

Abstract
We report an unusual phenotype in a child with a clinical diagnosis of recessive Stargardt disease (STGD1) and two pathogenic variants in the ABCA4 gene. Typically, the diagnosis of early-onset STGD1 is challenging because children may present with a variety of fundus changes and a variable rate of progression. At the time of his initial visit, the 6-year-old boy presented with 20/200 OD and 20/150 OS, symmetrical mild foveal atrophy without flecks on fundus exam, and foveal hypoautofluorescence surrounded by a homogeneous hyperautofluorescent background on wide-field fundus autofluorescence (WF-FAF). Over four years of follow-up, the retinal atrophy continued to progress, resulting in two well-defined and concentric hyperautofluorescent rings: one ring located at the posterior pole and the other located around the peripapillary region. Visual acuity also deteriorated to counting fingers at 4ft OD and 20/500 OS. To the best of our knowledge, this phenotype has not been previously described with the ABCA4 gene.
Keywords: ABCA4, fundus autofluorescence, macular degeneration, retinal degeneration, retinal imaging, Stargardt disease
INTRODUCTION
Recessive Stargardt disease (STGD1) is a juvenile-onset macular dystrophy caused by pathogenic variants in the ABCA4 gene and is the most common inherited macular dystrophy. (1, 2) The clinical diagnosis of early-onset STGD1 is challenging because children may have a variety of presentations, such as macular atrophy with macular or peripheral flecks, macular atrophy without flecks, flecks without atrophy, or normal fundus appearance. (3, 4) Fundus autofluorescence (AF) also demonstrates variable patterns, including multiple or localized low-signal areas with surrounding heterogeneous or homogeneous backgrounds that ultimately progress to widespread chorioretinal atrophy. (3–6)
In 2012, Escher et al reported an unusual appearance of fundus AF in patients with autosomal dominant retinitis pigmentosa due to a p.G56R mutation in NR2E3 of a double concentric autofluorescent ring on 55° field fundus AF. (7) Later in 2014, using wide-field fundus AF (WF-FAF), one of those patients was found to have not a double, but a triple concentric autofluorescent ring. (8) Trichonas et al reported the presence of a hyperautofluorescent ring in patients with RHO, USH2A, CEP290, RPGRIP1 and RPGR mutations, and he also noted the presence of a double hyperautofluorescent ring on WF-FAF in patients with a mutation in the USH2A gene.(9)
We describe the formation and progression of two concentric hyperautofluorescent rings over four years of follow-up in a child with a clinical diagnosis of STGD1 and two pathogenic variants in the ABCA4 gene. WF-FAF demonstrated a hyperautofluorescent ring around the optic nerve in addition to an outer hyperautofluorescent ring surrounding the posterior pole. To the best of our knowledge, this phenotype has not been previously described with the ABCA4 gene.
CASE REPORT
A 6-year-old boy of Taiwanese and Korean descent was referred for a six-month history of progressive visual loss in both eyes (OU). Other relevant past medical or ocular history was negative. His paternal uncle had a diagnosis of STGD and had previously undergone genetic testing that identified two pathogenic variants in the ABCA4 gene. Best-corrected visual acuity (BCVA) for the patient was 20/200 OD and 20/150 OS, and fundus examination showed signs of mild foveal atrophy without flecks. (Figure 1) Goldmann visual field (GVF) testing revealed central scotomata OU. Full-field ERG (ffERG), compliant with the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV), showed mildly reduced scotopic responses OU and normal photopic responses OD (photopic responses were not evaluated in OS due to patient discomfort). WF-FAF (Optos 200 Tx, Optos PLC, Dunefermline, United Kingdom) showed foveal hypoautofluorescence surrounded by a homogeneous hyperautofluorescent background OU. (Figure 2) SD-OCT (Spectralis, Heidelberg Engineering, Heidelberg, Germany) showed foveal atrophy, hyperreflective dots in the outer retinal layers, and foveal ellipsoid zone disruption that was halted at the border of the peripapillary hyperautofluorescent ring, showing peripapillary retinal preservation. (Figure 3)
Figure 1.
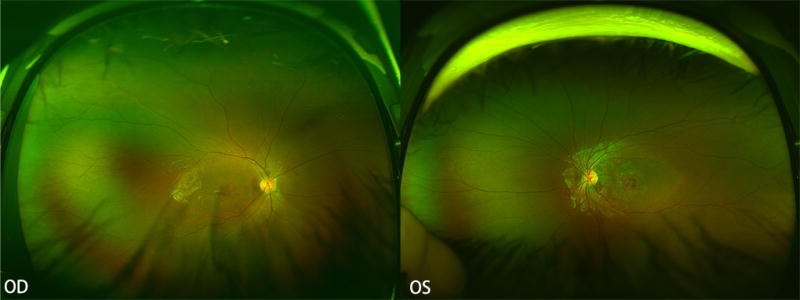
WF fundus photography at baseline shows central atrophy without flecks in both eyes.
Figure 2.
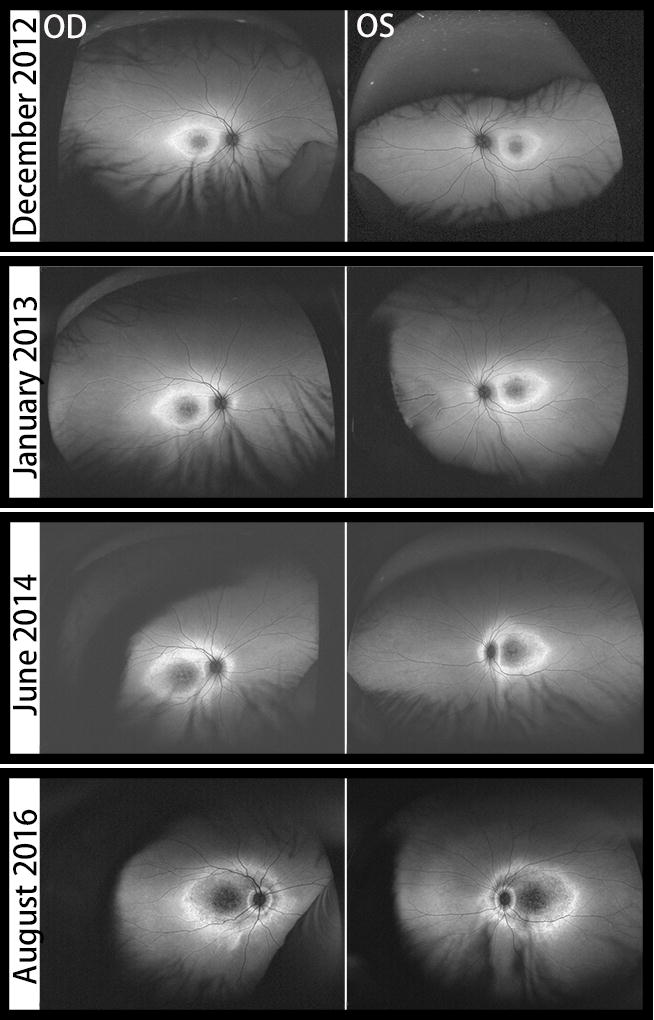
WF-FAF images of both eyes over four years of follow-up. At baseline, AF imaging demonstrates foveal hypoautofluorescence with surrounding hyperautofluorescent background. Overtime, a well-defined hyperautofluorescent ring develops around the posterior pole followed by subsequent atrophy, creating a gap that allows a second inner hyperautofluorescent ring around the peripapillary region to emerge.
Figure 3.
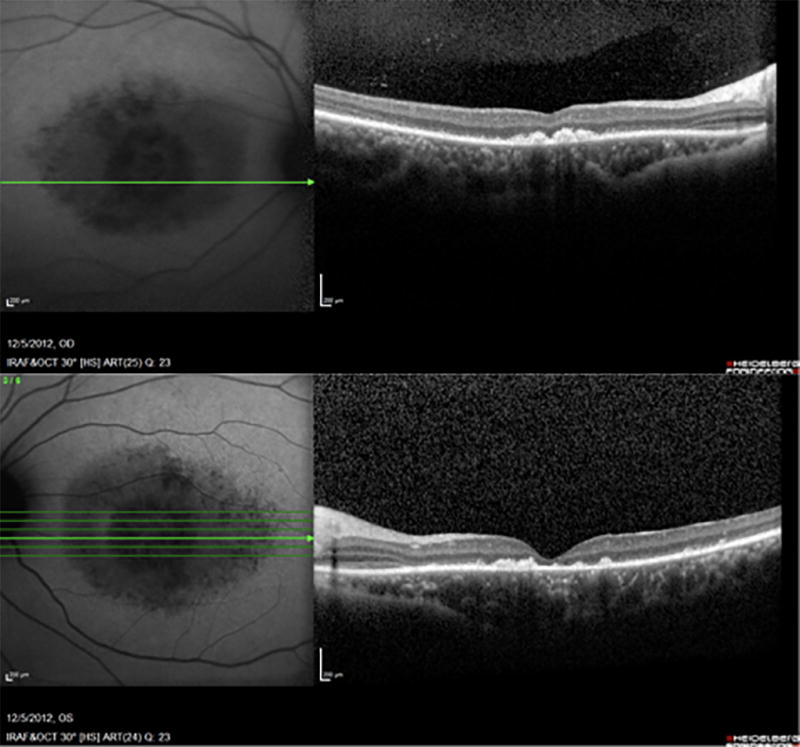
SD-OCT of both eyes at baseline shows foveal atrophy with disruption of the ellipsoid zone. Note that the ellipsoid zone is preserved nasally, demonstrating peripapillary retinal sparing.
Genetic testing was ordered, and two likely pathogenic variants in the ABCA4 gene were found: 1) IVS23-2A>G (c.3523-2A>G), a novel variant predicted to affect splicing, inherited maternally, and 2) p.L750P c.2249T>C, a novel missense variant predicted probably damaging by the clinical testing laboratory, inherited paternally. In silico analysis also supported this (Polyphen-2=probably damaging; SIFT=damaging). The patient’s paternal uncle presented with macular atrophy and carried one of the same ABCA4 variants as the patient (p.L750P:c.2249T>C) along with a previously-reported pathogenic variant in ABCA4 (c.4748T>C:p.Leu1583Pro). (10) Additionally, one variant of unknown significance (VUS) was identified in the patient’s RPGR gene (p.L428V c.1282C>G), inherited maternally. This VUS was predicted to be probably damaging by the clinical testing laboratory. In silico analysis also supported this (Polyphen-2=probably damaging; SIFT=damaging). However, the variant has been detected in 9/6464 alleles (including four males) in the ExAC database. This relatively high allele frequency suggests that the variant is unlikely to be pathogenic. In addition, all 9 alleles were detected in individuals of East Asian descent, suggesting that the c.1282C>G variant may be a benign variant in this population.
Over four years of follow-up, the patient’s BCVA progressively decreased to counting fingers at 4 feet OD and 20/500 OS. Over this same time, GVF showed enlargement of the central scotomata and constriction of the peripheral isopters. However, due to the young age of the patient, responses were inconsistent and unreliable. On WF-FAF, the hyperautofluorescent background gradually expanded centrifugally towards the vascular arcades and around the peripapillary region, with concomitant hypoautofluorecence of the encircled area. As a result, a well-defined hyperautofluorescent ring was formed around the entire posterior pole, and subsequent atrophy created a gap, allowing a second inner hyperautofluorescent ring around the peripapillary region to develop.
The temporal region of the peripapillary ring formed first, followed by the superior, nasal and inferior regions of the ring in both eyes over the four years of follow-up. It is noteworthy that the area between the edges of the peripapillary ring and the optic nerve head remained unaffected, reflecting peripapillary sparing. (Figure 3)
DISCUSSION
We describe the formation of two well-defined hyperautofluorescent rings in a child with two pathogenic variants in the ABCA4 gene: one ring circumscribing the posterior pole and the other circumscribing the peripapillary region encircled by the first. We believe that these findings represent an uncommon phenotype in a childhood-onset form of STGD1.
The posterior pole/parafoveal hyperautofluorescent ring seen in this case is a non-specific manifestation that can occur in several retinal dystrophies (11–13), including STGD1 (14), and likely results from abnormal parafoveal accumulation of lipofuscin.(14, 15) A hyperautofluorescent ring separates central affected retina from peripheral preserved retina or vice versa, depending on the gene, and thus usually reflects disease progression. (11) In this case, the outer ring encircled the affected posterior pole, because changes started in the fovea and progressed centrifugally, as expected for the ABCA4-STGD1 spectrum of disease. In contrast, in the retinitis pigmentosa (RP) spectrum, a ring generally progresses from the mid-periphery towards the macula. In patients with RP, a hyperautofluorescent ring encircles preserved areas. (12, 13, 16)
To our knowledge, the development and expansion of a hyperautofluorescent ring around the peripapillary region has not been previously reported in patients with STGD1. The hyperautofluorescent expansion around the optic nerve leading to a peripapillary hyperautofluorescent ring with outer hypoautofluorescent transformation most likely suggests a centrifugal pattern of progression, as seen in the parafoveal ring (4, 17) However, as opposed to the parafoveal ring, this peripapillary ring encircles an unaffected area, as demonstrated by a normal autofluorescence and preserved ellipsoid zone on SD-OCT, reflecting peripapillary sparing. Peripapillary sparing has been previously described, but not in association with a peripapillary hyperautofluorescent ring on fundus AF (18–20). Instead, it has been demonstrated on fluorescein angiography (FA), SD-OCT and FAF. Jayasundera et al demonstrated the peripapillary sparing as a dark choroidal ring on fluorescein angiography (FA) in the presence of diffuse posterior pole hyperfluorescence resulting from advanced disease. (20) Burke et al demonstrated a relative sparing of outer nuclear and outer photoreceptor layers at the peripapillary region using SD-OCT in patients with STGD1 extending outside the macula in contrast with normal controls. (18) Using fundus AF, Cideciyan et al demonstrated parapapillary sparing as an annulus of normal AF measuring at least 0.6mm in diameter around the optic nerve head in all disease stages. (19)
The presence of two concentric autofluorescent rings has been previously reported in NR2E3-linked autosomal dominant retinitis pigmentosa.(7) In this study, one constricted inner perifoveal hyperautofluorescent ring and one outer hyperautofluorescent ring located outside the vascular arcade were described. (7) WF-FAF was performed on one of these patients in a subsequent study, which revealed a third and outermost concentric autofluorescent ring (8). Trichonas et al also reported the presence of two hyperautofluorescent rings in USH2A-related retinitis pigmentosa. In this case, there was diffuse macular hyperautofluorescence and a ring of hyperautofluorescence. (9) Although the topography of these rings differed from the ones described in our patient, they were consistent with progressive photoreceptor loss and associated with advanced disease, similar to the child described in our report.
The two ABCA4 variants identified in our patient are likely to be disease- causing and sufficient to cause disease, considering the phenotype (peripapillary sparing, early-onset and rapid progression) and the fact that patient’s paternal uncle, who also has two pathogenic variants in ABCA4, has a similar fundus appearance and the same clinical diagnosis, although his disease is less severe. Although the VUS in the RPGR gene is likely to be benign, it is unknown what effect, if any, it may have on the patient’s fundus findings. Mutations in the RPGR gene that cause early involvement of the macula with sparing of the periphery are generally located at the 3’ end of the gene, rather than in exons 1–15, where our patient’s variant was detected. In addition, mutations in the RPGR gene that cause early macular involvement are usually associated with a cone-rod phenotype, and the age of onset is usually later, ranging from the second to the fourth decade.(21, 22) Therefore, due to the location of the patient’s VUS and age of onset, the RPGR variant is unlikely to be associated with his phenotype.
In addition to its reported presence in NR2E3-related RP, the hyperautofluorescent peripapillary ring and perimacular hyperautofluorescent ring (a double hyperautofluorecent ring) has now also been seen in patients with mutations in the ABCA4 gene. It is still not clear what this pattern represents. Prospective studies are necessary to demonstrate functional changes associated with this finding. Of note, the peripapillary area remained spared in our patient. This is a hallmark of ABCA4-related disease and has the potential to act as a preferred retinal locus in visual rehabilitation.
Footnotes
Color versions of one or more figures in the article can be found online at http://www.tandfonline.com/iopg.
References
- 1.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature genetics. 1997;15(3):236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, Brucker AJ, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Human molecular genetics. 2004;13(5):525–34. doi: 10.1093/hmg/ddh048. [DOI] [PubMed] [Google Scholar]
- 3.Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122(2):326–34. doi: 10.1016/j.ophtha.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, et al. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015;122(2):335–44. doi: 10.1016/j.ophtha.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 5.McBain VA, Townend J, Lois N. Progression of retinal pigment epithelial atrophy in stargardt disease. American journal of ophthalmology. 2012;154(1):146–54. doi: 10.1016/j.ajo.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Strauss RW, Ho A, Munoz B, Cideciyan AV, Sahel JA, Sunness JS, et al. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology. 2016;123(4):817–28. doi: 10.1016/j.ophtha.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Escher P, Tran HV, Vaclavik V, Borruat FX, Schorderet DF, Munier FL. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa. Investigative ophthalmology & visual science. 2012;53(8):4754–64. doi: 10.1167/iovs.11-8693. [DOI] [PubMed] [Google Scholar]
- 8.Escher P, Vaclavik V, Munier FL, Tran HV. Presence of a Triple Concentric Autofluorescence Ring in NR2E3-p.G56R-Linked Autosomal Dominant Retinitis Pigmentosa (ADRP) Investigative ophthalmology & visual science. 2016;57(4):2001–2. doi: 10.1167/iovs.16-19459. [DOI] [PubMed] [Google Scholar]
- 9.Trichonas G, Traboulsi EI, Ehlers JP. Ultra-widefield fundus autofluorescence patterns in retinitis pigmentosa and other retinal dystrophies. Ophthalmic genetics. 2017;38(1):98–100. doi: 10.3109/13816810.2015.1137328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukui T, Yamamoto S, Nakano K, Tsujikawa M, Morimura H, Nishida K, et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Investigative ophthalmology & visual science. 2002;43(9):2819–24. [PubMed] [Google Scholar]
- 11.Robson AG, Michaelides M, Saihan Z, Bird AC, Webster AR, Moore AT, et al. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Documenta ophthalmologica Advances in ophthalmology. 2008;116(2):79–89. doi: 10.1007/s10633-007-9087-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robson AG, Tufail A, Fitzke F, Bird AC, Moore AT, Holder GE, et al. Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina (Philadelphia, Pa) 2011;31(8):1670–9. doi: 10.1097/IAE.0b013e318206d155. [DOI] [PubMed] [Google Scholar]
- 13.Popovic P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2005;243(10):1018–27. doi: 10.1007/s00417-005-1186-x. [DOI] [PubMed] [Google Scholar]
- 14.Sodi A, Bini A, Passerini I, Forconi S, Menchini U, Torricelli F. Different patterns of fundus autofluorescence related to ABCA4 gene mutations in Stargardt disease. Ophthalmic surgery, lasers & imaging : the official journal of the International Society for Imaging in the Eye. 2010;41(1):48–53. doi: 10.3928/15428877-20091230-09. [DOI] [PubMed] [Google Scholar]
- 15.von Ruckmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. The British journal of ophthalmology. 1995;79(5):407–12. doi: 10.1136/bjo.79.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robson AG, Saihan Z, Jenkins SA, Fitzke FW, Bird AC, Webster AR, et al. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. The British journal of ophthalmology. 2006;90(4):472–9. doi: 10.1136/bjo.2005.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cukras CA, Wong WT, Caruso R, Cunningham D, Zein W, Sieving PA. Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Archives of ophthalmology (Chicago, Ill : 1960) 2012;130(2):171–9. doi: 10.1001/archophthalmol.2011.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke TR, Rhee DW, Smith RT, Tsang SH, Allikmets R, Chang S, et al. Quantification of peripapillary sparing and macular involvement in Stargardt disease (STGD1) Investigative ophthalmology & visual science. 2011;52(11):8006–15. doi: 10.1167/iovs.11-7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cideciyan AV, Swider M, Aleman TS, Sumaroka A, Schwartz SB, Roman MI, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investigative ophthalmology & visual science. 2005;46(12):4739–46. doi: 10.1167/iovs.05-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayasundera T, Rhoades W, Branham K, Niziol LM, Musch DC, Heckenlively JR. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. American journal of ophthalmology. 2010;149(4):656–60.e2. doi: 10.1016/j.ajo.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Sharon D, Bruns GA, McGee TL, Sandberg MA, Berson EL, Dryja TP. X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Investigative ophthalmology & visual science. 2000;41(9):2712–21. [PubMed] [Google Scholar]
- 22.Tee JJ, Smith AJ, Hardcastle AJ, Michaelides M. RPGR-associated retinopathy: clinical features, molecular genetics, animal models and therapeutic options. The British journal of ophthalmology. 2016;100(8):1022–7. doi: 10.1136/bjophthalmol-2015-307698. [DOI] [PubMed] [Google Scholar]