

Abstract
A 64-year-old woman had fragility fractures which caused her to have gross deformities and confined her to bed. These were initially ascribed to vitamin D deficiency. However, despite correction of the deficiency, she did not improve. A review of previous records already showed glucosuria in the absence of diabetes, but this finding was overlooked. Eight years into the disease, it was realised that the glucosuria despite normal blood sugar could also mean that the patient was losing other substances needed for proper bone formation. Further investigations showed hypophosphataemia, renal phosphate wasting, hypokalaemia, mild metabolic acidosis, alkaline urine pH, hypouricaemia and aminoaciduria, all compatible with a proximal renal tubular defect (Fanconi syndrome). The fragility fractures were due to poor bone mineralisation because of hypophosphataemia induced by the inability of the kidneys to conserve phosphorus.
Keywords: calcium and bone, renal medicine, fluid electrolyte and acid-base disturbances, orthopaedics
Background
Fragility fractures lead to immobility and limit an individual’s ability to travel, a consequence that our patient finds most troublesome. Here, we present her condition that has been undiagnosed for 8 years. The delay in diagnosis is probably because the disease was ascribed to vitamin D deficiency alone despite the other findings that could not be explained by this nutritional deficiency. More importantly, not seeing the significance of glucosuria, which was present from the start despite the absence of diabetes, kept the diagnosis unrecognised for so long.
In this case, it was the finding of glucosuria in the absence of diabetes that led our current team of doctors to think that there could be a proximal tubular defect that could explain the glucosuria, hypophosphataemia and all other abnormalities. This paper is being reported to highlight that the key to diagnosis of metabolic bone disease lies not just in the usual testing for calcium, parathyroid hormone or vitamin D, but can be deduced by meticulously looking at a simple urinalysis.
Case presentation
A 64-year-old Filipino woman was admitted for recurrent fractures. Starting at the age of 56, she experienced difficulty in standing, developed a change in gait and noted loss of dentition. This was followed by recurrent, multiple fractures, resulting in deformities and inability to ambulate at age 61 (figure 1). These fractures, which involved the left arm and left thigh, were spontaneous and were not a result of trauma. Medical history was unremarkable save for an episode of hypertension during pregnancy. No similar illness was present in her family members. She used to work as a textile chemist for 28 years, being exposed to hydrogen peroxide and chromium-containing indigo dyes.
Figure 1.
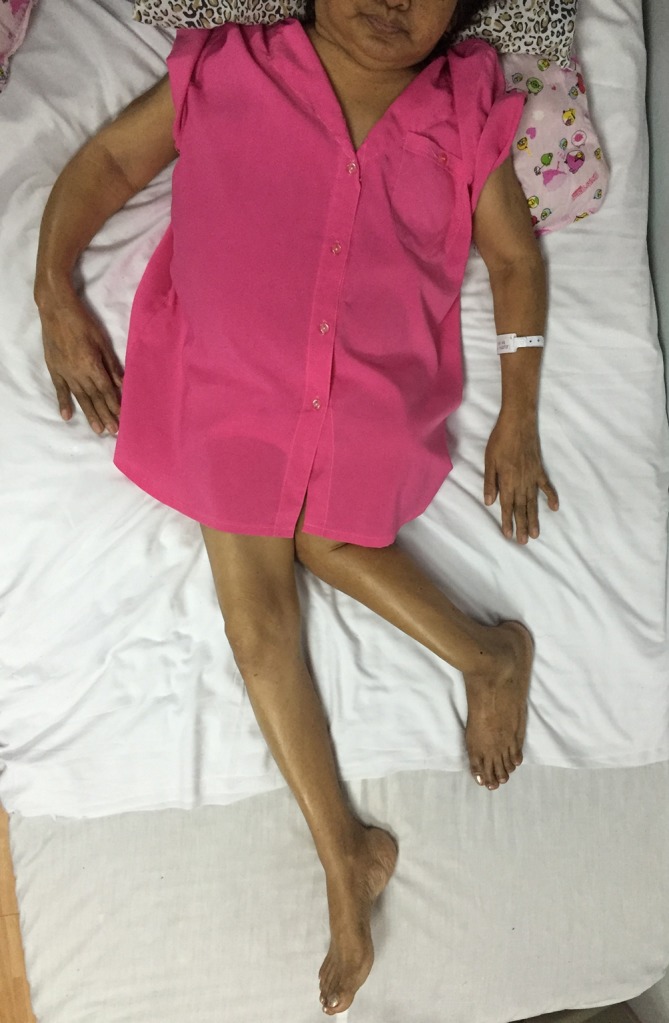
Patient is confined in bed in this supine position. Note the valgus deformity and the shorter appearance of the left lower extremity and the bowing deformity of the right forearm.
She had consulted various physicians during this 8-year period and was told to have osteoporosis from low vitamin D levels. The fractures, however, were recurrent despite intake of calcium, vitamin D and a bisphosphonate. She was admitted in our institution for completion of workup with the aim of arriving at a definite diagnosis.
On physical examination, she was confined to bed, but was awake, conversant, not in distress, not in pain and with normal vital signs. She was nearly edentulous with only three remaining teeth (three mandibular incisors). Her right forearm had a bowing deformity (figure 2). Her left thigh was shorter and stouter compared with the right. It was also internally rotated. The left leg was markedly shorter than the right and exhibited a valgus deformity (figure 3). There was no chest deformity. The costochondral junctions were not prominent.
Figure 2.
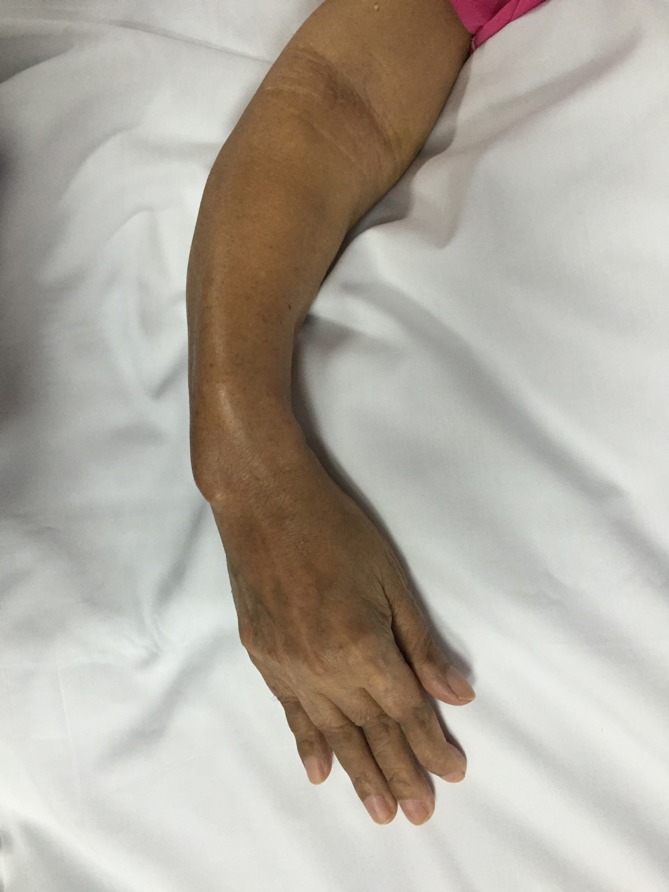
Bowing deformity of the right forearm.
Figure 3.
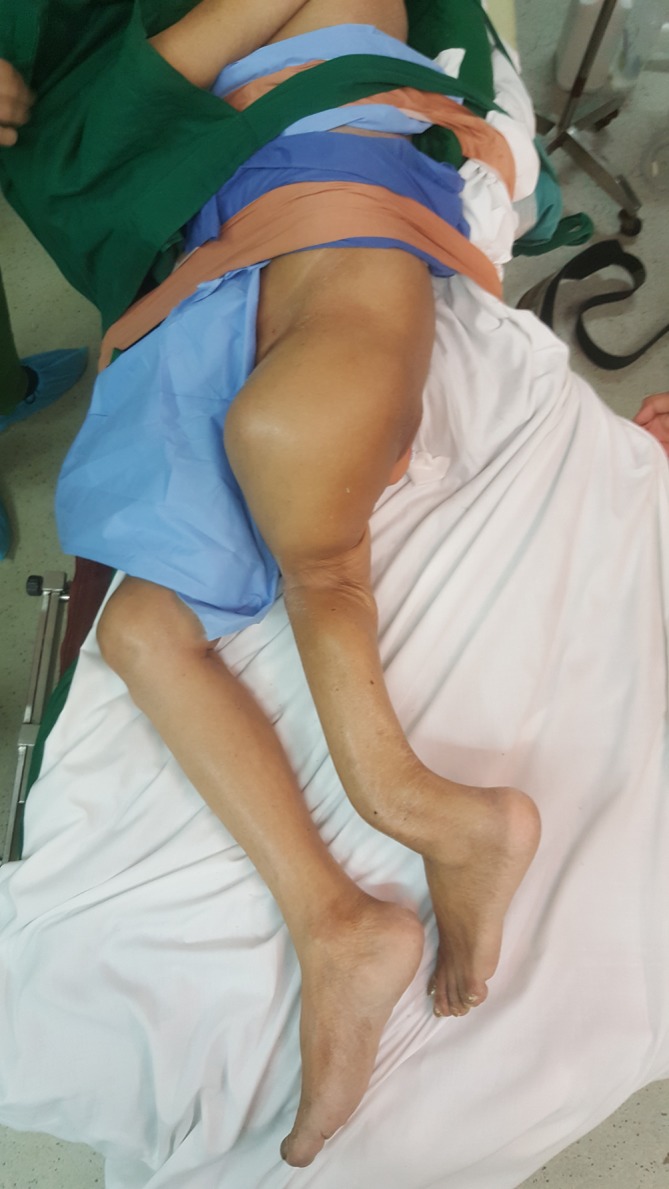
The left thigh is shorter than the right as evidenced by the asymmetric levels of the knees. The left thigh is also internally rotated. The left leg is also shorter than the right and has a gross deformity near the ankle.
Investigations
Skeletal survey X-rays showed a generalised decrease in bone density and demineralisation with marked thinning of the cortices of the visualised bones. Pertinent findings were as follows.
Skull: lucent and lytic defects in left parietal bone, marked loss of dentition (figure 4).
Spine: leftward deviation of the thoracolumbar spine, rightward deviation of the lower lumbar spine, compression deformities in the thoracolumbar and lumbosacral spine.
Upper extremities: fractures in the right proximal humerus (figure 5), midshaft of the left humerus with volar angulation (figure 6), segmental fracture of the right radius and ulna with dorsal angulation. Looser zones (insufficiency fractures) were seen in the right proximal humerus (figure 5) and left radius and ulna (figure 7).
Lower extremities: fracture of the left proximal femur with superomedial displacement of the distal fragment and the absence of calcified or mineralised callus, valgus deformity of the left knee (figure 8).
The epiphysial and metaphyseal regions are well developed with normal configuration. Growth lines are seen in the long bones. Lateral bowing and slight increase in the width of long bones were also seen.
Figure 4.
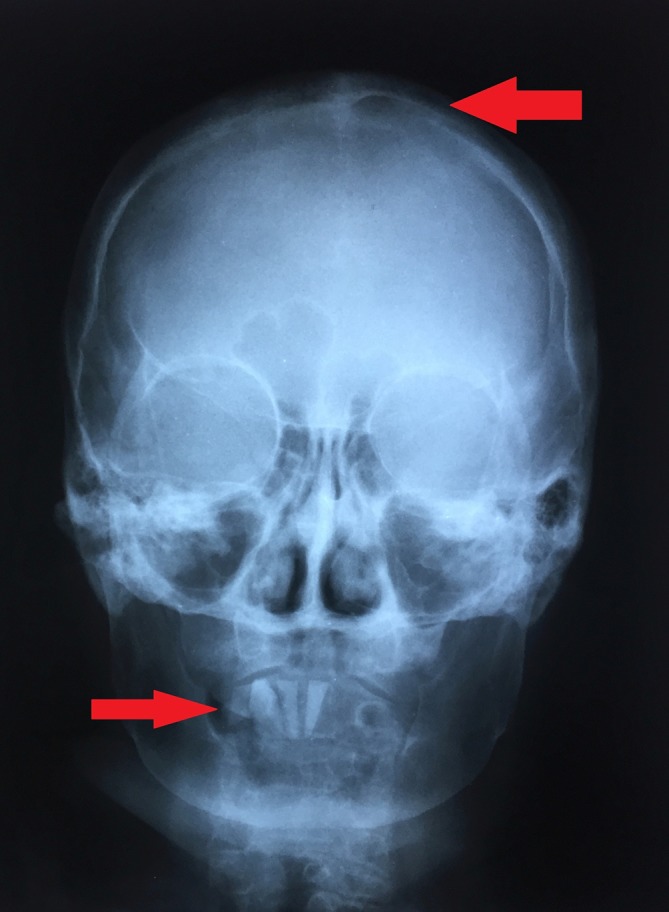
Skull X-ray shows a lytic lesion of the left parietal bone (top arrow) and marked loss of dentition with only three mandibular incisors remaining (bottom arrow).
Figure 5.
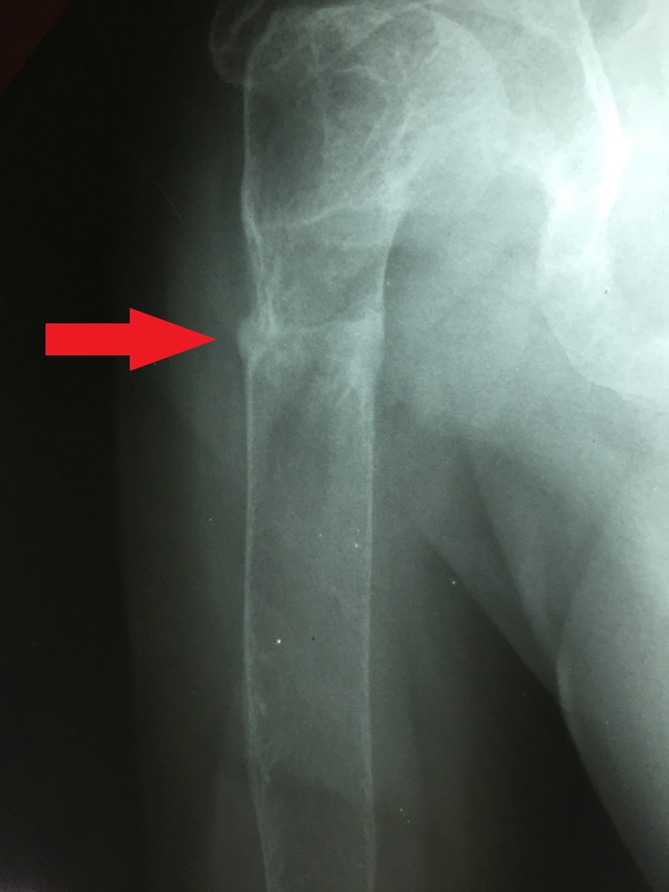
Insufficiency fracture or Looser zone (arrow) in the proximal right humerus.
Figure 6.
Fracture in the midshaft of the left humerus (arrow) resulting to volar angulation.
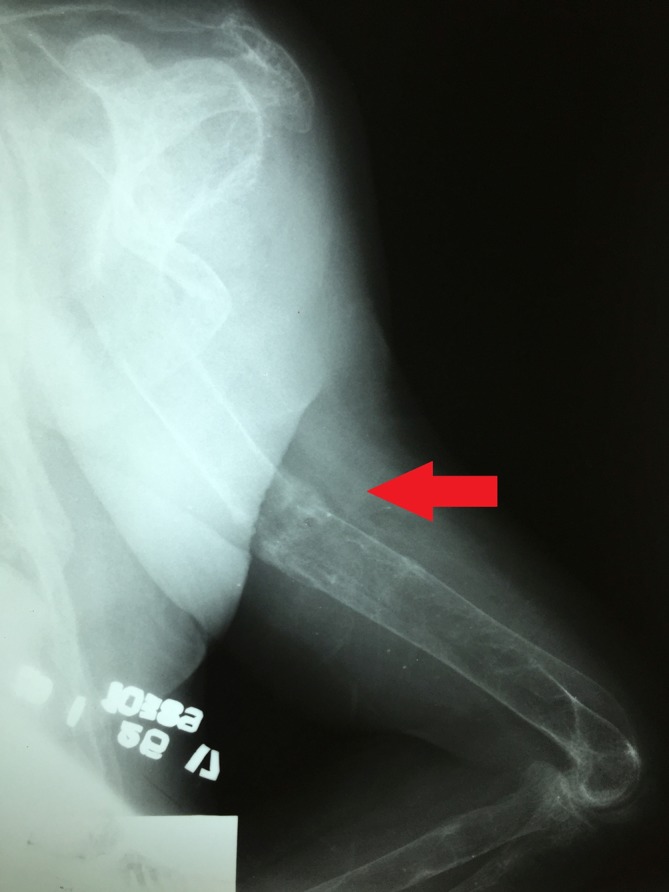
Figure 7.
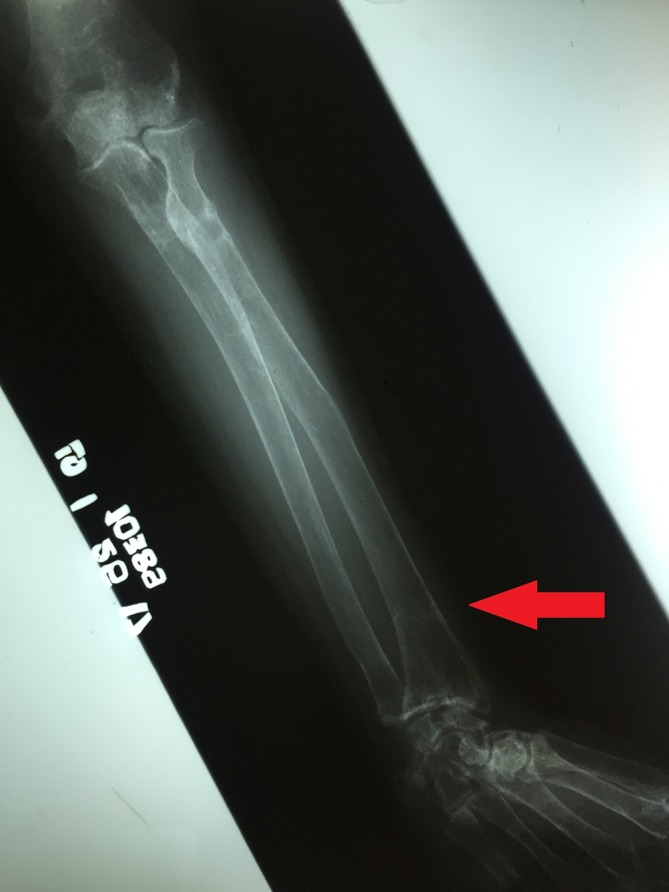
Insufficiency fracture or Looser zone (arrow) in the distal left radius.
Figure 8.
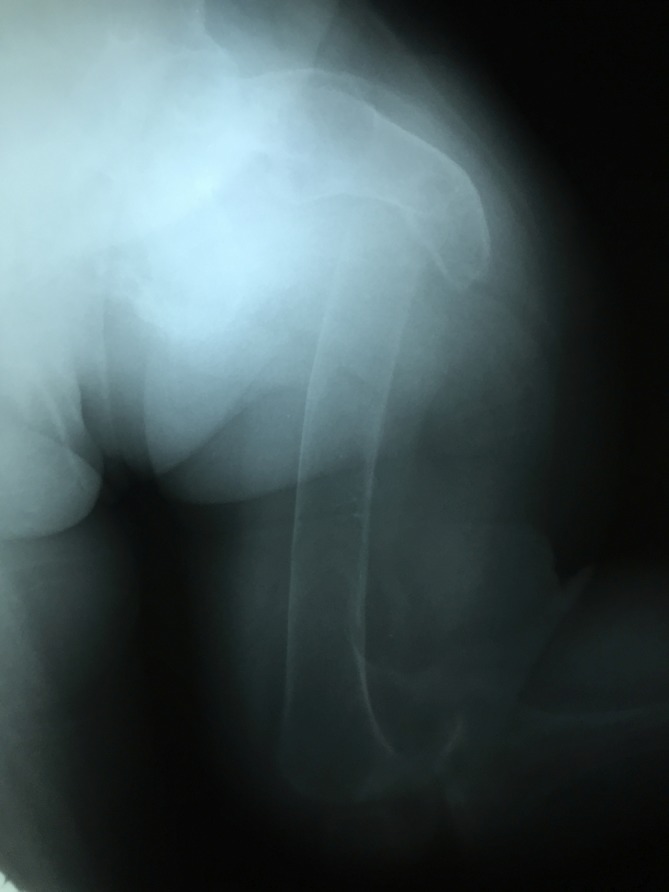
Fracture of the proximal left femur with flexion of the proximal segment and varus displacement and internal rotation of the distal lower extremity. There are signs of atrophic non-union at the fracture line.
A review of the various laboratory tests done over the preceding 8 years showed low to normal serum calcium (1.9 and 2.5 mmol/L) and low to low normal serum phosphorus (0.58, 0.68, 0.83 and 0.87 mmol/L), attributed to the low 25-OH vitamin D level (17 ng/mL). Intact parathyroid hormone was normal at 18.527 ng/mL.
With a daily intake of 3000 IU of vitamin D, her serum 25-OH vitamin D improved to 39 ng/mL (in 2014) then 63 ng/mL (in 2016), already within the normal range. Despite this and previous intake of a bisphosphonate, her fractures became recurrent.
Three previous urinalyses done in 2008, 2013 and 2016 already showed varying degrees of glucosuria (+1 to +3) despite the patient not having diabetes. The patient was also not taking a sodium glucose cotransporter 2 inhibitor, a diabetes medication that works by promoting urinary glucose excretion. In addition, urine was alkaline with a pH of 6.0–7.5 (table 1).
Table 1.
Urinalysis findings from 2008 to 2017
| Feature | Normal values | August 2008 | June 2013 | August 2016 | January 2017 (this admission) |
| Colour | Yellow | Light yellow | |||
| Turbidity | Clear | Hazy | Clear | ||
| Albumin | Negative | Trace | Negative | +2 | +1 |
| Ketones | Negative | Negative | Trace (0.5 mmol/L) | ||
| Glucose | Negative | +1 | +3 | +3 | +3 (28 mmol/L) |
| pH | 6 | 7 | 7.5 | 7 | |
| Specific gravity | 1.020 | 1.012 | |||
| Red blood cells | 0–9 μ/L | 1–2 | 4 | ||
| White blood cells | 0–22 μ/L | 3–6 | 13 | ||
| Epithelial cells | 0–13 μ/L | 9 | |||
| Casts | 308 |
Laboratory investigations during this admission (2017) confirmed the presence of glucosuria (+3) despite a normal fasting blood sugar of 5.1 mmol/L and a normal HbA1c of 5.2%. Other urine characteristics were pH of 7.0 and +1 albumin (table 1). With these urine abnormalities, our present team of doctors suspected that there could be a proximal renal tubular defect that could explain her condition.
Further investigations confirmed hypophosphataemia and revealed hypokalaemia, hypouricaemia and normal anion gap (anion gap of 11.6) metabolic acidosis. Serum calcium, creatinine, parathyroid hormone and thyroid function tests were normal. Serum alkaline phosphatase was elevated (table 2).
Table 2.
Blood chemistry and arterial blood gas results in 2017
| Results | Normal values | |
| Blood chemistry | ||
| Fasting blood sugar | 5.1 | 4.1–5.9 mmol/L |
| Haemoglobin A1c | 5.2 | <5.7% |
| Creatinine | 97–107 | 46–92 µmol/L |
| Sodium | 144 | 135–145 mmol/L |
| Potassium | 2.9–5.1 | 3.5–5.1 mmol/L |
| Chloride | 114 | 98–107 mmol/L |
| Calcium | 2.62 | 2.10–2.55 mmol/L |
| Albumin | 43 | 35–50 g/L |
| Ionised calcium | 1.20 | 1.15–1.33 mmol/L |
| Phosphorus | 0.62–0.84 | 0.81–1.49 mmol/L |
| Magnesium | 1.0–1.1 | 0.70–1.00 mmol/L |
| Uric acid | 0.092 | 0.149–0.369 mmol/L |
| Intact parathyroid hormone | 19.9 | 10–65 pg/mL |
| Alkaline phosphatase | 718 | 38–126 IU/L |
| Free thyroxine | 12.2 | 11–24 pmol/L |
| Thyroid stimulating hormone | 0.71 | 0.3–3.8 IU/mL |
| Serum chromium | 0.72 | <0.82 µg/L |
| Whole blood lead | 1.90 | <25 µg/dL |
| Arterial blood gas | ||
| pH | 7.375 | 7.35–7.45 |
| Partial pressure of carbon dioxide | 31.20 | 35–45 mm Hg |
| Partial pressure of oxygen | 94.5 | 80–100 mm Hg |
| Bicarbonate | 18.40 | 22–28 mEq/L |
| Base excess | −5.1 | |
| Oxygen saturation % | 97.1 | |
| Total carbondioxide | 19.4 | |
Chromatographic analysis of urine showed markedly elevated levels of many amino acids, ranging from 4× to 67×, the upper limit of their corresponding normal levels. Table 3 shows the different amino acids and their respective concentrations in urine. The presence of generalised aminoaciduria confirms the inability of the kidneys to reabsorb these amino acids as these are normally reabsorbed in the renal proximal convoluted tubule.
Table 3.
Amino acid concentration in urine determined by chromatography
| Amino acid | Measured amount | Reference values (µmol/mmol creatinine) |
| 1-Methyl-histidine | 84 | 5–125 |
| 3-Methyl-histidine | 48 | 8–75 |
| Alanine | 1204 | 16–68 |
| Arginine | 135 | <5 |
| Asparagine | 322 | |
| Aspartic acid | <6 | 2–7 |
| Beta aminobutyric acid | 71 | <91 |
| Citrulline | 269 | <4 |
| Cystine | 180 | 3–17 |
| Glutamic acid | <6 | <12 |
| Glutamine | 2107 | 20–76 |
| Glycine | 1353 | 43–173 |
| Histidine | 559 | 26–153 |
| Hydroxyproline | 164 | <13 |
| Isoleucine | 53 | <4 |
| Leucine | 117 | 2–11 |
| Lysine | 727 | 7–58 |
| Methionine | 21 | 2–16 |
| Ornithine | 218 | <5 |
| Phenylalanine | 194 | 2–19 |
| Proline | 379 | <9 |
| Serine | 499 | 21–50 |
| Taurine | 149 | 16–180 |
| Threonine | 439 | 7–29 |
| Tyrosine | 258 | 2–23 |
| Valine | 302 | 3–13 |
Ultrasound showed bilateral renal parenchymal disease and nephrolithiasis on the right.
To demonstrate renal phosphate wasting, the fractional excretion of filtered phosphate and the tubular maximum reabsorption of phosphate corrected for glomerular filtration rate (TmP/GFR) were computed using the following formulae1
Our computations showed a high fractional excretion of phosphate at 0.26 (normal 0.10–0.20) and a low TmP/GFR at 0.54 mmol/L (reference range for sex and age: 0.87–1.40). These mean that there is low renal tubular reabsorption of phosphate or inappropriate renal phosphate wasting. Renal phosphate wasting then led to chronic hypophosphataemia and this is the proximate cause of osteomalacia and recurrent fractures.
Dual-energy X-ray absorptiometry (DXA) bone densitometry (table 4) showed low bone mineral density (BMD) with T-scores of −5.2,–4.2, −4.9 and −9.6 at the lumbar spine, right femoral neck, right total hip and left radius, respectively. In addition, the respective Z-scores were also low at −3.7,–2.7, −4.0, and −7.9, compatible with a secondary cause for low BMD.
Table 4.
Dual-energy X-ray absorptiometry bone densitometry results (February 2017)
| BMD* (g/cm2) | T-score | Z-score | |
| Lumbar spine L1–L4 | 0.476 | −5.2 | −3.7 |
| Right femur (neck) | 0.426 | −4.2 | −2.7 |
| Right total hip | 0.338 | −4.9 | −4.0 |
| Left radius (1/3) | 0.112 | −9.6 | −7.9 |
*BMD was not measured in the left hip because the presence of a fracture leads to a distorted anatomy and inappropriate region of interest. Thus, measured BMD will be inaccurate.
BMD, bone mineral density.
Differential diagnosis
The differential diagnoses for these bone deformities include Paget’s disease, osteogenesis imperfecta, and osteomalacia from various causes.
Paget’s disease has a distinct radiological pattern not seen in our patient’s X-rays. Osteogenesis imperfecta presents with multiple fractures starting childhood and not during adulthood as in this patient’s case. For this same reason, the genetic causes of osteomalacia such as X-linked recessive hypophosphataemic osteomalacia (Dent’s disease and Lowe’s syndrome) are unlikely.
Acquired causes of osteomalacia include vitamin D deficiency, tumour-induced osteomalacia and Fanconi syndrome. Vitamin D deficiency was the initial impression of the previous doctors with whom she consulted, hence the initial treatment with vitamin D. However, the recurrence of the fractures despite correction of serum 25-OH vitamin D and the other renal findings suggest another disease entity. Tumour-induced osteomalacia is due to a tumour that secretes a phosphaturic hormone called fibroblast growth factor 23. Such tumours may be difficult to find, and this diagnosis is hard to exclude. This can explain the hypophosphataemia and the renal phosphate wasting but it could not account for the other renal findings, specifically the glucosuria, aminoaciduria, metabolic acidosis and alkaline urine.
Fanconi syndrome, named after Swiss paediatrician Guido Fanconi, is a disorder of proximal renal tubule transport. It is characterised by the urinary loss of phosphates, amino acids, glucose and other ions such as bicarbonate and uric acid.2 These substances are normally reabsorbed in the proximal renal tubule. Our patient had majority of these features: phosphaturia, generalised amino aciduria, normoglycaemic glycosuria and metabolic acidosis with persistently alkaline urine (urine pH 7–7.5). The renal phosphate wasting then led to chronic hypophosphataemia which then led to osteomalacia.
Treatment
Aside from removal of the offending agent, if known, there is no specific treatment for this condition. Medical management involves replacing the substances lost in the urine.3 For this patient, we started her on potassium citrate 1080 mg/tablet, two tablets three times a day and a multivitamin+phosphorus tablet preparation containing 200 mg of dicalcium phosphate per tablet, five tablets per day. We maintained her vitamin D at 3000 IU per day. With this regimen, serum potassium and phosphorus levels were maintained within the normal range, with her latest determinations at 3.71 mmol/L and 0.91 mmol/L, respectively.
Outcome and follow-up
After 1 year of phosphorus supplementation and maintaining serum phosphorus levels within the normal range, corrective surgery was done. Multiple osteotomies were done to correct malangulation and rotation of the femur and tibia, combined with hamstring tendon releases and hip adductor release to increase knee and hip ranges of motion. Intramedullary implants with pelvic band were used to stabilise the reduction (figure 9). These procedures have afforded acute straightening of her long bones with around 30° of residual knee flexion tightness (figure 10). She is being monitored for fracture healing to eventually allow physical therapy for independent transfers and mobility.
Figure 9.
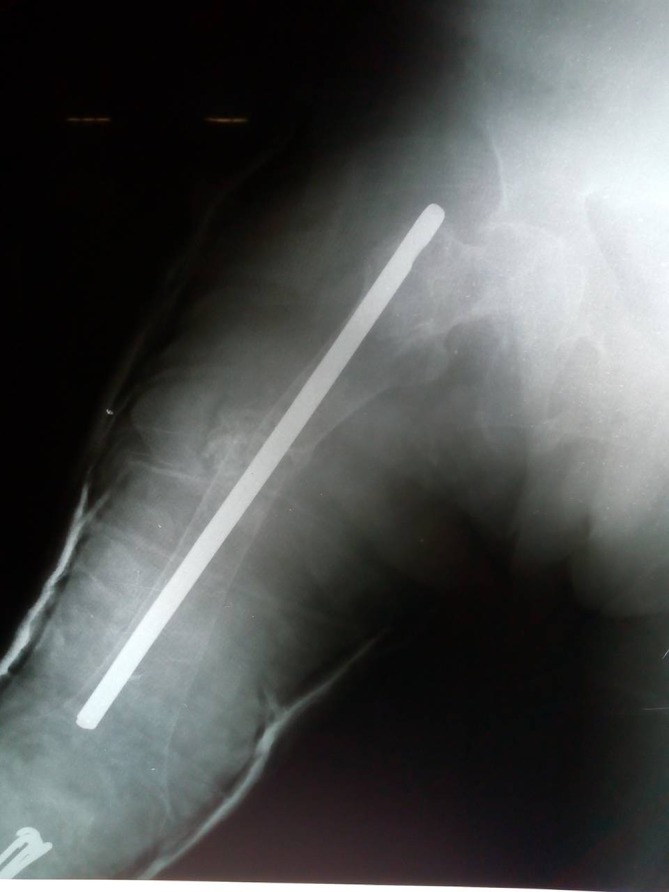
Intramedullary rod placement has straightened the left femur which was previously fractured into three segments.
Figure 10.
Corrective surgery has afforded acute straightening of long bones on the left lower extremity, with around 30° of residual knee flexion tightness.
Discussion
The association of glucosuria and hypophosphataemia in bone disease as seen in Fanconi syndrome has long been known. In fact, this entity has been called hypophosphataemic glycosuric rickets and glycosuric osteopathy in publications in the 1940–1950s.4 5 The lesson learnt more than 60 years ago seems to have been forgotten as several doctors our patient has seen in the course of 8 years were not able to tie up these findings together.
Fanconi syndrome has many aetiologies. It can be congenital or induced by drugs or heavy metals, particularly lead. The patient denies intake of drugs known to cause this syndrome such as aminoglycoside and tetracycline antibiotics, anticonvulsants, cancer medications like ifosfamide, cisplatin/carboplatin and imatinib and antiretroviral nucleotide and non-nucleotide reverse transcriptase inhibitors.3 Lead poisoning is unlikely since her whole blood lead level is normal at 1.9 μg/L.
Since the patient has a known exposure to chromium in her previous work as a textile chemist, we surmised if this could be the cause. Chromium, in its trivalent form Cr (III), is commonly present in commercial applications like tanning. Chromium is an essential trace metal, generally of low toxicity, and poorly absorbed by the gastrointestinal tract. It is excreted renally, thus making the kidney a target organ. However, no studies have found any association between Cr(III) and kidney disease.6 Her serum chromium level turned out to be normal at 0.72 μg/L (normal<0.82), excluding this as a cause.
Although Fanconi syndrome may result from drugs and heavy metal toxicity, most adult cases are due to urinary secretion of a monoclonal immunoglobulin light chain that usually forms crystals inside renal proximal tubular cells. Messiaen et al described 11 cases of adult Fanconi syndrome secondary to light chain gammopathy. Seven patients presented with typical clinical and pathological features of Fanconi syndrome and monoclonal gammopathy of undetermined significance (MGUS).2 Similar to our patient, the diagnosis of Fanconi syndrome remained unrecognised with a mean time from onset of symptoms to diagnosis of Fanconi syndrome of 3.1 years in their series. Fanconi syndrome was identified at the same time as the plasma cell dyscrasia in all but two patients whose diagnosis of monoclonal gammopathy preceded the diagnosis of Fanconi syndrome.2
Sudhaker Rao et al7 reports on two patients with osteomalacia confirmed by iliac bone histomorphometry associated with reduced renal tubular phosphate reabsorption and other features of Fanconi syndrome from light chain proteinuria secondary to myeloma in one and a subclinical lymphocytic leukaemia in the other. Both patients received oral phosphate therapy which resulted in complete histological correction of osteomalacia. One patient also received calcitriol. Myeloma was treated with prednisone and cyclophosphamide for 6 months, while the patient with subclinical blood dyscrasia did not receive treatment aside from the phosphate replacement. In the same article, Sudhaker Rao also reviewed 13 other similar cases of osteomalacia and Fanconi syndrome with light chain proteinuria. Light chain excretion was attributed to myelomatosis in nine, amyloidosis in two and idiopathic in another two. In all cases, Fanconi syndrome was already present at the time the blood dyscrasia was diagnosed.7
A serum and urine protein electrophoresis done on our patient was unremarkable. The aetiology of her Fanconi syndrome remains unknown, but we will continue to be on the look-out for MGUS, myeloma, other blood dyscrasias and malignancies.
Patient’s perspective.
I have been bedridden since the age of 61. My mental faculties, senses and spirit, however, are not affected. It has been depressing since the bone on my left thigh cracked and twisted.
My bones have already been severely affected before a correct diagnosis was reached.
I was deprived of my chances to travel.
We have an upcoming 50th anniversary alumni homecoming in 2018. I wish that my fractures would be corrected by then so I can attend the event and see my classmates. I do not want my classmates to see that I am bedridden.
Learning points.
The occurrence of fragility fractures should prompt workup for metabolic bone disease.
Metabolic bone disease should not just be ascribed to vitamin D deficiency, especially if there are findings that could not be explained by vitamin D deficiency alone.
Investigation for metabolic bone disease is not just determining calcium, parathyroid hormone or vitamin D but can be deduced by meticulously looking at a simple urinalysis.
The finding of glucosuria in the absence of diabetes should make us consider a proximal renal tubular defect (Fanconi syndrome).
In Fanconi syndrome, the renal phosphate wasting that leads to chronic hypophosphataemia results in osteomalacia and fractures.
Footnotes
Contributors: LFA wrote the first draft of the manuscript. MASS provided significant revisions and contributions to the manuscript. NSO and CTDD gave inputs to the manuscript. All were involved in the care of the patient being presented here and gave valuable inputs during the decision-making process. All approved the final version of the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem 1998;35 Pt 2:201–6. 10.1177/000456329803500203 [DOI] [PubMed] [Google Scholar]
- 2.Messiaen T, Deret S, Mougenot B, et al. . Adult Fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Medicine 2000;79:135–54. 10.1097/00005792-200005000-00002 [DOI] [PubMed] [Google Scholar]
- 3.Hall AM, Bass P, Unwin RJ. Drug-induced renal Fanconi syndrome. QJM 2014;107:261–9. 10.1093/qjmed/hct258 [DOI] [PubMed] [Google Scholar]
- 4.Linder GC, Bull GM. Hypophosphataemic glycosuric rickets (Fanconi syndrome). Clin Proc 1949;8:1–30. [PubMed] [Google Scholar]
- 5.Kuhlencordt F. [Glycosuric osteopathy: so-called Fanconi syndrome in adults]. Ergeb Inn Med Kinderheilkd 1958;9:622–55. [PubMed] [Google Scholar]
- 6.Barceloux DG. Chromium. J Toxicol Clin Toxicol 1999;37:173–94. 10.1081/CLT-100102418 [DOI] [PubMed] [Google Scholar]
- 7.Rao DS, Parfitt AM, Villanueva AR, et al. . Hypophosphatemic osteomalacia and adult Fanconi syndrome due to light-chain nephropathy. Another form of oncogenous osteomalacia. Am J Med 1987;82:333–8. [DOI] [PubMed] [Google Scholar]