

Abstract
Cytochrome (CYP) 450 isoenzymes are the basic enzymes involved in Phase I biotransformation. The most important role in biotransformation belongs to CYP3A4, CYP2D6, CYP2C9, CYP2C19 and CYP1A2. Inhibition and induction of CYP isoenzymes caused by drugs are important and clinically relevant pharmacokinetic mechanisms of drug interaction. Investigation of the activity of CYP isoenzymes by using phenotyping methods (such as the determination of the concentration of specific substrates and metabolites in biological fluids) during drug administration provides the prediction of negative side effects caused by drug interaction. In clinical practice, the process of phenotyping of CYP isoenzymes and some endogenous substrates in the ratio of cortisol to 6β-hydroxycortisol in urine for the evaluation of CYP3A4 activity has been deemed to be a quite promising, safe and minimally invasive method for patients nowadays.
Keywords: cytochrome CYP450, drug interaction, drug metabolism, phenotyping
Introduction
It is well known that efficacy and safety of drugs used in clinical practice mainly depend on drug concentration near target molecules. This concentration usually correlates with plasma drug concentration, which depends on absorption, distribution and elimination processes. Elimination includes biotransformation and/or excretion of a drug (Figure 1). The main enzymes of oxidation reaction in Phase I are cytochrome (CYP) 450 isoenzymes, alcohol dehydrogenase, aldehyde dehydrogenase and aminooxidase.1 Recently, a number of studies devoted to endogenous and exogenous markers of CYP expression and activity were published. The results of those investigations could be promising in the clinical practice for the prediction of the patient’s personal reaction to medication and negative drug interactions. Diczfalusy et al2 reported that 4β-hydroxycholesterol (4β-OHC), which is formed by CYP3A4 from cholesterol, is a promising endogenous marker for the prediction of negative drug interactions with inducers of CYP3A enzymes or CYP3A inhibitors such as ritonavir or itraconazole. In this review, the authors also established a relationship between the concentration of 4β-OHC and the number of active CYP3A5*1 alleles showing that 4β-OHC was formed not only by CYP3A4 but also by CYP3A5 and thus proved that the above-mentioned correlations depend on gender as well (women had higher concentration of 4β-OHC in comparison with men). 4β-OHC has half-life of 17 days, and this is the reason why, in short-term studies, exogenous markers such as midazolam or quinine probes may be superior; but in long-term studies, 4β-OHC is a sensitive marker of CYP3A activity especially to assess drug interactions caused by induction and inhibition of CYP.2 CYP3A4 converts cortisol CYP3A4 to 6β-hydroxycortisol (6β-OHC), which is used as an endogenous marker in urine for the determination of CYP3A activity. CYP3A4 also converts cholesterol to another marker 4β-OHC.2 Mårde Arrhén et al3 compared the ratio of 4β-OHC to cholesterol in plasma with the ratio of 6β-OHC to cortisol in urine after the administration of rifampicin for 2 weeks, and after the analysis of obtained data, they concluded that the abovementioned markers give similar results about CYP activity. The concentration of cortisol and its metabolites in the body is flexible and depends on chronobiological rhythms; that is why the use of the ratio of 6β-OHC to cortisol is more rational for the precision prediction of CYP3A4 induction by rifampicin.3 In their comparison of endogenous 4β-OHC with midazolam as markers for CYP3A4 induction by rifampicin, Björkhem-Bergman et al4 showed that the 4β-OHC ratio is comparable with midazolam clearance as a marker of CYP3A4 induction, and each may be used to evaluate CYP3A4 induction in clinical trials evaluating drug–drug interactions for new drugs. These data are in good agreement with the data of Kasichayanula et al,5 which showed that changes in the plasma of 4β-OHC can be utilized as a surrogate for midazolam pharmacokinetics after the administration of multiple doses of potent CYP3A inducers (rifampicin). There is a more limited dynamic range for 4βHC for the assessment of potential CYP3A inhibitors. This review also confirmed the opinion of previous authors that 4β-OHC is a valuable marker for the assessment of potential CYP3A inducers in early drug development and particularly in drug interaction.5,6 This review is devoted to the recent data about CYP isoenzyme and some new opportunities for the prediction of negative drug interaction in vivo.
Figure 1.
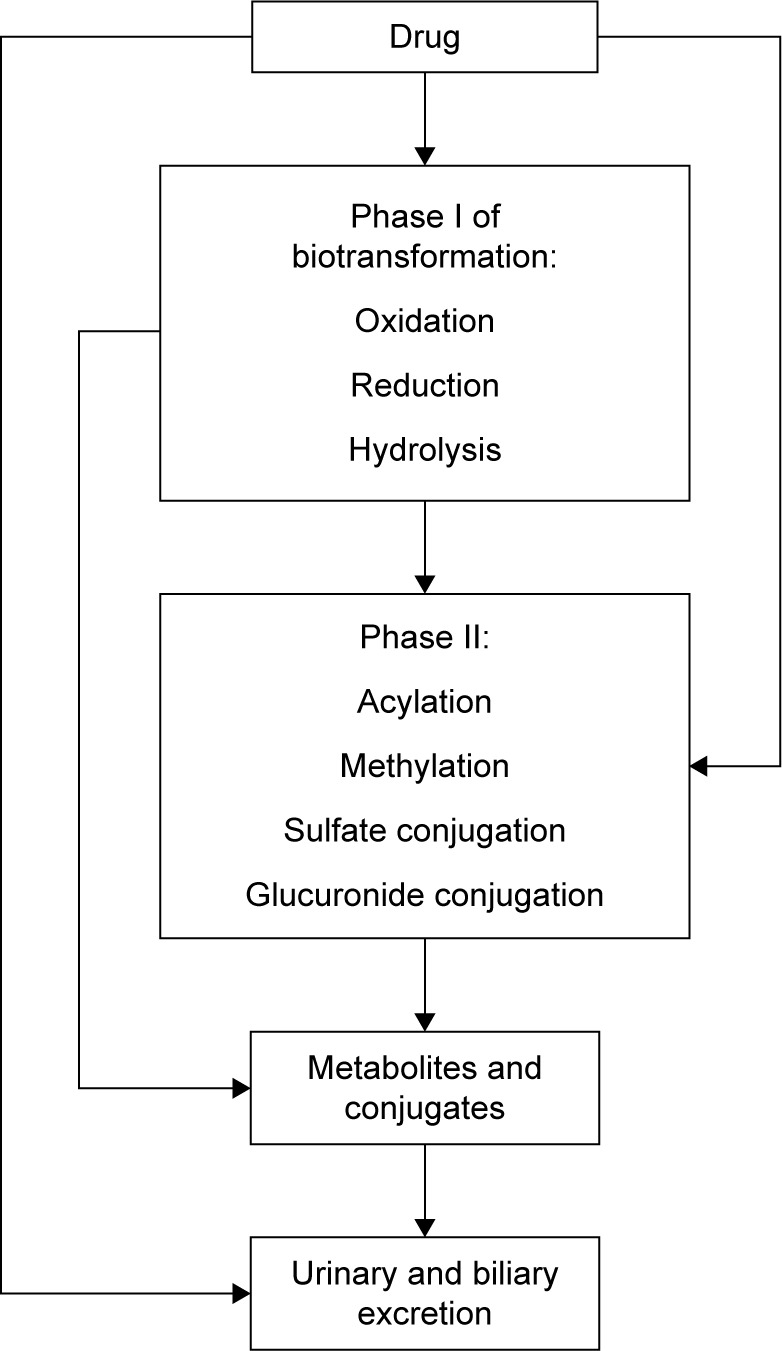
Phases of biotransformation.
Role of CYP isoenzymes in drug metabolism
The major biotransformation enzyme is CYP, which has more than 1,000 isoenzymes, of which five (CYP3A4, CYP2D6, CYP2C9, CYP2C19 and CYP1A2) metabolize 90% of all drugs (Table 1).7 CYP is a group of enzymes first isolated from liver microsomes of rats separately by Klingenberg8 and Garfinkel.9 Nomenclature of CYP includes family (CYP2, CYP3, etc.), subfamily (CYP2D, CYP3A, etc.) and the name of definite isoenzyme (CYP2D6, CYP3A4, etc.).10 CYP can be found in hepatocytes as well as in other cells of the body. According to the analysis of 200 most commonly prescribed drugs in the US (2002), about 73% of drugs are subject to metabolism, of which about 75% of the drugs are metabolized by CYP.11 Type CYP3A isoenzyme metabolizes 46% of drugs, type CYP2C9 isoenzyme metabolizes 16% of drugs, type CYP2C19 + CYP2D6 isozymes metabolize 12% of drugs, type CYP1A isoenzyme metabolizes 9% of drugs and type CYP2B6 + CYP2E1 isoenzymes metabolize 2% of drugs.12 Substrate specificity is an inherent characteristic of CYP isozymes, ie, they have the ability to bind and transform molecules with specific shape, charge and hydrophilic/hydrophobic characteristics.13,14 Some of the CYP isozymes have substrate stereospecificity, for example, CYP2C9 metabolizes S-warfarin (more active enantiomer of warfarin) and R-warfarin is metabolized by CYP1A2 and CYP3A4.15,16
Table 1.
The relative content of CYP isoenzymes in the liver, their basic share and their participation in the metabolism of drugs
| CYP isoenzyme | Content in the liver (%) | Contribution to metabolism (% metabolizing drugs) |
|---|---|---|
| 1A2 | ~13 | 8.2 |
| 2C | ~18 | 15.8 (2C8, 2C9) |
| 2D6 | Up to 2.5 | 18.8 (2C18, 2C19) |
| 3A4 | Up to 28 | 34.1 |
Note: Data from Kasichayanula et al.5
Abbreviation: CYP, cytochrome.
Most isoenzymes demonstrate broad substrate specificity, ie, each isozyme is able to metabolize a wide spectrum of xenobiotics, including drugs1 (Table 2). The activity of CYP isoenzymes may vary over a wide range after the exposure of inducers or inhibitors, thereby altering the metabolism of substrates of isoenzymes, which could result in drug–drug interactions.17 Isoenzyme CYP3A4 metabolizes approximately 40%–50% of the drugs used in clinical practice, including calcium slow channel blockers,18 macrolide antibiotics,19 statins (simvastatin, lovastatin, atorvastatin)20 and some of the “new” oral anticoagulants from the group of direct factor Xa inhibitors (rivaroxaban, apixaban).21 The most important inducers of CYP3A4 are carbamazepine,22 phenobarbital,23 phenytoin,24 rifampicin25 and St John’s wort extract.26 Some antifungal agents from the azole group (ketoconazole, itraconazole),27 protease inhibitors (indinavir, nelfinavir, ritonavir)28 and clarithromycin29 are the most important CYP3A4 inhibitors.
Table 2.
Typical substrates of basic CYP isozymes
| CYP isozyme | Substrates |
|---|---|
| CYP1A2 | Clozapine, caffeine, paracetamol, theophylline, phenacetin, R-warfarin |
| CYP2C9 | Hexobarbital, zidovudine, losartan, paracetamol, testosterone, tolbutamide, phenytoin, celecoxib, S-warfarin |
| CYP2C19 | Hexobarbital, diazepam, zidovudine, omeprazole, pantoprazole, testosterone, phenytoin, R-warfarin, S-warfarin |
| CYP2D6 | Haloperidol, dextromethorphan, codeine, metoprolol, nortriptyline, paracetamol, pravastatin, propafenone |
| CYP3A4 | Alprazolam, atorvastatin, vincristine, halothane, hydrocortisone, zidovudine, carbamazepine, codeine, cortisol, caffeine, lidocaine, lovastatin, midazolam, nifedipine, paracetamol, tacrolimus, tamoxifen, testosterone, phenytoin, cyclosporine, cyclophosphamide, erythromycin, R-warfarin, S-warfarin |
CYP2C9 is the main enzyme involved in the metabolism of many nonsteroidal anti-inflammatory drugs (NSAIDs): celecoxib,30 diclofenac,31 ibuprofen,32 lornoxicam,33 meloxicam,34 naproxen,35 S-warfarin,22 many antidiabetic sulfonylureas (glibenclamide,36 glimepiride,37 glipizide,38 tolbutamide39), angiotensin II receptor blockers (irbesartan,40 losartan41), fluvastatin,42 phenytoin,38,43 tamoxifen44 and cyclophosphamide.45,46 The most important inducer of CYP2C9 is rifampicin.47 Fluconazole and amiodarone are significant inhibitors of CYP2C9. CYP2C19 is the main metabolic enzyme for proton pump inhibitors, which in turn are the inhibitors of this isoenzyme (the so-called autoinhibitors).48,49 This enzyme is induced by carbamazepine, prednisolone and rifampicin.50,51 CYP2D6 metabolizes up to 20% of drugs, including tricyclic antidepressant amitriptyline, neuroleptics and β-adrenoblockers. Codeine is metabolized by CYP2D6 into the active metabolite of morphine.52 CYP2D6 is inhibited by fluoxetine,53 quinidine54 and bupropion.55 Unlike other isozymes, CYP2D6 has no verified inductors,56 but isoenzyme activity increases in the gestation period.57 However, there are controversial data about its weak induction by dexamethasone and rifampicin.58 CYP1A2 has no endogenous substrates. It metabolizes mainly xenobiotics, including theophylline, caffeine and acetaminophen. Autoinducers of CYP1A2 are polycyclic aromatic hydrocarbons (the main component of inhaled tobacco smoke), which by means of CYP1A2 are transformed into carcinogenic compounds.59 Besides, components of tobacco, smoke, grilled meal60 and broccoli61 are inductors of CYP1A2. Ciprofloxacin and fluvoxamine inhibit CYP1A2.62
Gene polymorphism in many CYP isozymes may be responsible for the interindividual differences in the speed of biotransformation of drugs and certain drug–drug interactions.63 The presence of single-nucleotide polymorphism (SNP) in the gene encoding a particular isozyme may lead to the synthesis of enzymes with altered activity, which in turn changes the pharmacokinetics of drugs metabolized by this isoenzyme. In clinical practice, it is possible to conduct pharmacogenetic testing of some drugs to identify genetic polymorphisms.42 This allows to predict the pharmacological response to these drugs, which in turn increases the efficacy and safety of drug therapy.50 CYP isoenzymes play a pivotal role in the metabolism of many drugs used in clinical practice. The changes in isoenzymes activity are the basis of drug–drug interaction at the level of biotransformation; thus, it is important to study the effect of drugs on CYP isoenzymes for safe pharmacotherapy.50
Inhibition of CYP isoenzymes
The inhibition of CYP isoenzymes is a particular mechanism of clinically significant drug interaction (Figure 2). A decrease in the metabolism of drugs which are substrates for the abovementioned enzyme leads to increased concentration of these drugs in plasma and exerts toxic effects.50,64 Inhibition of isoenzymes could be reversible as well as irreversible. The reversible mechanisms of inhibition can be competitive, noncompetitive and uncompetitive. In the type of competitive reversible inhibition, the drug inhibitor and drug substrate compete for the active site of isoenzyme; therefore, this type of inhibition could be overcome by increasing the concentration of drug substrate.65 Mechanism of noncompetitive reversible inhibition is due to the binding of inhibitor with the nonfunctional part of CYP isoenzyme, which changes the conformation of active site and prevents its binding to the drug substrate; therefore, this type of inhibition cannot be reversed by increasing the concentration of drug substrate. Noncompetitive inhibition of CYP isoenzymes is observed upon binding of the inhibitor to isoenzyme–substrate complex.66
Figure 2.

Schematic of drug–drug interaction between substrate and inhibitor of CYP isoenzyme.
Note: Data from Ritter et al.72
Abbreviation: CYP, cytochrome.
There are two types of irreversible inhibition: true irreversible inhibition and quasi-irreversible inhibition. Under true irreversible inhibition, the inhibitor or its intermediate metabolite covalently binds to the heme of CYP isoenzyme, thereby inactivating it. A strong noncovalent bond is formed between inhibitor and CYP isoenzyme in the case of quasi-irreversible inhibition. During irreversible inhibition, the recovery time of isoenzyme activity depends on the time needed for the synthesis of a new isoenzyme. Inhibitors of CYP isoenzymes (Table 3) may be classified in vivo according to the degree of inhibitory activity of drug substrates of the isoenzymes. The use of potent inhibitors result in more than five times increase in area under the curve (AUC) of drug substrate (a decrease in clearance of >80%); moderate inhibitors increase the AUC of drug substrate by two to five times (a decrease in clearance up to 50%–80%) and weak inhibitors are able to increase the AUC of drug substrate by 1.25–2 times (a decrease in clearance up to 20%–50%).67
Table 3.
Inhibitors of major CYP isozymes
| CYP isozyme | Potent inhibitors | Moderate inhibitors | Weak inhibitors |
|---|---|---|---|
| 1A2 | Ciprofloxacin, enoxacin, fluvoxamine | Zileuton, mexiletine, methoxsalen, felbamate, thiabendazole, phenylpropanolamine | Allopurinol, acyclovir, verapamil, disulfiram, caffeine, norfloxacin, propafenone, propranolol, ticlopidine, famotidine, cimetidine, echinacea extract |
| 2C9 | Amiodarone, miconazole, oxandrolone, fluconazole | Voriconazole, zafirlukast, capecitabine, co-trimoxazole (sulfamethoxazole + trimethoprim), metronidazole, sulfinpyrazone, tigecycline, fluvastatin, fluvoxamine, etravirine | |
| 2C19 | Fluconazole, fluvoxamine, ticlopidine | Fluoxetine, moclobemide, omeprazole, omeprazole, voriconazole | Armodafinil, carbamazepine, cimetidine, ethinyl estradiol, etravirine, somatotropin, felbamate, ketoconazole |
| 2D6 | Bupropion, paroxetine, fluoxetine, quinidine | Duloxetine, terbinafine, cinacalcet | Amiodarone, vemurafenib, verapamil, gefitinib, hydralazine, hydroxychloroquine, desvenlafaxine, diltiazem, diphenhydramine, imatinib, methadone, oral contraceptives, propafenone, ranitidine, ritonavir, sertraline, telithromycin, febuxostat, celecoxib, cimetidine, echinacea extract, escitalopram |
| 3A4 | Voriconazole, grapefruit juice (high concentration), itraconazole, ketoconazole, clarithromycin, lopinavir, nefazodone, posaconazole, ritonavir, ritonavir, saquinavir, telaprevir, telithromycin, conivaptan | Aprepitant, verapamil, grapefruit juice (normal concentration), darunavir, diltiazem, imatinib, ritonavir, fluconazole, fosamprenavir, ciprofloxacin, erythromycin | Alprazolam, amiodarone, amlodipine, atorvastatin, bicalutamide, zileuton, isoniazid, nilotinib, oral contraceptives, ranitidine, ranolazine, ticagrelor, tipranavir, fluvoxamine, fluoxetine, ciclosporin, cilostazol, cimetidine, an extract of goldenseal Canadian, Ginkgo biloba extract |
Note: Data from Wu et al.52
Abbreviation: CYP, cytochrome.
Induction of CYP isoenzymes
The absolute increase in the number and/or catalytic activity of isoenzymes and the associated reduction in the concentration of drug, which are substrate for CYP isoenzymes, are a result of the induction of isoenzymes. In most cases, the induction of isozymes is clinically expressed with the decrease in the pharmacological effects. In some cases, the induction of CYP isoenzymes results in the increase in pharmacological effects (in the case of the formation of active metabolites) and even in toxic effects. For example, the induction of CYP2E1 resulted in increased metabolism of acetaminophen and increased formation of hepatotoxic metabolite N-acetyl-p-benzoquinone imine.68
The most important mechanism for the induction of CYP isozymes is the interaction of inducer with specific intracellular receptors that in fact are transcription regulator proteins (pregnane-X-receptor, androstane constitutive receptor, aryl-hydrocarbonate receptor, etc.),69 thus forming a receptor–inductor complex. This complex penetrates into the cell nucleus and acts on the regulatory region of the gene that results in increased expression of the gene encoding CYP. There are mechanisms of induction, which are not associated with exposure to specific receptors. For example, the induction of CYP2E1 is associated with posttranscriptional stabilization of molecules of this isoenzyme.70 Based on the degree of induction of biotransformation of drug substrates in vivo, inducers are classified into three types: potent (≥80% reduction in AUC), moderate (50%–80% reduction in AUC) and weak (20%–50% reduction in AUC).67 Typical inductors are summarized in Table 4. Usually, it takes several days for the development of drug–drug interactions between drug inducer of CYP isoenzyme and drug substrate of this isozyme, because the mechanism of induction of CYP isoenzymes includes the induction of gene transcription encoding and subsequent synthesis.
Table 4.
Inductors of major CYP isozymes
| CYP isozyme | Potent inducers | Moderate inducers | Weak inducers |
|---|---|---|---|
| 1A2 | Montelukast, tobacco smoke, phenytoin | Moricizine, omeprazole, phenobarbital | |
| 2C9 | Arbamazepine, rifampicin | Aprepitant, bosentan, phenobarbital, St John’s wort extract | |
| 2C19 | Rifampicin, S-mephenytoin | Artemisinin | |
| 2D6 | Inducers are not identified | Inducers are not identified | |
| 3A4 | Avasimibe, carbamazepine, rifampicin, phenytoin, St John’s wort extract | Bosentan, modafinil, nafcillin, etravirine, efavirenz, lurasidone | Aprepitant, armodafinil, pioglitazone, prednisolone, refinamid, echinacea extract |
Note: Data from Wu et al.52
Abbreviation: CYP, cytochrome.
Legal status of studies on the effect of drugs on biotransformation enzymes
The most important step in the safety assessment of new and already registered drugs is a comprehensive evaluation of their potential side effects.71 The demographic aging of the population and increase in the number of drugs used by patients (including nonprescription drugs) have enhanced the risk of drug–drug interactions.60 Extensive study is being conducted worldwide to identify potential risk of negative drug–drug interaction between new and registered drugs, and effective measures have been introduced to reduce the risk of side effects (dose adjustment, additional therapeutic monitoring, etc.).72 Usually, in vitro studies are conducted before in vivo studies. The aim of screening investigation in vitro is to determinate the usefulness of studies in vivo. The Food and Drug Administration (FDA) has recommended the inclusion of instructions for medical use of drugs with information on the possible inhibitory or inducing effect on the biotransformation enzymes in the section describing pharmacokinetics. In the European Union also, pharmacokinetic studies have been designed to assess the influence of new drug(s) on the effects of well-known drug(s).67 The results of studies of side effects of new drug(s) could be used to predict their interaction with other drug(s) on the basis of the identified mechanisms of interaction.
Prediction of impact of drugs on CYP isoenzymes in vivo
The study of potential side effects of new drugs includes conducting studies on the effects of new drugs on the pharmacokinetics of marker substrates of CYP isoenzymes, extrapolation of the results of such study to other drugs, substrates of isozymes and, in some cases, the study of specific combinations of drugs to be able to work out specific recommendations on combined use of drugs.60 Marker substrates are substances that are metabolized primarily by one CYP isoenzyme (Figure 3). The purpose of the review for the investigation of influence of drugs on the marker substrates of CYP isoenzymes was to determine the presence or absence of any effect on CYP isoenzymes as well as to observe the magnitude of such influence, so as to classify the drugs that exhibit inductive/inhibitory properties in three groups: potent, moderate and weak. It is noted that Table 5 does not list all substrates that could be used to assess the effects of new drugs on the activity of CYP isoenzymes in vivo. For some CYP isozymes, methods for assessing the activity were developed. The special feature of those methods is that they do not require the administration of any xenobiotics. For example, in one of the guidelines for pharmaceutical companies, it was recommended to study biotransformation and transporters of new drugs for the evaluation of CYP3A4 activity in vivo by using the ratio of endogenous cortisol to one of its metabolites, 6β-OHC, which is formed exclusively as a result of CYP3A4 activity73 (Figure 4).
Figure 3.

Schematic of drug–drug interaction between the inductor and the substrate of CYP isoenzyme.
Note: Data from Ritter et al.72
Abbreviation: CYP, cytochrome.
Table 5.
Examples of sensitive substrates of basic CYP isoenzymes and substrates with a narrow therapeutic range
| CYP isozyme | Sensitive substratea | Substrates with a narrow therapeutic range |
|---|---|---|
| 1A2 | Alosetron, duloxetine, caffeine, melatonin, ramelteon, tacrine, tizanidine | Theophylline, tizanidine |
| 2C9 | Celecoxib | Warfarin, phenytoin |
| 2C19 | S-mephenytoin, clobazam, lansoprazole, omeprazole | S-mephenytoin |
| 2D6 | Atomoxetine, venlafaxine, desipramine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine | Pimozide, thioridazine |
| 3A4 | Alfentanil, aprepitant, budesonide, buspirone, vardenafil, dasatinib, darifenacin, darunavir, dronedarone, indinavir, quetiapine, conivaptan, lovastatin, lopinavir, lurasidone, maraviroc, midazolam, nisoldipine, saquinavir, sildenafil, simvastatin, sirolimus, ticagrelor, tipranavir, tolvaptan, triazolam, felodipine, fluticasone, everolimus, eletriptan, eplerenone | Alfentanil, astemizole, dihydroergotamine, pimozide, sirolimus, tacrolimus, terfenadine, fentanyl, quinidine, cisapride, cyclosporine, ergotamine |
Notes:
The sensitive substrate is a substrate with AUC values more than fivefold increase in the combined use with an inhibitor of CYP isoenzymes. Data from Wadelius et al.57
Abbreviations: AUC, area under the curve; CYP, cytochrome.
Figure 4.

Metabolic conversion of cortisol.
Note: Data from Gray et al.85
Abbreviations: DHF, 20β-DHF (20β-dihydrocortisol); DHE, 20β-DHE(20β-dihydrocortisone); THF, 3α,5β-THF (3α,5β-tetrahydrocortisol); THE, 3α,5β-THE (3α,5β-tetrahydrocortisone); OHC, 6 β-OHC(6 β-hydroycortisol); OHE, 6 β-OHE(6 β-hydroycortisone).
The past study showed that the dynamics of endogenous activity of CYP3A4 metabolic markers, including the ratio of 6β-OHC to cortisol in urine, correlate with midazolam clearance dynamics under the induction or inhibition of CYP3A4 in healthy volunteers; therefore, the ratio of 6β-OHC to cortisol in urine could be used to evaluate the activity of CYP3A4 in vivo.74 Formation clearance of the sum of 6β-OHC and 6β-hydroxycortisone was shown to detect moderate and potent inhibition of CYP3A4 in vivo.75 CYP2D6 activity may be evaluated using pinoline urinary metabolic ratio (6-methoxy-1,2,3,4-tetrahydro-β-carboline/6-hydroxy-1,2,3,4-tetrahydro-β-carboline).76 To estimate CYP2C19 activity in vivo, omeprazole plasma hydroxylation index (omeprazole/5-hydroxyomeprazole) was introduced.77 Losartan test could be used to assess the activity of CYP2C9. This test is based on the determination of concentration of losartan and its active metabolite, E-3174, which is produced mainly by CYP2C9 activity.78 According to in vitro studies, E-3174 is also formed by the action of CYP3A4,79,80 but in vivo studies with therapeutic doses of losartan showed no significant contribution to losartan CYP3A4 metabolism.81 The predominant contribution of CYP2C9 to losartan metabolism was confirmed indirectly by a decrease in the AUC of E-3174 when it was used in combination with moderate inhibitor CYP2C9 (fluconazole), while no change in AUC of E-3174 was observed when losartan was used in combination with a strong CYP3A4 inhibitor (itraconazole).47 In many studies, losartan has been safely used as a marker substrate for phenotyping CYP2C9 activity.82–84 Relative clinical safety of losartan and reliability of losartan test allow its usage in clinical studies dedicated to the investigations of the influence of new drugs on CYP2C9 activity in vivo.
Conclusion and future perspectives
Prediction of negative drug interaction is one of the most important and difficult objectives of clinical pharmacology. CYP isoenzymes are the basic enzymes involved in Phase I biotransformation. The use of CYP isoenzymes, cortisol to 6β-OHC ratio, losartan test and other tests used for the estimation of the activity of CYP so as to investigate the influence of new drugs can yield new and prospective findings related to the prediction of negative drug interaction, which in turn can provide new insights for researchers and clinicians.
Acknowledgments
Gjumrakch Aliev’s work was supported by a grant from the Russian Science Foundation (directed design, synthesis and study of biological activity of multitarget compounds as innovative drugs for the treatment of neurodegenerative diseases: PHΦ 14-23-00160Π). Gjumrakch Aliev is also grateful for the equipment of Center for Collective Use, Institute of Physiologically Active Compounds of Russian Academy of Sciences Chernogolovka, Russia, that was used for this review.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Diczfalusy U, Nylén H, Elander P, Bertilsson L. 4β-Hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br J Clin Pharmacol. 2011;71(2):183–189. doi: 10.1111/j.1365-2125.2010.03773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mårde Arrhén Y, Nylén H, Lövgren-Sandblom A, Kanebratt KP, Wide K, Diczfalusy U. A comparison of 4β-hydroxycholesterol: cholesterol and 6β-hydroxycortisol: cortisol as markers of CYP3A4 induction. Br J Clin Pharmacol. 2013;75(6):1536–1540. doi: 10.1111/bcp.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Björkhem-Bergman L, Bäckström T, Nylén H, et al. Comparison of endogenous 4β-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab Dispos. 2013;41(8):1488–1493. doi: 10.1124/dmd.113.052316. [DOI] [PubMed] [Google Scholar]
- 5.Kasichayanula S, Boulton DW, Luo W-L, et al. Validation of 4β-hydroxycholesterol and evaluation of other endogenous biomarkers for the assessment of CYP3A activity in healthy subjects. Br J Clin Pharmacol. 2014;78(5):1122–1134. doi: 10.1111/bcp.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leil TA, Kasichayanula S, Boulton DW, LaCreta F. Evaluation of 4β-hydroxycholesterol as a clinical biomarker of CYP3A4 drug interactions using a Bayesian mechanism-based pharmacometric model. CPT Pharmacometrics Syst Pharmacol. 2014;3(6):e120. doi: 10.1038/psp.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinto N, Dolan ME. Clinically relevant genetic variations in drug metabolizing enzymes. Curr Drug Metab. 2011;12(5):487–497. doi: 10.2174/138920011795495321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingenberg M. Pigments of rat liver microsomes. Arch Biochem Biophys. 1958;75(2):376–386. doi: 10.1016/0003-9861(58)90436-3. [DOI] [PubMed] [Google Scholar]
- 9.Garfinkel D. Studies on pig liver microsomes. I. Enzymic and pigment composition of different microsomal fractions. Arch Biochem Biophys. 1958;77(2):493–509. doi: 10.1016/0003-9861(58)90095-x. [DOI] [PubMed] [Google Scholar]
- 10.Amal AIO, Murry DJ. Pharmacogenetics of the cytochrome P450 enzyme system: review of current knowledge and clinical significance. J Pharm Pract. 2007;20(3):206–218. [Google Scholar]
- 11.Thunell S, Pomp E, Brun A. Guide to drug porphyrogenicity prediction and drug prescription in the acute porphyrias. Br J Clin Pharmacol. 2007;64(5):668–679. doi: 10.1111/j.0306-5251.2007.02955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams JA, Hyland R, Jones BC, et al. Drug–drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 13.Roy J. Introduction to the Pharmaceutical Sciences. Elsevier; 2011. [Google Scholar]
- 14.Sirim D, Widmann M, Wagner F, Pleiss J. Prediction and analysis of the modular structure of cytochrome P450 monooxygenases. BMC Struct Biol. 2010;10:34. doi: 10.1186/1472-6807-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73(1):67–74. doi: 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 16.Hosokawa M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules. 2008;13(2):412–431. doi: 10.3390/molecules13020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teo YL, Ho HK, Chan A. Metabolism-related pharmacokinetic drug–drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br J Clin Pharmacol. 2015;79(2):241–253. doi: 10.1111/bcp.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory potencies of 1,4-dihydropyridine calcium antagonists to P-glycoprotein-mediated transport: comparison with the effects on CYP3A4. Pharm Res. 2000;17(10):1189–1197. doi: 10.1023/a:1007568811691. [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues AD, Roberts EM, Mulford DJ, Yao Y, Ouellet D. Oxidative metabolism of clarithromycin in the presence of human liver microsomes. Major role for the cytochrome P4503A (CYP3A) subfamily. Drug Metab Dispos. 1997;25(5):623–630. [PubMed] [Google Scholar]
- 20.Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999;84(3):413–428. doi: 10.1016/s0163-7258(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 21.Heidbuchel H, Verhamme P, Alings M, et al. Updated European Heart Rhythm Association Practical Guide on the use of non-vitamin K antagonist anticoagulants in patients with non-valvular atrial fibrillation. Europace. 2015;17(10):1467–1507. doi: 10.1093/europace/euv309. [DOI] [PubMed] [Google Scholar]
- 22.Bertilsson L, Tybring G, Widén J, Chang M, Tomson T. Carbamazepine treatment induces the CYP3A4 catalysed sulphoxidation of omeprazole, but has no or less effect on hydroxylation via CYP2C19. Br J Clin Pharmacol. 1997;44(2):186–189. doi: 10.1046/j.1365-2125.1997.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohno M, Motojima K, Okano T, Taniguchi A. Induction of drug- metabolizing enzymes by phenobarbital in layered co-culture of a human liver cell line and endothelial cells. Biol Pharm Bull. 2009;32(5):813–817. doi: 10.1248/bpb.32.813. [DOI] [PubMed] [Google Scholar]
- 24.Fleishaker JC, Pearson LK, Peters GR. Phenytoin causes a rapid increase in 6 beta-hydroxycortisol urinary excretion in humans – a putative measure of CYP3A induction. J Pharm Sci. 1995;84(3):292–294. doi: 10.1002/jps.2600840305. [DOI] [PubMed] [Google Scholar]
- 25.Backman JT, Olkkola KT, Neuvonen PJ. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther. 1996;59(1):7–13. doi: 10.1016/S0009-9236(96)90018-1. [DOI] [PubMed] [Google Scholar]
- 26.Rahimi R, Abdollahi M. An update on the ability of St. John’s wort to affect the metabolism of other drugs. Expert Opin Drug Metab Toxicol. 2012;8(6):691–708. doi: 10.1517/17425255.2012.680886. [DOI] [PubMed] [Google Scholar]
- 27.Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56(6 pt 1):601–607. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 28.Eagling VA, Back DJ, Barry MG. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J Clin Pharmacol. 1997;44(2):190–194. doi: 10.1046/j.1365-2125.1997.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiyoshi T, Ito M, Murase S, et al. Mechanism-based inhibition profiles of erythromycin and clarithromycin with cytochrome P450 3A4 genetic variants. Drug Metab Pharmacokinet. 2013;28(5):411–415. doi: 10.2133/dmpk.dmpk-12-rg-134. [DOI] [PubMed] [Google Scholar]
- 30.Chan AT, Zauber AG, Hsu M, et al. Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gastroenterology. 2009;136(7):2127–2136. doi: 10.1053/j.gastro.2009.02.045. e2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morin S, Loriot MA, Poirier JM, et al. Is diclofenac a valuable CYP2C9 probe in humans? Eur J Clin Pharmacol. 2001;56(11):793–797. doi: 10.1007/s002280000240. [DOI] [PubMed] [Google Scholar]
- 32.Berka K, Hendrychová T, Anzenbacher P, Otyepka M. Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J Phys Chem A. 2011;115(41):11248–11255. doi: 10.1021/jp204488j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Y, Zhang Y, Wang Y, et al. Role of CYP2C9 and its variants (CYP2C9*3 and CYP2C9*13) in the metabolism of lornoxicam in humans. Drug Metab Dispos. 2005;33(6):749–753. doi: 10.1124/dmd.105.003616. [DOI] [PubMed] [Google Scholar]
- 34.Chesné C, Guyomard C, Guillouzo A, Schmid J, Ludwig E, Sauter T. Metabolism of meloxicam in human liver involves cytochromes P4502C9 and 3A4. Xenobiotica. 1998;28(1):1–13. doi: 10.1080/004982598239704. [DOI] [PubMed] [Google Scholar]
- 35.Bae J-W, Kim J-H, Choi C-I, et al. Effect of CYP2C9*3 allele on the pharmacokinetics of naproxen in Korean subjects. Arch Pharm Res. 2009;32(2):269–273. doi: 10.1007/s12272-009-1232-z. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y-F, Chen X-Y, Guo Y-J, Si D-Y, Zhou H, Zhong D-F. Impact of cytochrome P450 CYP2C9 variant allele CYP2C9*3 on the pharmacokinetics of glibenclamide and lornoxicam in Chinese subjects. Acta Pharm Sin. 2005;40(9):796–799. [PubMed] [Google Scholar]
- 37.Niemi M, Cascorbi I, Timm R, Kroemer HK, Neuvonen PJ, Kivistö KT. Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002;72(3):326–332. doi: 10.1067/mcp.2002.127495. [DOI] [PubMed] [Google Scholar]
- 38.Kidd RS, Straughn AB, Meyer MC, Blaisdell J, Goldstein JA, Dalton JT. Pharmacokinetics of chlorpheniramine, phenytoin, glipizide and nifedipine in an individual homozygous for the CYP2C9*3 allele. Pharmacogenetics. 1999;9(1):71–80. doi: 10.1097/00008571-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Kirchheiner J, Bauer S, Meineke I, et al. Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics. 2002;12(2):101–109. doi: 10.1097/00008571-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Chen G, Jiang S, Mao G, et al. CYP2C9 Ile359Leu polymorphism, plasma irbesartan concentration and acute blood pressure reductions in response to irbesartan treatment in Chinese hypertensive patients. Methods Find Exp Clin Pharmacol. 2006;28(1):19–24. doi: 10.1358/mf.2006.28.1.962773. [DOI] [PubMed] [Google Scholar]
- 41.McCrea JB, Cribb A, Rushmore T, et al. Phenotypic and genotypic investigations of a healthy volunteer deficient in the conversion of losartan to its active metabolite E-3174. Clin Pharmacol Ther. 1999;65(3):348–352. doi: 10.1016/S0009-9236(99)70114-1. [DOI] [PubMed] [Google Scholar]
- 42.Tutton R. Pharmacogenomic biomarkers in drug labels: what do they tell us? Pharmacogenomics. 2014;15(3):297–304. doi: 10.2217/pgs.13.198. [DOI] [PubMed] [Google Scholar]
- 43.Veronese ME, Mackenzie PI, Doecke CJ, McManus ME, Miners JO, Birkett DJ. Tolbutamide and phenytoin hydroxylations by cDNA-expressed human liver cytochrome P4502C9. Biochem Biophys Res Commun. 1991;175(3):1112–1118. doi: 10.1016/0006-291x(91)91680-b. [DOI] [PubMed] [Google Scholar]
- 44.Coller JK, Krebsfaenger N, Klein K, et al. The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol. 2002;54(2):157–167. doi: 10.1046/j.1365-2125.2002.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ekhart C, Doodeman VD, Rodenhuis S, Smits PHM, Beijnen JH, Huitema ADR. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics. 2008;18(6):515–523. doi: 10.1097/FPC.0b013e3282fc9766. [DOI] [PubMed] [Google Scholar]
- 46.Griskevicius L, Yasar U, Sandberg M, et al. Bioactivation of cyclophosphamide: the role of polymorphic CYP2C enzymes. Eur J Clin Pharmacol. 2003;59(2):103–109. doi: 10.1007/s00228-003-0590-6. [DOI] [PubMed] [Google Scholar]
- 47.Rana R, Chen Y, Ferguson SS, Kissling GE, Surapureddi S, Goldstein JA. Hepatocyte nuclear factor 4{alpha} regulates rifampicin-mediated induction of CYP2C genes in primary cultures of human hepatocytes. Drug Metab Dispos. 2010;38(4):591–599. doi: 10.1124/dmd.109.030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaukonen KM, Olkkola KT, Neuvonen PJ. Fluconazole but not itraconazole decreases the metabolism of losartan to E-3174. Eur J Clin Pharmacol. 1998;53(6):445–449. doi: 10.1007/s002280050405. [DOI] [PubMed] [Google Scholar]
- 49.Wedemeyer R-S, Blume H. Pharmacokinetic drug interaction profiles of proton pump inhibitors: an update. Drug Saf. 2014;37(4):201–211. doi: 10.1007/s40264-014-0144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hodgson E. A Textbook of Modern Toxicology. 4th ed. Wiley; [Accessed March 27, 2018]. webpage on the Internet. Available from: http://eu.wiley.com/Wiley-CDA/WileyTitle/productCd-047046206X.html. [Google Scholar]
- 51.Bibi Z. Role of cytochrome P450 in drug interactions. Nutr Metab. 2008;5:27. doi: 10.1186/1743-7075-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Wu X, Yuan L, Zuo J, Lv J, Guo T. The impact of CYP2D6 polymorphisms on the pharmacokinetics of codeine and its metabolites in Mongolian Chinese subjects. Eur J Clin Pharmacol. 2014;70(1):57–63. doi: 10.1007/s00228-013-1573-x. [DOI] [PubMed] [Google Scholar]
- 53.Preskorn SH, Shah R, Neff M, Golbeck AL, Choi J. The potential for clinically significant drug–drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5–12. doi: 10.1097/00131746-200701000-00002. [DOI] [PubMed] [Google Scholar]
- 54.O’Hara GE, Philippon F, Gilbert M, et al. Combined administration of quinidine and propafenone for atrial fibrillation: the CAQ-PAF study. J Clin Pharmacol. 2012;52(2):171–179. doi: 10.1177/0091270011399574. [DOI] [PubMed] [Google Scholar]
- 55.Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206–1227. doi: 10.1016/s0149-2918(08)80047-1. [DOI] [PubMed] [Google Scholar]
- 56.Haertter S. Recent examples on the clinical relevance of the CYP2D6 polymorphism and endogenous functionality of CYP2D6. Drug Metabol Drug Interact. 2013;28(4):209–216. doi: 10.1515/dmdi-2013-0032. [DOI] [PubMed] [Google Scholar]
- 57.Wadelius M, Darj E, Frenne G, Rane A. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400–407. doi: 10.1016/S0009-9236(97)90118-1. [DOI] [PubMed] [Google Scholar]
- 58.Rae JM, Johnson MD, Lippman ME, Flockhart DA. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther. 2001;299(3):849–857. [PubMed] [Google Scholar]
- 59.Ayari I, Fedeli U, Saguem S, Hidar S, Khlifi S, Pavanello S. Role of CYP1A2 polymorphisms in breast cancer risk in women. Mol Med Rep. 2013;7(1):280–286. doi: 10.3892/mmr.2012.1164. [DOI] [PubMed] [Google Scholar]
- 60.Fontana RJ, Lown KS, Paine MF, et al. Effects of a chargrilled meat diet on expression of CYP3A, CYP1A, and P-glycoprotein levels in healthy volunteers. Gastroenterology. 1999;117(1):89–98. doi: 10.1016/s0016-5085(99)70554-8. [DOI] [PubMed] [Google Scholar]
- 61.Hakooz N, Hamdan I. Effects of dietary broccoli on human in vivo caffeine metabolism: a pilot study on a group of Jordanian volunteers. Curr Drug Metab. 2007;8(1):9–15. doi: 10.2174/138920007779315080. [DOI] [PubMed] [Google Scholar]
- 62.Karjalainen MJ, Neuvonen PJ, Backman JT. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin Pharmacol Toxicol. 2008;103(2):157–165. doi: 10.1111/j.1742-7843.2008.00252.x. [DOI] [PubMed] [Google Scholar]
- 63.Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496–526. doi: 10.1016/j.pharmthera.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Feghali M, Venkataramanan R, Caritis S. Pharmacokinetics of drugs in pregnancy. Semin Perinatol. 2015;39(7):512–519. doi: 10.1053/j.semperi.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ogu CC, Maxa JL. Drug interactions due to cytochrome P450. Proc (Bayl Univ Med Cent) 2000;13(4):421–423. doi: 10.1080/08998280.2000.11927719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fowler S, Zhang H. In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: current status on methodologies and their utility for predicting drug–drug interactions. AAPS J. 2008;10(2):410–424. doi: 10.1208/s12248-008-9042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prueksaritanont T, Chu X, Gibson C, et al. Drug–drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 2013;15(3):629–645. doi: 10.1208/s12248-013-9470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michaut A, Moreau C, Robin M-A, Fromenty B. Acetaminophen-induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int. 2014;34(7):e171–e179. doi: 10.1111/liv.12514. [DOI] [PubMed] [Google Scholar]
- 69.Tompkins LM, Wallace AD. Mechanisms of cytochrome P450 induction. J Biochem Mol Toxicol. 2007;21(4):176–181. doi: 10.1002/jbt.20180. [DOI] [PubMed] [Google Scholar]
- 70.Azzalis LA, Fonseca FLA, Simon KA, et al. Effects of ethanol on CYP2E1 levels and related oxidative stress using a standard balanced diet. Drug Chem Toxicol. 2012;35(3):324–329. doi: 10.3109/01480545.2011.619192. [DOI] [PubMed] [Google Scholar]
- 71.Brewer L, Williams D. Clinically relevant drug–drug and drug–food interactions. Pharm Med. 2013;27(1):9–23. [Google Scholar]
- 72.Ritter J, Lewis L, Mant T, Ferro A. A Textbook of Clinical Pharmacology and Therapeutics. 5th ed. Boca Raton, Florida: CRC Press; 2008. [Google Scholar]
- 73.Рекомендации от 05.02.2009 – regmed.ru [webpage on the Internet] [Accessed March 27, 2018]. Available from: http://www.regmed.ru/Content/Doc.aspx?id=26a9128c-ee32-4469-9c64-5c666339049e.
- 74.Shin KH, Choi MH, Lim KS, Yu KS, Jang IJ, Cho JY. Evaluation of endogenous metabolic markers of hepatic CYP3A activity using metabolic profiling and midazolam clearance. Clin Pharmacol Ther. 2013;94(5):601–609. doi: 10.1038/clpt.2013.128. [DOI] [PubMed] [Google Scholar]
- 75.Peng CC, Templeton I, Thummel KE, Davis C, Kunze KL, Isoherranen N. Evaluation of 6β-hydroxycortisol, 6β-hydroxycortisone, and a combination of the two as endogenous probes for inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2011;89(6):888–895. doi: 10.1038/clpt.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang X-L, Shen H-W, Yu A-M. Pinoline may be used as a probe for CYP2D6 activity. Drug Metab Dispos. 2009;37(3):443–446. doi: 10.1124/dmd.108.025056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Niioka T, Uno T, Sugimoto K, Sugawara K, Hayakari M, Tateishi T. Estimation of CYP2C19 activity by the omeprazole hydroxylation index at a single point in time after intravenous and oral administration. Eur J Clin Pharmacol. 2007;63(11):1031–1038. doi: 10.1007/s00228-007-0331-3. [DOI] [PubMed] [Google Scholar]
- 78.Yasar U, Forslund-Bergengren C, Tybring G, et al. Pharmacokinetics of losartan and its metabolite E-3174 in relation to the CYP2C9 genotype. Clin Pharmacol Ther. 2002;71(1):89–98. doi: 10.1067/mcp.2002.121216. [DOI] [PubMed] [Google Scholar]
- 79.Stearns RA, Chakravarty PK, Chen R, Chiu SH. Biotransformation of losartan to its active carboxylic acid metabolite in human liver microsomes. Role of cytochrome P4502C and 3A subfamily members. Drug Metab Dispos. 1995;23(2):207–215. [PubMed] [Google Scholar]
- 80.Yun CH, Lee HS, Lee H, Rho JK, Jeong HG, Guengerich FP. Oxidation of the angiotensin II receptor antagonist losartan (DuP 753) in human liver microsomes. Role of cytochrome P4503A(4) in formation of the active metabolite EXP3174. Drug Metab Dispos. 1995;23(2):285–289. [PubMed] [Google Scholar]
- 81.Yang S-H, Cho Y-A, Choi J-S. Effects of ticlopidine on pharmacokinetics of losartan and its main metabolite EXP-3174 in rats. Acta Pharmacol Sin. 2011;32(7):967–972. doi: 10.1038/aps.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Andrés F, Sosa-Macías M, Llerena A. A rapid and simple LC-MS/MS method for the simultaneous evaluation of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 hydroxylation capacity. Bioanalysis. 2014;6(5):683–696. doi: 10.4155/bio.14.20. [DOI] [PubMed] [Google Scholar]
- 83.Dorado P, Gallego A, Peñas-Lledó E, Terán E, Llerena A. Relationship between the CYP2C9 IVS8-109A>T polymorphism and high losartan hydroxylation in healthy Ecuadorian volunteers. Pharmacogenomics. 2014;15(11):1417–1421. doi: 10.2217/pgs.14.85. [DOI] [PubMed] [Google Scholar]
- 84.Sekino K, Kubota T, Okada Y, et al. Effect of the single CYP2C9*3 allele on pharmacokinetics and pharmacodynamics of losartan in healthy Japanese subjects. Eur J Clin Pharmacol. 2003;59(8–9):589–592. doi: 10.1007/s00228-003-0664-5. [DOI] [PubMed] [Google Scholar]
- 85.Gray J. A textbook of clinical pharmacology and therapeutics, 5th edition. Clin Pharmacol Ther. 2009;85(2):125–126. [Google Scholar]