 0.1, and 0.05 μm. Dopamine uptake was terminated after 5 min (room temperature), or 10 min in the case of D421N, by washing in ice-cold uptake buffer. Nonspecific binding was determined using mock-transfected cells.
Sodium and chloride dose-response curves were obtained by equimolar titration of 200 mm NaCl in substitution buffer (250 mm Tris/HEPES, 50 mm potassium gluconate, 12 mm magnesium gluconate, 12 mm calcium gluconate, pH 7.4) against choline chloride and sodium gluconate to obtain final Na+ and Cl− concentrations of 200, 130, 80, 40, 20, 10, 5, and 0 mm. These solutions were supplemented with 5 mm d-glucose, 1 mm ascorbic acid, and 1 μm catechol-O-methyltransferase inhibitor. Before initiating dopamine uptake, transfected COS-7 cells were washed and equilibrated for 10 min in the denoted ion buffers. [3H]Dopamine uptake was carried out for 5 min (WT, A559V, E602G, and R615C) or 10 min for the functionally impaired variants (I312F, T356M, and D421N), using a 6.4 μm mixture of unlabeled dopamine and [3H]dopamine. Uptake was terminated by washing twice in ice-cold wash buffer. Nonspecific binding was determined using mock-transfected cells.
To evaluate Zn2+ regulation of dopamine uptake, Zn2+ was added at the indicated concentrations 5 min before uptake was initiated by addition of [3H]dopamine (1:200 dilution, ∼10 nm). Uptake was terminated after 10 min at room temperature by washing in ice-cold uptake buffer. Nonspecific binding was determined in the presence of 100 μm nomifensine.
MTSET accessibility
Changes in the conformational equilibria of I312F, T356M, D421N, and A559V were investigated in a substituted cysteine accessibility assay, as described previously (48, 55). Briefly, I312F, D421N, T356M, or A559V mutations were introduced into a MTSET-insensitive construct, DAT-E2C, in which endogenous cysteines at position Cys-90 and Cys-306 were substituted by alanine, as well as into a DAT-E2C I159C background, which is sensitive to inactivation by the membrane-impermeable cysteine-reactive agent, MTSET, in a conformation-dependent manner (48). Transiently transfected COS-7 cells were washed in uptake buffer and incubated for 10 min with 0.5 mm freshly prepared MTSET or vehicle. Cells were washed twice in uptake buffer before dopamine uptake (10 min, room temperature with 1:200 diluted [3H]dopamine, ∼10 nm) was carried out as described above. The effect of Zn2+ (10 μm) was evaluated by incubating cells 5 min with 10 μm Zn2+ prior to the MTSET treatment.
[3H]CFT binding
DAT-binding capacity and affinity were quantified by [3H]CFT/CFT competition binding, as described previously (25). Briefly, transiently transfected COS-7 cells, expressing either DAT WT or one of the disease-associated mutants, were seeded in 24 wells. Cells were washed once in ice-cold uptake buffer. Unlabeled CFT was added in the indicated concentrations, followed by addition of [3H]CFT (200-fold diluted, 76.6 Ci/mmol (PerkinElmer Life Sciences), ∼5 nm), after which cells were incubated ∼100 min at 5 °C to allow binding equilibrium. To terminate the assay, the cells were washed twice in ice-cold binding buffer.
Following all uptake and binding experiments, cells were lysed in 1% SDS and transferred to 24-well counting plates (PerkinElmer Life Sciences), and radioactivity was measured with a Wallac MicroBeta® TriLux Liquid Scintillation Counter (PerkinElmer Life Sciences).
Electrophysiology on oocytes
I-V curves and voltage-clamped trace recordings were generated as described previously (25, 41). WT hDAT or mutant constructs were expressed in oocytes using the pFROG vector (25, 75). Following linearization, cRNA was transcribed using the mMESSAGEmMACHINE kit (Ambion), and defoliculated Xenopus oocytes (Ecocide Bioscience) were injected with the purified RNA. Oocytes were maintained in KULORI buffer (88 mm NaCl, 1 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 5 mm HEPES/Tris, pH 7.4) at 18 °C. DAT-associated currents were measured using a conventional two-electrode voltage clamp. The oocytes were penetrated by two micropipettes, backfilled with 2 m KCl, and voltage-clamped using a Dagan CA-1B clamp (Dagan Corp.). The membrane potential was measured relative to an extracellular Ag/AgCl electrode that was connected to the recording chamber via a 2% agarose, 2 m KCl bridge. Recordings were digitized using Digidata 1440A (Molecular Devices) controlled with pClamp 10 (Axon Instruments). I-V curves were recorded by clamping the oocytes at potentials between −100 and +40 mV, using 20-mV incremental steps (250 ms) and returning to a holding potential of −60 mV before each step. For trace recordings, oocytes were voltage-clamped at −60 mV. Recording buffers were applied by gravity perfusion, using a standard NaCl solution (130 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 1 mm HEPES/Tris, pH 7.4) as reference. All ion-substituted buffers were kept isotonic. The substrate-induced current was defined as INaCl, DA − INaCl, wash, and cocaine-sensitive currents were derived as I − Icocaine. Unless otherwise stated, dopamine and cocaine were applied at 100 μm.
Quantification and statistics
GraphPad Prism 7.0 software (GraphPad Software, San Diego) was applied for statistical analysis and data fitting. Saturation uptake data were fitted by Michaelis-Menten kinetics to obtain uptake kinetic parameters, Km and Vmax. Competition uptake and binding experiments were fitted by nonlinear regression with variable slope to derive pIC50 values. Ion dependence uptake curves were fitted by a one-site–specific binding model to determine Ki. Statistical analyses were performed on the pIC50 values, and the mean pIC50 values and S.E. were applied to calculate the IC50 values and S.E. interval. Unless otherwise noted, ordinary one-way ANOVA with Dunnett's post test was applied to analyze for statistical significance.
Author contributions
F. H., T. F. A., and U. G. conceptualization; F. H., T. F. A., and F. B. data curation; F. H., T. F. A., C. J. L., and U. G. formal analysis; F. H., C. J. L., and U. G. supervision; F. H. and U. G. funding acquisition; F. H., F. B., and T. F. A. investigation; F. H., T. F. A., and F. B. methodology; F. H. and U. G. writing-original draft; F. H. and U. G. project administration; F. H., T. F. A., F. B., C. J. L., and U. G. writing-review and editing; F. B. validation; U. G. resources.
Supplementary Material
Acknowledgments
We gratefully acknowledge Pernille Emilie Petersen and Anette Denker Kaas for excellent technical support.
This work was supported by Danish Council for Independent Research–Medical Sciences Grants DFF-4183-00571 (to F. H.), DFF 4004-00097B (to U. G.), Lundbeck Foundation R181-2014-3090 (to F. H.), National Institutes of Health Grant P01 DA 12408, and the Novo Nordisk Foundation NNF16OC0023104. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- ADHD
- attention deficit hyperactivity disorder
- DAT
- dopamine transporter
- ASD
- autism spectrum disorder
- ANOVA
- analysis of variance
- CFT
- 2β-carbomethoxy-3β-(4-fluorophenyl)tropane
- MTSET
- [2-(trimethylammonium)ethyl]-methanethiosulfonate
- TM
- transmembrane domain
- SCN
- thiocyanate
- ADE
- anomalous dopamine efflux
- DA
- dopamine
- hDAT
- human DAT.
References
- 1. Sullivan P. F., Daly M. J., and O'Donovan M. (2012) Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551 10.1038/nrg3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Del Campo N., Chamberlain S. R., Sahakian B. J., and Robbins T. W. (2011) The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol. Psychiatry 69, e145–e157 10.1016/j.biopsych.2011.02.036 [DOI] [PubMed] [Google Scholar]
- 3. Dichter G. S., Damiano C. A., and Allen J. A. (2012) Reward circuitry dysfunction in psychiatric and neurodevelopmental disorders and genetic syndromes: animal models and clinical findings. J. Neurodev. Disord. 4, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cousins D. A., Butts K., and Young A. H. (2009) The role of dopamine in bipolar disorder. Bipolar Disord. 11, 787–806 10.1111/j.1399-5618.2009.00760.x [DOI] [PubMed] [Google Scholar]
- 5. Iversen S. D., and Iversen L. L. (2007) Dopamine: 50 years in perspective. Trends Neurosci. 30, 188–193 10.1016/j.tins.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 6. Owen M. J., Sawa A., and Mortensen P. B. (2016) Schizophrenia. Lancet 388, 86–97 10.1016/S0140-6736(15)01121-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howes O. D., and Kapur S. (2009) The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr. Bull. 35, 549–562 10.1093/schbul/sbp006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wray N. R., Lee S. H., Mehta D., Vinkhuyzen A. A., Dudbridge F., and Middeldorp C. M. (2014) Research review: polygenic methods and their application to psychiatric traits. J. Child Psychol. Psychiatry 55, 1068–1087 10.1111/jcpp.12295 [DOI] [PubMed] [Google Scholar]
- 9. Iossifov I., O'Roak B. J., Sanders S. J., Ronemus M., Krumm N., Levy D., Stessman H. A., Witherspoon K. T., Vives L., Patterson K. E., Smith J. D., Paeper B., Nickerson D. A., Dea J., Dong S., et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lai M.-C., Lombardo M. V., and Baron-Cohen S. (2014) Autism. Lancet 383, 896–910 10.1016/S0140-6736(13)61539-1 [DOI] [PubMed] [Google Scholar]
- 11. Martin J., O'Donovan M. C., Thapar A., Langley K., and Williams N. (2015) The relative contribution of common and rare genetic variants to ADHD. Transl. Psychiatry 5, e506 10.1038/tp.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gratten J., Wray N. R., Keller M. C., and Visscher P. M. (2014) Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat. Neurosci. 17, 782–790 10.1038/nn.3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cross-Disorder Group of the Psychiatric Genomics Consortium. (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee S. H., Ripke S., Neale B. M., Faraone S. V., Purcell S. M., Perlis R. H., Mowry B. J., Thapar A., Goddard M. E., Witte J. S., Absher D., Agartz I., Akil H., Amin F., et al. (2013) Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 10.1038/ng.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fromer M., Pocklington A. J., Kavanagh D. H., Williams H. J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D. M., Carrera N., Humphreys I., Johnson J. S., Roussos P., Barker D. D., et al. (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams N. M., Franke B., Mick E., Anney R. J., Freitag C. M., Gill M., Thapar A., O'Donovan M. C., Owen M. J., Holmans P., Kent L., Middleton F., Zhang-James Y., Liu L., Meyer J., et al. (2012) Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am. J. Psychiatry 169, 195–204 10.1176/appi.ajp.2011.11060822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robinson E. B., St Pourcain B., Anttila V., Kosmicki J. A., Bulik-Sullivan B., Grove J., Maller J., Samocha K. E., Sanders S. J., Ripke S., Martin J., Hollegaard M. V., Werge T., Hougaard D. M., et al. (2016) Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet. 48, 552–555 10.1038/ng.3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanders S. J., He X., Willsey A. J., Ercan-Sencicek A. G., Samocha K. E., Cicek A. E., Murtha M. T., Bal V. H., Bishop S. L., Dong S., Goldberg A. P., Jinlu C., Keaney J. F 3rd, Klei L., Mandell J. D., et al. (2015) Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 10.1016/j.neuron.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., and Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 10.1124/pr.108.000869 [DOI] [PubMed] [Google Scholar]
- 20. Gainetdinov R. R. (2008) Dopamine transporter mutant mice in experimental neuropharmacology. Naunyn-Schmiedeberg's Arch. Pharmacol. 377, 301–313 [DOI] [PubMed] [Google Scholar]
- 21. Schmitt K. C., Rothman R. B., and Reith M. E. (2013) Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J. Pharmacol. Exp. Ther. 346, 2–10 10.1124/jpet.111.191056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gizer I. R., Ficks C., and Waldman I. D. (2009) Candidate gene studies of ADHD: a meta-analytic review. Hum. Genet. 126, 51–90 10.1007/s00439-009-0694-x [DOI] [PubMed] [Google Scholar]
- 23. Reynolds G. P., McGowan O. O., and Dalton C. F. (2014) Pharmacogenomics in psychiatry: the relevance of receptor and transporter polymorphisms. Br. J. Clin. Pharmacol. 77, 654–672 10.1111/bcp.12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Opmeer E. M., Kortekaas R., and Aleman A. (2010) Depression and the role of genes involved in dopamine metabolism and signalling. Prog. Neurobiol. 92, 112–133 10.1016/j.pneurobio.2010.06.003 [DOI] [PubMed] [Google Scholar]
- 25. Hansen F. H., Skjørringe T., Yasmeen S., Arends N. V., Sahai M. A., Erreger K., Andreassen T. F., Holy M., Hamilton P. J., Neergheen V., Karlsborg M., Newman A. H., Pope S., Heales S. J., Friberg L., et al. (2014) Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J. Clin. Invest. 124, 3107–3120 10.1172/JCI73778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grünhage F., Schulze T. G., Müller D. J., Lanczik M., Franzek E., Albus M., Borrmann-Hassenbach M., Knapp M., Cichon S., Maier W., Rietschel M., Propping P., and Nöthen M. M. (2000) Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1). Mol. Psychiatry 5, 275–282 10.1038/sj.mp.4000711 [DOI] [PubMed] [Google Scholar]
- 27. Mazei-Robison M. S., Couch R. S., Shelton R. C., Stein M. A., and Blakely R. D. (2005) Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology 49, 724–736 10.1016/j.neuropharm.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 28. Sakrikar D., Mazei-Robison M. S., Mergy M. A., Richtand N. W., Han Q., Hamilton P. J., Bowton E., Galli A., Veenstra-Vanderweele J., Gill M., and Blakely R. D. (2012) Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J. Neurosci. 32, 5385–5397 10.1523/JNEUROSCI.6033-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamilton P. J., Campbell N. G., Sharma S., Erreger K., Herborg Hansen F., Saunders C., Belovich A. N., NIH ARRA Autism Sequencing Consortium, Sahai M. A., Cook E. H., Gether U., McHaourab H. S., Matthies H. J., Sutcliffe J. S., and Galli A. (2013) De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 18, 1315–1323 10.1038/mp.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cartier E., Hamilton P. J., Belovich A. N., Shekar A., Campbell N. G., Saunders C., Andreassen T. F., Gether U., Veenstra-Vanderweele J., Sutcliffe J. S., Ulery-Reynolds P. G., Erreger K., Matthies H. J., and Galli A. (2015) Rare autism-associated variants implicate syntaxin 1 (STX1 R26Q) phosphorylation and the dopamine transporter (hDAT R51W) in dopamine neurotransmission and behaviors. EBioMedicine 2, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bowton E., Saunders C., Reddy I. A., Campbell N. G., Hamilton P. J., Henry L. K., Coon H., Sakrikar D., Veenstra-VanderWeele J. M., Blakely R. D., Sutcliffe J., Matthies H. J., Erreger K., and Galli A. (2014) SLC6A3 coding variant Ala559Val found in two autism probands alters dopamine transporter function and trafficking. Transl. Psychiatry 4, e464 10.1038/tp.2014.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O'Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geschwind D. H., and Flint J. (2015) Genetics and genomics of psychiatric disease. Science 349, 1489–1494 10.1126/science.aaa8954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mazei-Robison M. S., Bowton E., Holy M., Schmudermaier M., Freissmuth M., Sitte H. H., Galli A., and Blakely R. D. (2008) Anomalous dopamine release associated with a human dopamine transporter coding variant. J. Neurosci. 28, 7040–7046 10.1523/JNEUROSCI.0473-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowton E., Saunders C., Erreger K., Sakrikar D., Matthies H. J., Sen N., Jessen T., Colbran R. J., Caron M. G., Javitch J. A., Blakely R. D., and Galli A. (2010) Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J. Neurosci. 30, 6048–6057 10.1523/JNEUROSCI.5094-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ng J., Heales S. J., and Kurian M. A. (2014) Clinical features and pharmacotherapy of childhood monoamine neurotransmitter disorders. Paediatr. Drugs 16, 275–291 10.1007/s40272-014-0079-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurian M. A., Li Y., Zhen J., Meyer E., Hai N., Christen H. J., Hoffmann G. F., Jardine P., von Moers A., Mordekar S. R., O'Callaghan F., Wassmer E., Wraige E., Dietrich C., Lewis T., et al. (2011) Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 10, 54–62 10.1016/S1474-4422(10)70269-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., and Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mazei-Robison M. S., and Blakely R. D. (2005) Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology 49, 737–749 10.1016/j.neuropharm.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 40. Sonders M. S., Zhu S. J., Zahniser N. R., Kavanaugh M. P., and Amara S. G. (1997) Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J. Neurosci. 17, 960–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borre L., Andreassen T. F., Shi L., Weinstein H., and Gether U. (2014) The second sodium site in the dopamine transporter controls cation permeation and is regulated by chloride. J. Biol. Chem. 289, 25764–25773 10.1074/jbc.M114.574269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ingram S. L., Prasad B. M., and Amara S. G. (2002) Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 5, 971–978 10.1038/nn920 [DOI] [PubMed] [Google Scholar]
- 43. Meinild A. K., Sitte H. H., and Gether U. (2004) Zinc potentiates an uncoupled anion conductance associated with the dopamine transporter. J. Biol. Chem. 279, 49671–49679 10.1074/jbc.M407660200 [DOI] [PubMed] [Google Scholar]
- 44. Sulzer D., Sonders M. S., Poulsen N. W., and Galli A. (2005) Mechanisms of neurotransmitter release by amphetamines: a review. Prog. Neurobiol. 75, 406–433 10.1016/j.pneurobio.2005.04.003 [DOI] [PubMed] [Google Scholar]
- 45. Norregaard L., Frederiksen D., Nielsen E. O., and Gether U. (1998) Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 17, 4266–4273 10.1093/emboj/17.15.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Y., Hasenhuetl P. S., Schicker K., Sitte H. H., Freissmuth M., and Sandtner W. (2015) Dual action of Zn2+ on the transport cycle of the dopamine transporter. J. Biol. Chem. 290, 31069–31076 10.1074/jbc.M115.688275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Y., Mayer F. P., Hasenhuetl P. S., Burtscher V., Schicker K., Sitte H. H., Freissmuth M., and Sandtner W. (2017) Occupancy of the zinc-binding site by transition metals decreases the substrate affinity of the human dopamine transporter by an allosteric mechanism. J. Biol. Chem. 292, 4235–4243 10.1074/jbc.M116.760140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loland C. J., Grånäs C., Javitch J. A., and Gether U. (2004) Identification of intracellular residues in the dopamine transporter critical for regulation of transporter conformation and cocaine binding. J. Biol. Chem. 279, 3228–3238 10.1074/jbc.M304755200 [DOI] [PubMed] [Google Scholar]
- 49. Loland C. J., Norregaard L., Litman T., and Gether U. (2002) Generation of an activating Zn2+ switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transport cycle. Proc. Natl. Acad. Sci. U.S.A. 99, 1683–1688 10.1073/pnas.032386299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmitt K. C., and Reith M. E. (2011) The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS One 6, e25790 10.1371/journal.pone.0025790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hong W. C., and Amara S. G. (2010) Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J. Biol. Chem. 285, 32616–32626 10.1074/jbc.M110.150565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sensi S. L., Paoletti P., Bush A. I., and Sekler I. (2009) Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10, 780–791 10.1038/nrn2734 [DOI] [PubMed] [Google Scholar]
- 53. Hamilton P. J., Shekar A., Belovich A. N., Christianson N. B., Campbell N. G., Sutcliffe J. S., Galli A., Matthies H. J., and Erreger K. (2015) Zn2+ reverses functional deficits in a de novo dopamine transporter variant associated with autism spectrum disorder. Mol. Autism 6, 8 10.1186/s13229-015-0002-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krishnamurthy H., and Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 10.1038/nature10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen J. G., and Rudnick G. (2000) Permeation and gating residues in serotonin transporter. Proc. Natl. Acad. Sci. U.S.A. 97, 1044–1049 10.1073/pnas.97.3.1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pereira D. B., and Sulzer D. (2012) Mechanisms of dopamine quantal size regulation. Front. Biosci. 17, 2740–2767 10.2741/4083 [DOI] [PubMed] [Google Scholar]
- 57. Jones S. R., Gainetdinov R. R., Jaber M., Giros B., Wightman R. M., and Caron M. G. (1998) Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc. Natl. Acad. Sci. U.S.A. 95, 4029–4034 10.1073/pnas.95.7.4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mick E., and Faraone S. V. (2009) Family and genetic association studies of bipolar disorder in children. Child Adolesc. Psychiatr. Clin. N. Am. 18, 441–453 10.1016/j.chc.2008.11.008 [DOI] [PubMed] [Google Scholar]
- 59. Serretti A., and Mandelli L. (2008) The genetics of bipolar disorder: genome 'hot regions,' genes, new potential candidates and future directions. Mol. Psychiatry 13, 742–771 10.1038/mp.2008.29 [DOI] [PubMed] [Google Scholar]
- 60. Sáiz P. A., García-Portilla M. P., Arango C., Morales B., Arias B., Corcoran P., Fernández J. M., Alvarez V., Coto E., Bascarán M. T., Bousoño M., Fañanas L., and Bobes J. (2010) Genetic polymorphisms in the dopamine-2 receptor (DRD2), dopamine-3 receptor (DRD3), and dopamine transporter (SLC6A3) genes in schizophrenia: data from an association study. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 26–31 10.1016/j.pnpbp.2009.09.008 [DOI] [PubMed] [Google Scholar]
- 61. Pardiñas A. F., Holmans P., Pocklington A. J., Escott-Price V., Ripke S., Carrera N., Legge S. E., Bishop S., Cameron D., Hamshere M. L., Han J., Hubbard L., Lynham A., Mantripragada K., Rees E., et al. (2016) Common schizophrenia alleles are enriched in mutation-intolerant genes and maintained by background selection. BioRxiv 10.1101/068593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh T., Walters J. T. R., Johnstone M., Curtis D., Suvisaari J., Torniainen M., Rees E., Iyegbe C., Blackwood D., McIntosh A. M., Kirov G., Geschwind D., Murray R. M., Di Forti M., Bramon E., et al. (2017) The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 49, 1167–1173 10.1038/ng.3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Samocha K. E., Robinson E. B., Sanders S. J., Stevens C., Sabo A., McGrath L. M., Kosmicki J. A., Rehnström K., Mallick S., Kirby A., Wall D. P., MacArthur D. G., Gabriel S. B., DePristo M., Purcell S. M., et al. (2014) A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 10.1038/ng.3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bustamante M. L., Herrera L., Gaspar P. A., Nieto R., Maturana A., Villar M. J., Salinas V., and Silva H. (2017) Shifting the focus toward rare variants in schizophrenia to close the gap from genotype to phenotype. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 663–670 10.1002/ajmg.b.32550 [DOI] [PubMed] [Google Scholar]
- 65. Rickhag M., Hansen F. H., Sørensen G., Strandfelt K. N., Andresen B., Gotfryd K., Madsen K. L., Vestergaard-Klewe I., Ammendrup-Johnsen I., Eriksen J., Newman A. H., Füchtbauer E. M., Gomeza J., Woldbye D. P., Wörtwein G., et al. (2013) A C-terminal PDZ domain-binding sequence is required for striatal distribution of the dopamine transporter. Nat. Commun. 4, 1580 10.1038/ncomms2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mergy M. A., Gowrishankar R., Davis G. L., Jessen T. N., Wright J., Stanwood G. D., Hahn M. K., and Blakely R. D. (2014) Genetic targeting of the amphetamine and methylphenidate-sensitive dopamine transporter: on the path to an animal model of attention-deficit hyperactivity disorder. Neurochem. Int. 73, 56–70 10.1016/j.neuint.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kovtun O., Sakrikar D., Tomlinson I. D., Chang J. C., Arzeta-Ferrer X., Blakely R. D., and Rosenthal S. J. (2015) Single-quantum-dot tracking reveals altered membrane dynamics of an attention-deficit/hyperactivity-disorder-derived dopamine transporter coding variant. ACS Chem. Neurosci. 6, 526–534 10.1021/cn500202c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang K. H., Penmatsa A., and Gouaux E. (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327 10.1038/nature14431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Beuming T., Kniazeff J., Bergmann M. L., Shi L., Gracia L., Raniszewska K., Newman A. H., Javitch J. A., Weinstein H., Gether U., and Loland C. J. (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 11, 780–789 10.1038/nn.2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Loland C. J., Desai R. I., Zou M. F., Cao J., Grundt P., Gerstbrein K., Sitte H. H., Newman A. H., Katz J. L., and Gether U. (2008) Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol. Pharmacol. 73, 813–823 [DOI] [PubMed] [Google Scholar]
- 71. Tavoulari S., Rizwan A. N., Forrest L. R., and Rudnick G. (2011) Reconstructing a chloride-binding site in a bacterial neurotransmitter transporter homologue. J. Biol. Chem. 286, 2834–2842 10.1074/jbc.M110.186064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rudnick G., Krämer R., Blakely R. D., Murphy D. L., and Verrey F. (2014) The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction. Pflugers Arch. 466, 25–42 10.1007/s00424-013-1410-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Neale B. M., Kou Y., Liu L., Ma'ayan A., Samocha K. E., Sabo A., Lin C. F., Stevens C., Wang L. S., Makarov V., Polak P., Yoon S., Maguire J., Crawford E. L., Campbell N. G., et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 10.1038/nature11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W. W., Hegde M., Lyon E., Spector E., Voelkerding K., Rehm H. L., and ACMG Laboratory Quality Assurance Committee. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Günther W., Lüchow A., Cluzeaud F., Vandewalle A., and Jentsch T. J. (1998) ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U.S.A. 95, 8075–8080 10.1073/pnas.95.14.8075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gorlov I. P., Gorlova O. Y., Frazier M. L., Spitz M. R., and Amos C. I. (2011) Evolutionary evidence of the effect of rare variants on disease etiology. Clin. Genet. 79, 199–206 10.1111/j.1399-0004.2010.01535.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.