

Abstract
Continuing review of studies approved by the Ethics Committees (ECs) involves review of the progress of the study, annual reports, protocol deviations/violations, serious adverse event monitoring, and on-site monitoring. International and national regulations and guidelines for continuing review state that it is an opportunity for the EC to be assured that risks to subjects are minimized and is are reasonable in relation to anticipated benefits if any to the subjects and the knowledge it will generate. There are several barriers (e.g. lack of workforce, lack of training of members for conducting onsite review, and poor infrastructure) for ECs to do ongoing review of projects approved by them. Industry is an important stakeholder for the research enterprise in India and strongly advocates that ECs should at a minimum have pragmatic standard operating procedures for continuing review/monitoring of studies initially approved. ECs which deal with larger volume of studies with well-functioning secretariat, appropriately trained EC members and funding should definitely conduct onsite review/monitoring in addition to the ongoing review.
Keywords: Ethics Committees, ongoing review, onsite review
INTRODUCTION
The purpose of continuing review of research studies approved by an Ethics Committees (ECs) is to monitor the progress of the study which was previously approved; not only for the changes but to ensure continued protection of the rights and welfare of research subjects. According to the current framework of regulatory approvals in India, the Institutional ECs (IECs) or institutional review boards (IRBs) have a responsibility to ensure that the clinical trial is conducted, data generated, documented, and reported in compliance with the study protocol, Schedule Y (2005),[1] Indian Good Clinical Practice (GCP) Guidelines (2001);[2] as well as all applicable statutory provisions of Drugs and Cosmetics Act and Rules (1940 and its amendments).[3] Schedule Y (2005) of the Drugs and Cosmetics Act and Rules (1940) in the section on clinical trials states the following as one of the roles and responsibilities of IECs “ECs should make, at appropriate intervals, an ongoing review of the trials for which they review the protocol(s).[1] Such a review may be based on the periodic study progress reports furnished by the investigators and/or monitoring and internal audit reports furnished by the sponsor and/or by visiting the study sites. In the case of academic research, the revised regulations [4] state that no permission for conduct of clinical trial intended for academic purposes in respect of approved drug formulation shall be required for any new indication or new route of administration or new dose or new dosage form where - (a) the trial is approved by the EC; and (b) subject to the provisions of sub-rule 5, the data generated is not intended for submission to licensing authority. Given this change in Regulatory orientation with regards to Academic research, it is logical to extrapolate that academic research will be governed under the provisions of the National Ethical Guidelines for Biomedical and Health Research involving human participants (2017).[5] The European Medicine Agency,[6] International Council on Harmonisation-GCP,[7] and the United States Food and Drug Administration (USFDA) guidelines [8] recommend that IRBs should conduct continuing review of each on-going trial at intervals appropriate to the degree of risk to human subjects. The National Accreditation Board for Hospitals and Healthcare Providers (NABH), Quality Council of India,[9] in consultation with various stakeholders, has formulated accreditation standards for clinical trial sites, ECs, and investigators. NABH has also trained assessors for accrediting ECs. Subsection 1.4.5 of the Accreditation standards states that monitoring of trials shall be done to ensure equitable selection of subjects, with special attention to vulnerable and high-risk subjects. The Ministry of Health and Family Welfare, India, has granted approval to making the accreditation of ECs, involved in supervision of clinical trials, mandatory with effect from January 1, 2018.[10]
IMPORTANCE OF ONGOING REVIEW IN HUMAN SUBJECT PROTECTION
To protect human participants in research, institutions must establish policies and mechanisms for the protection of human research participants. Irrespective of whether research occurs in medical institutions or private institutions/clinics, human subject protection can only be ensured if the IEC/IRB monitors the studies it has approved. Douglass et al.[11] concluded that an active monitoring program can detect deviations from the approved protocol not disclosed in the annual report. The EC of a tertiary level medical institution conducted seven site visits during 2008–2009 using a standardized format to monitor adherence to protocol and the informed consent process. The monitoring identified issues related to informed consent (6/7), protocol deviation (5/7), and reporting of study progress to the IEC (3/7), recruiting additional participants without IEC approval (2/7), reporting of serious adverse events (SAEs) (1/7).[12] Inspections carried out by the competent authority from 2011 to 2016 which involved ECs across India (data on file; collation of inspection findings of studies and ECs inspected by CDSCO over 2013-2016) observed that at several sites ongoing review was not carried out by EC as per Point (5), (ii) of Schedule of D and C Rules. The inspection findings also noted that EC members never visited sites, no queries/concern was raised in case of multiple protocol deviations and noncompliance when reported, ECs seem to function in isolation and that ECs have little or no communication with the regulatory agency or other ECs. To ensure the safety and well-being of participants, as well as to ascertain that potential risks have not altered, International and Indian guidelines also recommend site visits as one of the methods for continuing review by IECs.[13,14]
WHAT PREVENTS INSTITUTIONAL ETHICS COMMITTEES/INSTITUTIONAL REVIEW BOARDS FROM CARRYING OUT CONTINUOUS REVIEW/MONITORING?
Lack of administrative infrastructure, lack of a clear framework for undertaking monitoring,[15] difficulty in motivating members to conduct audits of ongoing studies,[16] lack of workforce, lack of training of EC members on how to conduct monitoring and inadequate funds are identified as major hurdles for conducting active site monitoring.[17] Most IECs spend a substantial amount of time in reviewing and approving protocols and reserve some time for passive monitoring but almost none for site visits.
Many IECs restrict themselves to passive monitoring of ongoing studies which includes reviewing data such as SAE reports,[17] protocol violations,[18] progress reports, and protocol amendments at prespecified regular intervals according to the guidelines.[19] If IECs have to look into human subject protection in its entirety, then they need to conduct active monitoring which requires IEC/IRB members to visit study site where studies approved by them are ongoing. Apart from prespecified standard operating procedures (SOPs) which will enable sites to conduct on-site monitoring visits, another useful tool could be a brief checklist that can be used at the site to record observations.
PROCESS FOR CONTINUING REVIEW/MONITORING
Given the lack of clarity on how the ECs should conduct ongoing review, it would be best to fall back on established regulations/guidance (e.g., USFDA and ICMR) on continuing review after the initial approval. USFDA's regulations [8] require an IRB to develop and follow written procedures for - (i) conducting continuing review of research at intervals appropriate to the degree of risk, but not less than once a year, (ii) determining which clinical investigations require review more often than annually, (iii) determining which clinical investigations need verification from sources other than the clinical investigator that no material changes in the research have occurred since the previous IRB review, and (iv) ensuring prompt reporting to the IRB of changes in research activity and for ensuring that changes in approved research, during the period for which IRB approval has already been given, may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects. According to the ICMR Guidelines (2017),[5] there should be mechanisms and policies for monitoring research in the domains of data capture, management, conflicts of interest, reporting of scientific misconduct, and appropriate initial and continuing training of researchers and EC members. ICMR Guidelines clearly state that ongoing research should be reviewed at regular intervals, at least once a year, (or more often, if deemed necessary depending on the level of risk) or as may be specified in the SOP of the EC and at the time of according approval, and as indicated in the communication letter.
These data usually include SAE reports, progress reports, reviews of protocol deviations/violations, amendments of protocol, and related documents submitted by the investigators as recommended under national and international guidelines and legislation.[13,14] The ECs can conduct monitoring/continuing review as “routine” or “for cause,” and this must be decided at a full committee meeting.
The checklist proposed by the author based on inspection and audit findings of ECs aims to ensure that the IEC/IRB member focuses on a limited set of areas that have direct bearing on the subject safety protection in the study. An example of such a checklist that can be used by ECs is given in Table 1.
Table 1.
On site monitoring checklist for Institutional Ethics Committees/institutional review boards
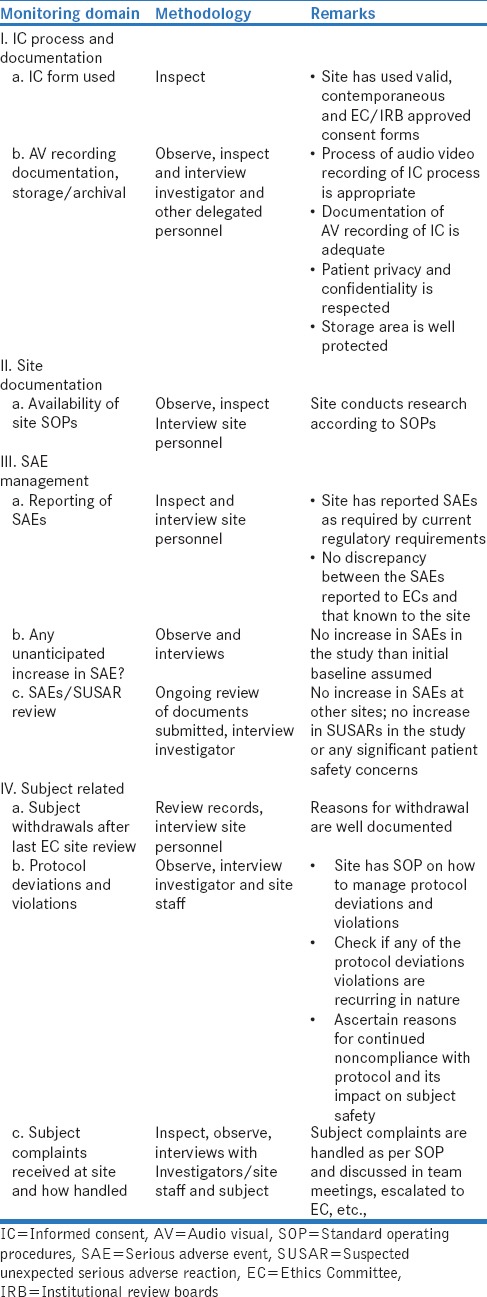
The checklist or a modified version based on ECs requirement also incorporate marks for each item, and that can also be a useful way to assess the change in site performance from one on-site monitoring visit to the next. It also can give the IEC/IRB the confidence that the site is taking efforts to improve in critical areas related to subject safety. The EC/IRB should also communicate to the site (especially the investigator) the findings of the monitoring team and have clearly identified areas of improvement for the site.
CONCLUSION
On-site monitoring of an approved study by the IEC/IRB ascertains the ethical conduct of clinical research and ensures that safety and wellbeing of the study participants are taken care. It also indirectly, ensures quality assurance and continued education of research staff and most importantly ensures that there are no breaches or lapses in the integrity of data. Depending on the degree of risk to the participants, the nature of the study, the vulnerability of the study participants and duration of the study, IEC may choose to review the study with more active monitoring. Hence, onsite monitoring by the IEC ensures the goal of human subject protection is maintained in research done at the site.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.2005. [Last accessed on 2018 Mar 20]. Available from: http://www.cdsco.nic.in/html/D&C_Rules_Schedule_Y.pdf .
- 2.CDSCO. Good Clinical Practice Guidelines. 2001. [Last accessed on 2018 Mar 20]. Available from: http://www.rgcb.res.in/wp.content/uploads/2014/07/Good.Clinical.Practice.Guideline.pdf.
- 3.Ministry of Health and Family Welfare, Department of Health. Drugs and Cosmetics Act 1940 and Rules 1945. New Delhi: Government of India; 2005. [Last accessed on 2018 Mar 20]. Drugs and Cosmetics Act 1940 and Rules 1945. Available from: http://www.cdsco.nic.in/writereaddata/2016Drugs%20and%20Cosmetics%20Act%201940%20 and %20Rules%201945.pdf . [Google Scholar]
- 4.G.S.R 313 (E) dated 16th March 2016 for Academic Clinical Study no Permission from DCG (I) Office is Required for Approved Drugs. [Last accessed on 2018 Mar 20]. Available from: http://www.cdsco.nic.in/writereaddata/GSR%20313%20(E)%20dated%2016_03_2016.pdf .
- 5.Indian Council of Medical Research. Ethical Guidelines for Biomedical Research in Human Participants. New Delhi: ICMR; 2017. [Last accessed on 2018 Mar 20]. Available from: http://www.icmr.nic.in/guidelines/ICMR_Ethical_Guidelines_2017.pdf . [Google Scholar]
- 6.Reflection Paper on Ethical and GCP Aspects of Clinical Trials of Medicinal Products for Human use Conducted Outside of the EU/EEA and Submitted in Marketing Authorization Applications to the EU Regulatory Authorities. [Last accessed on 2018 Mar 20]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2012/04/WC500125437.pdf .
- 7.International Council on Harmonization. Guideline for Good Clinical Practice E6 (R1), Current Step 4 Version. 1996. [Last accessed on 2018 Mar 20]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf .
- 8.Institutional Review Board (IRB) Written Procedures: Guidance for Institutions and IRBs. [Last accessed on 2018 Mar 20]. Available from: https://www.fda.gov/RegulatoryInformation/Guidances/ucm126420.htm.
- 9.National Accreditation Board for Hospitals and Healthcare Providers (NABH) Accreditation Standards for Clinical Trial in India Ethics Committee, Investigator, and Clinical Trial Site. 2015. [Last accessed on 2018 Mar 20]. Available from: http://www.cdsco.nic.in/writereaddata/finalAccreditation%20Standards.pdf.
- 10.NABH has Now Started Accepting Application for Accreditation of Ethics Committee Program with Refer to Letter no. QCI/NABH/CDSCO/15. [Last accessed on 2018 Mar 20]. Available from: http://www.nabh.co/Announcement/Accreditation_ECApproval.pdf.
- 11.Douglass AJ, Jarvis A, Bloore S. Monitoring of health research by ethics committees. N Z Med J. 1998;111:79–81. [PubMed] [Google Scholar]
- 12.Shetty YC, Marathe P, Kamat S, Thatte U. Continuing oversight through site monitoring: Experiences of an institutional ethics committee in an Indian tertiary-care hospital. Indian J Med Ethics. 2012;9:22–6. doi: 10.20529/IJME.2012.006. [DOI] [PubMed] [Google Scholar]
- 13.International Ethical Guidelines for Biomedical Research Involving Human Subjects. Council for International Organization of Medical Sciences (CIOMS) in Collaboration with World Medical Organization. Geneva: World Health Organization; 2002. [Last accessed on 2018 Mar 20]. Available from: http://www.cioms.ch/publications/laout_guide2002/pdf. [PubMed] [Google Scholar]
- 14.World Health Organization. Operational Guidelines for Ethics Committees that Review Biomedical Research. Geneva: World Health Organization; 2000. [Last accessed on 2018 Mar 20]. Available from: http://www.who.int/tdr/publications/publications/. [Google Scholar]
- 15.Pickworth E. Should local research ethics committees monitor research they have approved? J Med Ethics. 2000;26:330–3. doi: 10.1136/jme.26.5.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis S, Sule P, Bughediwala M, Pandya V, Sinha S. Ethics committees and the changed clinical research environment in India in 2016: A perspective! Perspect Clin Res. 2017;8:17–21. doi: 10.4103/2229-3485.198555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tripathi RK, Marathe PA, Kapse SV, Shetty YC, Kamat SK, Thatte UM, et al. Serious adverse events reports: Analysis and outcome of review by an institutional ethics committee of a tertiary care hospital in Mumbai, India. J Empir Res Hum Res Ethics. 2016;11:267–73. doi: 10.1177/1556264616654809. [DOI] [PubMed] [Google Scholar]
- 18.Jalgaonkar SV, Bhide SS, Tripathi RK, Shetty YC, Marathe PA, Katkar J, et al. An audit of protocol deviations submitted to an institutional ethics committee of a tertiary care hospital. PLoS One. 2016;11:e0146334. doi: 10.1371/journal.pone.0146334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith T, Moore EJ, Tunstall-Pedoe H. Review by a local medical research ethics committee of the conduct of approved research projects, by examination of patients' case notes, consent forms, and research records and by interview. BMJ. 1997;314:1588–90. doi: 10.1136/bmj.314.7094.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]