

Abstract
In humans, six α(1,3)-fucosyltransferases (α(1,3)-FTs: FT3/FT4/FT5/FT6/FT7/FT9) reportedly fucosylate terminal lactosaminyl glycans yielding Lewis-X (LeX; CD15) and/or sialyl Lewis-X (sLeX; CD15s), structures that play key functions in cell migration, development, and immunity. Prior studies analyzing α(1,3)-FT specificities utilized either purified and/or recombinant enzymes to modify synthetic substrates under nonphysiological reaction conditions or molecular biology approaches wherein α(1,3)-FTs were expressed in mammalian cell lines, notably excluding investigations using primary human cells. Accordingly, although significant insights into α(1,3)-FT catalytic properties have been obtained, uncertainty persists regarding their human LeX/sLeX biosynthetic range across various glycoconjugates. Here, we undertook a comprehensive evaluation of the lactosaminyl product specificities of intracellularly expressed α(1,3)-FTs using a clinically relevant primary human cell type, mesenchymal stem cells. Cells were transfected with modified mRNA encoding each human α(1,3)-FT, and the resultant α(1,3)-fucosylated lactosaminyl glycoconjugates were analyzed using a combination of flow cytometry and MS. The data show that biosynthesis of sLeX is driven by FTs-3, -5, -6, and -7, with FT6 and FT7 having highest potency. FT4 and FT9 dominantly biosynthesize LeX, and, among all FTs, FT6 holds a unique capacity in creating sLeX and LeX determinants across protein and lipid glycoconjugates. Surprisingly, FT4 does not generate sLeX on glycolipids, and neither FT4, FT6, nor FT9 synthesizes the internally fucosylated sialyllactosamine VIM-2 (CD65s). These results unveil the relevant human lactosaminyl glycans created by human α(1,3)-FTs, providing novel insights on how these isoenzymes stereoselectively shape biosynthesis of vital glycoconjugates, thereby biochemically programming human cell migration and tuning human immunologic and developmental processes.
Keywords: glycobiology, fucosyltransferase, mesenchymal stem cells (MSCs), mRNA, mass spectrometry (MS), glycoengineering, Lewis-X, modified mRNA, sialyl Lewis-X, E-selectin ligand
Introduction
The overwhelming majority of proteins and lipids in mammalian cells are glycosylated, and distinct glycan determinants regulate critical aspects of cell biology. Assembly of glycans on N-linked glycoproteins and on glycolipids is initiated in the endoplasmic reticulum, whereas O-glycosylation of proteins is initiated in the Golgi. In each case, glycan extension occurs by stepwise addition of monosaccharide units via the action of glycosyltransferases, type II integral membrane enzymes that stereo- and regiospecifically link the relevant monosaccharide to the pertinent substrate(s) (known as glycan “acceptors”). In humans, the terminal fucosylated lactosaminyl glycans known as Lewis-X (LeX)5 (also called “CD15”: Gal-β(1,4)-[Fuc-α(1,3)]-GlcNAc-R) and “sialyl Lewis-X” (sLeX) (also called “CD15s”: NeuAc-α(2,3)-Gal-β(1,4)-[Fuc-α(1,3)]-GlcNAc-R) bear major biological significance. These trisaccharide (LeX) and tetrasaccharide (sLeX) structures are displayed on the cell surface on both glycoproteins and glycolipids, and, in each case, the final step in their assembly requires α(1,3)-linked fucose modifications of N-acetylglucosamine (GlcNAc) within respective unsialylated or sialylated terminal Type 2 lactosamine (LacNAc) acceptors, i.e. Gal-β(1,4)-GlcNAc-R or NeuAc-α(2,3)-Gal-β(1,4)-GlcNAc-R (Fig. S1). Importantly, sLeX can only be created by fucosylation of sialylated LacNAc, as there is no mammalian sialyltransferase that can place sialic acid in α(2,3)-linkage to Gal in LeX to create sLeX. Thus, the biosynthesis of LeX and sLeX in each case critically pivots on fucose addition. This reaction is programmed by glycosyltransferases known as α(1,3)-fucosyltransferases (α(1,3)-FTs), which, in humans, constitute a family of six Golgi isoenzymes: FT3, FT4, FT5, FT6, FT7, and FT9.
Display of LeX and sLeX are each very tightly regulated among mammalian cells (1), indicating that they each serve highly specialized biology. LeX is well-known to mediate a variety of important cellular functions in development and immunity. In mice, LeX is known as stage-specific embryonic antigen-1 (SSEA-1); it serves as a major marker of murine (but not human) embryonic stem cells (2, 3), and its expression is necessary for compaction of the morula (4). Importantly, in both mice and humans, LeX is a marker for neural stem cells (5–8), and LeX-bearing glycoconjugates mediate neural stem cell proliferation by activating the Notch signaling pathway (9). LeX is immunomodulatory, serving as one of the main glycans recognized by DC-SIGN (CD209), a C-type lectin (i.e. requiring Ca2+ for ligand binding) expressed by dendritic cells (10). Conspicuously, human (but not mouse) myeloid leukocytes express LeX, and its expression in hematopoiesis is a hallmark of myeloid-specific lineage differentiation. Moreover, LeX is characteristically expressed on Reed-Sternberg cells in Hodgkin's lymphoma (11), and is displayed on certain human vascular and central nervous system malignancies (e.g. gliomas) in which it is considered an indicator of cancer stem cells (12, 13).
Although expression of LeX has garnered significant scientific interest, even more attention has been directed to sLeX as this glycan is the prototypical binding determinant for a family of C-type lectins called “selectins” that includes the endothelial molecule known as E-selectin (CD62E) (14). Binding of E-selectin to sLeX-bearing glycoconjugates on circulating cells is critical to enable the deceleration of the flowing cells onto the endothelial surface, which is the key first step in cell migration. In all mammals, E-selectin is constitutively expressed in microvessels in the bone marrow and skin and is inducibly expressed in endothelial beds at inflammatory sites in response to the cytokines tumor necrosis factor and interleukin-1 (15). Expression of cell-surface sLeX is therefore a prerequisite for extravasation of all mammalian leukocytes and for migration of mammalian hematopoietic stem/progenitor cells to marrow (15–17). Importantly, whereas sLeX plays a critical role in controlling leukocyte and hematopoietic stem/progenitor cell migration, aberrant expression of this tetrasaccharide by human malignant cells is a critical mediator of cancer metastasis (18–20). In addition to sLeX, E-selectin can also bind to other α(1,3)-fucosylated sialyllactosamines known as “VIM-2” (also known as CD65s) (21, 22) (in which fucose is α(1,3)-linked to GlcNAc within the penultimate LacNAc unit of a terminal polylactosaminyl glycan, i.e. NeuAc-α(2,3)-Gal-β(1,4)-GlcNAc-β(1,3)-Gal-β(1,4)-[Fuc-α(1,3)]-GlcNAc-R) and “difucosyl-sLeX” (in which fucose is α(1,3)-linked to GlcNAc within both the ultimate and penultimate LacNAc units, i.e. NeuAc-α(2,3)-Gal-β(1,4)-[Fuc-α(1,3)]-GlcNAc-β(1,3)-Galβ(1,4)-[Fuc-α(1,3)]-GlcNAc-R) (see Fig. S1) (21).
In light of the conspicuously restricted cell expression patterns and the vital roles of LeX and sLeX in human cell biology, it is important to understand how the various α(1,3)-FTs shape the intracellular biosynthesis of these glycan determinants in humans. Extensive efforts in the 1980s and 1990s led to the identification and molecular cloning of all six human α(1,3)-FUTs that can catalyze creation of LeX and/or sLeX (FUT3 (23), FUT4 (24, 25), FUT5 (26), FUT6 (27), FUT7 (28), and FUT9 (29)). These studies functionally characterized the enzymes as isolated proteins, analyzing their biosynthetic properties under non-physiologic ex vivo reaction conditions. Further studies employed a variety of molecular biology approaches in which cell-surface fucosylated glycans were evaluated after transfecting various mammalian cell lines with individual α(1,3)-FUT genes (30, 31), in some cases in combination with biochemical approaches whereby α(1,3)-FT activity was measured in the lysates of such cells using synthetic oligosaccharide (32–34) and glycolipid (35, 36) acceptors. This body of work provided key catalytic insights and yielded working classifications of the product specificities of the α(1,3)-FUT gene family, specificities that have been generally recognized as fact for the past quarter century (1, 37) (see Table 1).
Table 1.
Summary of human α(1,3)-fucosyltransferase substrate specificities based on current literature
The symbols indicate the ability of the enzyme to create the indicated structure with: +, low; ++, moderate; and +++, high potency. The – indicates inability to create the indicated structure, and (+) indicates controversy exists regarding product formation.
| FUT | Chromosomal localization | Classification |
Ex vivo |
Cell line-based |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LeX | sLeX | VIM-2 | Di-Fuc-sLeX | LeX | sLeX | VIM-2 | Di-Fuc-sLeX | |||
| FUT3 | 19p13.3 | Lewis | + | ++ | NAa | NA | ++ | ++ | + | + |
| FUT4 | 11q21 | Myeloid | +++ | + | ++ | − | ++ | (+) | +++ | − |
| FUT5 | 19p13.3 | − | + | + | ++ | NA | + | + | + | + |
| FUT6 | 19p13.3 | Plasma | ++ | +++ | + | ++ | ++ | +++ | (+) | + |
| FUT7 | 9q34.3 | Leukocyte | − | +++ | − | ++ | − | +++ | − | + |
| FUT9 | 6q16 | CNS, PBL | +++ | − | + | NA | +++ | − | (++) | − |
a NA indicates data not available.
There remain two overarching caveats regarding our current understanding of the human α(1,3)-FT isoenzymes that modify terminal lactosaminyl glycans: (i) their ex vivo chemistry may not reflect their Golgi chemical biology; and (ii) their chemical biology within immortal cell lines, and, in particular, immortal non-human cell lines, can differ significantly from that within human primary cells. To ascertain how these enzymes contribute to synthesis of LeX and sLeX in context of human cell biology, in this study we systematically evaluated the capability of all six human α(1,3)-FTs to generate sLeX and LeX, as well as the related structures VIM-2 and difucosyl sLeX, in primary cultures of human mesenchymal stem cells (hMSCs). hMSCs are an ideal model cell system to study the products of the human α(1,3)-FT isoenzymes because they are cultured as primary cells, have immense clinical significance (e.g. they are precursors of bone-forming osteoblasts and have potent immunomodulatory properties), and they can be readily transfected using Lipofectamine-based reagents (38, 39). Importantly, these cells are known to natively display sialylated terminal Type 2 lactosaminyl glycans (thus possessing glycosyltransferases necessary to create this structure), but are natively deficient in LeX and sLeX expression (38, 40). Accordingly, we transfected hMSCs with modified-mRNA encoding the relevant human α(1,3)-FTs (i.e. FT3, FT4, FT5, FT6, FT7, and FT9) to generate a controlled pulse of the pertinent FT activity in these cells. Across multiple donor hMSCs, the resulting cell-surface glycan determinants were examined using the complementary approaches of flow cytometry and mass spectrometry (MS) analysis. Our findings provide new insights on the biosynthetic properties of this important class of glycosyltransferases, yielding novel perspectives on how these enzymes shape expression of functionally significant α(1,3)-fucosylated glycan moieties on both glycoproteins and glycolipids in human cell biology.
Results
Human MSCs do not express sLeX or LeX determinants but construct both sialylated and “neutral” (unsialylated) Type 2 lactosamines
To elucidate the native lactosaminyl glycan “signature” of hMSCs, we performed gene expression studies of glycosyltransferases, in concert with flow cytometry of cell-surface lactosaminyl glycan display using a combination of antibody and lectin probes, in multiple hMSC cultures derived from marrow obtained from healthy donors. We first assessed the gene expression levels in hMSCs of glycosyltransferases required for construction of Type 2 lactosaminyl glycans: (i) the enzymes required for initiating O- and N-glycosylations that display terminal lactosamines, respectively, the O-glycan “core 2” branch-initiating enzyme (C2GnT-1) and the “complex-type” N-glycan-initiating enzyme (MGAT1); (ii) the galactosyltransferase that predominates in Type 2 lactosamine synthesis (β4GalT1); (iii) the three human α(2,3)-sialyltransferases (STs) that are capable of terminally sialylating a Type 2 lactosamine (ST3Gal-3, -4 and -6) (Fig. 1A); and (iv) The six members of the human α(1,3)-FT family (FT3, -4, -5, -6, -7, and -9) that can fucosylate GlcNAc within terminal Type 2 lactosaminyl glycans (Fig. 1B). In addition, as an internal control for evaluation of fucosyltransferase gene expression by qRT-PCR, we measured transcript levels for the ubiquitously expressed enzyme FT8, which places fucose in α(1,6)-linkage on the innermost (core) GlcNAc of N-glycans. We observed that hMSCs expressed appreciable transcript levels of all glycosyltransferases tested except for the α(1,3)-FUTs. Notably, FUT4 transcripts were detectable but at extremely low levels, while transcripts for the remaining α(1,3)-FUTs (i.e. FUT3, -5, -6, -7, and -9) were absent (Fig. 1B). However, hMSCs are not uniformly deficient in fucosyltransferases, as prominent transcript levels for FUT8 were observed (Fig. 1B). Collectively, these data indicate that hMSCs express the requisite enzymes to create lactosaminyl and sialylated lactosaminyl glycans on both N-glycans and O-glycans, but lack α(1,3)-FTs required for the synthesis of LeX and sLeX, and the related sialofucosylated structures VIM-2 and difucosyl sLeX. These data therefore supported the notion that this clinically relevant, primary human cell type would serve as a fitting model system to assess the in vivo substrate specificities of the human α(1,3)-FT isoenzymes.
Figure 1.

hMSCs express lactosaminyl and sialylated lactosaminyl glycans, but do not express the fucosylated (sialyl) lactosamines Lewis X (Lex) or sialyl Lewis X (sLex). A, quantitative RT-PCR was performed to measure human MSC gene expression levels of glycosyltransferases required for lactosamine synthesis (β4GalT1, n = 3), initiation of complex N-glycans (MGAT1, n = 7), and construction of core 2 O-glycans (C2GnT-1, n = 3), as well as the three human α(2,3)-sialyltransferases that can terminally sialylate a Type 2 lactosamine (n = 4). B, quantitative RT-PCR was performed to measure hMSC gene expression levels of the six human α(1,3)-fucosyltransferases (FUT3, -4, -5, -6, -7, -9) and the core α(1,6)-fucosyltransferase FUT8 (for each, minimum of n = 4). C, MALII lectin binding to untreated or sialidase-treated hMSCs was evaluated by flow cytometry (n = 5; paired t test, ***, p < 0.001; error bars represent S.D.). D, cell-surface levels of unsialylated lactosamines on untreated and sialidase-treated hMSCs, as measured by flow cytometry for ECA lectin (n = 5; paired t test, **, p = 0.01; error bars represent S.D.). E, cell-surface levels of LeX (measured by antibody HI98; light gray bars) and sLeX (measured by antibody CSLEX1; dark gray bars), on native (untreated) hMSCs or after exofucosylation (exo MSC) with purified FT6 enzyme. Unstained hMSCs are also shown to indicate background of detection (n = 4; paired t test, **, p < 0.01 between untreated and FT6 exo MSC). Individual data points are depicted, with error bars representing S.D.
To evaluate the native cell-surface expression of both unsialylated and sialylated terminal Type 2 lactosaminyl glycans, two complementary approaches were undertaken: (i) lectin staining and (ii) analysis of products of FT6-mediated exofucosylation. The lectin ECA binds to unsialylated Type 2 lactosamines, whereas the lectin MALII detects sialic acid α(2,3)-linked to galactose (41) (and is also reactive with certain sulfated glycans (41, 42)). There was significant baseline binding with both ECA and MALII lectin probes. In initial studies, we observed that treatment with the broad-specificity sialidase from Arthrobacter ureafaciens yielded decreased MALII binding and increased ECA binding (Fig. 1, C and D). To further assess the presence of α(2,3)-sialylated Type 2 lactosamines on hMSCs, we treated these cells with an α(2,3)-specific sialidase (sialidase from Macrobdella decora (43)), and evaluated binding of the lectins MALII, ECA, and SNA (a lectin that binds specifically to α(2,6)-linked sialic acid). Upon α(2,3)-sialidase treatment, MALII lectin binding was reduced, ECA lectin binding was increased sharply, and SNA lectin binding remained unchanged (Fig. S2A). Collectively, these results provided evidence that α(2,3)-sialylated Type 2 lactosamines are present on the hMSC surface. As predicted by results of α(1,3)-FUT transcript analysis, flow cytometry of cell-surface glycans revealed that, natively, hMSCs do not express sLeX (measured by mAb CSLEX1) or LeX (measured by mAb HI98) determinants (Fig. 1E, untreated, Fig. S2B). Upon exogenous fucosylation of these cells with FT6 enzyme and GDP fucose, production of both LeX and sLeX was observed (Fig. 1E, FT6 exo MSC, Fig. S2B). Altogether, these results confirmed that although hMSCs lack expression of α(1,3)-fucosylated lactosaminyl glycans, both unsialylated and sialylated terminal Type 2 lactosamine acceptors are present on the cell surface that can be converted to LeX and sLeX, respectively, by exofucosylation with a pertinent α(1,3)-FT.
Construction of modRNA encoding the six human α(1,3)-fucosyltransferases
To analyze the enzymatic product specificities of the human α(1,3)-FTs in the context of primary human cells, we utilized modified-mRNA (modRNA), which is non-permanent and nongenome integrative, and can be readily transfected directly into hMSCs (38). Once inside the cell, the modRNA is recognized by the cell's endogenous translational machinery, and protein synthesis ensues. The resulting protein then undergoes normal post-translational folding and processing, and subsequently becomes localized to its normal position within the cell. We synthesized modRNAs encoding the six human α(1,3)-fucosyltransferases (i.e. creating modRNAs FUT3, -4, -5, -6, -7, and -9), transfected them individually into hMSCs, and probed for the presence of surface LeX and/or sLeX by flow cytometry. In initial experiments, the α(1,3)-FUT modRNAs generated appreciable levels of LeX and/or sLeX with the exception of FUT4, which displayed little or no activity; despite confirming sequence integrity as previously reported in GenBankTM (accession number: BC136374), and controlling for modRNA integrity and transfection efficiency, we consistently observed no evidence of fucosylated products using multiple independent FUT4 modRNA preparations.
FUT4 modRNA transfection provides direct evidence that only “short” FUT4 RNA creates functional FT4
The original report of cloning of human FT4 indicated that two transcript variants of FUT4 (originally named “ELAM-1 ligand fucosyltransferase” (ELFT)) exist: ELFT (indicated here as “FUT4SHORT,” which encodes a 405-amino acid protein) and a putative longer isoform (ELFT-L; indicated here as “FUT4LONG”) (25). Although the molecular basis of these two transcript sizes has not been defined (i.e. alternative splicing of the primary longer transcript or an alternative transcription initiation site embedded within the gene sequence 5′ to the “short” ELFT gene product), it is recognized that, compared with ELFT, ELFT-L contains an extended 3′ UTR, a different 5′ UTR, as well as an alternative ATG start site yielding a coding sequence corresponding to 530 amino acids (Fig. 2). However, the putative ATG start site for ELFT-L is not embedded among any of the nucleotides typical of the consensus Kozak sequence, casting doubt on the translational capacity of this longer RNA transcript (25). Despite this fact, genome databases (e.g. GenBank accession number BC136374), have consistently annotated ELFT-L as the authentic FUT4 mRNA coding sequence, and thus this sequence was chosen for all initial experiments. However, as noted above, hMSC transfection with FUT4LONG-modRNA did not yield fucosylated products on the cell surface, and we thus tested whether transfection with FUT4SHORT-modRNA could yield enzymatic function. To this end, we performed transfection in hMSCs using FUT4SHORT-modRNA versus FUT4LONG-modRNA, and, as can be seen in Fig. 2, FUT4SHORT-modRNA produced robust levels of cell-surface LeX, whereas FUT4LONG-modRNA did not produce LeX. Although this variability of the human FUT4 RNA is not well-recognized, our findings corroborate the results of the initial human FT4 cloning report that suggested that ELFT (FUT4SHORT) encodes functional FT4 (25).
Figure 2.

Modified mRNA encoding the short isoform of FUT4 (FUT4SHORT) enforces CD15 expression in transfected hMSCs, whereas the long isoform of FUT4 (FUT4LONG) has no effect. A, schematic diagram of long and short FUT4 isoforms including 5′ and 3′ UTR, start codons (ATG), and stop codons. B, schematic diagram of modRNA constructs including the coding sequence from the long and short FUT4 isoforms. C, Lewis X (CD15) levels were measured by flow cytometry on hMSCs transfected 2 days prior with FUT4LONG modRNA or FUT4SHORT modRNA. Statistics performed were: one-way ANOVA (p < 0.0001) plus Dunnett's post-test to compare all stained samples with unstained control (**, p < 0.01). Individual data points are depicted, with error bars representing S.D.
Because the qRT-PCR primer sets used initially to probe FUT4 transcripts in hMSCs were embedded within the “common” FUT4 coding sequence (i.e. amplifying a 123-bp fragment within the coding region common to both FUT4LONG and FUT4SHORT) (Table S1), we analyzed for FUT4LONG (i.e. ELFT-L) transcripts using a 5′ primer that detects a 150-bp amplicon upstream of (and not overlapping with) the core FUT4 sequence (Table S1). Plasmid containing FUT4LONG cDNA was used as positive control, as was RNA isolated from the human promyelocytic cell line HL60 (which is known to express both FUT4 transcripts) (25, 44). qRT-PCR of HL60 cells confirmed the presence of the FUT4LONG transcript and the common FUT4 transcript (Fig. 3A), with amplicon sizes verified by gel electrophoresis of the PCR products (Fig. 3B). In hMSCs, a low transcript level was detected with FUT4common-primers, whereas no product was detected with FUT4LONG-primers (Fig. 3A). These findings indicate that only the short form of FUT4 is expressed in hMSCs, although neither LeX nor sLeX product was detectable on the cell surface by flow cytometry.
Figure 3.
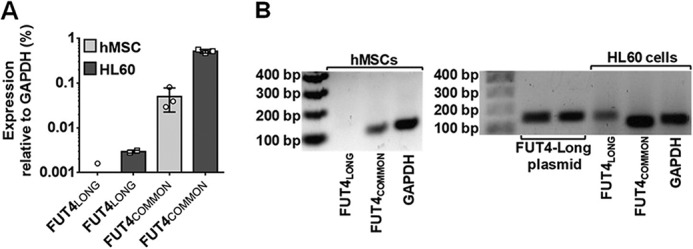
MSCs natively express low levels of FUT4SHORT transcript, whereas FUT4LONG transcript is absent. A, quantitative RT-PCR was performed to measure the transcript level of the FUT4LONG isoform in hMSCs (light gray bars; n = 3 independent hMSC lines) compared with HL60 cells used as positive control (dark gray bars; n = 2 independent measurements for FUT4LONG, and three independent measurements for FUT4COMMON). Individual data points are depicted, with error bars representing S.D. B, representative gel images display amplicon size detected by both FUT4LONG and FUT4COMMON primers in hMSCs (left panel) and in positive control samples (right panel). Numbers on the left of each image present size of DNA fragment in bp (bp).
modRNA-driven expression of α(1,3)-FTs reveal unique product specificities
We transfected hMSCs derived from multiple marrow donors with each of the α(1,3)-FUTs (n = 15 donors, for FUT-3, -5, -6, -7, -9; n = 9 donors, for FUT4SHORT), and 48 h later measured the resulting fucosylated lactosaminyl glycans using monoclonal antibodies recognizing the terminally α(1,3)-fucosylated lactosaminyl glycans LeX and sLeX, and the related internally (penultimate) α(1,3)-fucosylated lactosaminyl glycans VIM-2, as well as the determinant containing both terminal and penultimate α(1,3)-fucose modifications known as difucosyl sLeX (Fig. 4, Fig. S3). All six of the α(1,3)-FTs tested were able to fucosylate Type 2 lactosamines to create LeX and/or sLeX, although clearly distinct enzymatic specificities were observed (Fig. 4). With the exception of FT9, all other α(1,3)-FTs were able to terminally fucosylate a sialylated Type 2 lactosamine, with FT6 and FT7 having the highest potency, FT3 and FT5 having moderate capability, and FT4 showing very limited capacity to create sLeX (Fig. 4A, Fig. S3A). Conversely, FT9 and FT4 were most potent in terminally fucosylating an unsialylated Type 2 lactosamine to create LeX. FT3, FT5, and FT6 had moderate ability, and FT7 did not create LeX (Fig. 4B, Fig. S3B). Notably, FT3 and FT5 were the only FTs that could internally fucosylate a sialylated polylactosamine, creating the VIM-2 glycan determinant (Fig. 4C, Fig. S3C). FT3, FT5, FT6, and FT7 were all capable of fucosylating both the penultimate and terminal lactosamines of a sialylated poly-lactosamine, thereby creating the difucosyl-sLeX structure (Fig. 4D, Fig. S3D). Consistent with earlier reports (Table 1), FT4 and FT9 do not create any detectable difucosyl sLeX, but, in contrast to prior findings, FT6 did so with the highest potency of all the α(1,3)-FTs.
Figure 4.

Comparative analysis of human α(1,3)-FT product specificities by modified mRNA transfection of hMSCs, measured by flow cytometry. A, sLeX (measured by antibody CSLEX1) and B, LeX (measured by antibody HI98) levels measured on the cell surface. Non-transfected (NT) hMSCs and matched FUT-3, -5, -6, -7, and -9–transfected hMSCs were compared relative to each other (n = 15), while FUT4–transfected hMSCs were compared separately along with a partially overlapping set of matched NT controls (n = 9). Statistics performed were repeated measures of ANOVA (p < 0.0001 for both A and B) followed by a Tukey post-test, comparing all columns with each other. Symbol above each bar compares to NT. For sLeX, FUT9 is significantly lower than FUT-3, -5, -6, and -7 (p < 0.001), and for LeX, FUT7 is significantly lower than FUT3, FUT5, FUT6, and FUT9 (p < 0.001). C, cell-surface VIM-2 levels (paired analysis, n = 5). Statistics performed were repeated measures of ANOVA (p < 0.001) plus Dunnett's post-test, comparing all modRNA-transfected samples with control (NT). Symbols above each bar compare to NT. D, cell-surface difucosyl sLeX levels were measured by antibody FH6 binding (paired analysis, n = 3). Statistics performed were repeated measures of ANOVA (p < 0.001) plus Dunnett's post-test, comparing all modRNA-transfected samples with control (NT). Symbol above each bar compares to NT, n.s. = p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001. Individual data points are depicted, with error bars representing S.D.
The results of flow cytometry studies are summarized in Fig. 6A. Importantly, we observed strict consistency in this expression profile across all hMSC cultures, indicating that this panel identifies a “lactosaminyl glycosignature” of this human cell type. While our findings generally parallel the results of earlier in vitro assays, as well as the data derived from transfection studies of both non-human and human cell lines (Table 1), there are several divergences between our dataset and the prior findings: the VIM-2 glycan is not created by FT4, FT6, or FT9; there was modest but consistently detectable production of sLeX following FUT4SHORT-modRNA transfection; and, although FT6 does not create VIM-2, it creates significant amounts of difucosyl sLeX.
Figure 6.
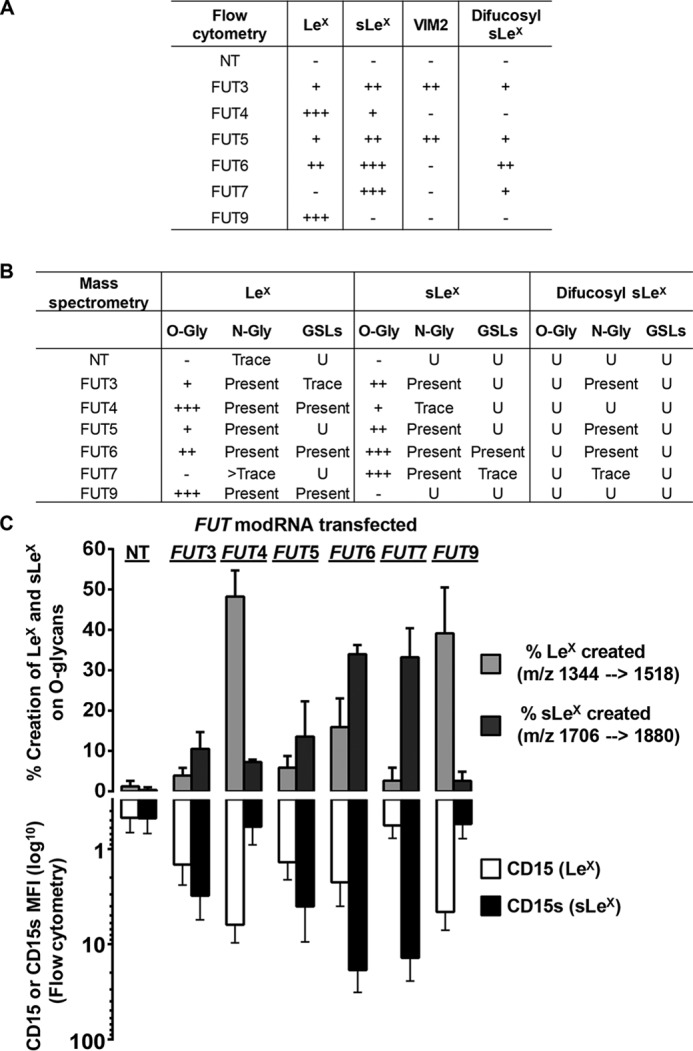
Summary and comparison of fucosylated glycan products generated by the human α(1,3)-FTs using flow cytometry of cell surface and MS analysis of fractionated cellular glycans. A, summary table of all flow cytometry data. Within a given column (i.e. a specific determinant), the relative range for that epitope created by various FTs is: insignificant amounts created on the cell surface (−); and comparatively low (+), moderate (++), or high (+++) levels created on the cell surface. NT, non-transfected. B, summary table of all MS data. Glycans were fractionated into N-glycans, O-glycans, and GSLs and analyzed separately for LeX, sLeX, and difucosyl-sLeX. N-Glycan and GSL analysis was not quantitative and results are presented as present, trace, or undetected (U). O-Glycan analysis was semi-quantitative (Fig. S6), and was classified as (−) insignificant, (+) low, (++) moderate, and (+++) high. C, comparison of LeX and sLeX creation on O-glycans (measured by LC-MS, n = 3 to 6) and on the cell surface (measured by flow cytometry, n = 9 to 15) after transfection with FUT modRNA. Error bars represent S.D.
Glycomic analysis of modRNA-transfected hMSCs
Flow cytometry analysis using monoclonal antibodies provides very useful information regarding the relative amounts of fucosylated lactosaminyl glycan determinants created by α(1,3)-FTs but is not able to distinguish their display on different scaffolds, such as O-linked versus N-linked glycans of protein, or glycosphingolipids (GSLs). However, identification of these various structures can be achieved by glycan fractionation followed by mass spectrometry (MS), and, furthermore, ion trap sequential MS (MSn) glycan analysis can provide detailed assessment of the component monosaccharide composition; this information complements and cross-validates the molecular identification of the relevant lactosaminyl glycan determinants detected by flow cytometry. To perform MS, cell lysates were prepared from different hMSC cultures derived from multiple donors at 48 h after transfection with individual FUT modRNAs. O-Glycans, N-glycans, and GSL-glycans were sequentially released from lysates and isolated. These fractions were then interrogated by various MS techniques to determine the presence or absence of specific fucosylated structures.
Reduced and permethylated N-glycan samples were subjected to electrospray ionization by direct infusion into an ion trap mass spectrometer. Fig. 5 presents a representative mass spectra and sequential MSn spectra of the N-glycans of nontransfected (Fig. 5A) and FUT6-transfected (Fig. 5B) hMSCs. The positive mode mass spectra can only determine the overall glycan compositions in these samples. To assess the synthesis of LeX/sLeX formation in N-glycans, detailed interrogation is required due to the presence of multiple isomeric structures for any sialofucosylated composition. We chose to focus on an N-glycan composition found in all samples, a composition with an m/z 1322 that is doubly-charged, and has the configuration, NeuAc1Hex2HexNAc2dHex1 + Man3GlcNAc2, corresponding to a biantennary, monosialylated, and monofucosylated structure. In this ion, the fucose (“dHex”) can be positioned either on the core GlcNAc (i.e. the α(1,6)-fucosylation catalyzed by FT8), or on the unsialylated LacNAc antenna (i.e. LeX or H type 2 (“H2,” the blood group O antigen on a Type 2 lactosamine)), or on the sialylated LacNAc antenna (i.e. sLeX) (Fig. S4), and, thus, the m/z 13222+ ions could contain one or more of the following isomeric possibilities: core fucosylation, LeX, the H2 structure, or sLeX. Using ion-trap MSn we could perform detailed structural analysis, and thus identify which of these isomers represent this composition. Specifically, the fucosylated fragment ions of interest include m/z 490 (core GlcNAc fucosylation), m/z 660 (terminal fucosylated LacNAc, i.e. Lex and/or H2), m/z 1021 (sLeX tetrasaccharide), and 646 (a desialylated sLeX, generating an LeX fragment) (Fig. S4). The MS2 spectra of the m/z 13222+ ions from nontransfected (Fig. 5C) and FUT6-transfected (Fig. 5D) hMSCs indicate that non-transfected hMSCs contain the core fucosylation fragment, but negligible antennal fucosylation. The FUT6-transfected sample on the other hand contains detectable fucosylated LacNAc fragments, both terminal (m/z 660) and internal (m/z 646), although these ions are much less abundant than the core-fucosylated fragment. MS2 is not sufficient to determine whether H2 or LeX yields the terminally fucosylated fragment (m/z 660). Thus, MS3 analysis was done to definitively characterize the m/z 660 ion (Fig. 3E) from the FUT6-transfected hMSCs. This spectrum, as described in an earlier article (45), empirically identifies the presence of LeX in the FUT6-transfected hMSC sample. Fig. 5F shows the MS4 spectrum of the internal LacNAc fragment (m/z 646). For this analysis, the sialofucosylated tetrasaccharide (m/z 1021) was isolated first, followed by fragmentation into the internal fuc-LacNAc trisaccharide (m/z 646). This spectrum and fragmentation pathway empirically identifies the sLeX motif (45). Identical analyses were done to identify these structures in hMSCs transfected with FUTs-3, -4, -5, -7, and -9. In addition, in an effort to assess for the presence of difucosyl sLeX, we selected the m/z 16342+ ion; this composition is representative of three lactosamine units, one sialic acid, three mannose, two GlcNAc, and two fucose molecules. This composition could be contained within both triantennary and biantennary structures, with the former containing a LacNAc on each antenna and the latter containing one antenna containing a single LacNAc and the other containing two contiguous LacNAc units (a “diLacNAc” motif). By selecting the putative sLeX-LeX heptasaccharide fragment (difucosyl sLeX) and then obtaining an MS3 spectrum, we could then assess for the presence of this motif (Fig. S4F). The results indicate that core α(1,6)-fucosylation is uniformly detectable in hMSCs, whereas sLeX is not detectable among native hMSCs and its creation is unique to specific fucosyltransferases (Fig. 6B). By MS analysis, traces of LeX were observed only on N-glycans of native hMSCs, which may reflect the endogenous low-level expression of FUT4SHORT transcripts, and, notably, this modest level of LeX expression was not detectable by flow cytometry. In contrast, although VIM-2 determinants were readily identified by flow cytometry in FUT3 and FUT5 transfectants, the MSn fragmentation profiles representative of this structure could not be definitively detected on any glycoconjugate sample from any transfectant. The incongruity between flow cytometry and MS detection of VIM-2 expression is explained by the fact that MS identification of this structure requires isolation of the hexasaccharide fragment with subsequent dissociation, and the lability of linkages in this structure, coupled with its apparent low abundance and the presence of other isobaric ions in these samples, prevented a confident MS assignment.
Figure 5.

Overall MS and sequential MSn spectra of the N-glycans of nontransfected and FUT6 modRNA-transfected hMSCs. Reduced and permethylated N-glycan samples were interrogated by direct infusion electrospray ionization in an ion trap mass spectrometer. A and B, positive mode mass spectra of (A) nontransfected (NT) and (B) FUT6 modRNA-transfected (FUT6) hMSC sample. C, MS2 spectra of the m/z 13222+ ion from NT hMSCs. D, MS2 spectra of the m/z 13222+ ion from FUT6 hMSCs. The insets in panels C and D represent the zoomed in view of the m/z 450–680 region. The m/z 490 ion, i.e. the core-fucosylated reduced GlcNAc is captured in the zoomed pane for abundance comparison purposes. E, MS3 fragmentation of the terminal fuc-LacNAc (m/z 660) consistent with LeX. F, MS4 fragmentation of the internal fuc-LacNAc (m/z 646) consistent with sLeX.
Because O-glycan fractions generally have fewer sample components than N-glycans and also are generally small-sized glycans, their greater polarity relative to permethylated N-glycans or glycosphingolipids makes reversed-phase LC-MS a useful approach to obtain relative quantitative data. By selecting relevant mass ranges, extracted ion chromatograms can be used to provide data informing both glycan structure and relative quantity. Since VIM-2 and difucosyl sLeX were not detected on O-glycans by direct infusion MS analysis, using LC-MS we focused on ions corresponding to simple branched O-glycan structures containing LeX or sLeX, or the corresponding unfucosylated acceptor structures (Fig. S6 and S7). By comparing the relative amounts of the product and acceptor, an approximate percent created could be estimated, enabling a semi-quantitative measurement of LeX and sLeX creation on O-glycans (Fig. 6, B and C). When LeX and sLeX creation on O-glycans was thus calculated, and compared with the flow cytometry data measured from the same samples, the data were strikingly similar (Fig. 6C). Besides cross-validating the specificity, sensitivity, and qualitative and quantitative utility of both approaches for detection of LeX and sLeX, the close correlation between results from flow cytometry and MS analysis of O-glycans indicates that, for each transfectant, there was no preferential skewing of terminal fucosylation away from O-glycan acceptors. However, a major difference is found in expression of difucosyl sLeX, which is clearly measurable by flow cytometry but MS indicates presence of this structure solely on N-glycans. Importantly, FT6 and FT7 can each construct difucosyl sLeX but not VIM-2; thus, for creation of difucosyl sLeX, it is most likely that FTs-3, -5, -6, and -7 first fucosylate the terminal GlcNAc (thereby creating sLeX), followed by fucosylation of the internal GlcNAc, preferentially on N-glycans.
For MS analysis of glycosphingolipids, two clusters of peaks were chosen for detailed analysis: m/z 1747, which would contain the fucosyltetraosylceramides (including LeX), and m/z 1066, which would contain the doubly-charged sialofucosylated tetraosylceramide (including sLeX) (Fig. S5). MSn was then performed on each of these m/z ranges to identify the specific fucosylated glycans present (if any) for each FUT modRNA transfectant. As summarized in Fig. 6B, LeX was detected in FUT4-, FUT6-, and FUT9-transfected hMSCs, which corresponds to the three fucosyltransferases that resulted in the highest levels of LeX by flow cytometry. Notably, despite the ability to produce LeX and sLeX on glycoproteins, FUT5-transfectants did not make any detectable LeX or sLeX on GSLs, and FUT3-transfectants made only a trace of LeX on GSLs, suggesting a unique substrate specificity and/or subcellular localization pattern of FT3 and FT5 compared to other FTs. Importantly, sLeX bearing GSLs were detected prominently only in FUT6-transfected hMSCs, indicating that FT6 is either specialized to create sLeX on GSLs or has unique accessibility to GSLs inside the Golgi.
Collectively, the MS results indicate that, with exception of trace amounts of LeX on N-glycans, all other types of terminally fucosylated LacNAc are not natively expressed on hMSC glycoconjugates. Moreover, both unsialylated and sialylated acceptors are natively distributed among N-glycans, O-glycans, and GSLs in hMSCs. Notably, hMSCs exclusively possess Type 2 lactosamines (Galβ(1,4)-GlcNAc), as empirical MSn analysis of LacNAcs showed no evidence of Type 1 (Galβ(1,3)-GlcNAc) structures. Moreover, we assessed production of sialyl Lewis-a (sLea) among the various hMSC-FUT transfectants by measuring binding of antibody CA19-9. In sharp contrast to the marked increases observed in HI98-reactivity (LeX) and CSLEX1-reactivity (sLeX), the FUT transfectants (particularly as might be expected from the specificities of FT3 and FT5 for Type 1 lactosamine acceptors) did not yield significant CA19-9 reactivity (sLea), thus indicating paucity of Type 1 lactosamine acceptors in hMSCs.
Discussion
To understand cellular glycobiology, it is imperative to elucidate the molecular effectors of glycan biosynthesis, the glycosyltransferases. However, elucidating the authentic drivers programming assembly of a particular glycan moiety of interest is challenging because of the following reasons: (i) in contrast to nucleic acids and proteins, glycan construction is not template-driven, it is directed by the sequential action(s) of glycosyltransferases that modify relevant acceptors in a stereospecific fashion; (ii) glycosyltransferase isoenzymes typically display functional redundancy (i.e. glycosyltransferases commonly create more than one glycan product, and the same glycan structure may be generated by many glycosyltransferases); and (iii) glycosyltransferase expression patterns vary in a species-specific and cell-specific fashion. Thus, to determine whether a certain glycosyltransferase can mediate construction of a given glycan, the relevant enzyme property can be analyzed using ex vivo or in vivo systems. However, because ex vivo studies of glycosyltransferases provide insights only on what these enzymes are capable of producing under the defined reaction conditions utilized, it is imperative to undertake studies in living cells to reveal their true biologic activity.
To date, our understanding of the product specificities of human α(1,3)-FTs is largely based on ex vivo studies. Such studies employed purified or recombinant α(1,3)-FTs, or extracts of cells expressing these enzymes, to catalyze fucose addition in cell-free reactions targeting synthetic oligosaccharide substrates presented on artificial reaction surfaces, each present at supraphysiologic concentrations. These studies also typically utilized extremely high levels of manganese (Mn2+) as a co-factor (i.e. as high as 40 mm, whereas, human physiologic manganese levels are <20 nm), raising fundamental questions about the relevance of these findings to native human cell biology (46). Thus, although such experimental systems can define the catalytic properties of an isolated enzyme, it cannot be assumed that the data derived therefrom can be extrapolated to cell biology. However, prior studies investigating the in vivo glycobiology of human α(1,3)-FTs utilized a wide variety of immortalized mammalian cell lines (predominantly non-human, e.g. CHO, COS, and BHK cells) as hosts for human α(1,3)-FUT transfection(s), and evaluated product profiles by flow cytometry of the cell surface. These diverse cell line-based studies provided broad insights that could only tentatively predict the α(1,3)-FT product specificities within primary human cells. Thus, the non-physiological systems employed in prior in vitro studies and the studies involving a wide range of cell/animal models have raised questions regarding the translatability of these findings to human cell biology, especially in the context of primary human cells.
In this study, we evaluated the impact of FT3, FT4, FT5, FT6, FT7, and FT9 on expression of LeX and sLeX moieties, as well as the structurally related VIM-2 and difucosyl sLeX determinants, on both protein-based and lipid-based glycoconjugates in primary cultures of hMSCs. Besides these six α(1,3)-FTs, two other α(1,3)-FTs, known as FT10 and FT11, have been identified. These enzymes were not evaluated as they reportedly modify GlcNAc only within the chitobiose core of N-glycans (47) (a modification not typical of mammalian cells). A recent study in FT9-knockout mice has claimed a possible role for FT10 in creation of LeX on a uniquely restricted set of N-glycan acceptors found only in mouse neural progenitor cells, but, importantly, a contributory role for FT4 was not formally excluded in that model system as FT4/9 double knockout mice were not studied (48).
We employed transfection of modRNA encoding the relevant α(1,3)-FT transcripts to enable assessment of the relative “in-Golgi” capacity of these isoenzymes to program expression of α(1,3)-fucosylated terminal lactosaminyl glycans. Our results are consistent with prior findings from both ex vivo and cell line-based studies (reviewed in Table 1) with regard to the capacity of the various α(1,3)-FTs to create LeX: FT4 and FT9 in hMSCs are most potent at fucosylating unsialylated Type 2 lactosamines, with FT6 demonstrating moderate capability, FT3 and FT5 showing weak ability, and FT7 having little to no ability. However, we observed key differences from historical data regarding the capacity of the various α(1,3)-FTs to produce sLeX and, also, VIM-2 and difucosyl sLeX. Similar to earlier studies, we found that FT6 and FT7 have the highest potency in fucosylating sialylated Type 2 lactosamines (thereby creating sLeX), followed by FT3 and FT5, whereas FT9 is unable to generate any significant levels of sLeX. Notably, whereas prior studies using the human prostate cancer cell line PC-3 (49) and using Chinese hamster ovary (CHO) cell lines (50, 51) each indicated that transfection of human FUT4 yields readily detectable levels of sLeX, we observed very limited production of sLeX by FT4 in hMSCs. In terms of VIM-2 expression, although MS was unable to resolve this structure within the various hMSC transfectants, flow cytometry analysis using anti-VIM-2 mAb clearly indicated that FT3 and FT5 are the only human α(1,3)-FTs that construct this glycan determinant on hMSCs. Again, key differences exist between our results and those of prior cell line-based studies indicating that VIM-2 could be created in CHO cells by FUT4 (24) and FUT6 (35) transfection, in Jurkat cells (a human lymphoblastic cell line) by FUT4 transfection (52), and in Namalwa cells (a human lymphoma cell line) following FUT9 transfection (53). Other cell line studies have also indicated that human FT9 can create the VIM-2 determinant on CHO Lec29 cells (54) and HEK293T cells (a human embryonic kidney line) (55), with commensurate induction of E-selectin binding in each (54, 55). Additionally, although FT3 and FT5 were the only enzymes that created VIM-2 in hMSCs, transfection of FUT3, FUT5, FUT6, and FUT7 resulted in cell-surface expression of difucosyl sLeX. The finding that difucosyl sLeX is created in FUT6 and FUT7 transfectants in the absence of measurable VIM-2 production suggests that these enzymes modify a sialylated di-LacNAc by first fucosylating the terminal GlcNAc to create an sLeX structure, then act to convert this structure to difucosyl sLeX via the addition of a fucose to the penultimate GlcNAc. In other words, these enzymes do not appear to make an intermediate VIM-2 structure, because they have a dominant preference for installing a fucose at the terminal GlcNAc. FT3 and FT5 are the only enzymes that can create VIM-2 in hMSCs, and, importantly, appear to have a relatively balanced production of both VIM-2 and sLeX determinants. Collectively, these aforementioned variations in products created by α(1,3)-FTs as reported using mammalian cell lines and as we have observed in primary hMSCs can be reconciled by considering Golgi dynamics, including factors such as glycosyltransferase localization within specific Golgi microdomains (56) and/or cell-specific variation(s) in Golgi topography that impact glycan formation. Additionally, the accessibility of acceptor glycans within such microdomains, and intra-Golgi dynamics of competing glycosyltransferases and of glycosidases that can modify the same glycan acceptor, could alone and in combination amend the product profile(s) for any given glycosyltransferase within a given cell. In addition to such variations in Golgi dynamics, it is important to consider how phylogenetic disparities impact our understanding of the glycobiology of the α(1,3)-FTs. In this regard, an early study of Fut7 KO mice identified this α(1,3)-FT as key to creation of sLeX displayed on selectin ligands, emphasizing its role in regulation of leukocyte homing (57). Subsequently, it was determined that FT4 also had a role in this process, since Fut4/Fut7 double knockout mice yielded more severe leukocyte homing deficits (58–60). Importantly, while FT4, FT7, and FT9 are expressed in both rodents and primates, three of the six human α(1,3)-FUTs (FUT3, FUT5, and FUT6) arose from a (relatively) recent gene duplication event in primates and are not present in the rodent genome (61). Our study shows that FT3, FT5, and FT6, the members of the human α(1,3)-FUT gene family without functional homologs in mice, can each produce all three forms of terminally fucosylated lactosamines: LeX, sLeX, and difucosyl sLeX. Among these “primate FTs,” the FT3 and FT5 isoenzymes display the broadest product profiles as they can also generate the internally fucosylated VIM-2 epitope. In contrast, FTs-4, -7, and -9 exhibit the most restricted product specificities. Taken together, the stark contrast in α(1,3)-FT repertoire between human and murine systems emphasizes the importance of undertaking studies in human cells using human glycosyltransferases to appropriately understand human glycobiology.
Fucosylated terminal lactosaminyl glycans can be displayed on glycoprotein N-glycans or O-glycans or on GSLs. In prior studies of α(1,3)-FT product specificities, the carrier scaffold “preferences” were commonly not analyzed, although it is well-recognized that the scaffolds play important roles in modulating the function of the glycan moiety. For example, E-selectin ligand motifs (i.e. sLeX, VIM-2, and difucosyl sLeX) on O-glycans and N-glycans participate in initiating E-selectin-mediated tethering and rolling of human myeloid leukocytes (62), however, when presented on glycolipids, they also contribute measurably to stabilizing this interaction and critically facilitate the slow rolling of leukocytes on the endothelial cells (63). Using MS, we could comprehensively delineate the glycoconjugate repertoire of fucosylated terminal lactosaminyl glycans, distinguishing whether LeX, sLeX, and difucosyl sLeX were displayed on glycoprotein N-glycans, O-glycans, or GSLs for each α(1,3)-FUT transfectant. Our data indicate that, generally, LeX production by α(1,3)-FTs showed no significant preference(s) for one scaffold versus another, with the exception of FT5, which does not create LeX on GSLs (Fig. 6B). However, for sLeX, major variations in scaffold preference were observed: FT6 is the only α(1,3)-FT specialized to construct sLeX on all three types of glycoconjugate scaffolds, and, contrary to a prior report indicating that human FUT4 transfection in CHO cells robustly creates sLeX preferentially on glycolipids (51), flow cytometry did not detect any significant level of sLeX expression on hMSC FUT4 transfectants; MS analysis of FT4 products revealed no sLeX on glycolipids and only trace sLeX on O- and N-glycans, whereas FT4 generated LeX robustly on all types of glycoconjugates. Additionally, in hMSCs, difucosyl sLeX was detected solely on N-glycans of FUT3, FUT5, FUT6, and FUT7 transfectants, but others have reported that this structure resides abundantly on GSLs (64).
Our study presents the first systematic approach not only to operationally define the human α(1,3)-FTs within the context of a primary cell, but also to elucidate how these various isoenzymes shape lactosaminyl glycan biosynthesis within a given cell type. Importantly, the pattern of glycans and their associated glycoconjugates produced by each FT was uniformly consistent among the numerous marrow-derived hMSC cultures for that distinct FUT modRNA transfectant, indicating that modRNA transfection yields reproducibly functional glycosyltransferases that operate with high fidelity and, additionally, that hMSCs possess a characteristic and well-defined lactosaminyl glycan “glycosignature.” The in-depth characterization of product specificities of the individual human α(1,3)-FTs provided by our work now offers a useful foundation for designing novel strategies for utilizing these glycosyltransferases, individually or in combination, to custom-engineer fucosylated lactosaminyl glycans for further elucidation of the role(s) of these structures in cell biology and for potential therapeutic purposes. In particular, hMSCs have drawn immense attention for clinical applications, as they natively possess considerable immunomodulatory properties, support tissue regeneration, and can themselves differentiate into various cell types essential for normal physiology (e.g. osteoblasts). Notably, although our results identify several differences in the human α(1,3)-FT product profiles between previously tested cell lines and hMSCs, many of our results broadly conform to the earlier findings. These disparities are reflective of the differences in the underlying biology of the cell types. Thus, while we recognize that the observed hMSC product profiles of the fucosyltransferases cannot be used as a broad standard per se, our data highlight a very important aspect of glycobiology, which we believe cannot be overstated: one must not overlook host cell biology nor the nonphysiologic nature of in vitro assays in establishing algorithms predicting glycosyltransferase product profiles in vivo.
Materials and methods
Isolation and culture of hMSCs
Human cells were obtained and used in accordance with the Declaration of Helsinki and with the procedures approved by Dana Farber/Harvard Cancer Center (DF/HCC) Institutional Review Board (IRB). Bone marrow cells from normal human donors were obtained from discarded bone marrow filter sets by flushing with PBS plus 10 units/ml of heparin (Hospira, Lake Forest, IL). The cells were then gradiented using Ficoll-Histopaque 1.077 (Sigma) and the mononuclear fraction was collected. The cells were suspended in MSC media, consisting of Dulbecco's modified Eagle's medium low glucose (1 g/liter) supplemented with 10% fetal bovine serum from select lots (Atlanta Biologicals, Atlanta, GA), 100 units/ml of penicillin, and 100 units/ml of streptomycin. Approximately 5 × 108 cells were seeded into T-175 tissue culture flasks and incubated overnight in a humidified incubator at 37 °C, 20% O2, 5% CO2. The next day, all nonadherent cells were removed and the flasks were rinsed several times with PBS. The adherent cells were cultured in fresh MSC media, with a full media change twice per week. When the cells approached 80% confluence, cells were harvested with 0.05% trypsin, 0.5 mm EDTA, washed with PBS for experimental use and/or passaged by diluting 3- to 5-fold in fresh MSC media. Cells were cultured for a maximum of 6 passages before discarding.
Quantitative RT-PCR
Total cellular RNA was purified from bone marrow-derived hMSCs, using RNeasy microkit (Qiagen, Hilden, Germany) as per the manufacturer's instructions. Total RNA was reverse transcribed to synthesize first strand cDNA using a iScript cDNA synthesis kit (Bio-Rad) with heat cycles as follows: 25 °C for 10 min, 42 °C for 60 min, and 85 °C for 5 min. For some experiments, a SuperScript VILO cDNA conversion kit (Invitrogen) was used for first strand synthesis using identical thermal cycling conditions. Quantitative real time PCR was performed with specific primers to amplify glycosyltransferase genes (Table S1) using the SYBR Select master mix (Applied Biosystems, Foster City, CA) and a StepOne Plus PCR detection system (Applied Biosystems). PCR for individual genes were performed in triplicate with each reaction containing 1% (v/v) of the total cDNA obtained. The following cycling condition was employed: initial activation at 95 °C for 20 s, followed by 40 cycles of 95 °C for 10 s, and 60 °C for 30 s. Post-amplification, melt curve analysis was performed to ensure primer binding specificity according to the following conditions: 95 °C for 15 s, 60 °C for 1 min, 3 °C increment every 15 s to reach 95 °C. Gene expression levels were evaluated relative to glyceraldehyde-3-phosphate dehydrogenase using comparative CT methods. In some cases, agarose gel electrophoresis was performed with the PCR product to confirm the size of the amplicon. To this end, PCR products were resolved in a 1.3% agarose gel with 0.2–0.5 μg/ml of ethidium bromide. DNA fragments were visualized and imaged using UV illumination (Alphaimager EC imaging system, Alpha-Innotech, San Leandro, CA).
Cloning of FUTs
Full-length human fucosyltransferase cDNAs were purchased from Open Biosystems (GenBank accession numbers for FUT-3, -4, -5, -6, -7, and -9 cDNAs are BC108675, BC136374, BC140905, BC061700, BC074746, BC036101, respectively) and amplified by PCR using HiFi Hotstart (KAPA Biosystems, Wilmington, MA). PCR products were then subcloned into the pORFIN plasmid (65), which contains the T7 promoter, 5′ UTR and 3′ UTR required for eventual modified mRNA production, using a Quick Ligation Kit (New England Biolabs, Ipswich, MA). The PCR products were first separated using an agarose gel. The products with expected size were gel-purified using QIAquick Gel Extraction Kit (Qiagen) and sequenced to check for mutations before ligation into pORFIN. PCR primer sequences used for cDNA amplification were as follows: FUT3 forward primer: AAAAGCGGCCGCCATGGATCCCCTGGGTGCA, reverse primer: AAAAAGATCTTCAGGTGAACCAAGCCGCT; FUT4 long form forward primer: TGAGGCGCTTGTGGGGC, reverse primer: AAAAGGATCCTCACCGCTCGAACCAGCTG; FUT4 short form forward primer: TGGGGGCACCGTGGGGCT, reverse primer: AAAAGGATCCTCACCGCTCGAACCAGCTG; FUT5 forward primer: AAAAGCGGCCGCCATGGATCCCCTGGGCCCA, reverse primer: AAAAAGATCTTCAGGTGAACCAAGCCGCTA; FUT6 forward primer: AAAAGCGGCCGCCATGGATCCCCTGGGCC, reverse primer: AAAAAGATCTTCAGGTGAACCAAGCCGCT; FUT7 forward primer: AAAACGGCCGCATGAATAATGCTGGGCACGGC, reverse primer: AAAAGGATCCTCAGGCCTGAAACCAACCCT; FUT9 forward primer: AAAAGCGGCCGCCATGACATCAACATCCAAAGG, reverse primer: AAAAGGATCCTTAATTCCAAAACCATTTCTCTAA.
Modified mRNA synthesis
Modified mRNAs (modRNAs) were synthesized as described previously (65). Briefly, human FUT cDNAs cloned in the pORFIN vector were amplified by PCR using HiFi Hotstart (KAPA Biosystems) to generate templates for in vitro transcription. 1.6 μg of the PCR products (including the human FUT open reading frames and 5′- and 3′-untranslated regions) were then used as templates for modRNA synthesis using the MEGAscript T7 kit (Ambion). 3′-0-Me-m7G(5′)ppp(5′)G ARCA cap analog (New England Biolabs), adenosine triphosphate, guanosine triphosphate (U. S. Biochemical Corp.), 5-methylcytidine triphosphate and pseudouridine triphosphate (TriLink Biotechnologies, San Diego, CA) were used for in vitro transcription. Synthesized modRNA product was purified using MEGAclear spin columns (Ambion), and aliquots were stored frozen for future use at −80 °C.
Modified mRNA transfection of hMSCs
modRNAs were transfected into hMSCs using Stemfect (Stemgent, Lexington, MA) as per the manufacturer's instructions. Briefly, 1 μg of modRNA and 2 μl of Stemfect reagent were individually diluted into Stemfect buffer, mixed together, and incubated for 15 min at room temperature. The mixture was then added to 1 × 106 recently harvested hMSCs that were suspended in 2 ml of MSC medium, and incubated at 37 °C for 1 h, inverting the tube at 15-min intervals. The cells were then diluted in MSC media supplemented with 200 ng/ml of interferon inhibitor (B18R, eBioscience) and cultured in tissue culture flasks for 2 additional days prior to harvest and downstream analysis.
FT6 exofucosylation and surface sialidase treatment of hMSCs
Human MSCs were harvested with 0.05% trypsin, 0.5 mm EDTA and washed twice with PBS. For sialidase treatment, cells were resuspended at 1 × 107 cells/ml in Hanks' balanced salt solution, 0.1% BSA (Sigma) and 0.1 units/ml of either A. ureafaciens neuraminidase (Sigma), or recombinant M. decora neuraminidase (Calbiochem), and incubated for 45 min at 37 °C. For exofucosylation, hMSCs were resuspended at 1 × 107 cells/ml in Hanks' balanced salt solution, 0.1% human serum albumin (Sigma), 20 mm HEPES (Gibco), 1 mm GDP-fucose (Carbosynth, Compton, UK), and 60 μg/ml of purified FT6 enzyme (38), and incubated for 1 h at 37 °C. After incubation, cells were washed with PBS and subject to downstream analysis.
Flow cytometry
Individual wells of 96-well plates were pre-loaded with 3 μl of anti-LeX/CD15-FITC (clone HI98, Biolegend, San Diego, CA), 3 μl of anti-sLeX/CD15s-A488 (clone CSLEX1, BD Pharmingen), 20 μl anti-CD65s-FITC (clone VIM-2, Bio-Rad), 1 μl of mAb FH6 (binding to difucosyl sLeX, Biolegend), 0.1 μl of ECA-FITC (Erythrina cristagalli lectin, binding to unsialylated type 2 lactosamine, Vector Labs, Burlingame, CA), 0.1 μl of SNA-FITC (Sambucus nigra lectin, binding to α(2,6)-linked sialic acid, Vector Labs), or 0.1 μl of MALII-biotin (Maackia amurensis lectin II, binding to α(2,3)-linked sialic acid, Vector Labs). hMSCs were harvested, washed, and resuspended in PBS plus 0.1% BSA at 1 × 106 cells/ml. 50 μl of cell suspension was added to each well and incubated at 4 °C for 30 min. The plate was washed with 200 μl of PBS + BSA per well. Cell pellets were resuspended in PBS + BSA (for directly conjugated antibodies), or secondary antibodies as follows: for FH6, 1 μl of FITC-conjugated goat anti-mouse Ig (Southern Biotech, Birmingham, AL); for MALII, 1 μl of streptavidin-FITC (Biolegend). After incubation for 20 min, the plate was washed again with 200 μl/well of PBS + BSA and resuspended in 200 μl of PBS. Fluorescence intensity was determined using a Cytomics FC 500 MPL flow cytometer (Beckman Coulter, Brea, CA).
Glycan release and processing
hMSCs were washed with PBS and cell pellets were frozen until use. Cell pellets were thawed, dried, and transferred to 13 × 100-mm glass test tubes. Lipids and glycosphingolipids were extracted with 2:1 HPLC-grade chloroform/methanol. The lipid extracts were dried down and purified and fractionated by silica solid phase extraction (Grace Alltech), as described previously (66). The dried protein pellet was digested with trypsin and chymotrypsin (Sigma), as described previously (67). Following heat-inactivation, N-glycans were released with N-glycanase (Prozyme, Hayward, CA) at 37 °C for 48 h. Released N-glycans were separated from peptides and O-glycopeptides via C18 solid phase extraction (Sep-Pak, Waters, Milford, MA), as described previously (68). Purified, released N-glycans were reduced via borane/ammonia reduction, as described earlier (69). The reduced N-glycan pool was further purified via graphitized carbon solid phase extraction (Envi-Carb, Supelco, Bellefonte, PA). O-Glycans were released from the dried peptide/O-glycopeptide fraction via reductive-elimination, as described earlier (70). Released O-glycans were purified via cation exchange and porous graphitized carbon solid phase extraction. Intact glycosphingolipid fractions, N-glycan fractions, and O-glycans were permethylated via spin column permethylation (Harvard Apparatus, Holliston, MA), as described earlier (71). Permethylated glycans were purified via liquid-liquid extraction with dichloromethane (EMD), 0.5 m aqueous sodium chloride (Fisher). Extracted permethylated glycoprotein glycans were dried and reconstituted in 50:50 HPLC-grade methanol/water for MS analysis. Intact permethylated glycosphingolipids were dried and reconstituted with 2:1 HPLC-grade methanol/water for MS analysis.
Mass spectrometry
Direct infusion MS experiments were performed using an LTQ (Thermo; ion-trap instrument) equipped with a Nanomate (Advion, Ithaca, NY). Each derived fraction (GSL, O-glycan, and N-glycan) was infused separately and analyzed independently. Normalized collision energy was set to 35% for the LTQ, with activation q set to 0.250 and activation time set to 30 ms. Peak selection for sequential disassembly was made manually; isolation widths were generally set to 2.0 m/z, and isolation was centered at m/z values ∼0.5–1.0 units higher than the monoisotopic peak to capture the entire isotopic envelope. All ions are sodium adducts, unless otherwise noted. Interpretation of mass spectral data sets was made manually. Where relevant and possible, and when standard materials and mass spectra were available, the unknown substructure spectra were compared with standard spectra, as described previously (45, 72). For each spectrum, normalization level and signal average time are indicated. Liquid chromatography-MS analysis of O-glycans was performed using a reversed-phase (C18) column (Thermo BDS Hypersil, 1 × 150 mm) with a 0.1% formic acid/acetonitrile gradient to elute the reduced and permethylated O-glycans. A Surveyor MS pump and Surveyor autosampler were employed in conjunction with a VelosPro mass spectrometer (all from Thermo). Peak areas of molecular ions were used for relative quantitation. Detailed structure analysis and confirmation were made by direct infusion MSn.
Author contributions
N. M., B. D., J. L., D. J. A., V. N. R., D. J. R., and R. S. designed experiments; N. M., B. D., J. L., and D. J. A. performed experiments and collected data, with N. M., B. D., and J. L. contributing equally to research efforts; N. M., B. D., J. L., D. J. A., D. J. R., and R. S. analyzed and interpreted data; N. M. and R. S. wrote the manuscript, with content contributions by B. D., J. L., and D. J. A.; R. S. conceived the research, supervised all investigative efforts, and approved the final manuscript.
Supplementary Material
Acknowledgments
We acknowledge the MGH Bone Marrow Transplant Cellular Processing Laboratory for providing discarded bone marrow harvest filters, and Dr. Steven Barthel for helpful scientific discussions.
D. J. R. is a founder of Moderna Therapeutics, Cambridge, MA, a company that is developing modified-mRNA therapeutics. According to National Institutes of Health policies and procedures, the Brigham & Women's Hospital has assigned intellectual property rights regarding cell-surface glycan engineering to R. S., and R. S. has licensed portions of this technology to an entity he has founded (Warrior Therapeutics, LLC). R. S.'s ownership interests were reviewed and are managed by the Brigham & Women's Hospital and Partners HealthCare in accordance with their conflict of interest policy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7 and Tables S1 and S2.
- LeX
- Lewis-X
- sLeX
- sialyl Lewis-X
- α(1,3)-FT
- α(1,3)-fucosyltransferase
- hMSC
- human mesenchymal stem cell
- modRNA
- modified-mRNA
- GSL
- glycosphingolipid
- qRT-PCR
- quantitative real-time PCR
- FUT
- fucosyltransferase
- ANOVA
- analysis of variance
- ST
- sialyltransferase
- FUT
- fucosyltransferase gene
- FT
- fucosyltransferase protein.
References
- 1. Sackstein R. (2009) Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: engineering a roadmap for cell migration. Immunol. Rev. 230, 51–74 10.1111/j.1600-065X.2009.00792.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gooi H. C., Feizi T., Kapadia A., Knowles B. B., Solter D., and Evans M. J. (1981) Stage-specific embryonic antigen involves α1 goes to 3 fucosylated type 2 blood group chains. Nature 292, 156–158 10.1038/292156a0 [DOI] [PubMed] [Google Scholar]
- 3. Solter D., and Knowles B. B. (1978) Monoclonal antibody defining a stage-specific mouse embryonic antigen (SSEA-1). Proc. Natl. Acad. Sci. U.S.A. 75, 5565–5569 10.1073/pnas.75.11.5565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fenderson B. A., Zehavi U., and Hakomori S. (1984) A multivalent lacto-N-fucopentaose III-lysyllysine conjugate decompacts preimplantation mouse embryos, while the free oligosaccharide is ineffective. J. Exp. Med. 160, 1591–1596 10.1084/jem.160.5.1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Capela A., and Temple S. (2002) LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron 35, 865–875 10.1016/S0896-6273(02)00835-8 [DOI] [PubMed] [Google Scholar]
- 6. Klassen H., Schwartz M. R., Bailey A. H., and Young M. J. (2001) Surface markers expressed by multipotent human and mouse neural progenitor cells include tetraspanins and non-protein epitopes. Neurosci. Lett. 312, 180–182 10.1016/S0304-3940(01)02215-7 [DOI] [PubMed] [Google Scholar]
- 7. Yanagisawa M., Taga T., Nakamura K., Ariga T., and Yu R. K. (2005) Characterization of glycoconjugate antigens in mouse embryonic neural precursor cells. J. Neurochem. 95, 1311–1320 10.1111/j.1471-4159.2005.03452.x [DOI] [PubMed] [Google Scholar]
- 8. Pruszak J., Ludwig W., Blak A., Alavian K., and Isacson O. (2009) CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells 27, 2928–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yagi H., Saito T., Yanagisawa M., Yu R. K., and Kato K. (2012) Lewis X-carrying N-glycans regulate the proliferation of mouse embryonic neural stem cells via the Notch signaling pathway. J. Biol. Chem. 287, 24356–24364 10.1074/jbc.M112.365643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Gisbergen K. P., Sanchez-Hernandez M., Geijtenbeek T. B., and van Kooyk Y. (2005) Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 201, 1281–1292 10.1084/jem.20041276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gruss H. J., and Kadin M. E. (1996) Pathophysiology of Hodgkin's disease: functional and molecular aspects. Baillieres Clin. Haematol. 9, 417–446 10.1016/S0950-3536(96)80019-9 [DOI] [PubMed] [Google Scholar]
- 12. Read T. A., Fogarty M. P., Markant S. L., McLendon R. E., Wei Z., Ellison D. W., Febbo P. G., and Wechsler-Reya R. J. (2009) Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 15, 135–147 10.1016/j.ccr.2008.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seidmann L., Anspach L., and Roth W. (2016) The embryo-placental CD15-positive “vasculogenic zones” as a source of propranolol-sensitive pediatric vascular tumors. Placenta 38, 93–99 10.1016/j.placenta.2015.12.013 [DOI] [PubMed] [Google Scholar]
- 14. Foxall C., Watson S. R., Dowbenko D., Fennie C., Lasky L. A., Kiso M., Hasegawa A., Asa D., and Brandley B. K. (1992) The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J. Cell Biol. 117, 895–902 10.1083/jcb.117.4.895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sackstein R. (2004) The bone marrow is akin to skin: HCELL and the biology of hematopoietic stem cell homing. J. Invest. Dermatol. 122, 1061–1069 10.1111/j.0022-202X.2004.09301.x [DOI] [PubMed] [Google Scholar]
- 16. Merzaban J. S., Burdick M. M., Gadhoum S. Z., Dagia N. M., Chu J. T., Fuhlbrigge R. C., and Sackstein R. (2011) Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood 118, 1774–1783 10.1182/blood-2010-11-320705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silva M., Fung R. K., Donnelly C. B., Videira P. A., and Sackstein R. (2017) Cell-specific variation in E-selectin ligand expression among human peripheral blood mononuclear cells: implications for immunosurveillance and pathobiology. J. Immunol. 198, 3576–3587 10.4049/jimmunol.1601636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Julien S., Ivetic A., Grigoriadis A., QiZe D., Burford B., Sproviero D., Picco G., Gillett C., Papp S. L., Schaffer L., Tutt A., Taylor-Papadimitriou J., Pinder S. E., and Burchell J. M. (2011) Selectin ligand sialyl-Lewis x antigen drives metastasis of hormone-dependent breast cancers. Cancer Res. 71, 7683–7693 10.1158/0008-5472.CAN-11-1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liang J. X., Liang Y., and Gao W. (2016) Clinicopathological and prognostic significance of sialyl Lewis X overexpression in patients with cancer: a meta-analysis. Onco Targets Ther. 9, 3113–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. St. Hill C. A. (2011) Interactions between endothelial selectins and cancer cells regulate metastasis. Front. Biosci. 16, 3233–3251 10.2741/3909 [DOI] [PubMed] [Google Scholar]
- 21. Handa K., Stroud M. R., and Hakomori S. (1997) Sialosyl-fucosyl poly-LacNAc without the sialosyl-Lex epitope as the physiological myeloid cell ligand in E-selectin-dependent adhesion: studies under static and dynamic flow conditions. Biochemistry 36, 12412–12420 10.1021/bi971181t [DOI] [PubMed] [Google Scholar]
- 22. Tiemeyer M., Swiedler S. J., Ishihara M., Moreland M., Schweingruber H., Hirtzer P., and Brandley B. K. (1991) Carbohydrate ligands for endothelial-leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. U.S.A. 88, 1138–1142 10.1073/pnas.88.4.1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kukowska-Latallo J. F., Larsen R. D., Nair R. P., and Lowe J. B. (1990) A cloned human cDNA determines expression of a mouse stage-specific embryonic antigen and the Lewis blood group α(1,3/1,4)fucosyltransferase. Genes Dev. 4, 1288–1303 10.1101/gad.4.8.1288 [DOI] [PubMed] [Google Scholar]
- 24. Lowe J. B., Kukowska-Latallo J. F., Nair R. P., Larsen R. D., Marks R. M., Macher B. A., Kelly R. J., and Ernst L. K. (1991) Molecular cloning of a human fucosyltransferase gene that determines expression of the Lewis X and VIM-2 epitopes but not ELAM-1-dependent cell adhesion. J. Biol. Chem. 266, 17467–17477 [PubMed] [Google Scholar]
- 25. Goelz S. E., Hession C., Goff D., Griffiths B., Tizard R., Newman B., Chi-Rosso G., and Lobb R. (1990) ELFT: a gene that directs the expression of an ELAM-1 ligand. Cell 63, 1349–1356 10.1016/0092-8674(90)90430-M [DOI] [PubMed] [Google Scholar]
- 26. Weston B. W., Nair R. P., Larsen R. D., and Lowe J. B. (1992) Isolation of a novel human α(1,3)-fucosyltransferase gene and molecular comparison to the human Lewis blood group α(1,3/1,4)-fucosyltransferase gene: syntenic, homologous, nonallelic genes encoding enzymes with distinct acceptor substrate specificities. J. Biol. Chem. 267, 4152–4160 [PubMed] [Google Scholar]
- 27. Weston B. W., Smith P. L., Kelly R. J., and Lowe J. B. (1992) Molecular cloning of a fourth member of a human α(1,3)-fucosyltransferase gene family: multiple homologous sequences that determine expression of the Lewis X, sialyl Lewis X, and difucosyl sialyl Lewis x epitopes. J. Biol. Chem. 267, 24575–24584 [PubMed] [Google Scholar]
- 28. Natsuka S., Gersten K. M., Zenita K., Kannagi R., and Lowe J. B. (1994) Molecular cloning of a cDNA encoding a novel human leukocyte α-1,3-fucosyltransferase capable of synthesizing the sialyl Lewis x determinant. J. Biol. Chem. 269, 16789–16794 [PubMed] [Google Scholar]
- 29. Kaneko M., Kudo T., Iwasaki H., Ikehara Y., Nishihara S., Nakagawa S., Sasaki K., Shiina T., Inoko H., Saitou N., and Narimatsu H. (1999) α1,3-Fucosyltransferase IX (Fuc-TIX) is very highly conserved between human and mouse: molecular cloning, characterization and tissue distribution of human Fuc-TIX. FEBS Lett. 452, 237–342 10.1016/S0014-5793(99)00640-7 [DOI] [PubMed] [Google Scholar]
- 30. Sasaki K., Kurata K., Funayama K., Nagata M., Watanabe E., Ohta S., Hanai N., and Nishi T. (1994) Expression cloning of a novel α1,3-fucosyltransferase that is involved in biosynthesis of the sialyl Lewis X carbohydrate determinants in leukocytes. J. Biol. Chem. 269, 14730–14737 [PubMed] [Google Scholar]
- 31. Kimura H., Shinya N., Nishihara S., Kaneko M., Irimura T., and Narimatsu H. (1997) Distinct substrate specificities of five human α-1,3-fucosyltransferases for in vivo synthesis of the sialyl Lewis X and Lewis X epitopes. Biochem. Biophys. Res. Commun. 237, 131–137 10.1006/bbrc.1997.7100 [DOI] [PubMed] [Google Scholar]
- 32. Cailleau-Thomas A., Coullin P., Candelier J. J., Balanzino L., Mennesson B., Oriol R., and Mollicone R. (2000) FUT4 and FUT9 genes are expressed early in human embryogenesis. Glycobiology 10, 789–802 10.1093/glycob/10.8.789 [DOI] [PubMed] [Google Scholar]
- 33. Niemelä R., Natunen J., Majuri M.-L., Maaheimo H., Helin J., Lowe J. B., Renkonen O., and Renkonen R. (1998) Complementary acceptor and site specificities of Fuc-TIV and Fuc-TVII allow effective biosynthesis of sialyl-TriLex and related polylactosamines present on glycoprotein counterreceptors of selectins. J. Biol. Chem. 273, 4021–4026 10.1074/jbc.273.7.4021 [DOI] [PubMed] [Google Scholar]
- 34. Nishihara S., Iwasaki H., Kaneko M., Tawada A., Ito M., and Narimatsu H. (1999) α1,3-Fucosyltransferase 9 (FUT9; Fuc-TIX) preferentially fucosylates the distal GlcNAc residue of polylactosamine chain while the other four α1,3FUT members preferentially fucosylate the inner GlcNAc residue. FEBS Lett. 462, 289–294 10.1016/S0014-5793(99)01549-5 [DOI] [PubMed] [Google Scholar]
- 35. Shetterly S., Jost F., Watson S. R., Knegtel R., Macher B. A., and Holmes E. H. (2007) Site-specific fucosylation of sialylated polylactosamines by α1,3/4-fucosyltransferases-V and -VI is defined by amino acids near the N terminus of the catalytic domain. J. Biol. Chem. 282, 24882–24892 10.1074/jbc.M702395200 [DOI] [PubMed] [Google Scholar]
- 36. Basu M., Hawes J. W., Li Z., Ghosh S., Khan F. A., Zhang B. J., and Basu S. (1991) Biosynthesis in vitro of SA-Lex and SA-diLex by α1–3-fucosyltransferases from colon carcinoma cells and embryonic brain tissues. Glycobiology 1, 527–535 10.1093/glycob/1.5.527 [DOI] [PubMed] [Google Scholar]
- 37. Becker D. J., and Lowe J. B. (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R 10.1093/glycob/cwg054 [DOI] [PubMed] [Google Scholar]
- 38. Dykstra B., Lee J., Mortensen L. J., Yu H., Wu Z. L., Lin C. P., Rossi D. J., and Sackstein R. (2016) Glycoengineering of E-selectin ligands by intracellular versus extracellular fucosylation differentially affects osteotropism of human mesenchymal stem cells. Stem Cells 34, 2501–2511 10.1002/stem.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Madeira C., Mendes R. D., Ribeiro S. C., Boura J. S., Aires-Barros M. R., da Silva C. L., and Cabral J. M. (2010) Nonviral gene delivery to mesenchymal stem cells using cationic liposomes for gene and cell therapy. J. Biomed. Biotechnol. 735349 10.1155/2010/735349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sackstein R., Merzaban J. S., Cain D. W., Dagia N. M., Spencer J. A., Lin C. P., and Wohlgemuth R. (2008) Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 14, 181–187 10.1038/nm1703 [DOI] [PubMed] [Google Scholar]
- 41. Geisler C., and Jarvis D. L. (2011) Letter to the Glyco-Forum: effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology 21, 988–993 10.1093/glycob/cwr080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bai X., Brown J. R., Varki A., and Esko J. D. (2001) Enhanced 3-O-sulfation of galactose in Asn-linked glycans and Maackia amurensis lectin binding in a new Chinese hamster ovary cell line. Glycobiology 11, 621–632 10.1093/glycob/11.8.621 [DOI] [PubMed] [Google Scholar]
- 43. Chou M.-Y., Li S.-C., and Li Y.-T. (1996) Cloning and expression of sialidase L, a NeuAcα2→3Gal-specific sialidase from the leech, Macrobdella decora. J. Biol. Chem. 271, 19219–19224 10.1074/jbc.271.32.19219 [DOI] [PubMed] [Google Scholar]
- 44. Kumar R., Potvin B., Muller W. A., and Stanley P. (1991) Cloning of a human α(1,3)-fucosyltransferase gene that encodes ELFT but does not confer ELAM-1 recognition on Chinese hamster ovary cell transfectants. J. Biol. Chem. 266, 21777–21783 [PubMed] [Google Scholar]
- 45. Ashline D. J., Hanneman A. J., Zhang H., and Reinhold V. N. (2014) Structural documentation of glycan epitopes: sequential mass spectrometry and spectral matching. J. Am. Soc. Mass Spectrom. 25, 444–453 10.1007/s13361-013-0776-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sackstein R. (2012) Re: ex vivo fucosylation improves human cord blood engraftment in NOD-SCID IL-2R null mice. Exp. Hematol. 40, 518–519 10.1016/j.exphem.2012.03.004 [DOI] [PubMed] [Google Scholar]
- 47. Mollicone R., Moore S. E., Bovin N., Garcia-Rosasco M., Candelier J.-J., Martinez-Duncker I., and Oriol R. (2009) Activity, splice variants, conserved peptide motifs, and phylogeny of two new α1,3-fucosyltransferase families (FUT10 and FUT11). J. Biol. Chem. 284, 4723–4738 10.1074/jbc.M809312200 [DOI] [PubMed] [Google Scholar]
- 48. Kumar A., Torii T., Ishino Y., Muraoka D., Yoshimura T., Togayachi A., Narimatsu H., Ikenaka K., and Hitoshi S. (2013) The Lewis X-related α1,3-fucosyltransferase, Fut10, Is required for the maintenance of stem cell populations. J. Biol. Chem. 288, 28859–28868 10.1074/jbc.M113.469403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barthel S. R., Wiese G. K., Cho J., Opperman M. J., Hays D. L., Siddiqui J., Pienta K. J., Furie B., and Dimitroff C. J. (2009) α1,3-Fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc. Natl. Acad. Sci. U.S.A. 106, 19491–19496 10.1073/pnas.0906074106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zöllner O., and Vestweber D. (1996) The E-selectin ligand-1 is selectively activated in Chinese hamster ovary cells by the α(1,3)-fucosyltransferases IV and VII. J. Biol. Chem. 271, 33002–33008 10.1074/jbc.271.51.33002 [DOI] [PubMed] [Google Scholar]
- 51. Huang M. C., Laskowska A., Vestweber D., and Wild M. K. (2002) The α(1,3)-fucosyltransferase Fuc-TIV, but not Fuc-TVII, generates sialyl Lewis X-like epitopes preferentially on glycolipids. J. Biol. Chem. 277, 47786–47795 10.1074/jbc.M208283200 [DOI] [PubMed] [Google Scholar]
- 52. Knibbs R. N., Craig R. A., Natsuka S., Chang A., Cameron M., Lowe J. B., and Stoolman L. M. (1996) The fucosyltransferase FucT-VII regulates E-selectin ligand synthesis in human T cells. J. Cell Biol. 133, 911–920 10.1083/jcb.133.4.911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakayama F., Nishihara S., Iwasaki H., Kudo T., Okubo R., Kaneko M., Nakamura M., Karube M., Sasaki K., and Narimatsu H. (2001) CD15 expression in mature granulocytes is determined by α1,3-fucosyltransferase IX, but in promyelocytes and monocytes by α1,3-fucosyltransferase IV. J. Biol. Chem. 276, 16100–16106 10.1074/jbc.M007272200 [DOI] [PubMed] [Google Scholar]
- 54. Patnaik S. K., Potvin B., and Stanley P. (2004) LEC12 and LEC29 gain-of-function Chinese hamster ovary mutants reveal mechanisms for regulating VIM-2 antigen synthesis and E-selectin binding. J. Biol. Chem. 279, 49716–49726 10.1074/jbc.M408755200 [DOI] [PubMed] [Google Scholar]
- 55. Buffone A. Jr., Mondal N., Gupta R., McHugh K. P., Lau J. T., and Neelamegham S. (2013) Silencing α1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J. Biol. Chem. 288, 1620–1633 10.1074/jbc.M112.400929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tu L., and Banfield D. K. (2010) Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. 67, 29–41 10.1007/s00018-009-0126-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Malýy P., Thall A., Petryniak B., Rogers C. E., Smith P. L., Marks R. M., Kelly R. J., Gersten K. M., Cheng G., Saunders T. L., Camper S. A., Camphausen R. T., Sullivan F. X., Isogai Y., Hindsgaul O., von Andrian U. H., and Lowe J. B. (1996) The α(1,3)-fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell 86, 643–653 10.1016/S0092-8674(00)80137-3 [DOI] [PubMed] [Google Scholar]
- 58. Homeister J. W., Daugherty A., and Lowe J. B. (2004) α(1,3)-Fucosyltransferases FucT-IV and FucT-VII control susceptibility to atherosclerosis in apolipoprotein E−/− mice. Arterioscler. Thromb. Vasc. Biol. 24, 1897–1903 10.1161/01.ATV.0000141844.28073.df [DOI] [PubMed] [Google Scholar]
- 59. Smithson G., Rogers C. E., Smith P. L., Scheidegger E. P., Petryniak B., Myers J. T., Kim D. S., Homeister J. W., and Lowe J. B. (2001) Fuc-TVII is required for T helper 1 and T cytotoxic 1 lymphocyte selectin ligand expression and recruitment in inflammation, and together with Fuc-TIV regulates naive T cell trafficking to lymph nodes. J. Exp. Med. 194, 601–614 10.1084/jem.194.5.601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Homeister J. W., Thall A. D., Petryniak B., Malý P., Rogers C. E., Smith P. L., Kelly R. J., Gersten K. M., Askari S. W., Cheng G., Smithson G, Marks R. M., Misra A. K., Hindsgaul O., von Andrian U. H., and Lowe J. B. (2001) The α(1,3)-fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity 15, 115–126 10.1016/S1074-7613(01)00166-2 [DOI] [PubMed] [Google Scholar]
- 61. Gersten K. M., Natsuka S., Trinchera M., Petryniak B., Kelly R. J., Hiraiwa N., Jenkins N. A., Gilbert D. J., Copeland N. G., and Lowe J. B. (1995) Molecular cloning, expression, chromosomal assignment, and tissue-specific expression of a murine α-(1,3)-fucosyltransferase locus corresponding to the human ELAM-1 ligand fucosyl transferase. J. Biol. Chem. 270, 25047–25056 10.1074/jbc.270.42.25047 [DOI] [PubMed] [Google Scholar]
- 62. Stolfa G., Mondal N., Zhu Y., Yu X., Buffone A. Jr, and Neelamegham S. (2016) Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and glycosphingolipids to human leukocyte-endothelium adhesion. Sci. Rep. 6, 30392 10.1038/srep30392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mondal N., Stolfa G., Antonopoulos A., Zhu Y., Wang S. S., Buffone A. Jr., Atilla-Gokcumen G. E., Haslam S. M., Dell A., and Neelamegham S. (2016) Glycosphingolipids on human myeloid cells stabilize E-selectin-dependent rolling in the multistep leukocyte adhesion cascade. Arteriosclerosis Thromb. Vasc. Biol. 36, 718–727 10.1161/ATVBAHA.115.306748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fukushi Y., Nudelman E., Levery S. B., Hakomori S., and Rauvala H. (1984) Novel fucolipids accumulating in human adenocarcinoma: III. a hybridoma antibody (FH6) defining a human cancer-associated difucoganglioside (VI3NeuAcV3III3Fuc2nLc6). J. Biol. Chem. 259, 10511–10517 [PubMed] [Google Scholar]
- 65. Mandal P. K., and Rossi D. J. (2013) Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat. Protoc. 8, 568–582 10.1038/nprot.2013.019 [DOI] [PubMed] [Google Scholar]
- 66. Garner B., Priestman D. A., Stocker R., Harvey D. J., Butters T. D., and Platt F. M. (2002) Increased glycosphingolipid levels in serum and aortae of apolipoprotein E gene knockout mice. J. Lipid Res. 43, 205–214 [PubMed] [Google Scholar]
- 67. Aoki K., Perlman M., Lim J. M., Cantu R., Wells L., and Tiemeyer M. (2007) Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 282, 9127–9142 10.1074/jbc.M606711200 [DOI] [PubMed] [Google Scholar]
- 68. Canis K., McKinnon T. A., Nowak A., Panico M., Morris H. R., Laffan M., and Dell A. (2010) The plasma von Willebrand factor O-glycome comprises a surprising variety of structures including ABH antigens and disialosyl motifs. J. Thromb. Haemost. 8, 137–145 10.1111/j.1538-7836.2009.03665.x [DOI] [PubMed] [Google Scholar]
- 69. Alley W. R. Jr., Madera M., Mechref Y., and Novotny M. V. (2010) Chip-based reversed-phase liquid chromatography-mass spectrometry of permethylated N-linked glycans: a potential methodology for cancer-biomarker discovery. Anal. Chem. 82, 5095–5106 10.1021/ac100131e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Carlson D. M. (1966) Oligosaccharides isolated from pig submaxillary mucin. J. Biol. Chem. 241, 2984–2986 [PubMed] [Google Scholar]
- 71. Kang P., Mechref Y., Klouckova I., and Novotny M. V. (2005) Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass. Spectrom 19, 3421–3428 10.1002/rcm.2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ashline D. J., Zhang H., and Reinhold V. N. (2017) Isomeric complexity of glycosylation documented by MSn. Anal. Bioanal. Chem. 409, 439–451 10.1007/s00216-016-0018-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.