

Abstract
Since the discovery of repeat-associated non-ATG (RAN) translation, and more recently its association with amyotrophic lateral sclerosis/frontotemporal dementia, there has been an intense focus to understand how this process works and the downstream effects of these novel proteins. RAN translation across several different types of repeat expansions mutations (CAG, CTG, CCG, GGGGCC, GGCCCC) results in the production of proteins in all three reading frames without an ATG initiation codon. The combination of bidirectional transcription and RAN translation has been shown to result in the accumulation of up to six mutant expansion proteins in a growing number of diseases. This process is complex mechanistically and also complex from the perspective of the downstream consequences in disease. Here we review recent developments in RAN translation and their implications on our basic understanding of neurodegenerative disease and gene expression.
Keywords: RAN translation, microsatellite expansion, SCA8, DM1, FXTAS, HD, C9-ALS/FTD, nucleocytoplasmic transport, dipeptide repeat, antisense
An expanding field: updates on RAN-associated disorders
The discovery of repeat associated non-ATG (RAN) translation in 2011 [1] has changed our understanding of the mechanisms of microsatellite expansion diseases and of protein translation. We previously reviewed the discovery of RAN translation and early research findings [2]. These previous studies demonstrated that RAN translation, coupled with bidirectional transcription, from expanded repeats is capable of producing a startling array of toxic expansion proteins. Here we focus on recent developments in RAN translation, its mechanistic underpinnings and efforts to understand the impact of RAN proteins in neurologic disease.
RAN translation and the resulting toxic homopolymeric proteins were first described in spinocerebellar ataxia type 8 and myotonic dystrophy type 1 [1]. This discovery was followed by descriptions of homopolymeric RAN proteins in fragile X tremor ataxia syndrome (FXTAS) [3] and dipeptide RAN proteins in C9of72 amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD)[4-8]. While research on the effects of RAN dipeptide repeat (DPR) proteins in C9-ALS/FTD has produced the largest amount of data and interest to date, RAN translation has been observed in other diseases and interest in its impact and mechanisms is growing.
RAN proteins in Huntington disease and other polyglutamine disorders
Huntington disease (HD), a progressive neurodegenerative disorder characterized by severe movement, cognitive and behavioral alterations, is caused by a CAG•CTG expansion in the HTT gene [9]. The mutant huntingtin mHTT protein, which contains a polyglutamine expansion tract, is expressed from the sense-strand as part of a large ATG-initiated open reading frame (ORF). While it is clear that the HD CAG•CTG expansion mutation causes disease [10], HD research has almost exclusively been focused on the mutant huntingtin (mHTT) polyglutamine expansion protein because it was thought to be the only mutant protein that could be expressed across the expansion mutation. The first examples of RAN translation were found to occur across expansion mutations located in non-coding gene regions (SCA8, DM1, C9orf72 ALS/FTD, and FXTAS). Because the HD expansion is much smaller and is located within a large canonical ORF that could inhibit RAN translation, it was unclear if RAN translation could also occur across the HD mutation.
In 2015, Bañez-Coronel et al. [11] answered this question and reported that four additional novel homopolymeric RAN expansion proteins (polyAla, polySer, polyLeu and polyCys) expressed from both sense and antisense transcripts accumulate in HD human autopsy brain samples. These HD-RAN proteins were prominently found brain regions showing neuronal loss, cell death and microglial activation, including the striatum. While HD-RAN proteins accumulate in brain regions that overlap sites of polyGln accumulation, including the caudate and putamen, HD-RAN protein staining is also found in several white matter regions in the absence of detectable polyGln accumulation [11] suggesting HD-RAN proteins play a role in previously reported HD white matter abnormalities [12-16]. Bañez-Coronel et al. [11] also observed robust RAN, but almost no detectable polyGln, accumulation within the severely affected cerebellar layers of juvenile onset HD cases. Cell culture studies using codon replacement constructs demonstrated that polySer, polyLeu and polyCys expansion proteins are toxic to neural cells independent of CUG and CAG RNA gain of function effects. Additional studies showed repeat length thresholds for polySer and polyAla protein accumulation at 35 and >52 repeats. Because 35 repeats is close to the disease threshold for HD, it is possible that polySer expression is a trigger of disease and that the longer repeats associated with juvenile onset HD result from the expression of a toxic cocktail containing multiple RAN and polyGln expansion proteins. It is also possible that CUG and CAG RNA gain of function effects contribute to HD [11].
Taken together these data suggest that RAN proteins contribute to HD and may be uniquely responsible for specific changes including white matter abnormalities and neurodegenerative changes in severely affected juvenile onset cases. Approaches to reduce the production or increase the turnover of the mHTT polyGln expansion protein have also been the focus of many studies of HD [17-21]. One recently described approach, which ameliorated behavioral phenotypes in HD mice, was to target the accumulation of mHTT and other SUMOylated proteins by decreasing levels of PIAS1, an E3 SUMO ligase that targets mHTT [22]. It will be interesting to determine if posttranslational modifications such as SUMOylation and ubiquitination [22-24] occur on the HD-RAN proteins and if these or other pathways also affect HD-RAN protein turnover.
The discovery that RAN translation occurs in HD raises the possibility that RAN proteins also contribute to eight other CAG polyglutamine expansion disorders. Consistent with this possibility, Scoles et al. [25] showed RAN proteins are produced using minigenes containing sequence flanking the CAG•CTG expansion mutation causing spinocerebellar ataxia type 2 (SCA2). These authors showed that SCA2 RAN protein accumulation is influenced by the sequence context 3’ of the repeat. Similarly, minigenes containing sequence upstream of the CAG repeat in SCA3, HD, DM1 or HDL2 also express RAN proteins [1]. Further research will be required to test if, similar to HD, RAN proteins are also found in patient tissues with these diseases. There are also additional regulatory connections between the polyglutamine disorders that affect protein translation. For example, the MID1-translation regulatory protein complex binds and regulates the translation of CAG expansions in the canonical ATG-initiated frame in a length dependent manner in HD and several SCAs [26]. It will be interesting to determine if this regulatory mechanism also influences RAN translation.
Fragile X tremor ataxia syndrome
In 2013, Todd et al. demonstrated that the CGG repeat expansion in FXTAS can, in cell culture, produce a poly-glycine (FMR-polyGly) and a poly-alanine (FMR-polyAla) protein with FMR-polyGly accumulating in fly, mouse and patient samples [3]. More recently this group demonstrated that homopolymeric proteins are also expressed in the antisense direction generating polyPro, polyArg, polyAla in cell culture with at least two of these proteins accumulating in human autopsy tissue [27]. Similar to previous results, cell culture studies show steady state levels of these proteins increase with repeat length.
Zu et al. [1] previously showed, even small changes in flanking sequence can influence RAN translation across CAG repeat expansions. The role of flanking sequence is further highlighted in studies of the Fragile X tremor ataxia syndrome (FXTAS) [27-29]. Kearse et al. [28] demonstrated that translation of the FMR-polyGly initiates preferentially at close-cognate start codons upstream of the repeat, in a repeat-length independent manner. In contrast, translation in the polyAla reading frame is repeat length dependent and initiates within the expanded repeat with no close-cognate initiation codon requirement. Additionally, translation in both reading frames was shown to be cap-, eIF4E- and eIF4A-dependent. The authors suggest these findings supports canonical pre-initiation complex loading and ribosome scanning and that ribosome stalling caused by secondary CGG RNA structure facilitates translation initiation in both the polyAla and polyGly reading frames [28]. In contrast, RAN translation in the polyArg frame was not detected yet interestingly translation in this frame could be detected in the absence of the CGG repeats. Taken together, these data highlight the role of frame specific effects of flanking sequence on protein translation across FXTAS CGG repeats.
In 2011, Zu et al. [1] showed by mass spectrometry that the N-terminal amino acid for the polyAla RAN protein expressed across expanded CAG repeats began with the amino acid alanine at various sites within the repeat tract. In contrast, mass spectrometry performed by Sellier et al. [29] on the FMR-polyGly protein showed that initiation occurs at an upstream ACG close cognate start codon and that the N-terminal amino acid is a methionine in mammalian cells. These data suggest a canonical protein initiation mechanism in the polyGly reading frame but with the use of a close cognate initiation codon. Ribosome profiling studies have led to the identification of thousands of additional putative upstream open reading frames (uORFs) that use similar close cognate codons [30,31]. It will be interesting to determine if translation of the FMR1 uORF in the +1 reading frame inhibits translation of FMRP in the major ORF, as has been shown for other uORFs [32,33]. Additionally, Sellier et al. [29] showed with conditional mouse models that neuronal expression of the FMRpolyGly protein, but not the CGGEXP RNA alone, decreases survival in mice and that both the polyGly repeat tract and the C-terminal sequence contribute to toxicity in cell culture. They also show that the C-terminal region of the FMR-polyGly protein interacts with and disrupts the nuclear lamina protein, LAP2β [29], which may underlie previous observations of disorganized nuclear lamina structure in FXTAS [34]. Taken together, these findings highlight the different roles that flanking sequences can play in both the expression and toxicity of repeat expansion proteins.
C9orf72 ALS/FTD
Since the discovery of the G4C2•G2C4 expansion in the C9orf72 gene as the most common known genetic cause of both ALS and FTD, research into disease mechanisms have focused on three main areas: 1) haploinsufficiency of the C9orf72 gene caused by decreased expression of expansion allele [35-40]; 2) RNA gain-of-function effects leading to sequestration of key RNA binding proteins [36,40-46]; and 3) protein gain-of-function effects caused by the RAN dipeptide repeat (DPR) proteins [4-8,36,37,46-49]. Comprehensive reviews on ALS and C9orf72 ALS/FTD have been previously written [50-54]. Here we will focus on recent developments on the effects of the RAN dipeptide proteins and outstanding questions and new tools that will allow a better understanding of their impact on disease.
A first basic set of questions is where and how does translation of the DPRs initiate and are there frame specific differences that may result in the use of both canonical and novel RAN translation mechanisms? Examination of the flanking sequences around the hexanucleotide repeat shows ATG initiation codons upstream of the antisense G2C4 repeats in both the ProlineArginine (PR) and GlycineProline (GP) reading frames (see Fig S3 in [6]). This finding raises the question of whether or not these upstream ATG initiation codons are used in these reading frames and, if so, how these N-terminal flanking sequences would affect the function and toxicity of these proteins. While it is not clear if a mixture of canonical translation and RAN translation occurs in the antisense direction, a mixture of canonical and RAN mechanisms in C9-ALS/FTD would have parallels to both HD [11] and FXTAS [28].
Additional sequence analysis of the C9orf72 repeat region predicts unique C-terminal flanking regions for five of the six RAN proteins [6]. Immunodetection of these C-terminal regions confirms that they are expressed and accumulate in C9 patient autopsy tissue [6]. While these unique C-terminal regions increase the complexity of the C9-RAN proteins beyond the DPR repeat motifs, C9-RAN protein toxicity studies have typically focused on the DPR repeat motifs alone or as a fusion protein with a C-terminal reporter. It will be important to broaden studies of C9-RAN protein toxicity to include both the DPR repeat motifs and the corresponding endogenous flanking sequences.
Numerous cellular targets and interacting partners of RAN DPR proteins have been identified including RNA-binding proteins, nucleoporin proteins, low-complexity domain proteins, ribosomal proteins, translation initiation and elongation factors [55-58]. Of particular interest to the field has been the direct interaction of C9orf72-RNA with nuclear pore complex (NPC) proteins [59] and the disruption of nucleocytoplasmic transport by C9-RAN protein [60-63] (see Haeusler et al. [50] and Boeynaems et al. [64] for reviews). Disruptions to the nucleocytoplasmic machinery are thought to play a role in the adult-onset neurodegeneration observed in ALS/FTD as the NPC has many long-lived structural components that can deteriorate during aging [65] and post-mitotic cells, such as neurons, cannot rely upon mitotic nuclear envelope disassembly for nuclear waste clearance. However, it is not yet clear if the NPC and nucleocytoplasmic transport disruptions in C9-ALS/FTD contribute directly to cellular pathogenesis, enhance age-related defects and/or exacerbate existing nuclear RNA loss-of-function and cytoplasmic protein gain-of-function mechanisms. Nevertheless, the connection between the NPC and nucleocytoplasmic transport defects extends beyond C9-ALS/FTD to amyloid-like protein aggregates in other diseases [66] as well as aggregates from other microsatellite diseases including DM1 [67], HD [66,68-70], DRPLA [71,72] and several polyglutamine expansion disorders [73,74].
To better understand the impact and biology of RNA gain of function and RAN protein effects in C9-ALS/FTD, four independent groups developed C9orf72 BAC transgenic mouse models. While mice from each of these models showed the expected molecular phenotypes of sense and antisense RNA foci and RAN protein accumulation, surprisingly these models had very different phenotypic outcomes. Models developed by O’Rourke et al. [75] and Peters et al. [76] did not develop phenotypic or neurodegenerative features of the disease. A third study by Jiang et al. [77] showed mice with 110 or 450 repeats did not develop behavioral, physiological or neuropathological evidence of motor neuron disease out to 18 months of age. However, deficits in spatial learning, anxiety and mild neuronal loss in the hippocampus without gliosis or TDP43 mislocalization were detected in lines with 450 repeats [77]. In contrast, BAC transgenic mice developed by Liu et al. [78] develop both the key phenotypic and molecular features of the disease. These C9-BAC mice show, in a repeat-length and expression dependent manner, features of both ALS and FTD including decreased survival, paralysis, muscle denervation, motor-neuron loss, anxiety-like behavior and cortical and hippocampal neurodegeneration. These mice express C9orf72 sense and antisense transcripts and RNA foci are found throughout the CNS. Additionally, RAN protein accumulation increases with age and disease severity and TDP-43 inclusions are found in degenerating brain regions in end-stage animals.
While the striking phenotypic differences between there models are puzzling, in the end they may provide fundamental insights into the onset and triggers of both ALS and FTD. Possible reasons for the phenotypic differences between the BAC mouse models include: 1) the absence of the full length gene and 3’ UTR regulatory regions in two models [76,77]; 2) possible cis modifiers differences on individual BAC transgenes; 3) differences in mouse genetic background - Liu et al. [78] used FVB whereas B6 backgrounds were used in the other studies [75-77]; 4) differences in spatial or temporal expression of both the sense and antisense transcripts. Regarding the last point, in the Liu model, sense foci were found throughout the CNS whereas antisense foci were found in regions that later showed neurodegeneration [78]. These data suggest that antisense transcripts or antisense RAN proteins may be especially pathogenic. These mice can be used tease apart the most important molecular mechanisms of this complex disease and test the efficacy of therapeutic strategies, not just by reversing one or more molecular markers of disease, but by reversing the disease itself.
Therapeutics
The demonstration of bidirectional transcription and RAN translation in a growing number of diseases highlights the potential pathogenic effects of both sense and antisense RNAs and RAN proteins. A challenge for the field is to understand the consequences of individual RAN proteins versus RNA gain of function mechanisms and the contributions of sense versus antisense genes to clarify the most important therapeutic targets. To date, research for most expansion diseases has focused on blocking the expression of the initially discovered “sense” gene [17,19,79-83]. Because most of microsatellite expansions are late onset, therapeutic benefit may be achieved by targeting sense transcripts alone, as this approach would lower the overall burden of expansion RNAs and RAN proteins. For example, antisense oligonucleotide (ASOs) treatment against the intronic region of the sense C9orf72 transcript in C9 BAC mice developed by Jiang et al. were shown to decrease both sense RNA foci and levels of poly-GA and poly-GP RAN proteins and the mild behavioral deficits in these mice [77]. While studies for C9orf72 suggest that targeting the sense transcript does not alter levels of antisense foci in patient cells or mouse models [77,84,85] decreases in sense expansion transcripts resulted in increased antisense transcript expression in another microsatellite expansion disorder, spinocerebellar ataxia type 7 (SCA7) [86]. Additional studies will be required to understand if targeting just one strand could potentially upregulate and exacerbate pathogenic effects from the other strand.
Alternative approaches have focused on targeting pathways downstream of the expansion RNA and RAN proteins, such as nucleocytoplasmic transport defects [59,61,62] or protein quality control processes that affect mutant protein aggregation and turnover [21-24,87]. These strategies may be effective but also may have unintended consequences. For example, small molecule and peptide inhibitors aimed at disrupting MBNL1 or other RNA binding proteins to CUG expansion RNAs in DM1 [88-93], may alleviate MBNL1 loss of function effects but may also increase the available pool of expansion transcripts for RAN translation.
Given the complex downstream consequences of microsatellite expansion mutations and their sense and antisense RNA and RAN protein products, preventing expression of the repeats is also an attractive strategy as it could block transcription and RAN protein production in both directions. Targeting of a C9orf72 transcript transcription factor, SUPT5H, in patient-derived cells reduced both sense and antisense RNA foci as well as poly-GP protein with little apparent effect of other transcripts [85]. Earlier experiments showed SPT5 expression in yeast is required for transcription through expanded CAG repeats [94] . Additional studies showed knockdown of Supt4h expression in Huntington’s mice results in decreased expression of mutant Htt mRNA, reduced Htt protein aggregates and phenotypic improvements [95]. It will be important to understand how blocking sense transcription affects antisense transcripts and the more recently discovered HD RAN proteins. Finally, the development of effective therapeutic strategies will require the use of models that accurately mimic the temporal/spatial expression patterns, the physiological levels of expansion RNAs and RAN proteins and the phenotypic changes found in patients.
Conclusions
RAN proteins have now been reported in five microsatellite expansion diseases [1,3-8,11], a number likely to grow with improved detection techniques. Animal and cell culture models are being used to determine the roles that RAN proteins play in disease and the mechanisms that regulate their translation. Understanding how RAN translation works and being able to modulate RAN protein expression will allow us to test the contribution of these proteins to disease and provides additional therapeutic targets. Because repetitive sequences make up approximately 50% of the human genome it is possible that RAN proteins may be widely expressed and have normal roles in cellular function.
Figure 1. Cellular consequences of expanded microsatellite repeats and repeat-associated non-ATG (RAN) translation.
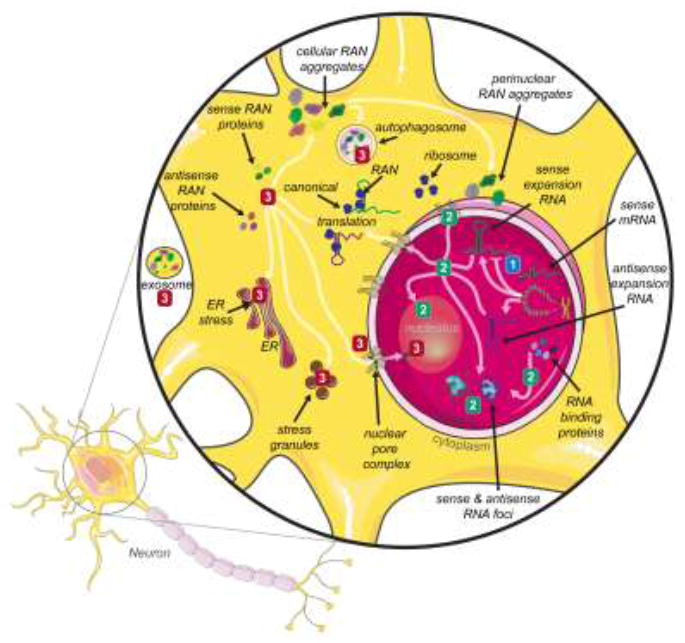
Although microsatellite repeats are expressed in a wide variety of cells, the majority of RAN proteins have been identified in the central nervous system (see box 1). While this generalized illustration utilizes a neuron, RAN proteins are found in a variety of CNS cell types (neurons, astrocytes, microglia, motorneurons, etc). Bidirectional transcription has been observed in a growing number of microsatellite repeat expansion diseases [96-99] resulting in the expression of both sense and antisense expansion transcripts. Three non-exclusive disease mechanisms have been proposed for the majority of microsatellite expansion diseases. (1) Loss-of-function (blue) – reduction in mRNA transcript resulting in reduced C9orf72 protein levels. The presence of expanded repeats in the DNA may result in epigenetic silencing and/or transcriptional inhibition that reduce the available levels of the gene’s canonical protein product. Protein loss-of-function has been proposed for the C9orf72 hexanucleotide repeats [35,39,100-102]. (2) RNA gain-of-function (green) – sequestration of RNA-binding proteins into nuclear foci by the expanded RNA repeats reduces the available RBP concentration and its normal cellular function. In DM1, CUG expansion RNAs sequester MBNL proteins from their normal splicing targets and MBNL loss-of-function leads to alternative splicing dysregulation and pathogenesis [37,52,59,62,63,103]. The expanded repeat transcript may also interact with, and alter the function of other cellular components, such as the interaction between G4C2 expansion RNA and proteins of the nuclear pore complex [59]. (3) Protein gain-of-function (red) – the production of up to six toxic RAN proteins from the sense and antisense expansion transcripts results in defects in cellular processes, including protein homeostasis. In C9-ALS/FTD, sense and antisense dipeptide RAN proteins have been shown to lead to nucleolar dysfunction, ER stress, stress granules, altered autophagy, transcellular transmission, disruption of nucleocytoplasmic defects and nuclear envelope defects [4,7,104,105].
Box 1. RAN proteins and human disease.
Spinocerebellar ataxia type 8 (SCA8: CAG•CTG)
A dominantly inherited slowly progressive ataxia with cerebellar degeneration that is characterized by gait and limb ataxia, nystagmus, and dysarthria. SCA8 is caused by expansion of a CAG•CTG repeat in the overlapping ATXN8OS and ATNX8 genes on chromosome 13. The RAN protein, SCA8-poly-Alanine, expressed from the antisense CAG expansion transcript, was detected in the cerebellar Purkinje cells of SCA8 mouse and human brains [1].
Myotonic Dystrophy Type 1 (DM1: CAG•CTG)
A dominantly inherited multisystemic neuromuscular disease that is characterized by myotonia, muscle wasting, cardiac defects, cataracts and central nervous system alterations. DM1 is caused by expansion of CAG•CTG repeat with the 3’ UTR of the DMPK gene on chromosome 19. The RAN protein, DM1-poly-Glutamine expressed from the antisense CAG expansion transcript was detected in myoblasts, skeletal muscle and blood of DM1 patients [1].
C9of72 amyotrophic lateral sclerosis and frontotemporal dementia (C9-ALS/FTD: GGGGCC•GGCCCC)
A dominantly inherited motor neuron (ALS) and/or temporal lobar degenerative (FTD) disease that is characterized by upper or lower motor neuron degeneration (ALS) and/or progressive changes in behavior, executive dysfunction and language impairment (FTD). C9-ALS/FTD is caused by an expansion of a GGGGCC•GGCCCC repeat within the 1st intron of the C9orf72 gene on chromosome 9. The RAN proteins, C9-poly-GlycineProline, C9-poly-GlycineArginine and C9-poly-GlycineAlanine expressed from the sense GGGGCC expansion transcript and C9-poly-AlanineProline, C9-poly-ProlineArginine and C9-poly-GlycineProline expressed from the antisense GGCCCC expansion transcript were detected in the brains, spinal cord and motor cortex of postmortem C9-ALS/FTD patients [4,5,7,8,49,104]. The C9-poly-GylcineProline RAN protein has also been detected in the CSF from C9-ALS/FTD patients [106].
Fragile X Tremor Ataxia Syndrome (FXTAS: CGG•CCG)
A dominantly inherited late-onset disease primarily affecting males characterized by tremor, ataxia, parkinsonism and cognitive decline. FXTAS is caused by premutation (55-200 repeats) expansion of a CGG•CCG repeat in the 5’ UTR of the FMR1 gene on the X chromosome. The RAN protein, FXTAS-poly-Glycine expressed from the sense CGG expansion transcript were detected in frontal cortex and hippocampal neurons in FXTAS postmortem brain tissues [3] and FXTAS-poly-Proline and FXTAS-poly-Alanine expressed from the antisense CCG expansion transcript [27] were detected in hippocampus, cortex and cerebellum of FXTAS postmortem tissue.
Huntington’s disease (HD: CAG•CTG)
A dominantly inherited progressive neurodegenerative disorder characterized by movement abnormalities, cognitive decline and psychiatric problems. HD is caused by CAG•CTG expansions in the 1st exon of the HTT gene on chromosome 4. The RAN proteins, HD-poly-Alanine and HD-poly-Serine expressed from the sense expansion transcript and HD-poly-Leucine and HD-poly-Cysteine expressed from the antisense expansion transcript were detected in striatum, frontal cortex and cerebellum of postmortem HD patients [11].
Acknowledgments
The authors wish to thank Dr. Maurice Swanson and Dr. Marina Scotti for their helpful comments and suggestions. This work was supported by the National Institutes of Health (RO1 NS098819, R01 NS040389 and P01 NS058901), Target ALS, CHDI, National Ataxia Foundation, ALS Association, Packard Foundation and the Muscular Dystrophy Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev. 2014;26:6–15. doi: 10.1016/j.gde.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 5.Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 6.Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 10.Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–216. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 11.Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL, et al. RAN Translation in Huntington Disease. Neuron. 2015;88:667–677. doi: 10.1016/j.neuron.2015.10.038. (••) This paper shows the first demonstration that RAN proteins are expressed across a large ATG-initiated open-reading frame and that four novel HD-RAN proteins accumulate in patient autopsy brains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herndon ES, Hladik CL, Shang P, Burns DK, Raisanen J, White CL., 3rd Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J Neuropathol Exp Neurol. 2009;68:250–261. doi: 10.1097/NEN.0b013e318198d320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohanna I, Georgiou-Karistianis N, Sritharan A, Asadi H, Johnston L, Churchyard A, Egan G. Diffusion tensor imaging in Huntington’s disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav. 2011;5:171–180. doi: 10.1007/s11682-011-9121-8. [DOI] [PubMed] [Google Scholar]
- 14.Fennema-Notestine C, Archibald SL, Jacobson MW, Corey-Bloom J, Paulsen JS, Peavy GM, Gamst AC, Hamilton JM, Salmon DP, Jernigan TL. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. 2004;63:989–995. doi: 10.1212/01.wnl.0000138434.68093.67. [DOI] [PubMed] [Google Scholar]
- 15.Paulsen JS, Nopoulos PC, Aylward E, Ross CA, Johnson H, Magnotta VA, Juhl A, Pierson RK, Mills J, Langbehn D, et al. Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull. 2010;82:201–207. doi: 10.1016/j.brainresbull.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, Yallapragada V, Rosenblatt A, Margolis RL, Aylward EH, Brandt J, et al. Regional white matter change in pre-symptomatic Huntington’s disease: a diffusion tensor imaging study. Psychiatry Res. 2005;140:55–62. doi: 10.1016/j.pscychresns.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Hu J, Liu J, Yu D, Chu Y, Corey DR. Mechanism of allele-selective inhibition of huntingtin expression by duplex RNAs that target CAG repeats: function through the RNAi pathway. Nucleic Acids Res. 2012;40:11270–11280. doi: 10.1093/nar/gks907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skotte NH, Southwell AL, Østergaard ME, Carroll JB, Warby SC, Doty CN, Petoukhov E, Vaid K, Kordasiewicz H, Watt AT, et al. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS One. 2014;9:e107434. doi: 10.1371/journal.pone.0107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Switonski PM, Szlachcic WJ, Gabka A, Krzyzosiak WJ, Figiel M. Mouse models of polyglutamine diseases in therapeutic approaches: review and data table. Part II. Mol Neurobiol. 2012;46:430–466. doi: 10.1007/s12035-012-8316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter C, Clemens LE, Müller AJ, Fallier-Becker P, Proikas-Cezanne T, Riess O, Metzger S, Nguyen HP. Activation of AMPK-induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology. 2016;108:24–38. doi: 10.1016/j.neuropharm.2016.04.041. [DOI] [PubMed] [Google Scholar]
- 22.Ochaba J, Monteys AM, O’Rourke JG, Reidling JC, Steffan JS, Davidson BL, Thompson LM. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington’s Disease-Associated Phenotypes In Vivo. Neuron. 2016;90:507–520. doi: 10.1016/j.neuron.2016.03.016. (••) This paper characterizes how alterations in a component of SUMOylation and protein quality control pathway, PIAS1, can attenuate the Huntington phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao T, Hong Y, Li XJ, Li SH. Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front Mol Neurosci. 2016;9:27. doi: 10.3389/fnmol.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scoles DR, Ho MH, Dansithong W, Pflieger LT, Petersen LW, Thai KK, Pulst SM. Repeat Associated Non-AUG Translation (RAN Translation) Dependent on Sequence Downstream of the ATXN2 CAG Repeat. PLoS One. 2015;10:e0128769. doi: 10.1371/journal.pone.0128769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griesche N, Schilling J, Weber S, Rohm M, Pesch V, Matthes F, Auburger G, Krauss S. Regulation of mRNA Translation by MID1: A Common Mechanism of Expanded CAG Repeat RNAs. Front Cell Neurosci. 2016;10:226. doi: 10.3389/fncel.2016.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krans A, Kearse MG, Todd PK. RAN translation from antisense CCG repeats in Fragile X Tremor/Ataxia Syndrome. Ann Neurol. 2016 doi: 10.1002/ana.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell. 2016;62:314–322. doi: 10.1016/j.molcel.2016.02.034. (••) This paper characterizes the translation requirements of the FMRpoly-Gly protein produced from the expanded CGG repeats of FXTAS and highlights the role of an upstream open-reading frame in FXTAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sellier C, Buijsen RA, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A, et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron. 2017 doi: 10.1016/j.neuron.2016.12.016. • This paper demonstrated that the requirement of an upstream close cognate initiation codon for translation of FMRpoly-G and characterized FMRpolyG protein toxicity in a mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sachs MS, Geballe AP. Downstream control of upstream open reading frames. Genes Dev. 2006;20:915–921. doi: 10.1101/gad.1427006. [DOI] [PubMed] [Google Scholar]
- 33.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arocena DG, Iwahashi CK, Won N, Beilina A, Ludwig AL, Tassone F, Schwartz PH, Hagerman PJ. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- 35.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35:1779.e1775–1779.e1713. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mori K, Lammich S, Mackenzie IR, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125:413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 43.Celona B, Dollen JV, Vatsavayai SC, Kashima R, Johnson JR, Tang AA, Hata A, Miller BL, Huang EJ, Krogan NJ, et al. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife. 2017;6 doi: 10.7554/eLife.19032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. (•) This study used AAV delivery to overexpress the sense G4C2 transcript and sense GP, GA, GR dipeptide proteins in mouse brains caused neurodegenerative changes in the cortex and cerebellum and TDP-43 pathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130:845–861. doi: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- 48.Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 49.Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383–395. doi: 10.1038/nrn.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen X, Westergard T, Pasinelli P, Trotti D. Pathogenic determinants and mechanisms of ALS/FTD linked to hexanucleotide repeat expansions in the C9orf72 gene. Neurosci Lett. 2017;636:16–26. doi: 10.1016/j.neulet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belzil VV, Katzman RB, Petrucelli L. ALS and FTD: an epigenetic perspective. Acta Neuropathol. 2016;132:487–502. doi: 10.1007/s00401-016-1587-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem. 2016;138(Suppl 1):145–162. doi: 10.1111/jnc.13623. [DOI] [PubMed] [Google Scholar]
- 55.Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016;25:1803–1813. doi: 10.1093/hmg/ddw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1620293114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 2016;167:774–788.e717. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell. 2016;167:789–802.e712. doi: 10.1016/j.cell.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. (••) This paper characterizes the link between C9orf72 hexanucleotide repeats with a key regulator of nucleocytoplasmic transport and shows small molecule or ASO targeting the repeats can rescue nuclear import defects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishiura H, Tsuji S. Epidemiology and molecular mechanism of frontotemporal lobar degeneration/amyotrophic lateral sclerosis with repeat expansion mutation in C9orf72. J Neurogenet. 2015;29:85–94. doi: 10.3109/01677063.2015.1085980. [DOI] [PubMed] [Google Scholar]
- 61.Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri G, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. (•) The paper utilizes an unbiased yeast-based genetic screen to identify modifiers of C9-polyProArg toxicity and identified components of nuclear pore complex pointing toward impaired nucleocytoplasmic transport as a pathogenic mechanism in C9-ALS/FTD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. (••) This paper developed a fly model of C9orf72 and through a modifier screen identified components of nuclear pore complex as genetic interactors that alter nuclear nucleocytoplasmic transport. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132:159–173. doi: 10.1007/s00401-016-1586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knockenhauer KE, Schwartz TU. The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell. 2016;164:1162–1171. doi: 10.1016/j.cell.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–176. doi: 10.1126/science.aad2033. [DOI] [PubMed] [Google Scholar]
- 67.Dansithong W, Jog SP, Paul S, Mohammadzadeh R, Tring S, Kwok Y, Fry RC, Marjoram P, Comai L, Reddy S. RNA steady-state defects in myotonic dystrophy are linked to nuclear exclusion of SHARP. EMBO Rep. 2011;12:735–742. doi: 10.1038/embor.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia J, Lee DH, Taylor J, Vandelft M, Truant R. Huntingtin contains a highly conserved nuclear export signal. Hum Mol Genet. 2003;12:1393–1403. doi: 10.1093/hmg/ddg156. [DOI] [PubMed] [Google Scholar]
- 69.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cornett J, Cao F, Wang CE, Ross CA, Bates GP, Li SH, Li XJ. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- 71.Yazawa I. Aberrant phosphorylation of dentatorubral-pallidoluysian atrophy (DRPLA) protein complex in brain tissue. Biochem J. 2000;351(Pt 3):587–593. [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi H, Egawa S, Piao YS, Hayashi S, Yamada M, Shimohata T, Oyanagi K, Tsuji S. Neuronal nuclear alterations in dentatorubral-pallidoluysian atrophy: ultrastructural and morphometric studies of the cerebellar granule cells. Brain Res. 2001;919:12–19. doi: 10.1016/s0006-8993(01)02986-9. [DOI] [PubMed] [Google Scholar]
- 73.Taylor J, Grote SK, Xia J, Vandelft M, Graczyk J, Ellerby LM, La Spada AR, Truant R. Ataxin-7 can export from the nucleus via a conserved exportin-dependent signal. J Biol Chem. 2006;281:2730–2739. doi: 10.1074/jbc.M506751200. [DOI] [PubMed] [Google Scholar]
- 74.Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci U S A. 2002;99:9310–9315. doi: 10.1073/pnas.152101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. (•) This paper describes a non-symptomatic C9orf72 BAC mouse model with RNA foci, RAN dipeptides and nucleolar stress and shows that these molecular features can be alleviated in primary cortical cultures by ASO treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. (•) This paper describes a C9orf72 BAC transgenic mouse model with molecular features of the disease, RNA foci and DPR proteins but no phenotypic or neurodegeneration features. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. (•) This paper highlights the gain-of-function role of the C9orf72 ALS/FTD hexanucleotide repeat expansion and characterizes a C9orf72 BAC mouse models that has RNA foci and DPR proteins and mild cognitive and neurodegenerative changes that are reversed with ASO treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. (••) This paper reports the first C9orf72 BAC mouse model that recapitulates the molecular, behavioral, and neurodegenerative features of both ALS and FTD. These mice show decreased survival, paralysis, muscle denervation, motor neuron loss, anxiety-like behavior, and cortical and hippocampal neurodegeneration. Antisense RNA foci accumulate in vulnerable brain regions, RAN proteins increase with age and disease and TDP-43 inclusions are found in end-stage animals. [DOI] [PubMed] [Google Scholar]
- 79.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110:E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keiser MS, Boudreau RL, Davidson BL. Broad Therapeutic Benefit After RNAi Expression Vector Delivery to Deep Cerebellar Nuclei: Implications for Spinocerebellar Ataxia Type 1 Therapy. Mol Ther. 2014;22:588–595. doi: 10.1038/mt.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Denovan-Wright EM, Davidson BL. RNAi: a potential therapy for the dominantly inherited nucleotide repeat diseases. Gene Ther. 2006;13:525–531. doi: 10.1038/sj.gt.3302664. [DOI] [PubMed] [Google Scholar]
- 83.Rodríguez-Lebrón E, Costa MoC, Costa M, Luna-Cancalon K, Peron TM, Fischer S, Boudreau RL, Davidson BL, Paulson HL. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol Ther. 2013;21:1909–1918. doi: 10.1038/mt.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, Prudencio M, Van Blitterswijk M, Belzil V, Couthouis J, et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science. 2016;353:708–712. doi: 10.1126/science.aaf7791. (••) These authors show that targeting the transcription elongation factor Spt4, previously known to decrease Huntingtin transcription, also decreases transcription of both sense and antisense C9 expansion transcripts. This study provides proof of principle data for a strategy to target both sense and antisense expansion transcripts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sopher BL, Ladd PD, Pineda VV, Libby RT, Sunkin SM, Hurley JB, Thienes CP, Gaasterland T, Filippova GN, La Spada AR. CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron. 2011;70:1071–1084. doi: 10.1016/j.neuron.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoskins JW, Ofori LO, Chen CZ, Kumar A, Sobczak K, Nakamori M, Southall N, Patnaik S, Marugan JJ, Zheng W, et al. Lomofungin and dilomofungin: inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res. 2014;42:6591–6602. doi: 10.1093/nar/gku275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. 2012;7:856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Childs-Disney JL, Parkesh R, Nakamori M, Thornton CA, Disney MD. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem Biol. 2012;7:1984–1993. doi: 10.1021/cb3001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ofori LO, Hoskins J, Nakamori M, Thornton CA, Miller BL. From dynamic combinatorial ‘hit’ to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res. 2012;40:6380–6390. doi: 10.1093/nar/gks298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.García-López A, Llamusí B, Orzáez M, Pérez-Payá E, Artero RD. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc Natl Acad Sci U S A. 2011;108:11866–11871. doi: 10.1073/pnas.1018213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu CR, Chang CR, Chern Y, Wang TH, Hsieh WC, Shen WC, Chang CY, Chu IC, Deng N, Cohen SN, et al. Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell. 2012;148:690–701. doi: 10.1016/j.cell.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 95.Cheng HM, Chern Y, Chen IH, Liu CR, Li SH, Chun SJ, Rigo F, Bennett CF, Deng N, Feng Y, et al. Effects on murine behavior and lifespan of selectively decreasing expression of mutant huntingtin allele by supt4h knockdown. PLoS Genet. 2015;11:e1005043. doi: 10.1371/journal.pgen.1005043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet. 2010;19:R77–82. doi: 10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 98.Ladd PD, Smith LE, Rabaia NA, Moore JM, Georges SA, Hansen RS, Hagerman RJ, Tassone F, Tapscott SJ, Filippova GN. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- 99.Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, Tapscott SJ. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell. 2005;20:483–489. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 100.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78:426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goodwin M, Swanson MS. RNA-binding protein misregulation in microsatellite expansion disorders. Adv Exp Med Biol. 2014;825:353–388. doi: 10.1007/978-1-4939-1221-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016;17:645–652. doi: 10.1016/j.celrep.2016.09.032. (••) This report shows evidence for cell-to-cell transmission of C9 DPR proteins which may explain the progressive nature of C9 ALS/FTD and pattern of DPR accumulation in the brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]