

Abstract
The most prevalent form of dementia in the elderly is Alzheimer's disease. A significant contributing factor to the progression of the disease appears to be the progressive accumulation of amyloid-β42 (Aβ42), a small hydrophobic peptide. Unfortunately, attempts to develop therapies targeting the accumulation of Aβ42 have not been successful to treat or even slow down the disease. It is possible that this failure is an indication that targeting downstream effects rather than the accumulation of the peptide itself might be a more effective approach. The accumulation of Aβ42 seems to affect various aspects of physiological cell functions. In this review, we provide an overview of the evidence that implicates Aβ42 in synaptic dysfunction, with a focus on how it contributes to defects in synaptic vesicle dynamics and neurotransmitter release. We discuss data that provide new insights on the Aβ42 induced pathology of Alzheimer's disease and a more detailed understanding of its contribution to the synaptic deficiencies that are associated with the early stages of the disease. Although the precise mechanisms that trigger synaptic dysfunction are still under investigation, the available data so far has enabled us to put forward a model that could be used as a guide to generate new therapeutic targets for pharmaceutical intervention.
Keywords: Alzheimer's disease, amyloid-β 42, synaptic vesicles, synaptic dysfunction, neurotransmitter release
Introduction
It was more than a hundred years ago when Alois Alzheimer described the pathology of a type of dementia that we now know as Alzheimer's disease (AD). AD accounts for almost 70% of dementia cases and is characterised by progressive memory loss and in advanced stages, impairments in language and behaviour (McKhann et al., 1984). The key phenotypic hallmarks of AD in post-mortem brain tissue were found to be extracellular deposits (or amyloid plaques) of a small peptide called amyloid beta (Aβ) and intracellular deposits (or neurofibrilliary tangles) of a hyperphosphorylated microtubule associated protein called tau. There are two forms of AD, the rare familial or early onset AD (EOAD) where the disease is diagnosed before the age of 65, and the common sporadic or late onset AD (LOAD) with the age of onset after 65 years of age. Even though there are specific genetic mutations that have been identified as causative factors for EOAD, the actual cause of the most common LOAD is still unknown, although there are several genetic risk factors associated with it. Despite the differences in prevalence and age of onset, it was realised that the dominant genetic mutations associated with EOAD shared a similar phenotype with LOAD: the accumulation of a small hydrophobic peptide called Aβ. Aβ peptides are small hydrophobic peptides composed of 39–43 amino acids that are generated by a physiological cellular process, the sequential proteolytic cleavage of the amyloid precursor protein (APP). APP is a type I, single-pass transmembrane protein with a large extracellular domain, a short cytoplasmic tail and is encoded by a single gene (Goldgaber et al., 1987; Kang et al., 1987; Tanzi et al., 1987). A conventional model describing the proteolytic processing of APP proposes that it can be processed by two distinct proteolytic pathways, the amyloidogenic and the non-amyloidogenic pathway (Andrew et al., 2016). In the non-amyloidogenic pathway, APP is cleaved at the cell surface within the Aβ domain by α-secretase, producing a secreted fragment (sAPPα) and a membrane bound C-terminal fragment (CTFα). Several members of the ADAM family of metalloproteases have been reported to possess α-secretase activity with ADAM10 being the major α-secretase that cleaves APP in the brain (Lammich et al., 1999; Kuhn et al., 2010; Prox et al., 2013). In the amyloidogenic pathway, APP proteolysis is initiated by β-secretase (BACE1), which cleaves APP in the endosome at the amino terminus of the Aβ domain (Vassar et al., 1999), producing a soluble secreted fragment (sAPPβ) and a membrane bound C-terminal fragment (CTFβ). CTFα and β are subsequently cleaved by the γ-secretase complex releasing the APP intracellular domain (AICD). In addition to the generation of AICD, cleavage of CTFα produces a small P3 fragment, whereas cleavage of CTFβ produces several Aβ peptides with the most prominent being Aβ40 and the longer, more hydrophobic Aβ42 (Takami et al., 2009). However, the discovery of additional proteins that also cleave APP to produce a multitude of biologically active fragments suggests that this view of APP processing is rather simplistic and that there is still a lot to learn (Andrew et al., 2016).
Aβ and the Pathophysiology of AD
The realisation that the overproduction of Aβ peptides is a causative factor for EOAD alongside the discovery that the amyloid plaques in patients with LOAD contained aggregates of Aβ peptides prompted the formation of the “amyloid cascade hypothesis”. According to this hypothesis, the formation of plaques generated by the accumulation and aggregation of Aβ peptides induce neurodegeneration, neuronal death and cognitive decline, and was initially considered the underlying cause of all types of AD (Hardy and Higgins, 1992). For many years the amyloid cascade hypothesis was central to AD research. However, it was realised that plaque densities correlate poorly with cognitive impairment, whereas increased levels of soluble Aβ oligomers correlate strongly with the severity of the disease and the extent of synaptic loss (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999). These findings shifted the attention from amyloid plaques to soluble Aβ oligomers and their contribution to the pathology of AD. Thus the initial hypothesis evolved, retaining the core element of a link between Aβ peptides and the progression of the disease. Since then, the biggest challenge in dementia research has been to understand the physiological role of Aβ oligomers and to discover their impact on the progression of AD. A search in the literature shows that several cellular processes are thought to be affected by Aβ. For example, it has been shown that Aβ peptides promote the generation of free radicals inducing cellular stress and loss of Ca2+ homeostasis, and accumulate in the mitochondria triggering dysfunction and cell death. Furthermore, it has also been shown that Aβ peptides interfere with the proteasome activity in neuronal cells, affect the regulation of cell signalling kinases or phosphatases and modify the cytoskeletal organisation of the cell (Selkoe and Hardy, 2016). In addition to these effects, oligomeric Aβ has also been shown to trigger synapse dysfunction and induce loss of synapses that characterise AD (Selkoe, 2002).
Current Available Therapies for AD
The prevalence of AD is increasing at an alarming rate, but unfortunately there is no available cure, and the few approved drugs so far can only be used for symptomatic treatment of the disease. To date, research for new drugs is predominately focused on four major neuropathological themes of AD: accumulation of Aβ as a result of its overproduction or defective clearance, synaptic dysfunction mediated either by acetylcholine (ACh) deficiency or glutamate excitotoxicity, neuroinflammation and finally, formation of NFTs.
A key target area for the development of new drugs for AD patients has been to regulate the levels of Aβ peptides. Various attempts have been made to develop agents that either interfere with the production or enhance the clearance of Aβ, reducing their load in the brain. For instance, several inhibitors for β or γ secretase have been developed in an effort to reduce the generation of Aβ peptides. Although not all clinical trials of β-secretase inhibitors have been completed, the clinical data so far are not encouraging either because the side effects and the toxicity of the compounds are severe, or because the compounds fail to meet the desired endpoints (Khan et al., 2017). Similarly, compounds targeting γ-secretase have also failed in clinical trials. Blocking its function has several side effects, most likely because of the many substrates that are cleaved by γ-secretase, but also due to the complexity of its activity. Nevertheless, new compounds with different specificities are still being examined in ongoing clinical trials (Khan et al., 2017).
In addition, immunotherapeutic approaches have also been attempted to remove the burden of Aβ from the brain. These approaches aim to either stimulate the immune system to produce antibodies to clear Aβ peptides (active immunotherapy), or to use Aβ-specific antibodies that are injected directly into the patient to stimulate clearing of Aβ peptides (passive immunotherapy). On the positive side, some of these efforts have been effective in reducing the levels of Aβ from the brain. Unfortunately, the primary clinical endpoint in slowing down the cognitive decline has not been met yet. Most recently, results were published from clinical trials for Solanezumab, one of the two major monoclonal antibodies currently tested in patients with Alzheimer's dementia. The results showed that the antibody did not meet its primary outcome of significantly slowing the cognitive decline in patients with advanced or mild AD (Honig et al., 2018).
Although these failures are disheartening, they can be used to obtain information for guidance in fine tuning the efforts to develop a successful approach. For example, to design antibodies, it might be important to consider that the hydrophobic Aβ peptides adopt several configurations that appear to have different physiological roles. For instance, Aβ oligomers are considered to be the toxic species of the peptide (Sevigny et al., 2016), whereas the monomeric form of Aβ is considered to be neutral or even neuroprotective (Giuffrida et al., 2009). If this is indeed the case, then an antibody that would selectively bind and remove only Aβ oligomers may be more beneficial than one that binds to both Aβ monomers and oligomers. Thus, a possible reason for the failure of Solanezumab in clinical trials could be its preference to bind to monomeric Aβ (Zhao et al., 2017) rather than the more toxic oligomeric forms. Interestingly, an antibody called Aducanumab has been reported to show selective binding to oligomeric rather than the monomeric forms of the peptide. This antibody is still undergoing phase III trials on patients displaying moderate to mild AD with the results expected in 2022.
Alternatively, significant efforts have been made to develop drugs targeting deficiencies in ACh neurotransmission and glutamate induced excitotoxicity. It is now evident that disruption in synaptic activity and loss of synapses is the earliest event in AD preceding the clinical manifestation of the disease or the accumulation of Aβ deposits in the brain and is induced, at least in part, by increased levels of Aβ oligomers (Lambert et al., 1998). According to the cholinergic hypothesis, a key contributor to the pathophysiology of AD is the loss of cholinergic neurons (Davies and Maloney, 1976). It has been proposed that this loss results in decreased levels of ACh in the hippocampus and in the cortex, resulting in cognitive deficits (Francis et al., 1999). Clinical data showed that enhancing cholinergic transmission in the brain of affected individuals can be effective in treating the symptoms of mild to moderate AD. To date, three such drugs (donepezil, rivastigmine and galanthamine) have been licenced to treat the symptoms of dementia but unfortunately, they are limited in scope. These drugs enhance ACh neurotransmission either by reducing the degradation of ACh through acting as inhibitors of acetylcholinesterase, or by enhancing the response to ACh (Hung and Fu, 2017). It is encouraging that these compounds have shown some efficacy in improving cognitive function and slowing the progression of the symptoms of mild to moderate AD (and occasionally of moderate to severe AD), but unfortunately the benefits are not long lasting. In addition, glutamate induced excitotoxicity has also been linked with the progression of AD. Glutamate induced excitotoxicity is the result of aberrant NMDA receptor activation due to increased levels of glutamate. Memantine (an NMDAR antagonist) is the first and only approved drug that targets NMDAR and prevents its activation. Memantine has shown good tolerability in patients and there is also substantial evidence that it delays the decline in cognitive functions but again, the effects are not long lasting (Graham et al., 2017; Khan et al., 2017). Other than that, there has been no significant drug approved for AD in more than a decade.
The failure of therapeutic approaches to display long-term beneficial results in patients with severe to moderate AD may also be an indication that a different, more focused approach might be needed. Since the only approved drugs target defects in cholinergic or glutamatergic synaptic activity, it is likely that a better understanding of the mechanisms that link oligomeric Aβ to synaptic dysfunction will be vital in uncovering novel and more specific targets that could be used to generate new therapeutic approaches.
Intraneuronal Aβ Peptides and Synaptic Dysfunction
The controversial results from clinical trials of candidate drugs targeting Aβ over the past years has prompted an ongoing re-evaluation of the importance of Aβ as a causative factor for AD (Herrup, 2015; Amanatkar et al., 2017). Even so, the neuronal dysfunction induced by Aβ is still considered a significant contributing factor of AD (Morris et al., 2014; Herrup, 2015; Selkoe and Hardy, 2016; Forner et al., 2017) and a lot of effort is still being put into understanding how it contributes to the progression of the disease. It is established that elevated levels of Aβ oligomers disrupt synaptic plasticity, triggering memory loss and cognitive deficits. One of the key events that shifted the attention from amyloid plaques to Aβ oligomers as contributors to AD was a report by Lambert et al who demonstrated that soluble oligomers of Aβ, rather than monomers or fibrils, triggered the loss of dendritic spines and that these oligomers could also interfere with synaptic function, disrupting NMDAR dependent long term potentiation (LTP) (Lambert et al., 1998). Since then, many groups have confirmed that Aβ oligomers disrupt NMDA dependent LTP and facilitate NMDA dependent long term depression (LTD) (Hsia et al., 1999; Walsh et al., 2002, 2005; Wang et al., 2002; Cleary et al., 2005; Hsieh et al., 2006; Klein, 2006; Shankar et al., 2007). Hence, a considerable amount of research over the past decade has focused on unravelling the mechanisms responsible for the defects in synaptic function that are mediated by Aβ oligomers.
Although these mechanisms are still not fully understood, it is now becoming apparent that at high concentrations, Aβ peptides interfere with glutamatergic neurotransmission affecting both, pre- and post-synaptic mechanisms. The discovery of Aβ peptides localised in spines of dissociated hippocampal neurons initially suggested it might directly affect post synaptic processes (Lacor et al., 2004). This led to the hypothesis that the effects of Aβ peptides in synaptic dysfunction could be a result of an agonist action on NMDARs (Molnar et al., 2004). This hypothesis was further supported by findings showing that Aβ was indeed localised at the post synaptic ends in brains of AD patients (Koffie et al., 2009). Further to being considered as an agonist for NMDARs, Aβ peptides were also detected inside neurons of AD patients but also in individuals without AD neuropathology (Grundke-Iqbal et al., 1989). Subsequently, it became clear that the intraneuronal accumulation of Aβ peptides preceded NFT or plaque formation and was closely linked to the pathogenesis of AD (Hartmann, 1999; Wilson et al., 1999; Gouras et al., 2000). Moreover, immunolabelling studies using antibodies specific for Aβ40 and Aβ42 showed that the majority of intraneuronal Aβ peptides (iAβ) in brain regions vulnerable to AD were the more toxic Aβ42 species rather than the less toxic Aβ40 (Gouras et al., 2000). The use of transgenic animal models for AD further established the presence of Aβ peptides inside neurons and in addition, revealed that levels of iAβ were at a dynamic equilibrium with extracellular pools, and that they contributed to the early memory deficits that characterise AD (Oddo et al., 2003, 2006; Billings et al., 2005). The discovery that iAβ oligomers were also present at the presynaptic nerve terminals of glutamatergic neurons in AD patients (Kokubo et al., 2005; Sokolow et al., 2012) put forward the idea that the synaptic deficits induced by Aβ could be triggered by presynaptic deficits. Further research revealed more intriguing details of the time and concentration dependent effects of Aβ peptides (Puzzo et al., 2008; Abramov et al., 2009; Parodi et al., 2010; Russell et al., 2012). It was shown that brief exposure of neurons to low concentrations of Aβ peptides could increase LTP as well as basic transmission, whereas higher concentrations and/or longer exposure times of neurons to Aβ decreased excitatory postsynaptic potential and inhibited NMDAR-dependent LTP (Puzzo et al., 2008). A possible explanation of these rather surprising findings, supported by a growing body of evidence, is that soluble Aβ oligomers affect the activity of NMDARs by gradually increasing the amount of glutamate at the synaptic cleft, in addition to exerting a direct agonist effect on NMDARs (Arias et al., 1995; Harris et al., 1995; Kabogo et al., 2008; Puzzo et al., 2008; Li et al., 2009, 2011). This hypothesis is supported by findings confirming that acute exposure of neurons to low concentrations of Aβ initially induced a presynaptic deficit, whereas prolonged exposure to higher concentrations was required to induce postsynaptic alterations (Parodi et al., 2010). Furthermore, it was also shown that the increase of glutamate release at synapses was specifically induced by iAβ (Parodi et al., 2010; Ripoli et al., 2014). We have also provided evidence that Aβ oligomers can be readily internalised and accumulate at the presynaptic terminals of glutamatergic neurons (Russell et al., 2012). Our results also showed that the enhancement of NT release in mature neurons exposed to low concentrations of Aβ oligomers correlated with the internalisation of the peptides and their localisation to the presynaptic compartment (Russell et al., 2012). Taken together, these data support the notion that neurotransmission may initially be affected through defects at the presynapse that are triggered by the gradual accumulation of iAβ. The subsequent aberrant release of glutamate may well be a significant contributing factor to the widespread synaptic dysfunction in AD that is characterised by excitotoxicity, desensitisation of glutamate receptors, inhibition of LTP and triggering of LTD. Interestingly, this bell-shaped effect of Aβ on synaptic function is also supported by fMRI scans on patients with high plaque load that either display symptoms of AD or are non-symptomatic. These scans showed enhanced synaptic activity at the hippocampus on non-symptomatic patients, whereas in patients with more advanced symptoms, fMRI scans revealed reduced synaptic activity as a result of synaptic depression (Sperling et al., 2010). Collectively, these data provide a link between accumulation of iAβ oligomers and early presynaptic defects in the mechanisms of NT release.
The Effect of Aβ Oligomers on Synaptic Vesicle (SV) Dynamics
In support of the hypothesis that Aβ peptides enhance neurotransmitter (NT) release, several reports demonstrated that key proteins which regulate either the interaction of SVs with the presynaptic membrane or the availability of SVs to participate in NT release are affected by Aβ peptides. For instance, it was shown that Aβ42 oligomers can directly interact either with proteins involved in SV docking and fusion that regulate NT release such as Syntaxin 1a (Stx1a) (Yang et al., 2015) and Synaptophysin (Syp1) (Russell et al., 2012), or can interfere with signalling mechanism that regulate the recovery and availability of SVs such as the regulation of dynamin (Kelly et al., 2005; Kelly and Ferreira, 2006, 2007) and of Synapsin1 (Snp1) (Liu et al., 2017; Marsh et al., 2017; Park et al., 2017).
Stx1a belongs to the family of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins. SNARE proteins are present on SVs and the target membrane, and form a tight complex whose primary function is to mediate SV docking and fusion at the presynaptic active zone. Using an in vitro vesicle mixing assay to monitor SNARE-mediated fusion of single vesicles, Yang and co-workers showed that binding of Aβ oligomers to the SNARE motif of Stx1a disrupts the formation of the SNARE complex and the fusion of vesicles (Yang et al., 2015). Surprisingly though, it did not affect the ability of the vesicles to dock to the target membrane. These results suggest that the interaction between Aβ oligomers and Stx1a affect the “zippering” of the SNARE complex rather than its initial formation. Moreover, these results also suggest that this interaction would reduce NT release. Since a reduction in NT release has been associated to chronic exposure of elevated Aβ levels, it would be interesting to further explore this interaction and examine how the docking and fusion of SVs is affected by Aβ over time in the context of a functional synapse containing all the regulatory components.
Another SV associated protein that was shown to directly interact with Aβ peptides is Syp1 (Russell et al., 2012). Syp1 is a glycoprotein localised on SVs, and although its precise function is still not fully understood, it has been shown to interact with a SNARE protein called Synaptobrevin 2 (VAMP2). The interaction between Syp1 and VAMP2 at the cell soma regulates the transport of VAMP2 containing SVs from the Golgi to the synapse. At the presynaptic compartment though, this interaction regulates the availability of VAMP2 to participate in the assembly of the SNARE complex during the formation of the fusion pore complex (Pennuto et al., 2003). The results from Russell et al demonstrated that a brief exposure of neurons to Aβ peptides results in their internalisation and binding to Syp1 at the presynapse, disrupting its interaction with VAMP2. Disruption of this interaction at the presynapse increases the availability of VAMP2 to participate in the formation of the SNARE complex and consequently, enhance NT release (Russell et al., 2012). In addition to VAMP2, Syp1 interacts with dynamin, regulating the endocytosis and recovery of SVs (Daly et al., 2000). Dynamin is a presynaptic protein involved in SV endocytosis, promoting the fission, pinching off and recycling of SVs (Yamashita et al., 2005). The levels and function of dynamin are regulated by its cleavage by calpain, and reduction of dynamin levels inhibits SV endocytosis and consequently their refill with NT (Kelly et al., 2005). Interestingly, prolonged exposure of neurons to high concentration of Aβ oligomers induces a reduction of dynamin by increasing its cleavage by calpain, resulting in the depletion of the readily releasable pool of SVs (Kelly et al., 2005; Kelly and Ferreira, 2006, 2007).
Further to the direct interactions between Aβ peptides and proteins on SVs, we recently showed that Aβ oligomers disrupt the regulation of SnpI (Marsh et al., 2017), which was also confirmed by a later independent study (Park et al., 2017). Snp1 belongs to a family of pre-synaptic phosphoproteins that are involved in synaptic fine-tuning and remodelling. Their main role is to regulate the availability of SVs to participate in NT release during neuronal activity through direct association/dissociation cycles (Cesca et al., 2010). Under resting conditions, they tether SVs to the cytoskeletal network, clustering them in the resting pool by interacting with SVs as well as the actin cytoskeleton (Cesca et al., 2010; Orenbuch et al., 2012). However, activity-dependent phosphorylation/dephosphorylation cycles at key residues of Snps induce their transient disassembly from the SVs (Jovanovic et al., 2001), releasing them from the resting pool and enabling their participation in NT release (Benfenati et al., 1989, 1991, 1992; Shupliakov et al., 2011). Neurons exposed to Aβ42 showed sustained levels of SnpI phosphorylation on Ser9 after neuronal activity due to increased calcium influx and calcium/calmodulin kinase IV (CaMKIV) activity (Marsh et al., 2017; Park et al., 2017), suggesting that the availability of SVs for NT release after neuronal activity is increased.
Collectively, the interaction of Aβ42 with proteins participating in the regulation of the SV life cycle and the deregulation of key molecules that orchestrate their availability, sheds some light into the cellular mechanisms underpinning the reported aberrant glutamate release in the early stages of AD (summarised in Figure 1).
Figure 1.
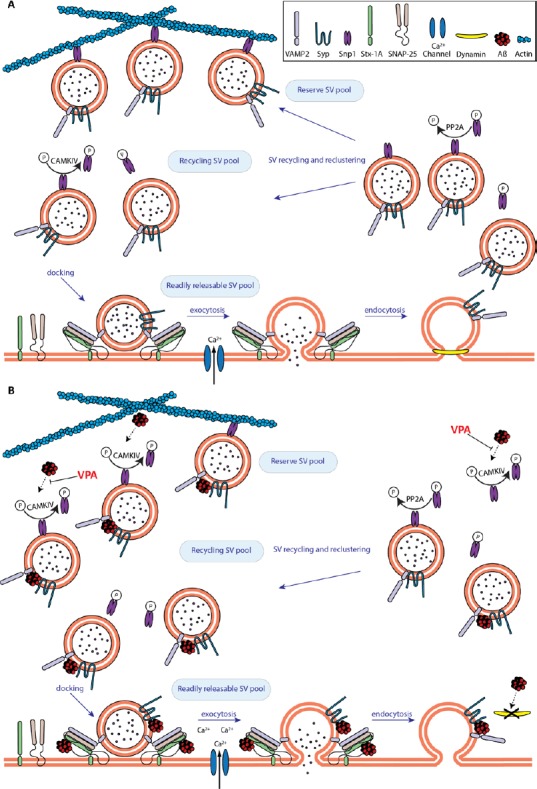
The early effects of amyloid beta (Aβ) on neurotransmission.
(A) Under physiological conditions, the amount of synaptic vesicles (SVs) participating in neurotransmitter (NT) release is tightly regulated. Upon neuronal activity, SVs are released from the reserve pool to participate in NT release via CAMKIV-induced Synapsin1 (Snp1) phosphorylation at Ser9. Released SVs dock via soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex assembly and then fuse with the membrane following calcium influx, releasing NT. SVs are pinched of the presynaptic membrane and endocytosed by dynamin1 and are either recycled or tethered back to the reserve pool after protein phosphatase 2A (PP2A)-mediated dephosphorylation of Snp1. (B) Aβ disrupts SV recycling and enhances NT release. Aβ increases the number of SVs available for docking due to sustained calcium/calmodulin kinase IV (CaMKIV)/Snp1 phosphorylation. SV docking is also enhanced due to Aβ-induced disruption of the synaptophysin (Syp1)/VAMP2 complex, enabling Synaptobrevin 2 (VAMP2) to participate in the formation of the SNARE complex. The increased number of docked SVs combined with an Aβ-mediated increase in calcium influx results in abberant NT release. Furthermore, deregulation of endocytosis due to a reduction of dynamin1 levels will eventually deplete the reserve pool of SVs. Notably, the deregulation of activity dependent phosphorylation of Snp1 can be restored by valproic acid (VPA).
A Hypothesis for the Progressive Synaptic Defects Induced by Aβ Peptides
The emerging data on the targets of Aβ at the presynapse, as well as the reported concentration and time dependent variations of the effects of Aβ peptides on NT release, allow us to put forward a working model to explain the progressive decline in synaptic function and cognitive deficits of AD patients.
If the synaptic defects during the early phase of AD is equivalent to the synaptic defects observed in neurons briefly exposed to low concentrations of Aβ peptides in vitro, then the early phase of AD would also be characterised by enhanced neurotransmission. The prolonged phosphorylation of Snp1 would contribute to the enhancement of neurotransmission by increasing the availability of SVs that would be able to dock to the active zone. Furthermore, disruption of the Syp1/VAMP2 complex on these vesicles would increase the accessibility of VAMP2 to the other SNARE proteins, promoting the formation of SNARE complexes and enhancing the probability of NT release. Finally, an Aβ-mediated increase of Ca2+ levels inside the presynapse would also enhance SV fusion and the release of glutamate. This aberrant release of glutamate would initially activate NMDARs, but the sustained levels of glutamate would eventually induce excitotoxicity. In the long term, the extensive use of SVs combined with defects in their endocytosis and recovery due to the inactivation of dynamin and sustained phosphorylation of SnpI, would gradually deplete these vesicles from the synapse, reducing synaptic activity. Indeed, depletion of the SV reserve pools after prolonged exposure to Aβ in neuronal cultures has been reported (Kelly et al., 2005; Parodi et al., 2010). Notably, a key mechanism to replenish SV pools is the transport of functional SVs from the Golgi to the presynapse. This transport is regulated at least in part, by the interaction between Syp1 and VAMP2 (Pennuto et al., 2003). Since binding of Aβ oligomers to Syp1 disrupts the Syp/VAMP2 complex, it is likely that sustained exposure to Aβ peptides would also compromise the trafficking and redistribution of SVs back to the presynaptic terminal, depleting the synapse of SVs. Although this hypothesis is yet to be conclusively addressed, there is evidence that SV mobility is affected by Aβ peptides. Park et al showed that exposure of neurons to Aβ reduces the activity dependent lateral dispersion of SVs, providing significant evidence that Aβ reduces SV mobility (Park et al., 2017). Although the authors suggested that the sustained phosphorylation of Snp1 might be the underlying cause for the inhibition of intersynaptic vesicular movements, we would like to propose that the disruption of the Syp1/VAMP2 complex by Aβ could well be a contributing factor to this inhibition. Collectively, the chronic combination of these effects would have a substantial impact on the gradual progression of synaptic dysfunction and ultimately loss of synapses, a key sign of AD pathology.
Targeting Presynaptic Defects for Therapeutic Interventions
Although there is an increasing body of evidence highlighting the significant contribution of Aβ peptides to the deregulation of NT release and to the dysfunction of SV dynamics, this area as a prospective therapeutic target has been largely overlooked. This is particularly surprising since defects in SV dynamics may be one of the earliest pathologies in AD and could serve as a target for crucial early intervention.
In support of such an alternative therapeutic approach, we recently showed that valproic acid (VPA) could be used to abrogate some of these early presynaptic defects (Marsh et al., 2017). VPA is a short-branched chain fatty acid and although its most common use is to treat epilepsy and bipolar disorder, several additional beneficial effects in the CNS have been reported. Studies on animal models suggest that VPA affects LTP and LTD and has therapeutic potential to combat AD (Zhang et al., 2003; Leng et al., 2008; Qing et al., 2008; Chang et al., 2010; Hu et al., 2011). Moreover, it has also been shown that it prevents Aβ42 induced reduction in SV recycling (Williams and Bate, 2016) and that it can induce clustering of Snp1 in developing neurons (Hall et al., 2002). We provided evidence showing that a possible mechanism by which VPA could be beneficial for AD patients, is by reversing the effects of Aβ on Snp1 phosphorylation in a concentration dependent manner (Marsh et al., 2017). Even though these data are based on an in vitro system, we believe they are promising. Indeed, the results from our in vitro study correlate well with in vivo data suggesting that VPA can improve cognition (Wu et al., 2016). Currently, we can only speculate how VPA abrogates the effects of Aβ and further research is needed to understand the details of how and where Aβ and VPA pathways intercept at the presynapse. Nevertheless, we believe this example highlights that a detailed understanding of the cellular mechanisms affected by Aβ can serve as the starting point in designing new therapies or adopting existing ones.
Conclusion
Despite AD being the focus of intense research, a treatment that will significantly benefit the people affected by the disease is not yet in sight. The inability of several approaches targeting Aβ peptides to demonstrate the expected effectiveness has sparked a debate in the scientific community regarding the validity of the amyloid cascade hypothesis. Although the contribution of Aβ peptides to the progression of the disease cannot be discounted, the failure to design an effective therapy by simply targeting Aβ peptides suggests that a different approach is needed. We believe that a detailed comprehension of the physiological role of Aβ peptides and their cumulative effect on the ageing brain will significantly aid the design of novel therapeutic approaches that target specific cellular mechanisms. For instance, recent research focused on understanding the mechanisms underlying Aβ effects on NT release has provided new insights into how this remarkable peptide affects synaptic function and neuronal communication. Based on this evidence, we have proposed that the synaptic dysfunction occurring in the early stages of AD involves a gradual, additive disruption of key aspects of SV dynamics due to the presence of iAβ at the presynapse, which also reveals new drug targets for therapeutic intervention. There is still a lot to learn, but increasing our knowledge of the different aspects of the biology of Aβ peptides will facilitate a more targeted and hopefully more efficient therapeutic approach.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Syed Faraz Kazim, Icahn School of Medicine at Mount Sinai, USA; Jaspreet Kalra, ISF College of Pharmacy, India.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Amanatkar HR, Papagiannopoulos B, Grossberg GT. Analysis of recent failures of disease modifying therapies in Alzheimer's disease suggesting a new methodology for future studies. Expert Rev Neurother. 2017;17:7–16. doi: 10.1080/14737175.2016.1194203. [DOI] [PubMed] [Google Scholar]
- Andrew RJ, Kellett KA, Thinakaran G, Hooper NM. A Greek tragedy: the growing complexity of alzheimer amyloid precursor Protein Proteolysis. J Biol Chem. 2016;291:19235–19244. doi: 10.1074/jbc.R116.746032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Arrieta I, Tapia R. beta-Amyloid peptide fragment 25-35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J Neurosci Res. 1995;41:561–566. doi: 10.1002/jnr.490410416. [DOI] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Greengard P. Computer modeling of synapsin I binding to synaptic vesicles and F-actin: implications for regulation of neurotransmitter release. Proc Natl Acad Sci U S A. 1991;88:575–579. doi: 10.1073/pnas.88.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P. Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol. 1989;108:1863–1872. doi: 10.1083/jcb.108.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Chieregatti E, Greengard P. Interaction of free and synaptic vesicle-bound synapsin I with F-actin. Neuron. 1992;8:377–386. doi: 10.1016/0896-6273(92)90303-u. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chang P, Chandler KE, Williams RS, Walker MC. Inhibition of long-term potentiation by valproic acid through modulation of cyclic AMP. Epilepsia. 2010;51:1533–1542. doi: 10.1111/j.1528-1167.2009.02412.x. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Daly C, Sugimori M, Moreira JE, Ziff EB, Llinas R. Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97:6120–6125. doi: 10.1073/pnas.97.11.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic impairment in Alzheimer's disease: a dysregulated symphony. Trends Neurosci. 2017;40:347–357. doi: 10.1016/j.tins.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham WV, Bonito-Oliva A, Sakmar TP. Update on Alzheimer's disease therapy and prevention strategies. Annu Rev Med. 2017;68:413–430. doi: 10.1146/annurev-med-042915-103753. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, George L, Tung YC, Kim KS, Wisniewski HM. Amyloid protein and neurofibrillary tangles coexist in the same neuron in Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:2853–2857. doi: 10.1073/pnas.86.8.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Brennan A, Goold RG, Cleverley K, Lucas FR, Gordon-Weeks PR, Salinas PC. Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci. 2002;20:257–270. doi: 10.1006/mcne.2002.1117. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harris ME, Carney JM, Cole PS, Hensley K, Howard BJ, Martin L, Bummer P, Wang Y, Pedigo NW Jr, Butterfield DA. beta-Amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for Alzheimer's disease. Neuroreport. 1995;6:1875–1879. doi: 10.1097/00001756-199510020-00013. [DOI] [PubMed] [Google Scholar]
- Hartmann T. Intracellular biology of Alzheimer's disease amyloid beta peptide. Eur Arch Psychiatry Clin Neurosci. 1999;249:291–298. doi: 10.1007/s004060050102. [DOI] [PubMed] [Google Scholar]
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Poole Hoffmann V, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321–330. doi: 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JP, Xie JW, Wang CY, Wang T, Wang X, Wang SL, Teng WP, Wang ZY. Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res Bull. 2011;85:194–200. doi: 10.1016/j.brainresbull.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Hung SY, Fu WM. Drug candidates in clinical trials for Alzheimer's disease. J Biomed Sci. 2017;24:47. doi: 10.1186/s12929-017-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabogo D, Rauw G, Amritraj A, Baker G, Kar S. beta-Amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices. Neurobiol Aging. 2008;31:1164–1172. doi: 10.1016/j.neurobiolaging.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;147:60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Vassar R, Ferreira A. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem. 2005;280:31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Corbett A, Ballard C. Emerging amyloid and tau targeting treatments for Alzheimer's disease. Expert Rev Neurother. 2017;17:697–711. doi: 10.1080/14737175.2017.1326819. [DOI] [PubMed] [Google Scholar]
- Klein WL. Synaptic targeting by A beta oligomers (ADDLS) as a basis for memory loss in early Alzheimer's disease. Alzheimers Dement. 2006;2:43–55. doi: 10.1016/j.jalz.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo H, Kayed R, Glabe CG, Yamaguchi H. Soluble Abeta oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer's disease brain. Brain Res. 2005;1031:222–228. doi: 10.1016/j.brainres.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin MB, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe D. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Zhang Y, Han L, He G, Xie W, Zhou Z, Jia Z. Regulation of neurotransmitter release by amyloid precursor protein through synapsin phosphorylation. Neurochem Res. 2017 doi: 10.1007/s11064-017-2418-2. doi: 10.1007/s11064-017-2418-2. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J, Bagol SH, Williams RSB, Dickson G, Alifragis P. Synapsin I phosphorylation is dysregulated by beta-amyloid oligomers and restored by valproic acid. Neurobiol Dis. 2017;106:63–75. doi: 10.1016/j.nbd.2017.06.011. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Soos K, Lengyel I, Penke B, Szegedi V, Budai D. Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport. 2004;15:1649–1652. doi: 10.1097/01.wnr.0000134471.06244.d2. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Orenbuch A, Shalev L, Marra V, Sinai I, Lavy Y, Kahn J, Burden JJ, Staras K, Gitler D. Synapsin selectively controls the mobility of resting pool vesicles at hippocampal terminals. J Neurosci. 2012;32:3969–3980. doi: 10.1523/JNEUROSCI.5058-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Na M, Kim JA, Lee U, Cho E, Jang M, Chang S. Activation of CaMKIV by soluble amyloid-beta1-42 impedes trafficking of axonal vesicles and impairs activity-dependent synaptogenesis. Sci Signal. 2017:10. doi: 10.1126/scisignal.aam8661. [DOI] [PubMed] [Google Scholar]
- Parodi J, Sepulveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem. 2010;285:2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Bonanomi D, Benfenati F, Valtorta F. Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol Biol Cell. 2003;14:4909–4919. doi: 10.1091/mbc.E03-06-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D’Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, Lammich S, Isbrandt D, Schweizer M, Horre K, De Strooper B, Saftig P. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci. 2013;33:12915–12928, 12928a. doi: 10.1523/JNEUROSCI.5910-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, He G, Ly PTT, Fox CJ, Staufenbiel M, Cai F, Zhang Z, Wei S, Sun X, Chen CH, Zhou W, Wang K, Song W. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med. 2008;205:2781–2789. doi: 10.1084/jem.20081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli C, Cocco S, Li Puma DD, Piacentini R, Mastrodonato A, Scala F, Puzzo D, D’Ascenzo M, Grassi C. Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci. 2014;34:12893–12903. doi: 10.1523/JNEUROSCI.1201-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P. Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One. 2012;7:e43201. doi: 10.1371/journal.pone.0043201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Haucke V, Pechstein A. How synapsin I may cluster synaptic vesicles. Semin Cell Dev Biol. 2011;22:393–399. doi: 10.1016/j.semcdb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, Gylys KH. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer's disease cortex. Neurobiol Dis. 2012;45:381–387. doi: 10.1016/j.nbd.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Williams RS, Bate C. An in vitro model for synaptic loss in neurodegenerative diseases suggests a neuroprotective role for valproic acid via inhibition of cPLA2 dependent signalling. Neuropharmacology. 2016;101:566–575. doi: 10.1016/j.neuropharm.2015.06.013. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Doms RW, Lee VM. Intracellular APP processing and A beta production in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:787–794. doi: 10.1097/00005072-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Wu P, Hong S, Zhong M, Guo Y, Chen H, Jiang L. Effect of Sodium Valproate on Cognitive Function and Hippocampus of Rats After Convulsive Status Epilepticus. Med Sci Monit. 2016;22:5197–5205. doi: 10.12659/MSM.898859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kim J, Kim Hye Y, Ryoo N, Lee S, Kim Y, Rhim H, Shin YK. Amyloid-β oligomers may impair SNARE-MEDIATED EXOCYTOSIS BY DIRECT BINDING TO SYNTaxin 1a. Cell Reports. 2015;12:1244–1251. doi: 10.1016/j.celrep.2015.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, Xiao C, Yu K, Ruan DY. Effects of sodium valproate on synaptic plasticity in the CA1 region of rat hippocampus. Food Chem Toxicol. 2003;41:1617–1623. doi: 10.1016/s0278-6915(03)00195-9. [DOI] [PubMed] [Google Scholar]
- Zhao J, Nussinov R, Ma B. Mechanisms of recognition of amyloid-beta (Abeta) monomer, oligomer, and fibril by homologous antibodies. J Biol Chem. 2017;292:18325–18343. doi: 10.1074/jbc.M117.801514. [DOI] [PMC free article] [PubMed] [Google Scholar]