

Abstract
Transposable elements are one of the main drivers of plant genome evolution. Transposon insertions can modify the gene coding capacity or the regulation of their expression, the latter being a more subtle effect, and therefore particularly useful for evolution. Transposons have been show to contain transcription factor binding sites that can be mobilized upon transposition with the potential to integrate new genes into transcriptional networks. Miniature inverted-repeat transposable elements (MITEs) are a type of noncoding DNA transposons that could be particularly suited as a vector to mobilize transcription factor binding sites and modify transcriptional networks during evolution. MITEs are small in comparison to other transposons and can be excised, which should make them less mutagenic when inserting into promoters. On the other hand, in spite of their cut-and-paste mechanisms of transposition, they can reach very high copy numbers in genomes. We have previously shown that MITEs have amplified and redistributed the binding motif of the E2F transcription factor in different Brassicas. Here, we show that MITEs have amplified and mobilized the binding motifs of the bZIP60 and PIF3 transcription factors in peach and Prunus mume, and the TCP15/23 binding motif in tomato. Our results suggest that MITEs could have rewired new genes into transcriptional regulatory networks that are responsible for important adaptive responses and breeding traits in plants, such as stress responses, flowering time, or fruit ripening. The results presented here therefore suggest a general impact of MITEs in the evolution of transcriptional regulatory networks in plants.
Keywords: peach, Prunus mume, tomato, transposable element, transcription regulation, evolution
Introduction
Transposable elements (TEs) are genetic mobile elements that constitute a variable but major fraction of virtually all eukaryote genomes. Their movement is an important source of mutations and can be a threat for the genomes, which have evolved sophisticated mechanisms, such as epigenetic silencing, to control TEs (Ito and Kakutani 2014; Sarkar et al. 2017). However, TE insertions can also generate new variability useful for evolution, and there is a growing list of examples of TEs insertions that are causative mutations of new selected phenotypes. Indeed, in plants TEs are considered together with polyploidy as the major mechanisms for plant evolution, both in the wild and under plant breeding (Olsen and Wendel 2013; Wendel et al. 2016; Vicient and Casacuberta 2017). TEs can contribute to genome evolution in many ways, and in particular they are a rich source of new genes and regulatory elements. In particular, TEs can contribute new transcriptional terminators, splicing signals, and promoters, as well as a number of transcriptional regulatory elements such as insulators, enhancers, and in general, transcription factor binding sites (TFBS) (Chuong et al. 2017; Oka et al. 2017). This endows TEs with the capacity to generate transcriptional variability that can in some cases be selected during evolution. In animals, many examples of TFBS contributed by TEs, and in particular by LTR retrotransposons, have been reported in the last few years (Kunarso et al. 2010; Chuong et al. 2017). In plants, LTR retrotransposons have also been shown to frequently overlap with potential enhancers (Oka et al. 2017). However, the amplification of TFBS by these elements has not ben reported yet. On the contrary, we have previously reported that a different type of TEs, the miniature inverted-repeat transposable elements (MITEs), has amplified and redistributed the E2F TFBS during Brassica evolution, and demonstrated that E2F binds in vivo to the TFBS located within MITEs (Hénaff et al. 2014).
MITEs are small nonautonomous DNA transposons that can be present in genomes at relatively high copy numbers (Feschotte et al. 2002). Together with LTR retrotransposons, and TRIMs (Gao et al. 2016) MITEs are the most prevalent type of TEs in plants were they are frequently found in the gene-rich euchromatic regions of the chromosomes (Casacuberta and Santiago 2003; Guermonprez et al. 2012). Although MITEs can be mobilized by related transposases by a cut-and-paste nonreplicative mechanism, they can also be amplified, by mechanisms that are still to be clarified, and attain very high copy numbers in genomes (Guermonprez et al. 2012). The small size of MITEs together with their excision capacity may make these elements particularly suited to generate new gene variability with a reduced risk of created deleterious mutations, in particular when inserting in gene regulatory regions. At the same time, their capacity to be amplified and reach high copy numbers render them capable of having a global impact on gene and genome evolution.
Peach (Prunus persica), is a well-characterized fruit crop species notable for its economic value and role as a model species for the Prunus genus. Peach is a diploid (2n = 2× = 16) species with a relatively small genome size (∼230 Mb). A high quality whole genome sequence from double haploid Lovell cultivar is available (Verde et al. 2017). TEs account for at least 30% of the genome space (Verde et al. 2013). In addition to the significant genome fraction that TEs account for, they have been shown to have generated important phenotypic variation in this species. As an example, LTR retrotransposon insertions are responsible for the yellow flesh phenotype (Falchi et al. 2013) and the glabrous phenotype of nectarines (Vendramin et al. 2014). This suggests that, although the genetic variability in peach is considered to be low (Velasco et al. 2016), TEs have been active and have generated new alleles in the recent evolution of this species. In an attempt to better annotate the MITE fraction of the peach genome, and to study their potential impact in peach genome evolution, we performed a dedicated annotation of peach MITEs and analyzed their possible contribution to the amplification and redistribution of transcription factor (TF) binding motifs. Our results show that MITEs have amplified several TF binding motifs during the evolution of peach and other Prunus genomes, which are linked to the regulation of stress responses and flowering time, two essential processes for plant adaptation, and also two key traits for crop breeding. Moreover, our work shows that MITEs have also amplified TF binding motifs related to fruit ripening in tomato and suggests that the capture, amplification, and redistribution of TF binding motifs by MITEs could have had a general impact in plant genome evolution.
Materials and Methods
Analysis of 10-nt Motif Frequencies
As the final purpose of the work was to look for the potential enrichment in MITEs of sequence motifs that could be bound by TFs, the length of the k-mer to be searched was decided on the basis of the length of the characterized plant TF binding motifs. The size distribution of the plant TF binding motifs as defined by the JASPAR database has a major peak between eight and ten nucleotides (nt), and a median at 11 nt. We therefore decided to look for 10-nt motif sequences. All the permutations of words of length 10 nt (410; 1,048,576 words) were searched against both strands of the peach genome. Microsatellites (word composed exclusively of 1-, 2-, or 3 nt-period repeats) were removed from analysis.
MITE Prediction and TF Binding Motif Search
MITE prediction was performed with MITE-Hunter (Han and Wessler 2010). About 262 and 241 consensus sequences, with its multiple sequence alignments (MSA) associated, were obtained for peach and P. mume, respectively. As MITE-Hunter manual recommends, manual inspection of every MSA, which included 60-nt flanking sequences, was performed in order to better define the ends of each predicted element and select only those for which clear borders could be defined. We retained 206 and 180 family consensus sequences for peach and P. mume, respectively. Stringent search for MITE copies for each consensus was performed with RepeatMasker (www.repeatmasker.org) with a cut-off of 250. The RepeatMasker output gff3 file was converted into a bed file and its coordinates were merged. RepeatMasker copies obtained with each consensus sequence were considered as members of the corresponding MITE family. In order to avoid including DNA transposons in the MITE annotation the sequence of each putative MITE was translated into six frame protein sequence and analyzed with hmmscan (version 3.1.b2, hmmer.org) against PFAM (Finn et al. 2016) the presence of transposases or any other TE domain.
Potential terminal-inverted repeat (TIR) sequences were defined by blasting all peach MITE elements against themselves and analyzing inverted-repeated sequences (E value < 10−5) within the first or last 200 nt starting at less than ten of the element ends.
The potential enrichment of each 10-nt word in the peach MITE fraction was analyzed by performing a Fisher’s exact test, and words enriched in the MITE fraction with a P value <0.05 were selected. In order to concentrate in words highly enriched in the MITE fraction, we identified those showing a frequency in this fraction exceeding three times the peach MITE genome coverage (12.54%). The highly enriched words (as well as their complementary words) were transformed to MEME motif format with iupac2meme (from MEME suite; Bailey et al. 2009) and the TF binding motifs identification was performed with TomTom from the MEME suite software with Plant Jaspar Database (JASPAR_CORE_2016_plants.meme) (Mathelier et al. 2016), with an e-value of <0.01. Five additional TF binding motifs used in a previous report on A. thaliana (Hénaff et al. 2014) were also added to the analysis (E2F, GBOX, Ibox, MSA, UP1). TFBS Jaspar Matrix from all predicted TF binding motifs (58) and additional TF binding motifs (5) were retrieved and degenerate sequences for each matrix was obtained with “convert-matrix” from RSAT (Medina-Rivera et al. 2015).
For both peach and P. mume a search for degenerate sequences in not-MITE and MITEs genome fractions was performed with “DNA-pattern” from RSAT. Each predicted TF binding motif in MITE was assigned to a MITE family. Significance of TF binding motifs enrichment in MITEs was calculated with a Fisher’s exact test with a P value < 0.05 for every TF binding motifs. In order to concentrate in TF binding motifs highly enriched in the MITE fraction, we identified those showing a frequency in this fraction exceeding three times the peach and P. mume MITE genome coverage.
Candidate Genes
Distances to genes/CDS were obtained using closest feature from bedops (Neph et al. 2012). Orthologous relationships between peach and P. mume proteins was computed with InParanoid 4.1 (Sonnhammer and Östlund 2015), with bootstrap (100 replicates) and no outgroup. Candidate UPR-involved genes were retrieved from (Silva et al. 2015). Genes showing altered transcription levels in rin and cnr mutants were retrieved from (Zhong et al. 2013). Candidate PIF3 regulated genes in Arabidopsis thaliana were obtained from Pfeiffer et al. (2014). Orthologous genes in peach and P. mume were retrieved from peach functional annotation.
Orthologous pairs between S. lycopersicum and S. tuberosum were retrieved from Phytozome v11 (Goodstein et al. 2012).
Use of P-MITE Predictions
A complete list of the 11 genomes and its respective P-MITE data analyzed in this work is available at supplementary table 1, Supplementary Material online, all of them downloaded in September 20, 2016 (Chen et al. 2014). The analyzed genomes were chosen to cover both big and small genomes, and both high and low MITE predicted coverage. We also included both cultivated crops and wild species and closely and more distantly related genomes. P-MITE database contains MITE annotations predicted using different MITE detection softwares, including MITE-Hunter, MITE Digger, and RPSB. MITE sequence coordinates were obtained with BLASTn (with options ungapped, max_target_seqs = 1, perc_identity = 100 and 100% sequence coverage) of each MITE element sequence against its genome and subsequently the coordinates were merged, as there was some degree of overlapping between the sequences retrieved. Sequences with Ns were discarded. The statistical significance of TF binding motifs enrichment in MITEs was calculated with a Fisher’s exact test with a P value < 0.05 for every TF binding motif in each species.
Results
A 10-nt Word Representation in Peach Genome
As a first unbiased step to analyze the impact of MITEs in amplifying sequences in peach, we searched the genome for all the possible combinations of 10-nt words. The expected frequency for a 10-nt word in a random genome of 227 Mb, which is the size of the published assembly of the peach genome (Verde et al. 2013), is 434 copies (probability of finding a 10-nt word of 2×9.54×10−7). However, as an important part of genome sequences has functional constraints, these figures may vary. The mean frequency of the 10-nt words was 425.7 copies per genome and the median was 244, with a maximum count number of 36,945 and some words not present at all in the genome (supplementary fig. 1, Supplementary Material online).
With respect to the words that are highly overrepresented, we analyzed the chromosomal distribution of the words that were present in >5,000 copies in the peach genome and found that they concentrate in a single location in each chromosome close to the potential centromere position (Verde et al. 2017) (fig. 1). This suggests that these sequences are part of peach centromere repeats. Indeed, the sequence of the already described 166-nt peach centromeric repeats (Melters et al. 2013) coincides with that of some of the highly prevalent short sequences here described (not shown).
Fig. 1.

—Distribution of 10-nt enriched words (>5,000 copies) along chromosomes. A 10-nt word frequency (% nt per 1 Mb bin) for words in MITEs (orange line) and outside of MITEs (blue line). Approximate centromere position is depicted as green bars.
The 10-nt Words Overrepresented in MITEs in Peach
In order to assess the impact of MITEs in the amplification of short sequence motifs, we performed a dedicated annotation of MITEs in the peach genome using MITE-Hunter (Han and Wessler 2010). MITE Hunter provided us with a list of potential MITEs that were clustered in 262 families, which were reduced to 206 after manual inspection and curation of the alignments (see Materials and Methods for details). We then used RepeatMasker (www.repeatmasker.org) to look for copies in the genome obtaining a total of 39,620 MITE sequences that account for 4.18% of the peach genome.
In order to look for possible 10-nt words potentially amplified by MITEs, we analyzed their potential enrichment in the MITE fraction by performing a Fisher’s exact test, and selected the words enriched in the MITE fraction with a P value <0.05. About 42,869 words (8.2% of the total) were overrepresented in MITEs to a variable extent. In order to concentrate on those highly overrepresented in MITEs, we selected those whose percentage of presence in the MITE fraction exceeded three times the MITE coverage (i.e., as MITEs account for 4.18% of the peach genome, a motif is considered as highly overrepresented in MITEs when 12.54% of its instances are found within MITEs). About 19,621 words, 3.75% of 10-nt sequence motifs present in the peach genome, are overrepresented in the MITE fraction (fig. 2).
Fig. 2.
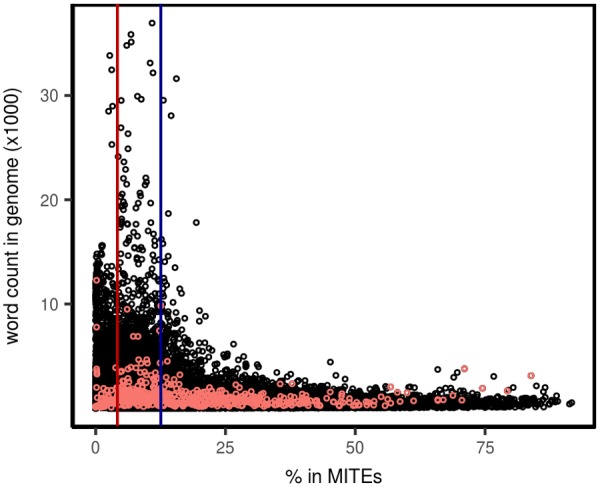
—10-nt word copy number in peach versus fraction of 10-nt word in MITEs. Red and blue lines indicates, respectively, fraction of MITEs in the genome (4.18%) and three times this value (12.54%). Red circles represent words in TIR of MITEs.
As shown earlier, the sequences represented >5,000 times in the peach genome concentrate in the centromere. Recently, it has been proposed that MITEs could also concentrate in the heterochromatic centromeres in Arabidopsis (Guo et al. 2017), in contrast with the accepted view of MITEs being more frequently found close to genes in euchromatic regions of chromosomes (Casacuberta and Santiago 2003). We therefore checked whether the sequences present in MITEs could contribute to the concentration close to the centromeres of the highly represented sequences. As it can be seen in figure 1, the 10-nt words highly represented in MITEs are almost evenly distributed along chromosomes and do not concentrate in the centromere, with a distribution that is compatible with the accepted distribution of MITEs in euchromatic regions.
MITEs frequently contain short TIR sequences that are bound by transposases of related transposons for mobilization (Feschotte et al. 2002; Loot et al. 2006) and as a consequence, MITE amplification may amplify short TIR sequences, which should be overrepresented in the MITE fraction of the genome. We therefore identified sequences corresponding to TIRs of the MITE families annotated in peach among the 10-nt sequences overrepresented in MITEs and found 527 sequences (2.6% of the sequences overrepresented in MITEs) corresponding to MITE TIRs (fig. 2).
MITEs are defective class II elements and, a part from the TIRs and possible subterminal sequences (Feschotte et al. 2002; Casacuberta and Santiago 2003; Loot et al. 2006), they do not contain other obvious functional sequences. Nevertheless, the amplification of a MITE family will imply the amplification of its internal sequence, which, irrespective of the role for the element itself, could have an impact on genes located in their vicinity. A particular case is the presence of TFBS within the MITE internal sequence, as we have recently shown for different MITE families in Brassica containing the E2F TFBS (Hénaff et al. 2014).
Presence of TF Binding Motifs in MITEs
In order to analyze the possible amplification of sequences showing similarity to known TF binding motifs by MITEs in peach, we analyzed if any of the 10-nt words highly (presence in the MITE fraction exceeded three times the MITE coverage) overrepresented in peach MITEs fits the consensus of plant TF binding motifs as defined in the JASPAR database (Mathelier et al. 2016). This analysis showed that the 19,621 10-nt sequences highly overrepresented in the MITE fraction matched 58 putative TF binding motifs as defined in the JASPAR database. As TF binding motifs are usually defined by a sequence profile rather than a single sequence, we used the corresponding consensus sequences to recalculate their distribution between the MITE fraction and the rest of the genome. Most of these degenerated sequences are not overrepresented in the MITE fraction, but 23 putative TF binding motifs are, 5 of them being highly overrepresented (i.e., presence in the MITE fraction exceeded three times the MITE coverage) (table 1 andsupplementary table 2, Supplementary Material online). These are the binding motifs of the bZIP transription factors bZIP60, TGA6, TGA2, and ABI5, as well as the TCP domain-containing factor Glyma 19g26560.1. Among them, the enrichment for a sequence fitting the TF binding motif of bZIP60 is the most striking, with almost half of the sequences fitting the consensus found within MITEs. The binding motifs for the other bZIP transcription factors partially overlap with that of bZIP60 (table 1) and therefore their prevalence within MITEs could simply be the result of the bZIP60 enrichment in these elements. We therefore decided to concentrate on the bZIP60 TF binding motif. An analysis of the MITEs than contain sequences fitting the bZIP60 binding motif showed that the vast majority of them (85.5%) belong to two closely related MITE families (peach_2_69252 + peach_3_60840) related to a PIF-Harbinger DNA transposon described in Repbase (Harbinger-1_Pp) (Jurka 2014) (supplementary fig. 2, Supplementary Material online). The peach genome contains only 43 copies of the complete Harbinger-1_Pp element, but contains 1,051 of the MITEs belonging to the peach_2_69252 and peach_3_60840 families. Interestingly these families show a high sequence conservation, in particular peach_2_69252 (supplementary fig. 3, Supplementary Material online), suggesting that they have been amplified recently during peach genome evolution.
Table 1.
Distribution of the Most Abundant Transcription Factor Binding Motifs in MITE in Peach and Prunus mume
| TF Name | TF Binding Motif Degenerate Sequence |
Prunus persica |
Prunus mume |
||||
|---|---|---|---|---|---|---|---|
| #Genome | #Mite | % in MITEa | #Genome | #Mite | % in MITEb | ||
| ABF3 | ACACGTGT | 3,200 | 367 | 11.47* | 3,538 | 387 | 10.94* |
| ABI5 | bgmCACGTGk | 12,932 | 1,716 | 13.27* | 11,297 | 1,459 | 12.91* |
| bZIP60 | TGACGTCA | 2,656 | 1,313 | 49.44* | 3,071 | 1,165 | 37.94* |
| bZIP910 | mTGACGT | 30,003 | 3,242 | 10.81* | 24,736 | 2,762 | 11.17* |
| Glyma19g26560.1 | gGGsCCCAC | 2,822 | 468 | 16.58* | 3,209 | 643 | 20.04* |
| MYB55 | ACCTAMCG | 3,956 | 280 | 7.08* | 3,006 | 145 | 4.82 |
| PIF3 | dgCCACGTGr | 2,667 | 275 | 10.31* | 2,848 | 567 | 19.91* |
| TCP5 | GGGACCAY | 7,254 | 785 | 10.82* | 6,933 | 642 | 9.26* |
| TGA2 | ACGTCAkC | 10,662 | 2,275 | 21.34* | 9,008 | 1,904 | 21.14* |
| TGA6 | kaTGACGTma | 1,167 | 308 | 26.39* | 1,429 | 255 | 17.84* |
Note.—Asterisk denotes significant enrichment in the MITE fraction (Fisher’s exact test, P value < 0.05). Frequencies in the MITE fraction exceeding three times the MITE coverage are highlighted in red.
MITE coverage: 4.18%.
MITE coverage: 4.1%.
Potential Impact of TFBS-TEs in Gene Regulation in Peach
Although in some plants such as maize enhancers can be located far from the transcriptional unit (Oka et al. 2017), in general plant promoters are supposed to be relatively compact with TFBS that effectively participate in gene regulation located relatively close to the transcribed gene unit. In order to start studying whether the potential TFBS located within MITEs (MITE-TFBS) could contribute directly to gene regulation in peach, we analyzed the distance between the bZIP60 TFBS, both located within MITEs or outside MITEs, and peach coding sequences. Our results show that almost one-third of the bZIP60 MITE-TFBS are located at <1 kb from an annotated coding sequence, and an half of them lay in the gene proximal upstream region (supplementary table 3, Supplementary Material online). This distribution is compatible with the already mentioned general prevalence of MITEs in genic regions but outside coding regions where their insertion could be deleterious. In any case, the high percentage of MITE-TFBS in proximal upstream regions could allow bZIP60 MITEs to participate in gene regulation.bZIP60 is a key transcription factor of the plant unfolded protein response (UPR), which is a set of signaling pathways that respond to stress in the endoplasmic reticulum triggered by biotic and abiotic stresses (Hollien 2013). Moreover, UPR also elicits seed stratification and bud dormancy in plants, and in particular in peach (Fu et al. 2014), and it has recently been shown in Arabidopsis that there is an important crosstalk between the UPR response and the light signal transduction (Nawkar et al. 2017). Peach, and in general the species of the Prunus genus, bloom early in spring, as compared with other fruit trees after a short dormant and chilling period, and differences in signaling pathways regulating dormancy, such as bZIP60, may be relevant for this important agronomic trait in these species.
We manually retrieved 320 peach genes belonging to gene families known to be involved in UPR in other plant species, such as chaperones, membrane-associated NAC transcription factors (Silva et al. 2015) and inspected them for the presence of putative bZIP60 TFBS at <1-kb upstream of the coding sequence (supplementary table 4, Supplementary Material online). Only seven genes have a bZIP60 TF binding motif in the proximal upstream region, which is not surprising as only a minor fraction of the 320 genes potentially involved in the UPR response are expected to be bZIP60 direct targets. In four out of the seven cases, the bZIP60 TFBS is contributed by a MITE.
Analysis of MITE-TFBS in Prunus mume
The results presented above suggest that MITEs have amplified the bZIP60 TF binding motifs during the evolution of the peach genome. In order to get more insight into this process, we have analyzed the possible amplification of TF binding motifs by MITEs in Prunus mume, a close relative to peach whose genome has already been sequenced (Zhang et al. 2012). To this end, we annotated MITEs in Prunus mume using the same approach and parameters used to annotate MITEs in peach. The number of MITEs identified (41,132) and the fraction of the Prunus mume genome they occupy (4.1%) is very similar to that of peach. Among the 58 TFBS analyzed, 23 are significantly enriched in the MITE fraction of the Prunus mume genome, and 7 of them are highly overrepresented in MITEs (table 1). About five of them coincide with those highly overrepresented in the MITE fraction of the peach genome, with numbers that are very similar between the two genomes (table 1), suggesting that the amplification occurred, at least in part, prior to the split of both species. Indeed, the analysis of the regions flanking MITEs containing one of the enriched TF binding motifs, that of bZIP60, shows that 40.1% of them are inserted at orthologous positions in peach and Prunus mume. In addition, in Prunus mume, the binding motif for the bZIP60 transcription factor is one of the most enriched in the MITE fraction. As already discussed, bZIP60 is an important regulator of stress responses in plants, and it has been shown to also regulate seed and bud dormancy in peach (Fu et al. 2014). Plant adaptation to different environments, as part of the speciation process, implies the adjustment of the stress responses to new and different stresses. The presence of stress-related TFBS within mobile modules such as MITEs may help to generate the variability needed in the stress response. In addition, Prunus species show important variability in bud dormancy and flowering time, Prunus mume being one of the first trees that blooms in early spring (Zhang et al. 2012), whereas peach flowers much later. We therefore analyzed the variability of the bZIP60 binding motifs located within or outside MITEs, and laying at <1 kb of a coding sequence, among the two Prunus genomes analyzed. Our data show that whereas 34.7% of the bZIP60 binding motifs found outside MITEs in the gene proximal upstream regions (<1 kb from the coding region) are common between the two genomes (i.e., they are found in the upstream region of orthologous genes), <6% of those located in MITEs and laying at the gene proximal upstream regions are common between peach and P. mume (fig. 3). Interestingly, whereas two out of the three bZIP60 TF binding motifs not located in MITEs that lay upstream of the genes selected as potentially participating in the UPR response are conserved in P. mume, none of those identified as contributed by MITEs is conserved (supplementary table 4, Supplementary Material online). This highlights the potential expression variability induced by the movement of TFBS-MITEs, which in the case of stress responses could be of particular interest.
Fig. 3.
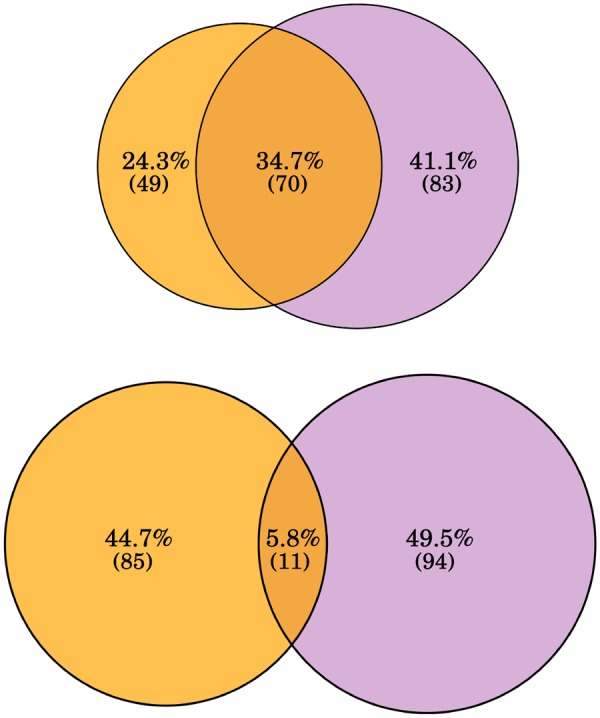
—Orthologous one-to-one genes which contain bZIP60 a binding motif at <1-kb upstream of CDS in peach and/or Prunus mume. The top diagram shows the bZIP60 binding motifs outside of MITEs and the bottom diagram depicts the bZIP60 binding motifs in MITEs.
Interestingly, in addition to the five TF binding motifs amplified by MITEs in peach, two other TF binding motifs seem to have been amplified in Prunus mume. In particular, MITEs seems to have amplified the binding site of PIF3 to a greater extend in Prunus mume as compared with peach. Although the binding motifs of the different PIF transcription factors are similar, and are related to the G-box sequence, the motif amplified in Prunus mume seems to better fit the binding site of PIF3 as defined in the JASPAR database specifically, as the other PIF TF binding motifs and the G-box TF binding motif do not seem to be highly overrepresented in the MITE fraction (supplementary table 2, Supplementary Material online). The vast majority (>90%) of PIF3 binding motifs within MITEs have been amplified by three close MITEs families related to Harbinger-1_Pp. These MITE families are overlap with those that have amplified the bZIP60 TF binding motif in peach and Prunus mume, and are present in 2,445 copies in Prunus mume genome. A close inspection of these families in both peach and Prunus mume showed that both the bZIP60 and the PIF3 TF binding motifs can be found in the same MITE element, this situation being more frequent in Prunus mume than in peach. The high sequence conservation of these families suggests that they amplified very recently during the evolution of peach and Prunus mume (supplementary fig. 4, Supplementary Material online).
PIFs plant transcription factors are involved in different light-regulated processes, and in particular in the regulation of dormancy and flowering in perennial plants (Shim et al. 2014). As already explained Prunus mume flowers much earlier than peach, and differences in the light-regulated transcriptional networks, and in particular on those related to dormancy and flowering are to be expected.
An important fraction of the MITEs containing a sequence fitting the PIF3 TF binding motif are located in the upstream proximal region (<1 kb away) of an annotated coding sequences in both peach (12.7%) and Prunus mume (22%) (supplementary table 5, Supplementary Material online). These include several genes that in Arabidopsis are known to be regulated by PIFs, such as two glycosyltransferases that may glycosylate anthocyanin and be therefore involved in flower pigmentation (Cheng et al. 2014) that contain a PIF3-MITE in their proximal upstream region in Prunus mume. In addition, the list also contains a number of genes potentially related to the regulation of flowering and flowering time (supplementary table 6, Supplementary Material online). These genes include, for example, the one coding for the Ultraviolet-B receptor UVR8, the one encoding a Phosphorylcholine cytidylyltransferase protein and the one encoding a subunit of the Nuclear Factor Y. UV-B regulates photomorphogenesis and flowering (Yin and Ulm 2017), the Nuclear Factor Y transcription factors initiates photoperiod-dependent flowering by cooperatively interacting with CONSTANS to drive the expression of FLOWERING LOCUS T (FT) (Siriwardana et al. 2016). The Phosphorylcholine cytidylyltransferase is an enzyme of the phosphatidylcholine (PC) biosynthesis pathway, and increased levels of PC induce flowering in Arabidopsis. Moreover, it has been shown that PC is specifically bound by the FT, a component of florigen that induces flowering (Nakamura et al. 2014).
MITEs Amplify TF Binding Motifs in Different Plants Species
The results presented above suggest that MITEs have the capacity to amplify and distribute TF binding motifs throughout the genome in different Prunus species, and suggest that MITEs could have had an important impact generating transcription regulation variability during the evolution of plant genomes in general. In order to start exploring this possibility, we undertook the analysis of the prevalence within MITEs of the 58 TF binding motifs here analyzed in other plant species. To this end, we took advantage of the plant MITE database P-MITE (Chen et al. 2014) that contains an annotation of MITEs in 41 genomes of plants. P-MITE also contains a MITE annotation of the peach genome, which allowed us to compare the peach MITE annotation here performed with that of P-MITE. The MITE coverage is slightly higher in the annotation here presented as compared with the one available from P-MITE (4.18% vs. 3.66%). The two MITE annotations are highly overlapping with 78.8% of the P-MITE elements and 79.3% of the P-MITE coverage included in the annotation here presented and 71.5% of MITEs and 69.4% of the MITE coverage from this annotation annotated in P-MITE. Although, the two MITE annotations were based on MITE-Hunter, both include different steps of manual curation of the MITE representatives obtained by this program and slightly different parameters to look for copies using RepeatMasker that may explain the small differences found. In spite of these small differences the two annotations are essentially overlapping. We therefore decided to use the P-MITE annotation of 11 plant species, including that of peach, to analyze whether any of the 58 TFBS here analyzed could have been amplified and mobilized by MITEs. The analysis of the distribution of the 58 TF binding motifs among the MITE and non-MITE fractions of these 11 genomes (supplementary table 7, Supplementary Material online) shows that in 10 out of the 11 genomes there are some sequences fitting TF binding motifs that are significantly overrepresented in the MITE fraction suggesting that MITEs have amplified TF binding motifs in all these genomes. The results obtained with the P-MITE annotation for peach are remarkably similar to those obtained with the MITE Hunter annotation here reported (compare supplementary table 2 and supplementary table 7, Supplementary Material online), confirming that the data obtained with the two approaches can be compared. Moreover, this analysis also shows the previously reported concentration of the E2F TF binding motifs in MITEs in Arabidopsis and A. Lyrata (Hénaff et al. 2014).
Interestingly, as already shown when analyzing the results obtained for peach and P. mume, or the aforementioned MITE-related amplification of the E2F TFBS in Brassica species, phylogenetically related species tend to show similar patterns of amplification. For example, both the tomato and potato genomes show a potential amplification by MITEs of sequences fitting the binding consensus for several TFs, including bZIP TFs such as ABF3, ABI5, and PIFs, three TFs that bind to G-box related TFBSs, with the G-box motif itself also being highly present in the MITE fraction (table 2 and supplementary table 7, Supplementary Material online). This suggests that G-box like TF binding motifs may have been amplified by MITEs during Solanum evolution and that this may endow G-box related transcriptional networks, such as light signal transduction networks, with a high plasticity in these species. As an example, we have analyzed the presence of ABF3 and ABI5 TF binding motifs in the proximal upstream region (<1 kb away) of orthologous genes in tomato and potato, and found that whereas ∼20% of the TF binding motifs laying outside MITEs are common, only 2.2% or 3.2% are shared for ABF3 and ABI5, respectively, when they are located within MITEs (fig. 4).
Table 2.
Distribution of the Most Abundant TF Binding Motifs (A) and Additional Motifs Analyzed (B) in Tomato and Potato
| (A) TF Name | TF Binding Motif Degenerate Sequence |
Solanum lycopersicum |
S. tuberosum |
||||
|---|---|---|---|---|---|---|---|
| #Genome | #Mite | % in MITEa | #Genome | #Mite | % in MITEb | ||
| ABF3 | ACACGTGT | 5,627 | 717 | 12.74* | 5,501 | 1,349 | 24.52* |
| ABI5 | bgmCACGTGk | 7,117 | 1,379 | 19.38* | 9,749 | 3,024 | 31.02* |
| bZIP911 | GrTGACGTGkmC | 230 | 136 | 59.13* | 100 | 5 | 5 |
| FHY3 | ywCACGCGCThw | 522 | 134 | 25.67* | 393 | 62 | 15.78* |
| MYB3 | dGGTAGGTara | 1,798 | 75 | 4.17 | 1,669 | 265 | 15.88* |
| OJ1058_F05.8 | mCACGTGk | 16,498 | 2,079 | 12.6* | 18,626 | 5,716 | 30.69* |
| PIF3 | dgCCACGTGr | 2,320 | 513 | 22.11* | 3,766 | 828 | 21.99* |
| PIF4 | CACGTGsc | 11,085 | 1,079 | 9.73* | 11,650 | 1,903 | 16.33* |
| TCP15/TCP23 | GGGCCCAC | 4,651 | 1,499 | 32.23* | 3,619 | 306 | 8.46* |
| (B) TF name | TF binding motif degenerate sequence | #Genome | #Mite | % in MITE | #Genome | #Mite | % in MITE |
| E2F | TTSSCGSSAA | 2525 | 48 | 1.90 | 2069 | 57 | 2.75 |
| Gbox | GCCACGT | 32741 | 5882 | 17.97* | 27804 | 4591 | 16.51* |
| Ibox | CTTATCC | 80750 | 3128 | 3.87* | 69964 | 8640 | 12.35* |
| MSA | GACCGTT | 26610 | 326 | 1.23 | 16359 | 524 | 3.20 |
| UP1 | GGCCCA | 112751 | 6376 | 5.65* | 111904 | 4677 | 4.18 |
Note.—Asterisk denotes significant enrichment in the MITE fraction (Fisher’s exact test, P value < 0.05). Frequencies in the MITE fraction exceeding three times the MITE coverage are highlighted in red.
MITE coverage: 3.15%.
MITE coverage: 4.65%.
Fig. 4.

—Orthologous one-to-one genes which contain an ABF3 (A) or an ABI5 (B) binding motif at <1-kb upstream of genes in tomato and/or potato. Top diagrams show the binding motifs outside of MITEs and the bottom diagram depicts the binding motifs in MITEs.
In spite of the similarity of the amplification patterns, tomato and potato also show clear differences, suggesting that MITEs could have also amplified TF binding motifs after speciation. This is the case of the bZIP911 TFBS and, more strikingly, that of the TCP15/23 TF. An analysis of the MITEs containing the TCP15/23 binding motifs in tomato shows that they are almost exclusively (96%) located in one MITE family related to the Mutator superfamily (DTM_Sol3), (Chen et al. 2014). This MITE family shows a high sequence conservation (supplementary fig. 5, Supplementary Material online), which suggest a recent amplification compatible with its specific presence in tomato.
The genome of tomato is more than four times bigger than that of peach, with much bigger intergenic regions. However, there is an important fraction of the TCP15/23 TF binding motifs contributed by MITEs (6.6%) located in the close proximity (<1 kb away) from tomato coding sequences, suggesting that they may contribute to gene regulation (supplementary table 8, Supplementary Material online). TCPs are TFs involved in different developmental processes in plants. In particular, in tomato some TCPs, including TCP15, are expressed during fruit development and ripening, and its expression is regulated by key ripening regulators RIN, CNR, and SlAP2a (Parapunova et al. 2014). This suggests that the amplification and mobilization of a sequence fitting the consensus of the TCP15/23 TFs could have had an important impact on the evolution of transcriptional networks that regulate fruit ripening in tomato. Indeed, among the 96 tomato genes that contain a TCP15/23 TF binding motif contributed by a MITE in the proximal upstream region, 12 have been reported to show altered transcription levels in mutants for the RIN and CNR transcription factors (Zhong et al. 2013), which are key regulators of fruit ripening (supplementary table 9, Supplementary Material online), suggesting that MITEs may have contributed to wire new genes into the ripening transcriptional network, modifying this process which is of paramount agronomic importance in this species.
Discussion
TEs are known to be an important source of genetic variability that can be used in evolution. In plants, an important number of TE causative mutations leading to phenotypes selected during evolution both in the wild and under plant breeding have been reported (Vicient and Casacuberta 2017). TEs are thus considered as one of the main drivers of plant genome evolution (Lisch 2013; Wendel et al. 2016). Among the different changes that TEs can induce, the modification of gene transcriptional regulation is probably one of the subtlest, and therefore, in most cases, one of the most useful for evolution. Indeed, the robustness of transcriptional regulation tolerates mutations without an immediate phenotypic consequence, and allows in the long run the creation of complex regulatory network topologies through neutral evolution (Payne and Wagner 2015). This should be particularly true for MITE-induced mutations, as MITEs are small elements that can even be excised, and therefore could more frequently lead to neutral mutations than other TEs. We have previously shown that MITEs have amplified and redistributed TF binding motifs for the E2F TF during the evolution of different Brassica species, and that these binding motifs within MITEs were bound in vivo by the E2F TF (Hénaff et al. 2014). Here, we show that MITEs could have amplified different TF binding motifs in different plant species. The analysis of 12 different plant genomes for the presence of 58 TF binding motifs in MITEs shows that in 11 out of the 12 plant genomes analyzed have at least one TF binding motif overrepresented in the MITE fraction. Related species tend to share MITE overrepresented TF binding motifs (see, e.g., the two Prunus species and the two Solanum species analyzed), suggesting MITE-related TF binding motif amplification predating the split of the two species. However, there are also many species-specific MITE-related amplifications, even when considering the Prunus and the Solanum related species, suggesting that the amplification of TF binding motifs by MITEs could happen at different stages of the genome evolution within a species.
Our results show that two related families of Harbinger MITEs have amplified the TF binding motif for the bZIP60 TF during Prunus evolution, and as a consequence, both peach and Prunus mume have an important percentage of these TF binding motifs sitting within MITEs. This may endow the bZIP60 transcriptional network a high plasticity. Indeed, we show here that whereas the bZIP60 TFBS located in the proximal upstream regions of genes laying outside MITEs are essentially conserved between the two species, those within MITEs are much more variable. These variability of genes containing bZIP60 TF binding motifs in their upstream proximal region induced by MITEs could modify the responses of these two species to the stimuli transduced by bZIP60 TFs. Interestingly bZIP60 is a key regulator of signaling pathways of biotic and abiotic stresses (Hollien 2013) and bud dormancy, in particular in peach (Fu et al. 2014). The type of stresses a plant species needs to face depend on the environmental conditions in which the plants are grown, and therefore are expected to differ from species to species. The response to these changing environments should also evolve and differ between different species. Mobilizing key TF binding motifs through MITEs could help to evolve these networks in a rapid and efficient way. Similarly, bud dormancy is a key strategy to ensure blooming in the appropriate environmental conditions and different species have different dormancy periods and blooming times. The species of the Prunus genus bloom early in spring, as compared with other fruit trees. However, there are important differences between Prunus mume, which is one of the first trees that blooms in early spring (Zhang et al. 2012), and peach, that flowers much later, and differences in signaling pathways regulating dormancy, such as bZIP60, may be relevant to explain these differences. Interestingly, our results show that in Prunus mume, the amplification of bZIP60 TF binding motif has been accompanied by the amplification of the binding sites of PIF (and in particular PIF3) TFs, which also participates in the regulation of dormancy and flowering in perennial plants. This amplification by a subset of the elements that amplified the bZIP60 TF binding motif seems to have happened in Prunus mume after the split from the common ancestor of this species and peach. The analysis of the genes that contain this new PIF3 binding motif contributed by MITEs in their proximal upstream region shows that many of them are related to flowering regulation, strongly suggesting that MITEs could have rewired these genes into the PIF3 transcriptional network in Prunus mume, affecting the regulation of flowering time in this early blooming species. It seems therefore that MITEs could have not only affected transcriptional networks that are important for plant adaptation to the environment but also to networks that regulate important agronomic traits that have been subjected to intensive breeding, as it is the case of flowering time in Prunus. Indeed, the results here presented also show that MITEs could also have contributed to the recent evolution of tomato, and in particular to the evolution of the fruit ripening transcriptional network, one of the most important targets of tomato breeding. The amplification by MITEs of the TCP15/23 TF binding motif in tomato, and the presence of these MITEs in the proximal upstream region of genes known to be regulated by key ripening regulators, strongly suggests that MITEs could have played an important role in the evolution of the ripening transcriptional network in tomato.
MITE are among the smallest and more highly repetitive TEs in plant genomes, this, together with their general association to genes and their capacity to be excised, makes them the ideal vector for mobilizing TF binding motifs and creating new regulatory networks or testing the inclusion of new genes into the existing ones. Although molecular biology and genetic approaches will be needed to unambiguously demonstrate the impact of the TFBS-MITE insertions here described in the regulation of the genes located close to them, the work here presented provides for the first time strong indications for a general role of MITEs in the evolution of transcription networks in plants.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Spanish Ministerio de Economia y Competitividad (grant numbers AGL2013-43244-R and AGL2016-78992-R). We acknowledge financial support from the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa Program for Centers of Excellence in R&D” 2016–2019 (SEV-2015-0533). We are grateful to Elena Casacuberta, Lluïsa Espinás, Carlos Vicient, and Jason Argyris for their critical reading of the manuscript, and to Elena Monte and Soraya Pelaz for helpful discussions.
Literature Cited
- Bailey TL, et al. , 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37(Web Server issue):W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta JM, Santiago N.. 2003. Plant LTR-retrotransposons and MITEs: control of transposition and impact on the evolution of plant genes and genomes. Gene 311:1–11. [DOI] [PubMed] [Google Scholar]
- Chen J, Hu Q, Zhang Y, Lu C, Kuang H.. 2014. P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res. 42(Database issue):D1176–D1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, et al. , 2014. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 166(2):1044–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C.. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchi R, et al. , 2013. Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach. Plant J. 76(2):175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Zhang X, Wessler SR.. 2002. Miniature inverted-repeat transposable elements (MITEs) and their relationship with established DNA transposons In: Craig NL, et al., editors. Mobile DNA II. Washington (DC: ): ASM Press; p. 1147–1158. [Google Scholar]
- Finn RD, et al. , 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44(D1):D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XL, et al. , 2014. Roles of endoplasmic reticulum stress and unfolded protein response associated genes in seed stratification and bud endodormancy during chilling accumulation in Prunus persica. PLoS One 9(7):e101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li Y, Kim KD, Abernathy B, Jackson SA.. 2016. Landscape and evolutionary dynamics of terminal repeat retrotransposons in miniature in plant genomes. Genome Biol. 17:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein DM, et al. , 2012. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40(Database issue):D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermonprez H, Hénaff E, Cifuentes M, Casacuberta JM.. 2012. MITEs, miniature elements with a major role in plant genome evolution. In Grandbastien MA, Casacuberta JM, editors. Plant transposable elements. Springer-Verlag. p. 113–124.
- Guo C, Spinelli M, Ye C, Li QQ, Liang C.. 2017. Genome-wide comparative analysis of miniature inverted repeat transposable elements in 19 Arabidopsis thaliana ecotype accessions. Sci Rep. 7(1):2634.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wessler SR.. 2010. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38(22):e199.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hénaff E, et al. , 2014. Extensive amplification of the E2F transcription factor binding sites by transposons during evolution of Brassica species. Plant J. 77(6):852–862. [DOI] [PubMed] [Google Scholar]
- Hollien J. 2013. Evolution of the unfolded protein response. Biochim Biophys Acta 1833(11):2458–2463. [DOI] [PubMed] [Google Scholar]
- Ito H, Kakutani T.. 2014. Control of transposable elements in Arabidopsis thaliana. Chromosome Res. 22(2):217–223. [DOI] [PubMed] [Google Scholar]
- Jurka J. 2014. DNA transposons from the peach genome. Repbase Rep. 14:2389–2389. [Google Scholar]
- Kunarso G, et al. , 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 42(7):631–634. [DOI] [PubMed] [Google Scholar]
- Lisch D. 2013. How important are transposons for plant evolution? Nat Rev Genet. 14(1):49–61. [DOI] [PubMed] [Google Scholar]
- Loot C, Santiago N, Sanz A, Casacuberta JM.. 2006. The proteins encoded by the pogo-like Lemi1 element bind the TIRs and subterminal repeated motifs of the Arabidopsis emigrant MITE: consequences for the transposition mechanism of MITEs. Nucleic Acids Res. 34(18):5238–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, et al. , 2016. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44(D1):D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Rivera A, et al. , 2015. RSAT 2015: regulatory sequence analysis tools. Nucleic Acids Res. 43(W1):W50–W56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melters DP, et al. , 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14(1):R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, et al. , 2014. Arabidopsis florigen FT binds to diurnally oscillating phospholipids that accelerate flowering. Nat Commun. 5:3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawkar GM, et al. , 2017. HY5, a positive regulator of light signaling, negatively controls the unfolded protein response in Arabidopsis. Proc Natl Acad Sci U S A. 114(8):2084–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, et al. , 2012. BEDOPS: high-performance genomic feature operations. Bioinformatics 28(14):1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka R, et al. , 2017. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol. 18(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KM, Wendel JF.. 2013. A bountiful harvest: genomic insights into crop domestication phenotypes. Annu Rev Plant Biol. 64:47–70. [DOI] [PubMed] [Google Scholar]
- Parapunova V, et al. , 2014. Identification, cloning and characterization of the tomato TCP transcription factor family. BMC Plant Biol. 14:157.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JL, Wagner A.. 2015. Mechanisms of mutational robustness in transcriptional regulation. Front Genet. 6:322.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Shi H, Tepperman JM, Zhang Y, Quail PH.. 2014. Combinatorial complexity in a transcriptionally centered signaling hub in Arabidopsis. Mol Plant 7(11):1598–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Volff JN, Vaury C.. 2017. PiRNAs and their diverse roles: a transposable element-driven tactic for gene regulation? FASEB J. 31(2):436–446. [DOI] [PubMed] [Google Scholar]
- Shim D, et al. , 2014. A molecular framework for seasonal growth-dormancy regulation in perennial. Plants 1:14059.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva PA, et al. , 2015. Comprehensive analysis of the endoplasmic reticulum stress response in the soybean genome: conserved and plant-specific features. BMC Genomics 16:783.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriwardana CL, et al. , 2016. Nuclear factor Y, subunit A (NF-YA) proteins positively regulate flowering and act through flowering locus T. PLoS Genet. 12(12):e1006496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer ELL, Östlund G.. 2015. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 43(D1):D234–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco D, Hough J, Aradhya M, Ross-Ibarra J.. 2016. Evolutionary genomics of peach and almond domestication. G3 (Bethesda) 6:3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendramin E, et al. , 2014. A unique mutation in a MYB gene cosegregates with the nectarine phenotype in peach. PLoS One 9(3):e90574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I, et al. , 2013. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 45(5):487–494. [DOI] [PubMed] [Google Scholar]
- Verde I, et al. , 2017. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genomics 18(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicient CM, Casacuberta JM.. 2017. Impact of transposable elements on polyploid plant genomes. Ann Bot. 120(2):195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF, Jackson SA, Meyers BC, Wing RA.. 2016. Evolution of plant genome architecture. Genome Biol. 17:37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Ulm R.. 2017. How plants cope with UV-B: from perception to response. Curr Opin Plant Biol. 37:42–48. [DOI] [PubMed] [Google Scholar]
- Zhang Q, et al. , 2012. The genome of Prunus mume. Nat Commun. 3:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, et al. , 2013. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol. 31(2):154–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.