

α-Synuclein causes synaptic pathologies in several neurodegenerative diseases: Parkinson's disease (PD) is a neurodegenerative disease that impacts the lives of millions of people worldwide. A pathological hallmark of PD, as well as dementia with Lewy bodies (DLB) and several Alzheimer's disease variants, is the appearance of intracellular inclusions called Lewy bodies, which contain high levels of aggregated α-synuclein. α-Synuclein is a presynaptic protein that normally associates with synaptic vesicle membranes and regulates synaptic vesicle trafficking under physiological conditions (Calo et al., 2016). However, in familial PD, multiplication and several point mutations in the α-synuclein gene (SNCA) ultimately lead to toxic aggregation of the α-synuclein protein and subsequent degeneration of dopaminergic neurons in the substantia nigra, although other brain areas are also affected (Schulz-Schaeffer, 2010).
Recent studies indicate that toxic oligomerization and aggregation of α-synuclein also occurs in presynaptic boutons concurrently with or before Lewy body formation in both experimental models and human patients. When overexpressed, α-synuclein forms synaptic aggregates, which are associated with compromised neurotransmission in PD animal models (Nemani et al., 2010). These synaptic aggregates, rather than Lewy body prevalence, are highly correlated with greater cognitive decline in PD and DLB patients (Schulz-Schaeffer, 2010; Calo et al., 2016). Therefore, synaptic aggregation of α-synuclein likely plays a central role in triggering α-synucleinopathies, making the synapse a crucial place in which to study disease pathogenesis.
The classical model for α-synuclein toxicity suggests that misfolding of cytosolic monomeric α-synuclein leads to assembly of small toxic oligomers, followed by fibrillation, aggregation, and eventually Lewy body formation. In this model, the native form of α-synuclein is monomeric and possesses a random coil structure. However, recent studies challenge this model, suggesting that folded alpha-helical tetramers represent the native form of α-synuclein (Selkoe et al., 2014). By these reports, tetramers are more stable and resistant to aggregation compared to monomeric α-synuclein. Despite this debate, it is generally accepted that neurons naturally produce some monomeric, dimeric, and oligomeric forms of α-synuclein and that altering the balance of these molecular species contributes to disease pathogenesis. However, how each molecular species of α-synuclein impacts neuronal function, including synaptic function, is unknown.
Acute introduction of α-synuclein impairs clathrin-mediated synaptic vesicle endocytosis: In traditional overexpression models, it is difficult to control the oligomerization status of α-synuclein, making it challenging to determine which molecular species of α-synuclein are affecting synaptic processes and how. In addition, α-synuclein overexpression induces downregulation of several presynaptic proteins (Nemani et al., 2010), making it difficult to assess the direct impacts of α-synuclein itself. These particular issues can be overcome by utilizing an acute perturbation strategy where distinct molecular species of recombinant α-synuclein protein can be introduced directly to presynapses. We have taken advantage of this approach using the lamprey giant reticulospinal synapses, which provide an ideal model for these experiments because they allow for acute introduction of distinct α-synuclein species to presynapses in known quantities via axonal microinjection (Busch et al., 2014; Medeiros et al,. 2017). We thus began by acutely introducing excess monomeric human α-synuclein to lamprey synapses in concentrations ranging from 10–20 μM, which is 2–3 times the current estimates of endogenous α-synuclein at synapses and commensurate with overexpression levels observed in PD patients. Acute introduction of monomeric α-synuclein dramatically impaired synaptic vesicle endocytosis, as indicated by a loss of synaptic vesicles, which was compensated by larger plasma membrane extensions and greater numbers of endocytic profiles, including clathrin-coated intermediates (Figure 1) (Busch et al., 2014). Similarly, acute dialysis of monomeric α-synuclein and α-synuclein overexpression at the mammalian calyx of Held synapse also impaired synaptic vesicle endocytosis, with little or no effects on exocytosis (Xu et al., 2016).
Figure 1.
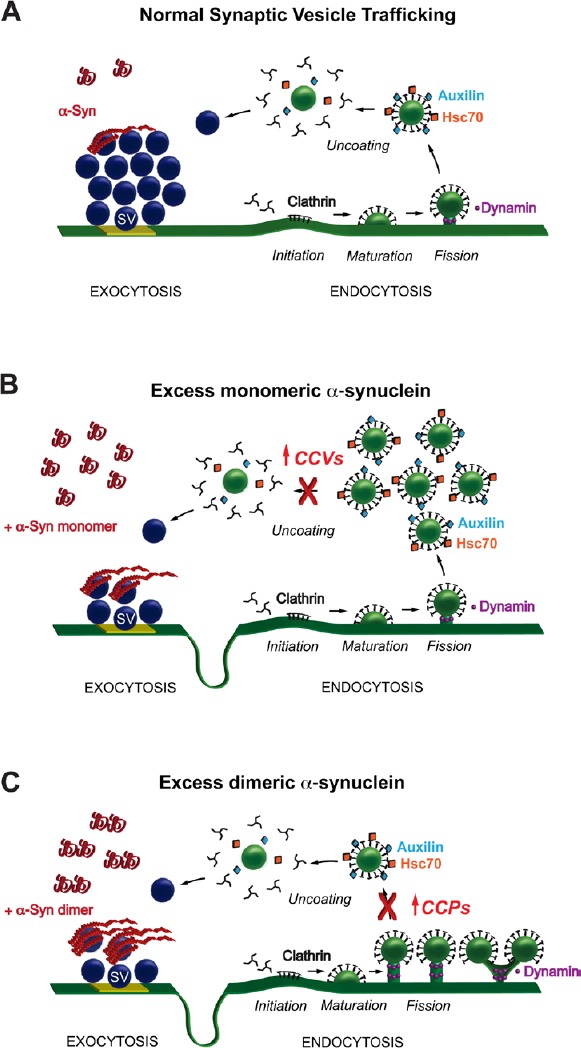
Monomeric and dimeric α-synuclein produce distinct effects on clathrin- mediated synaptic vesicle (SV) endocytosis.
(A) Model showing SV recycling and the role for clathrin-mediated endocytosis. For simplicity, other membrane recycling pathways (e.g., bulk endocytosis) are not shown. Under physiological conditions, α-synuclein localizes to the SV cluster and regulates exocytosis and endocytosis, though the exact mechanisms are unclear. Following SV exocytosis and neurotransmitter release, the vesicle membrane is recycled via clathrin-mediated endocytosis. The basic stages of clathrin-mediated endocytosis are: initiation of clathrin coat formation; maturation of the coated pit; vesicle fission; and clathrin uncoating. Once uncoated, the vesicle is returned to the cluster where it can be re-used in another bout of neurotransmitter release. (B) Acute introduction of monomeric α-synuclein to synapses results in a loss of SVs, extension of the plasma membrane, and build up clathrin intermediates, predominantly CCVs. Thus, monomeric α-synuclein inhibits SV recycling at least in part by impairing clathrin uncoating. (C) In contrast, introduction dimeric α-synuclein increases the numbers of CCPs that are still connected to the plasma membrane, indicating a defect in vesicle fission. Thus, α-synuclein monomers and dimers produce distinct effects on clathrin-mediated SV recycling.
We next set out to determine exactly how excess α-synuclein interfered with clathrin-mediated vesicle endocytosis at synapses. Clathrin-mediated endocytosis, which is a major route for recycling synaptic vesicles, progresses through several morphologically distinct stages (Figure 1A). Initiation of clathrin-coated pit (CCP) formation occurs when clathrin adaptor proteins recruit clathrin triskelions to the plasma membrane (Initiation). The CCP then matures through a process involving actin cytoskeleton and other proteins (Maturation). Next, the GTPase dynamin is recruited to the neck of the CCP, where it mediates vesicle fission (Fission). Once separated from the plasma membrane, the free clathrin-coated vesicle (CCV) is uncoated by the chaperone protein, heat shock cognate protein 70 (Hsc70), and its co-chaperone auxilin (Uncoating). Upon further investigation of the synaptic phenotype produced by increased levels of monomeric α-synuclein, we observed a selective increase in the numbers of free CCVs, indicating a defect in the process of clathrin uncoating (Figure 1B) (Medeiros et al., 2017). This uncoating defect is caused at least in part by sequestration of Hsc70 because when exogenous Hsc70 was added back to the synapse, the defects in CCV uncoating and vesicle endocytosis were ameliorated (unpublished).
Monomeric and dimeric α-synuclein produce distinct effects on clathrin-mediated synaptic vesicle endocytosis: We next wanted to determine whether increasing α-synuclein dimers produced a similar – or distinct - phenotype at synapses compared with monomeric α-synuclein. For these experiments, we used recombinant α-synuclein dimers that were generated by covalent linkage of two human α-synuclein molecules, which allowed us to begin distinguishing the synaptic effects caused by an imbalance of α-synuclein oligomers (Pivato et al., 2012). Biochemically, recombinant α-synuclein dimers exhibited in vitro properties, including folding, fibrillation, and lipid binding, that were similar to monomeric α-synuclein, albeit with slightly different efficacies (Pivato et al., 2012; Medeiros et al., 2017). Like monomeric α-synuclein, the α-synuclein dimers inhibited synaptic vesicle endocytosis and increased the number of clathrin-coated intermediates (Medeiros et al., 2017). However, unlike monomeric α-synuclein, the dimers increased the number of CCPs that were still attached to the plasma membrane (Figure 1C). These structures presented as single CCPs with atypical elongated necks or branched tubules ending in multiple clathrin-coated buds, indicating a defect in the earlier stage of vesicle fission. CCPs induced by α-synuclein dimers were also morphologically distinct from those induced by a dynamin GTP-ase inhibitor called Dynasore (Medeiros et al., 2017). These data suggest that the phenotype induced by α-synuclein dimers is not simply due to an inhibition of dynamin GTPase activity, though dynamin or other fission-associated proteins such as endophilin, amphiphysin or synaptojanin could still play a role. Notably, with α-synuclein dimers, the number of free CCVs was unchanged, suggesting that clathrin uncoating was relatively unaffected (Medeiros et al., 2017). Taken together, these acute perturbation studies at lamprey giant synapses contribute to a growing body of evidence indicating that increased levels of α-synuclein at synapses impairs vesicle endocytosis and recycling (Busch et al., 2014; Xu et al., 2016) and/or vesicle reclustering (Nemani et al., 2010). The lamprey studies also provide the first evidence that different molecular species of α-synuclein may have different impacts at the synapse, suggesting that they may have distinct molecular targets that function within the clathrin pathway.
Mutations in genes associated with clathrin-mediated endocytosis are linked to PD and parkinsonism: Our studies at lamprey synapses implicate clathrin-mediated endocytosis as a possible target of α-synuclein synaptic pathologies. Similarly, human genetics studies on PD are also pointing toward defects in the clathrin pathway as a possible underlying cause. For example, some patients with juvenile Parkinsonism carry a deletion in DNAJC6, the gene that encodes for auxilin, the Hsc70 co-chaperone that functions during CCV uncoating at synapses (Edvardson et al., 2012). In addition, increased expression of GAK, the ubiquitously expressed Hsc70 co-chaperone, is associated with risk of familial PD (Nagle et al., 2016). A missense mutation in synaptojanin 1 (R258Q), another protein that regulates clathrin uncoating at synapses, has also been linked to early onset parkinsonism (Krebs et al., 2013). Indeed transgenic mice carrying the synaptojanin I R258Q mutation accumulated CCVs within their synaptic boutons and exhibited impaired synaptic vesicle endocytosis, dystrophic axons, severe motor deficits, and increased death rate (Cao et al., 2017). Thus, a growing body of evidence from both animal models and human genetics indicates that defects in clathrin-mediated endocytosis are at least susceptibility factors in PD and parkinsonism, if not causal factors.
Going forward, it will be important to determine how higher molecular weight species and small toxic oligomers of α-synuclein impact synaptic vesicle trafficking and clathrin-mediated endocytosis at synapses. Understanding the molecular targets for each distinct α-synuclein species will be critical to these efforts. It will also be important to determine how the other human mutations associated with PD and parkinsonism affect synaptic structure and function, as has been done for the synaptojanin I R258Q mutation (Cao et al., 2017). Collectively, the current data suggest the possibility that synaptic deficiencies associated with increased levels of α-synuclein may be corrected by targeting aspects of the clathrin pathway. Determining how to ameliorate synaptic defects is a key step toward improving the symptoms in patients with α-synucleinopathies and many other related neurodegenerative diseases.
This research was supported by a grant from National Institutes of Health (NINDS/NIA R01NS078165 to JRM), research funds from the Marine Biological Laboratory (to JRM), and a research grant from Horizon 2020 Grant No. InCure EU Joint Programme-JPND (to LB).
This research was presented in American Society for Cell Biology in December 2016 and in Society for Neuroscience in November 2017.
Footnotes
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review report:
Reviewer: Andre Toulouse, University College Cork, Ireland.
Comments to authors: The perspective provides an overview of synaptic clathrin-mediated endocytosis and the impact of excess alpha-synuclein on the process.
References
- Busch DJ, Oliphint PA, Walsh RB, Banks SM, Woods WS, George JM, Morgan JR. Acute increase of alpha-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol Biol Cell. 2014;25:3926–3941. doi: 10.1091/mbc.E14-02-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo L, Wegrzynowicz M, Santivanez-Perez J, Grazia Spillantini M. Synaptic failure and alpha-synuclein. Mov Disord. 2016;31:169–177. doi: 10.1002/mds.26479. [DOI] [PubMed] [Google Scholar]
- Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA, Boassa D, Ellisman MH, Ryan TA, De Camilli P. Parkinson sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron. 2017;93:882–896.e5. doi: 10.1016/j.neuron.2017.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisan-Ruiz C. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros AT, Soll LG, Tessari I, Bubacco L, Morgan JR. α-Synuclein dimers impair vesicle fission during clathrin-mediated synaptic vesicle recycling. Front Cell Neurosci. 2017;11:388. doi: 10.3389/fncel.2017.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle MW, Latourelle JC, Labadorf A, Dumitriu A, Hadzi TC, Beach TG, Myers RH. The 4p16.3 Parkinson disease risk locus is associated with GAK expression and genes involved with the synaptic vesicle membrane. PLoS One. 2016;11:e0160925. doi: 10.1371/journal.pone.0160925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivato M, De Franceschi G, Tosatto L, Frare E, Kumar D, Aioanei D, Brucale M, Tessari I, Bisaglia M, Samori B, de Laureto PP, Bubacco L. Covalent alpha-synuclein dimers: chemico-physical and aggregation properties. PLoS One. 2012;7:e50027. doi: 10.1371/journal.pone.0050027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Dettmer U, Luth E, Kim N, Newman A, Bartels T. Defining the native state of alpha-synuclein. Neurodegener Dis. 2014;13:114–117. doi: 10.1159/000355516. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu XS, Sheng J, Zhang Z, Yue HY, Sun L, Sgobio C, Lin X, Peng S, Jin Y, Gan L, Cai H, Wu LG. α-Synuclein mutation inhibits endocytosis at mammalian central nerve terminals. J Neurosci. 2016;36:4408–4414. doi: 10.1523/JNEUROSCI.3627-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]