

Abstract
A porous Si (pSi) microparticle-based delivery system is investigated, and the intrinsic luminescence from the particles is employed as a probe to monitor the release of a model protein payload, bovine serum albumin (BSA). The microparticles consist of a core Si skeleton surrounded by a SiO2 shell. Two types of pSi are tested, one with smaller (10 nm) pores and the other with larger (20 nm) pores. The larger pore material yields a higher mass loading of BSA (3 vs 20%). Two different methods are used to load BSA into these nanostructures: the first involves loading by electrostatic physisorption, and the second involves trapping of BSA in the pSi matrix by local precipitation of magnesium silicate. Protein release from the former system is characterized by a burst release, whereas in the latter system, release is controlled by dissolution of the pSi/magnesium silicate matrix. The protein release characteristics are studied under accelerated (0.1 M aqueous KOH, 21 °C) and physiologically relevant (phosphate-buffered saline, pH 7.4, 37 °C) conditions, and the near-infrared photoluminescence signal from the pSi skeleton is monitored as a function of time and correlated with protein release and silicon dissolution. The thickness of the Si core and the SiO2 shell are systematically varied, and it is found that the luminescence signature can be tuned to provide a signal that either scales with protein elution or that changes rapidly near the end of useful life of the delivery system. Although payload release and particle dissolution are not driven by the same mechanism, the correlations between luminescence and payload elution for the various formulations can be used to define design rules for this self-reporting delivery system.
Keywords: fluorescence imaging, nanomaterials, oxidation, core–shell, quantum confinement, magnesium silicate, protein therapeutics
Graphical abstract
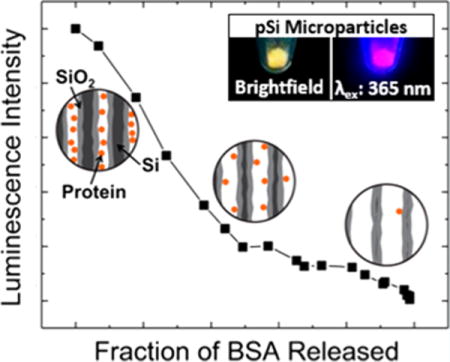
Hollow, or porous nanomaterials are attractive as drug-delivery vehicles because they can provide the advantages of sustained release, lower systemic toxicity, and tissue-specific targeting while protecting the drug payload from degradation.1–5 Inorganic materials such as porous silicon (pSi),6,7 nanoporous anodic alumina,8 and mesoporous silica9,10 have been exploited as materials to deliver drugs via many different administration routes; of interest in this study are intraocular, intramuscular, and subcutaneous delivery, where larger, depot-based formulations are typically used.11–13 Controlling the kinetics of drug release from such nanomaterials is crucial for optimal treatment, and it poses a substantial challenge due to patient-to-patient variability and other physiological characteristics that affect release kinetics.14,15 There is a particular need to monitor drug burden in developmental stages and during clinical trials, and indirect methods of drug quantification by sampling bodily fluids such as blood or urine are limited due to cost and complexity. Theranostic systems that are self-reporting, such that they indicate the status of the delivery vehicle in terms of local drug concentration or residual drug capacity,16–18 is a poorly developed area that could provide substantial benefits in personalized medicine.19,20
Compared to other systems, a unique dimension of pSi is that it contains intrinsic photoluminescent domains as part of the nanostructure. It is one of few semiconductor “quantum dot” materials that is nontoxic and degrades to nontoxic byproducts.21–24 For self-reporting drug-delivery applications, advantages of pSi include: (1) the photoluminescence appears at tissue-penetrating near-infrared wavelengths;24 (2) the measurement of light emission from pSi has little to no angular dependence or directional light scattering (unlike photonic crystals, for example);16,25 and (3) the long-lived (micro-seconds) excited state of pSi enables elimination of interfering background fluorescence by time-gated imaging.26,27 In this work, we explore the possibility of using this intrinsic luminescence from pSi particles to self-report carrier degradation and drug delivery.
Microparticles of pSi were prepared by anodic electrochemical etch of highly p-doped single-crystal silicon wafers in an electrolyte consisting of aqueous hydrofluoric acid (HF) and ethanol. The porous film was removed from the silicon substrate by electrochemical lift-off and subjected to ultrasonic fracture to form irregular microparticles of sizes ranging from 15 to 110 μm (Figure S1a, Supporting Information). The thickness and open porosity (obtained by spectroscopic liquid infiltration method, or SLIM),28 of the porous silicon layers prior to particle formation were ∼20 μm and ∼60–70%, respectively. Scanning electron microscope images revealed pores with diameters of 5–50 nm and wall thickness of ∼10 nm (Figure S1b,c, Supporting Information). In the present work, three types of core–shell luminescent Si–SiO2 structures were prepared by systematically varying the SiO2 shell thickness. This was achieved by controlling the duration of thermal oxidation (25, 35, or 45 min) at 700 °C (Figure 1a), designated “thin shell”, “medium shell”, and “thick shell”.29 Because the shell was formed by partial conversion of the crystalline Si walls to oxide, the longer oxidation times resulted in a simultaneously thicker oxide shell and a thinner Si core for a given pore wall thickness. This depletion of the Si core was evident in the visual appearance of the particles; the optical density progressively decreased with increasing oxidation time, ranging from dark brown for the as-etched particles to a light ochre color for material treated at 700 °C for 45 min. Material oxidized for 1 h at 800 °C had the pure white appearance of silica and showed no evidence of crystalline silicon by Raman spectroscopy. The growth of the oxide shell resulted in minimal changes in the overall pore size and porosity, as determined by nitrogen adsorption/desorption measurements (Figure S2, Supporting Information) and was accompanied by a shift in the emission maximum of photoluminescence to the blue (Table 1). The photoluminescence of porous Si originates from quantum confinement in the crystalline silicon skeleton,30 and the SiO2 shell serves to passivate the Si surface, increasing the photoluminescence quantum yield of the material.29,31–33 Consistent with extensive prior work with porous Si,33–37 the blue shift in the photoluminescence spectrum is attributed to a reduction in size of the quantum-confined Si features. The values of average thickness (d) of the emissive crystalline Si features in the skeleton were estimated from photoluminescence and Raman measurements. The photoluminescence data were fit to the inverse power law of eq 1, taken from published theoretical calculations on Si quantum dots:38,39
| (1) |
where EPL and Eg (Eg of Si = 1.12 eV) are the energies (in eV) of the emission maximum and of the silicon bandgap, respectively. The values of d are depicted in Figure 1a and listed in Table 1. The reported average sizes of the particles are consistent with Raman measurements, which displayed a shift of the 520 cm−1 band associated with the lattice optical phonon mode of crystalline Si to lower energy by between 5 and 20 cm−1 (Figure S3, Supporting Information), suggesting average Si feature sizes of 5 nm or smaller.40,41 Characteristic of nanophase silicon, this band was broadened on the low energy side relative to the porous Si starting material, indicative of some loss of crystallinity.42 Conversion of Si to silicon oxide was confirmed by Fourier-transform infrared (FTIR) spectroscopy (Figure S3, Supporting Information). After thermal treatment, the vibrational stretching modes from surface-bound Si–H species at ∼2100 cm−1 were no longer observable, and an intense band associated with Si–O stretching modes was observed at ∼1100 cm−1 indicating that all three particle formulations displayed a similar oxide surface chemistry.
Figure 1.
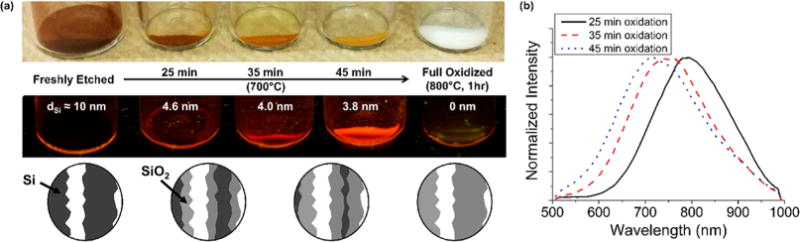
Thermal oxidation decreases the size of the Si skeletal core and increases the thickness of the SiO2 shell. (a) Top: Photographs of thermally treated porous silicon microparticles as a function of thermal oxidation time. The brownish color observed derives from the crystalline Si core, which is completely oxidized when the material is treated at a temperature of 800 °C for 1 h (“Full Oxidized”). Middle: The particles illuminated with UV (365 nm) light display differing degrees of luminescence intensity. Only the partially oxidized particles displayed detectable photoluminescence. Values of dSi indicate average size of the luminescent Si features, estimated from eq 1. Bottom: A schematic depicting the proposed nanostructural changes is shown below the relevant images; the crystalline Si core decreases in diameter and the SiO2 shell increases in thickness with increasing extent of oxidation. (b) Relative photoluminescence spectra of the pictured samples (λex = 365 nm, 500 nm long-pass filter). The photoluminescence spectra exhibit a blue shift as the Si core oxidizes, corresponding to a reduction in average size of the crystalline Si domains in the sample.
Table 1.
Characteristics of Protein-Loaded Porous Si–SiO2 Core–Shell Particles (Small Pores)
| sample
|
|||
|---|---|---|---|
| thin shell | medium shell | thick shell | |
| oxidation timea | 25 min | 35 min | 45 min |
| surface area (m2/g)b | 265 ± 30 | 267 ± 30 | 246 ± 30 |
| pore volume (cm3/g)c | 0.71 ± 0.08 | 0.68 ± 0.08 | 0.65 ± 0.08 |
| pore diameter (nm)b | 11.5 ± 1 | 11.9 ± 1 | 11.9 ± 1 |
| photoluminescence λmax (nm)d | 795 | 748 | 724 |
| Si feature size (nm)e | 4.6 | 4.0 | 3.8 |
| % BSA loaded by adsorptionf | 5.5 ± 0.7 | 5.2 ± 0.5 | 3.6 ± 0.3 |
| % BSA loaded by Mg silicate trappingg | 2.5 ± 0.2 | 2.5 ± 1 | 2.7 ± 0.8 |
Time that pSi samples (in microparticulate form) were oxidized in air at 700 °C to generate the SiO2 shell. Preparation parameters for the particles prior to oxidation were the same for all sample types.
Measured by nitrogen adsorption and determined using BET (Brunnauer–Emmett–Teller) analysis of the adsorption isotherms.
Measured by nitrogen adsorption and determined using BJH (Barret–Joyner–Halenda) analysis of the adsorption/desorption isotherms.
Measured with λex = 365 nm.
Average size of the luminescent Si features in the sample, estimated from PL spectrum using eq 1 (Eg of Si = 1.12 eV).
Mass percentage. BSA loaded from pH 4 acetate buffer by electrostatic adsorption. Each value represents mean ± standard deviation (n = 3).
Mass percentage. BSA loaded from pH 9 tris buffer in the presence of 1.3 M MgCl2(aq). Each value represents mean ± standard deviation (n = 3).
Next the luminescent core–shell nanomaterials were loaded with bovine serum albumin (BSA) as a surrogate for a biologic therapeutic. Two methods of loading the model drug payload within the pores were compared: the first was based on simple electrostatic adsorption, and the second was based on physical trapping of the protein (Figure 2). Adsorption loading was employed to represent a common release model, wherein the elution kinetics display an initial burst followed by a slower steady-state release. To promote strong electrostatic adsorption of BSA to the silicon oxide surface during loading, the solution pH was adjusted to 4, which is lower than the isoelectric point of BSA (pI = 4.8).43 These conditions have been shown to yield strong adsorption of BSA onto oxidized porous Si surfaces.44 In the second loading method, the loaded protein was trapped in the pores using localized deposition of magnesium silicate. This chemistry follows a previously reported approach involving the ability of added calcium ion to form a silicate precipitate that traps the drug payload within the pores.45 In the present case, we found that the alkaline earth metal magnesium behaved similarly to calcium in the ability of its dication to form an insoluble silicate salt. In this mechanism, the formation of magnesium silicate results from reaction of a relatively high concentration (1.3 M) of Mg2+ ion with silicic acid that is locally generated by dissolution of the SiO2 shell (at pH = 9) of the porous Si–SiO2 core–shell particles (Figure 2b).45–47 The presence of magnesium in the nanomaterials was confirmed in the scanning electron microscope images using energy dispersive spectroscopy EDS-SEM (not shown).
Figure 2.
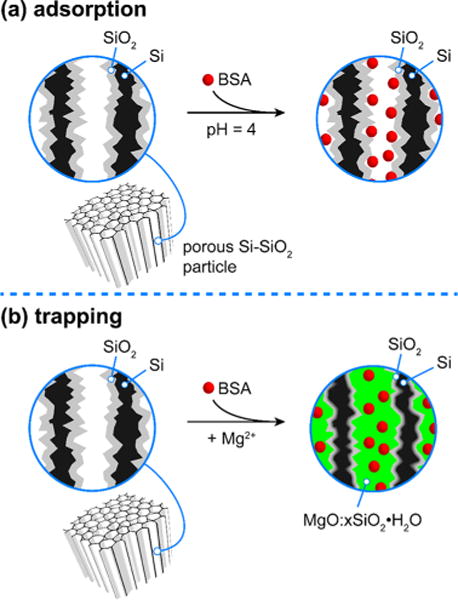
Schematics depicting the two approaches used to load the test protein bovine serum albumin (BSA). (a) Adsorption loading is performed at pH 4, which is lower than the isoelectric point of BSA. The cationic charge on the protein displays a strong electrostatic attraction to the negatively charged surface oxide of the particles. (b) Trapping of BSA is accomplished by loading the protein in the presence of high concentration of magnesium ion at pH 9. The orthosilicate product of SiO2 dissolution reacts with magnesium ions to form a stable silicate that immobilizes the protein within the pores.
We tested both methods of protein loading (electrostatic adsorption and magnesium silicate trapping) on two different types of pSi microparticles, one in which the average pore diameter was ∼10 nm (small pores) and another in which the average pore diameter was ∼20 nm (large pores). It was found that the “small pore” (Table 1) samples loaded substantially less BSA than the “large pore” samples (Table 2) with either loading method. For pSi particles of a given pore size, the mass loading of BSA showed no strong dependence on oxide shell thickness. Both loading methods preserved the photoluminescence of the original core–shell nanomaterial, with little change in the wavelength maximum or efficiency of photoluminescence.
Table 2.
Characteristics of Protein-Loaded Porous Si–SiO2 Core–Shell Particles (Large Pores)
| sample
|
|||
|---|---|---|---|
| thin shell | medium shell | thick shell | |
| oxidation timea | 15 min | 20 min | 30 min |
| surface area (m2/g)b | 239 ± 16 | 206 ± 30 | 237 ± 10 |
| pore volume (cm3/g)c | 1.04 ± 0.06 | 1.05 ± 0.04 | 1.05 ± 0.08 |
| pore diameter (nm)b | 21 ± 1 | 22 ± 0.2 | 21 ± 0.5 |
| photoluminescence λmax (nm)d | 823 | 783 | 747 |
| Si feature size (nm)e | 5.1 | 4.5 | 4.0 |
| % BSA loaded by adsorptionf | 19 ± 1.7 | 17 ± 1.1 | 16 ± 1.0 |
| % BSA loaded by Mg silicate trappingg | 29 ± 2.5 | 27 ± 3.6 | 33 ± 2.1 |
Time that pSi samples (in microparticulate form) were oxidized in air at 700 °C to generate the SiO2 shell. Preparation parameters for the particles prior to oxidation were the same for all sample types.
Measured by nitrogen adsorption and determined using BET (Brunnauer–Emmett–Teller) analysis of the adsorption isotherms.
Measured by nitrogen adsorption and determined using BJH (Barret–Joyner–Halenda) analysis of the adsorption/desorption isotherms.
Measured with λex = 365 nm.
Average size of the luminescent Si features in the sample, estimated from PL spectrum using eq 1 (Eg of Si = 1.12 eV).
Mass percentage. BSA loaded from pH 4 acetate buffer by electrostatic adsorption. Each value represents mean ± standard deviation (n = 3).
Mass percentage. BSA loaded from pH 9 tris buffer in the presence of 200 mM MgCl2(aq). Each value represents mean ± standard deviation (n = 3).
Next, the photoluminescent BSA-loaded formulations were tested under aqueous dissolution conditions. The mechanism of protein release can be split into two pathways, related to the method used to load the protein into the nanomaterial. A schematic depicting hypothetical release processes for the two classes of samples is given in Figure 3. For material that was loaded by the electrostatic adsorption process of Figure 2a, release of BSA can be considered to proceed primarily via desorption (Figure 3a). For material loaded by the trapping method, where the protein was trapped in the pSi matrix by precipitation of magnesium silicate (Figure 2b), we consider that BSA release is more closely tied to degradation of the carrier matrix (Figure 3b). To experimentally probe and discriminate these two processes, we performed accelerated dissolution experiments in 0.1 M aqueous KOH while monitoring the concentration of free protein released into solution, the concentration of dissolved silicon in solution, and the photoluminescence signal from the remaining particles as a function of time. Immediately after acquisition of each photoluminescence spectrum, the release solution was sampled and assayed for dissolved silicon/silicon dioxide (orthosilicate ion) and protein content using the molybdenum blue48,49 and bicinchoninic acid (BCA) assays, respectively.
Figure 3.
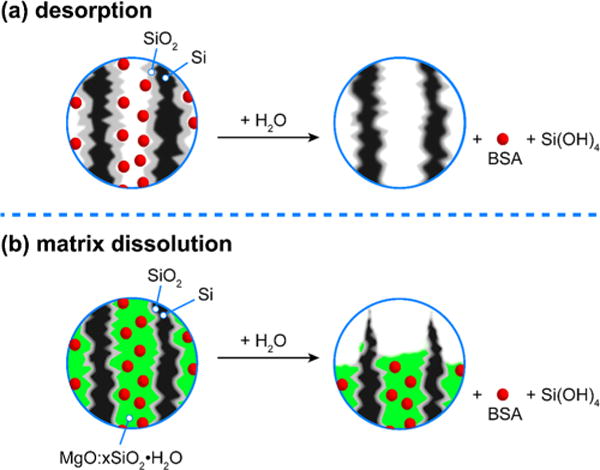
Schematics depicting the two proposed mechanisms for protein release from (a) adsorption-loaded porous Si–SiO2 particles and (b) matrix-trapped porous Si–SiO2 particles. Images depict cross sections of the mesopores with bovine serum albumin (BSA) protein depicted with red spheres. (a) With adsorption-loaded porous Si– SiO2, the protein is weakly bound to the matrix and it is released prior to degradation/dissolution of the photoluminescent Si core. A burst release is observed, and photoluminescence from the Si skeleton degrades on a slower time scale. (b) With matrix-trapped porous Si– SiO2, the protein is embedded in a magnesium silicate matrix. Because the protein is trapped in this insoluble magnesium silicate phase, the release of protein is more closely tied to degradation of the carrier. A more controlled release is observed, and photoluminescence from the Si skeleton degrades on a similar time scale.
The adsorption-loaded, small-pore particles (Figure 2a, 3a) all displayed an initial burst release of protein, and this was followed by a more steady and slow release with respect to time (Figure 4a) and to fraction of silicon dissolved (Figure 4b). This result is expected because at the high pH of the dissolution medium both the SiO2 surface and BSA are negatively charged, resulting in a strong electrostatic repulsion44,50 and a relatively rapid release of any surface-adsorbed protein. The rate of BSA release showed no statistically significant difference as a function of thickness of the SiO2 shell (Figure 4a), consistent with the expectation that the protein is predominantly surface-bound for the adsorption-loaded material.
Figure 4.
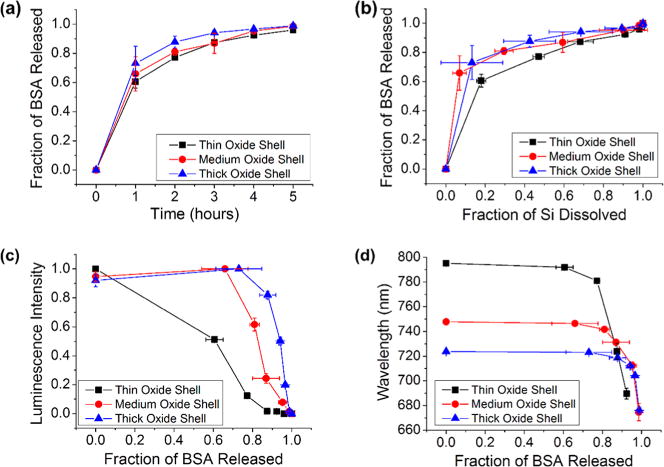
Correlations between protein released, silicon dissolved, and photoluminescence from the Si skeletal core for adsorption-loaded core–shell porous Si–SiO2 particles as they undergo dissolution in aqueous base (0.1 M KOH). Experiments for three core–shell porous Si–SiO2 structures are displayed in each plot: Traces designated “thin oxide shell”, “medium oxide shell”, and “thick oxide shell” correspond to pSi particles oxidized at 700 °C for 25, 35, and 45 min, respectively. These samples were all “small-pore” microparticles, prepared to contain ∼10 nm pores (Table 1). (a) Fraction of bovine serum albumin (BSA) released as a function of time. (b) Fraction of BSA released as a function of silicon dissolved. (c) Integrated photoluminescence intensity (in range 650–800 nm) from particles as a function of BSA released. (d) Wavelength of maximum photoemission from the Si cores as a function of fraction of BSA released.
In contrast to the observed time dependence of BSA release, luminescence intensity measured from the degrading particles showed a strong dependence on thickness of the SiO2 shell (Figure S4, Supporting Information). Photoluminescence from particle samples with the thinnest oxide shell decayed the most rapidly, and those with the thickest shell decayed the most slowly. The wavelength maximum of photoluminescence also tracked with extent of dissolution, with all samples displaying a pronounced blue shift with time (Figure S4e, Supporting Information). This observed blue shift is consistent with the quantum confinement model; as the silicon features become smaller, the emission energy of the quantum-confined semiconductor is expected to increase.30 The amount of silicon that dissolved per unit time, measured by molybdenum blue assay, was similar for the thin, medium, and thick shell materials (Figure S4c, Supporting Information). Whereas the molybdenum blue measurements represent the total amount of silicon that dissolved at a given time, including both the silicon oxide shell and the crystalline silicon cores, the photoluminescence measurements only report on the silicon cores. Thus the data are consistent with the interpretation that the thicker oxide shell takes more time to dissolve, and so it protects the luminescent silicon core for a longer period of time.
The samples with the thin oxide shell displayed photoluminescence signals that most closely matched the release of surface-adsorbed BSA (Figure 4c) whereas the loss of photoluminescence was delayed substantially relative to BSA release with samples containing the thicker oxide shells. Luminescence from samples with the thickest oxide shell showed no substantial change in intensity until ∼80% of BSA had been released into solution (Figure 4c). Because the protein was primarily surface-adsorbed and not entrained in the oxide matrix, this preparation released protein substantially in advance of the degradation of the pSi matrix. This relationship is most apparent in the correlation between dissolved silicon and BSA detected in solution (Figure 4b).
In contrast to the burst release seen with the adsorption-loaded particles, those prepared by physically trapping BSA in the pores generated a more gradual protein release profile. In the physical trapping method, BSA was simultaneously loaded into the pores and trapped by formation of a magnesium silicate precipitate (Figure 2b),45 and so the release of BSA is expected to be more closely tied to the dissolution of this matrix. Accordingly, this preparation displayed near zero-order release kinetics with respect to time (Figure 5a), with minimal burst release compared with adsorption-loaded particles (Figure 4a). The release of BSA also showed a more linear correlation to the appearance of dissolved silicon (Figure 5b) compared with adsorption-loaded particles (Figure 4b). The close correspondence of BSA release with appearance of silicon in solution is consistent with the matrix dissolution process depicted in Figure 3b.
Figure 5.
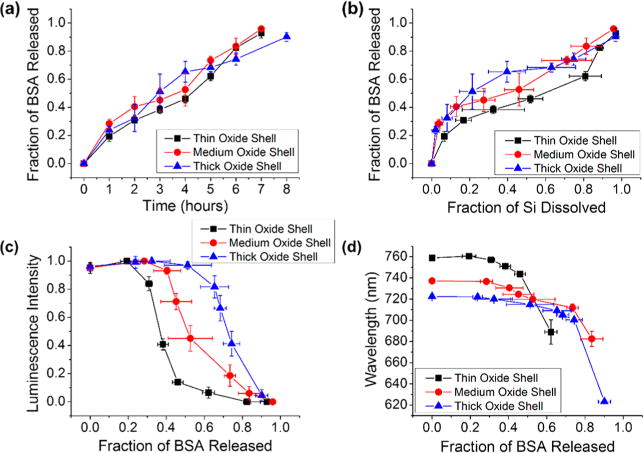
Correlations between protein released, silicon dissolved, and photoluminescence from the Si skeletal core for core–shell porous Si–SiO2 particles loaded with BSA via magnesium silicate-trapping chemistry as they undergo dissolution in aqueous base (0.1 M KOH) at room temperature. BSA protein was trapped in the porous Si–SiO2 particles by means of precipitation of magnesium silicate concomitant with protein loading as described in Figure 2. Traces designated “thin oxide shell”, “medium oxide shell”, and “thick oxide shell” correspond to pSi particles where the skeletal core was oxidized at 700 °C for 25, 35, and 45 min, respectively, prior to protein/magnesium silicate loading. These samples were all “small-pore” microparticles, prepared to contain ∼10 nm pores (Table 1). (a) Fraction of bovine serum albumin (BSA) released from the particles as a function of time. (b) Fraction of BSA released as a function of silicon dissolved. (c) Integrated photoluminescence intensity (in range 650–800 nm) from particles as a function of fraction of BSA released. (d) Wavelength of maximum photoemission from the Si cores as a function of fraction of BSA released. Error bars represent standard deviation (n = 3).
As seen with the adsorption-loaded material, photoluminescence intensity from the magnesium silicate-trapped material degraded as a function of time exposed to the dissolution medium (Figure S5, Supporting Information). However, degradation was delayed by the presence of the silicate coating. This was particularly apparent in the data from the pSi samples that contained the thinnest oxide shell; whereas photoluminescence intensity began to decay immediately upon immersion in the aqueous dissolution medium for the adsorption-loaded material (Figure S4a, Supporting Information), loss of photoluminescence intensity from the magnesium silicate-coated material was not apparent until after 2 h of immersion (Figure S5a, Supporting Information). As with the adsorption-loaded particles, photoluminescence was more persistent with increasing thickness of the SiO2 shell. The half-life for loss of photoluminescence intensity, defined as the time at which photoluminescence intensity reached half of its initial value during exposure to the aqueous dissolution solution, also scaled with thickness of the SiO2 shell for the magnesium silicate-trapped pSi particles: it was ∼3, ∼4, and ∼6 h for the thin, medium, and thick SiO2 shell, respectively (Figure S5a, Supporting Information).
The functional form of the protein release vs photoluminescence intensity relationship differed depending on the particular protein loading method employed and on the thickness of the SiO2 shell surrounding the Si skeleton of the pSi particles. Although decreases in photoluminescence intensity were related to protein release in all the formulations studied, the trapping-loaded core–shell particles yielded a relationship that could potentially be useful as a predictive tool in a drug-delivery system. Degradation of the photoluminescence signal was a direct result of dissolution of the SiO2-shell portion of the material, because photoluminescence originates from the quantum-confined silicon skeleton. The magnesium silicate matrix served to delay this process. In addition, the magnesium silicate coating suppressed the burst release of protein, substantially extending its release under the accelerated degradation conditions employed in this portion of the study. The correlation between photoluminescence intensity and BSA detected in solution showed that loss of photoluminescence was delayed relative to release of protein (Figure 5c). With the magnesium silicate-trapped particles, the relationship tended to follow a sigmoidal curve with respect to BSA released, where a pronounced drop in photoluminescence intensity was observed after a particular fraction of the BSA payload had been released. The inflection point of the curve scaled with the thickness of the SiO2 shell on the Si skeleton. Thus the material that had been prepared with the thin SiO2 shell had released 40% of its protein payload when the intensity of photoluminescence reached half of its initial value; material with the medium thickness SiO2 shell had released 50% of the protein at the half intensity point; and material with the thick SiO2 shell had released 70% of the protein at the half point (Figure 5c). The photoluminescence intensity measurement could therefore be used as a surrogate indication of the fraction of protein remaining in the nanomaterial. The emission wavelength also varied systematically with protein release, blue shifting with time (Figure S5b, Supporting Information) and with fraction of protein released (Figure 5d).
For the magnesium silicate trapping chemistry, the kinetics of photoluminescence loss and of protein release are almost certainly dictated by separate mechanisms. Whereas the loss of photoluminescence is related to dissolution of the electronically passivating SiO2 shell, the release of protein is probably more closely tied to the dissolution of the magnesium silicate phase that seals the protein in the pores. This can be inferred from the very rapid release of protein observed in the absence of the magnesium silicate chemistry (with the adsorption-loaded material). However, the SiO2 shell appears to influence the nature of the magnesium silicate sealant and its ability to load protein. It is noteworthy that, for the small-pore samples, protein loading using the magnesium silicate chemistry was approximately half that of the adsorption-loading process. This suggests that the dissolution of SiO2 needed to form the magnesium silicate sealant released surface-bound protein, reducing the total amount of protein that was trapped in the pores. In addition, the presence of the magnesium silicate sealant clearly influenced the rate of degradation of the photoluminescence signal. This is to be expected; the SiO2 shell should dissolve more slowly if it lies underneath an insoluble magnesium silicate coating. Although the photoluminescence decay kinetics depended on the thickness of the SiO2 shell, the kinetics were also influenced by the presence of the magnesium silicate sealant. We conclude that the rate of photoluminescence intensity loss from the silicon skeleton comprising the particles relies on multiple factors, although controlling the thickness of the SiO2 shell appears to be a convenient (if somewhat phenomenological) means to control the correlation between photoluminescence and protein release.
While the study described up to this point established some design principles that may be useful for self-reporting drug-delivery systems, it has several drawbacks that limit its translational relevance: (1) the loading of protein using either of the methods was quite low (2–5%) for most therapeutic applications; (2) the use of very high concentrations (>1 M) of Mg2+ in the magnesium silicate trapping method is likely to denature some proteins, reducing their therapeutic efficacy; and (3) the release kinetics measurements were performed under accelerated conditions using a very high pH that is physiologically unrealistic. In order to address these issues, we performed a set of studies using porous Si particles that had been engineered to load more drug under milder conditions, and we performed a longer-term (30 day) release study under more pysiologically relevant in vitro conditions. In order to increase the mass loading of protein in the microparticles, the electrochemical conditions for particle preparation were modified1 to approximately double the average pore diameter (Table 2, Figure S1 and S2 Supporting Information). These particles are referred to as the “large-pore” samples. The large-pore particles displayed significantly greater mass loading of BSA for both the adsorption and the Mg silicate trapping methods (∼17 and ∼30 wt %, respectively, Table 2), compared to the small-pore particles (∼5 and ∼3 wt %, respectively, Table 1). For the trapping chemistry, the Mg2+ ion concentration was lowered to 200 mM.
The long-term protein release study was carried out in phosphate-buffered saline (PBS) solution at pH 7.4 and a constant temperature of 37 °C. The data for magnesium silicate-trapped materials are presented in Figure 6 and the data for adsorption-loaded material are given in Figure S6, Supporting Information. Similar to the accelerated release study performed in alkaline solution, photoluminescence decayed more slowly from samples with the thicker oxide shells (Figure 6a and Figure S6a, Supporting Information). The magnesium silicate-trapped material demonstrated minimal burst release of BSA, and release of protein tracked fairly linearly with dissolution of silicon for all oxide shell thicknesses (Figure 6e). By contrast, the adsorption-loaded formulation displayed a pronounced burst that released 20% of the protein within the first 5 days of the 30-day experiment (Figure S6e, Supporting Information).
Figure 6.
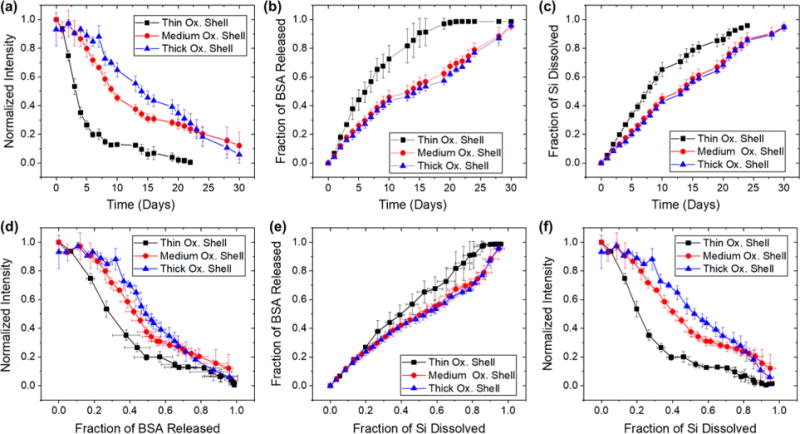
Correlations between photoluminescence from the Si skeletal core, protein released, silicon dissolved, and time for trapping-loaded core– shell porous Si–SiO2 particles as they undergo dissolution in aqueous PBS (pH = 7.4) at 37 °C. The porous Si–SiO2 particles were prepared with a larger average pore size (∼20 nm), in contrast to the samples of Figure 5 (∼10 nm pores), which resulted in a substantially greater level of protein loaded (see Table 2). Protein trapping was achieved by means of precipitation of magnesium silicate as described in Figure 2, using 200 mM Mg2+. Traces designated “Thin Ox. Shell”, “Medium Ox. Shell”, and “Thick Ox. Shell” correspond to pSi particles where the skeletal core was oxidized at 700 °C for 15, 20, and 30 min, respectively, prior to protein/magnesium silicate loading. (a) Integrated photoluminescence intensity (in wavelength range 600–800 nm) from particles as a function of time. (b) Fraction of bovine serum albumin (BSA) released from the particles as a function of time. (c) Fraction of silicon dissolved as a function of time. (d) Integrated photoluminescence intensity from particles as a function of fraction of BSA released. (e) Fraction of BSA released as a function of silicon dissolved. (f) Integrated photoluminescence intensity as a function of fraction of silicon dissolved. Error bars represent standard deviation (n = 3).
The long-term release studies showed that the magnesium silicate-trapped samples with a thin oxide shell released protein more rapidly (Figure 6b) and dissolved more rapidly (Figure 6c) than either of the medium- or thick-oxide shell materials. This difference was not apparent in the accelerated dissolution experiments that were performed in strong KOH solution (Figure 5a and Figure S5c, Supporting Information). Thus particles with a thin oxide shell appear to form the least stable construct with the magnesium silicate-trapping chemistry. By contrast, adsorption-loaded samples dissolved at the same rate under these conditions, regardless of the thickness of their oxide shell (Figure S6c, Supporting Information). Overall, the long-term release study performed under more physiologically relevant conditions showed more consistent tracking of photoluminescence intensity with BSA release for both loading types and across all oxide thicknesses, compared with the accelerated release study.
To be used as a self-reporting drug-delivery system, the luminescence signature would need to be monitored in vivo. Fluorescence methods in general do not lend themselves to in vivo monitoring due to the low optical transmittance and high optical scattering of live tissues. However, materials administered via subcutaneous, intramuscular, or intraocular injection, or via trans-dermal patch could conceivably be monitored by optical fluorescence. To assess the potential for such remote monitoring, the degradation/dissolution of empty pSi particles was monitored in vitro using a low-cost NIR imaging camera and a UV diode excitation source. Particles were subjected to accelerated dissolution conditions, and luminesecence intensity was quantified as integrated intensity as a function of time (Figure 7). The decay in luminescence intensity with time followed a similar trend as was observed with either adsorption-loaded (Figure S4a, Supporting Information) or magnesium silicate-sealed (Figure S5a, Supporting Information) particles.
Figure 7.
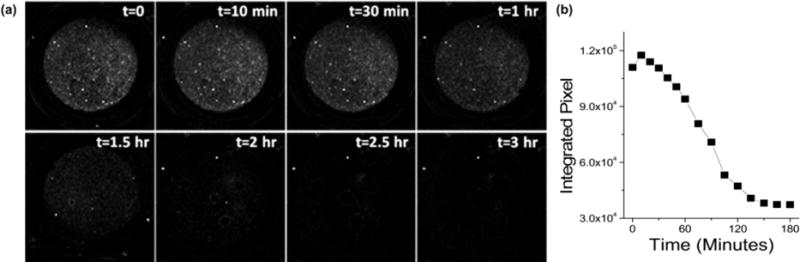
(a) Photoluminescence images of small-pore pSi particles (∼2 mg) dispersed in a microtiter well containing 2 M KOH (200 μL), displayed as a time series. Excitation source was a 365 nm light-emitting diode and images were obtained using a NIR camera fitted with a 600 nm long-pass filter. The porous Si particles were prepared by 35 min oxidation at 700 °C, corresponding to the “medium oxide shell” material of Table 1. (b) Plot of photoluminescence intensity, integrated as a sum of pixel intensities (ImageJ), as a function of time.
In conclusion, the present work demonstrated the use of intrinsic photoluminescence of porous silicon microparticles as a self-reporting feature for the degradation of the porous scaffold and the release of a protein payload into aqueous solution. We found that the time at which photoluminescence disappears can be predetermined by controlling the thickness of the protective SiO2 sheath surrounding the quantum-confined Si features. We compared these temporal profiles for particles that were loaded with protein via two different loading schemes—either by simple adsorption or by physical trapping. For protein that was trapped in the porous particles (via a magnesium silicate precipitation reaction), we found that the temporal profile of the decay in photoluminescence can be used to indicate the time at which the amount of protein remaining in the particles has reached a predetermined level. The magnesium silicate chemistry also suppresses burst release of protein, and it yields a zero-order release kinetic that can be tracked by the photoluminescence signal. We expect that the results of this work will be useful for remote monitoring of payload release for a variety of drug delivery or other controlled release applications.
EXPERIMENTAL SECTION
Materials and Chemicals
Silicon wafers, polished on the (100) face, boron-doped (p-type), resistivity <1.5 mΩ·cm were obtained from Virginia Semiconductor, Inc. or from Siltronix. Absolute ethanol (200 proof) was obtained from Rossville Gold Shield Chemicals. Potassium hydroxide (ACS grade) and sodium sulfite (ACS grade) were obtained from Fisher Scientific. Ammonium molybdate tetrahydrate (crystal, ACS grade) was obtained from Spectrum Chemicals. Magnesium chloride (6-hydrate, crystalline, ACS grade), concentrated sulfuric acid (ACS grade), and hydrofluoric acid (48% aqueous, ACS grade) were obtained from Macron Chemicals. Micro BCA protein assay kit (No. 23235) was obtained from Thermo-Fisher Scientific. Bovine serum albumin from porcine gastric mucosa (lyophilized powder, 32004500 units/mg protein), 4-(methylamino)-phenol hemisulfate salt (ACS grade, 99%), and oxalic acid dehydrate (ACS grade, >99%) were obtained from Sigma-Aldrich Chemicals.
Fabrication of Luminescent Porous Silicon (pSi) Micro-particles
Porous Si microparticles were prepared by electrochemical etch of highly boron-doped single-crystalline Si wafers as previously described.51 A wafer was mounted in a sealed etching cell that exposed a circular area of 60 cm2. The cell was fitted with a copper slab that served as a back contact for the Si wafer. For the small (∼10 nm) pore formulation, a 3:1 (v:v) 48% aqueous HF:absolute ethanol electrolyte was used and the wafer was etched at a constant current density of 70 mA/cm2 for 600 s. For the large (∼20 nm) pore formulation, a 1:1 (v:v) 48% aqueous HF:absolute ethanol electrolyte was used and the wafer was etched at a constant current density of 30 mA/cm2 for 720 s. These conditions generated a porous Si layer of thickness approximately 20 μm and porosity approximately 60% or 70% for the small-pore or large-pore formulations, respectively. The electrolyte was replaced with an electrolyte consisting of 1:20 (v:v) 48% aqueous HF:absolute ethanol, and the porous Si film was removed from the wafer by an electropolishing step consisting of a current density of 4.17 mA/cm2 applied for 550s. The freestanding porous Si layer was rinsed with absolute ethanol three times, removed by gentle agitation, and the fragments were placed in a sealed vial containing absolute ethanol (20 mL). The vial was suspended in an ultrasonic bath (1.9 L Ultrasonic Cleaner, No. 97043, VWR, inc.) and the porous layer was subjected to ultrasonic fragmentation for 20 min, resulting in a suspension of nano and microparticles. The nanoparticles were removed from the suspension by removing the supernatant after the microparticles were allowed to settle for 20 min in the vial. The microparticles were dried under vacuum for 1 h and then placed in a ceramic boat and inserted into the hot zone of a preheated tube furnace at 700 °C for 25, 35, or 45 min (for small-pore samples) or 15, 20, or 30 min (for large-pore samples) to activate photoluminescence. After heating for the desired times, the boat containing the particles was moved to the edge of the tube furnace to cool for 2 min, and it was then removed to a ceramic surface and allowed to cool to room temperature.
Characterization of pSi Particles
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were collected using a Thermo Scientific Nicolet 6700 FTIR instrument with a Smart iTR diamond ATR fixture. Raman spectra were collected using a Renishaw inVia Raman microscope with a 100 mW 532 nm laser excitation source. Nitrogen adsorption/desorption isotherms were recorded with a Micromeritics ASAP2020.
Adsorption Loading of Bovine Serum Albumin (BSA)
Porous silicon particles (5 mg) were placed in 5 mL Falcon tubes (Corning, inc.) and soaked in a 1.5 mL solution of BSA (8 mg/mL) in acetate buffer (pH 4) for 16 h while agitating on a spinning wheel. The particles were washed 5 times with 1 mL of acetate buffer and dried under vacuum for 1 h.
Loading by Silicate Trapping
Bovine serum albumin was loaded into the pSi particles from aqueous solutions containing a magnesium salt. The loading solution was adjusted to pH 9 with potassium hydroxide (KOH) and consisted of 1.0 mL of 12 mg/mL BSA in Tris buffer and 0.5 mL of 4 M (for the small-pore samples) or 600 mM (for the large-pore samples) MgCl2 in deionized water. The loading solution (1.5 mL) was added to 5 mg of particles in a Falcon tube (5 mL). The mixture was agitated on a spinning wheel for 16 h. After the particles were loaded, they were washed 5 times with 1 mL of Tris buffer and dried under vacuum for 1 h.
BSA Release and Luminescence Decay Studies
Loaded pSi particles (1–2 mg) were immersed in 0.1 M KOH (1.5 mL) in plastic cuvettes. Luminescence spectra (λex: 365 nm, 375 ± 5 nm band-pass filter) were collected hourly with an Ocean Optics QE-Pro TE cooled CCD spectrometer using a 515 nm long pass filter. The cuvettes were stirred constantly (via magnetic stir bar) during spectral acquisition to disperse the particles. The stirring was stopped after spectral acquisition, the particles were allowed to settle for 55 min, the supernatant release medium (1 mL) was collected for Si and protein assays, and the cuvette was then replenished with fresh 0.1 M aqueous KOH solution. The study was carried out until the photoluminescence spectrum was no longer detectable. After the last time point for data collection, the sample was left overnight to ensure complete dissolution of silicon prior to final assay. Concentration of protein was quantified using the commercial micro BCA assay kit following the protocol described by the vendor. Dissolved Si was quantified using molybdenum blue assay (see below). The total mass loading of BSA and mass of Si were quantified by summing the amounts measured at each time point during the release assay and the final measurement, after all pSi particles had been dissolved. The long-term (30 day) release studies were performed using the same protocol but using aqueous PBS at pH 7.4 as the dissolution medium, maintaining the temperature at 37 °C, and sampling/replacing 1.2 mL every 1–3 days.
Molybdenum Blue Assay
Dissolved silicon content was determined using the published molybdenum blue assay.49 Two solutions (A and B) were prepared for this assay. Solution A consisted of 16.2 mM ammonium molybdate and 0.72 M HCl. Solution B consisted of 0.16 M oxalic acid, 7.7 mM of 4-(methylamino)phenol hemisulfate, 6.3 mM of sodium sulfite, and 1.8 M of sulfuric acid. To quantify the silicic acid concentration, 100 μL of Solution A was added to 400 μL of each sample (prediluted by a factor of 100–1000) and allowed to react for 15 min. Then, 500 μL of Solution B was added to each sample and the mixture was allowed to react for 1 h. The optical absorbance at 810 nm of each sample was quantified with a UV–vis plate reader (Spectra Max Plus 384, Molecular Devices). A standard curve was prepared from a serially diluted standard silicon solution (Sigma-Aldrich Si Standard for ICP, TraceCERT, 1000 mg/mL), and the working range was 0.5–20 ppm in water and 1–10 ppm in PBS at pH 7.4 (diluted from stock 10 times).
In Vitro Imaging Studies
Luminescent porous silicon particles (35 min oxidation) were treated in 2 M KOH (2 mg pSi/200 μL KOH) in a 96-well plate (UV-transparent). A higher concentration of base was used in these accelerated aging experiments relative to the other experiments described in this paper in order to reduce the time duration of the experiments. Luminescence images were collected as a function of time using a Point Gray IR camera fitted with a 600 nm long pass filter and a 240 mm lens (λex: 365 nm). The automatic exposure settings on the camera were disabled to maintain constant image acquisition parameters during the experiment. The images were processed with ImageJ software (NIH) to quantify the luminescence intensity. Intensity of pixels was integrated within a selected square frame. The frame included the image of the entire well.
Supplementary Material
Acknowledgments
Funding
This material is based upon work supported by the National Science Foundation under Grant No. CBET-1603177 and by the Food and Drug Administration through grant 1U01FD005173-01, views expressed do not necessarily reflect the official policies of the Department of Health and Human Services; nor does any mention of trade names, commercial practices, or organization imply endorsement by the United States Government.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.7b09071.
Representative bright-field optical microscope images and plan-view SEM; characterization of porous silicon particles by nitrogen adsorption/desorption isotherms; ATR-FTIR spectra and Raman spectra; correlations of photoluminescence intensity, photoluminescence wave-length, and fraction of Si dissolved with time exposed to the accelerated dissolution conditions; and Correlations between photoluminescence from the Si skeletal core, protein released, silicon dissolved, and time for adsorption-loaded core-shell porous Si–SiO2 particles (PDF)
ORCID
Michael J. Sailor: 0000-0002-4809-9826
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare the following competing financial interest(s): MJS is a scientific founder of Spinnaker Biosciences, Inc., and has an equity interest in the company. Although this grant has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Spinnaker Biosciences, Inc., the research findings included in this particular publication may not necessarily relate to the interests of Spinnaker Biosciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
References
- 1.Kaasalainen M, Rytkönen J, Mäkilä E, Närvänen A, Salonen J. Electrostatic Interaction on Loading of Therapeutic Peptide GLP-1 into Porous Silicon Nanoparticles. Langmuir. 2015;31(5):1722–1729. doi: 10.1021/la5047047. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream. Science. 2004;303(5665):1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: Therapeutic applications developments. Clin Pharmacol Ther. 2008;83(5):761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 4.Canham LT. Bioactive Silicon Structure Fabrication Through Nanoetching Techniques. Adv Mater. 1995;7(12):1033–1037. [Google Scholar]
- 5.Santos HA. Porous Silicon for Biomedical Applications. Woodhead Publishing, Ltd; Cambridge, U.K.: 2014. [Google Scholar]
- 6.Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drug delivery applications. J Pharm Sci. 2008;97(2):632–653. doi: 10.1002/jps.20999. [DOI] [PubMed] [Google Scholar]
- 7.Anglin EJ, Cheng L, Freeman WR, Sailor MJ. Porous silicon in drug delivery devices materials. Adv Drug Delivery Rev. 2008;60(11):1266–1277. doi: 10.1016/j.addr.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Losic D, Simovic S. Self-ordered nanopore nanotube and platforms for drug delivery applications. Expert Opin Drug Delivery. 2009;6(12):1363–1381. doi: 10.1517/17425240903300857. [DOI] [PubMed] [Google Scholar]
- 9.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. Multifunctional inorganic nanoparticles for imaging targeting and drug delivery. ACS Nano. 2008;2(5):889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slowing II, Vivero-Escoto JL, Wu CW, Lin VSY. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Delivery Rev. 2008;60(11):1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L, Anglin E, Cunin F, Kim D, Sailor MJ, Falkenstein I, Tammewar A, Freeman WR. Intravitreal properties of porous silicon photonic crystals: a potential self-reporting intraocular drug delivery vehicle. Br J Ophthalmol. 2008;92:705–711. doi: 10.1136/bjo.2007.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zilony N, Tzur-Balter A, Segal E, Shefi O. Bombarding Cancer: Biolistic Delivery of therapeutics using Porous Si Carriers. Sci Rep. 2013;3 doi: 10.1038/srep02499. Article No. 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canham LT, Barrett CP, Cox TI, Wright PJ, Bowditch AP. Implants for administering substances and methods of producing implants. 7,763,277. US Patent No. 2010 Jul 27;
- 14.Tzur-Balter A, Shatsberg Z, Beckerman M, Segal E, Artzi N. Mechanism of erosion of nanostructured porous silicon drug carriers in neoplastic tissues. Nat Commun. 2015;6:6208. doi: 10.1038/ncomms7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barenholz Y. Doxil (R) - The first FDA-approved nano-drug: Lessons learned. J Controlled Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 16.Hou H, Nieto A, Belghith A, Nan K, Li Y, Freeman WR, Sailor MJ, Cheng L. A sustained intravitreal drug delivery system with remote real time monitoring capability. Acta Biomater. 2015;24:309–321. doi: 10.1016/j.actbio.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy JR, Weissleder R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv Drug Delivery Rev. 2008;60(11):1241–1251. doi: 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Delivery Rev. 2010;62(11):1052–1063. doi: 10.1016/j.addr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weissleder R, Schwaiger MC, Gambhir SS, Hricak H. Imaging approaches to optimize molecular therapies. Sci Transl Med. 2016;8:355ps16. doi: 10.1126/scitranslmed.aaf3936. [DOI] [PubMed] [Google Scholar]
- 20.Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic Nanomedicine. Acc Chem Res. 2011;44(10):1029–1038. doi: 10.1021/ar200019c. [DOI] [PubMed] [Google Scholar]
- 21.Nagesha DK, Whitehead MA, Coffer JL. Biorelevant calcification and non-cytotoxic behavior in silicon nanowires. Adv Mater. 2005;17(7):921. [Google Scholar]
- 22.Low SP, Williams KA, Canham LT, Voelcker NH. Evaluation of mammalian cell adhesion on surface-modified porous silicon. Biomaterials. 2006;27(26):4538–4546. doi: 10.1016/j.biomaterials.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Coffer JL, Whitehead MA, Nagesha DK, Mukherjee P, Akkaraju G, Totolici M, Saffie RS, Canham LT. Porous silicon-based scaffolds for tissue engineering and other biomedical applications. Phys Status Solidi A. 2005;202(8):1451–1455. [Google Scholar]
- 24.Park J-H, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradable luminescent porous silicon nano-particles for in vivo applications. Nat Mater. 2009;8:331–336. doi: 10.1038/nmat2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu EC, Andrew JS, Cheng L, Freeman WR, Pearson L, Sailor MJ. Real-time Monitoring of Sustained Drug Release using the Optical Properties of Porous Silicon Photonic Crystal Particles. Biomaterials. 2011;32:1957–1966. doi: 10.1016/j.biomaterials.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie YH, Wilson WL, Ross FM, Mucha JA, Fitzgerald EA, Macaulay JM, Harris TD. Luminescence and structural study of porous silicon films. J Appl Phys. 1992;71(5):2403–2407. [Google Scholar]
- 27.Joo J, Liu X, Kotamraju VR, Ruoslahti E, Nam Y, Sailor MJ. Gated Luminescence Imaging of Silicon Nanoparticles. ACS Nano. 2015;9(6):6233–6241. doi: 10.1021/acsnano.5b01594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal E, Perelman LA, Cunin F, Di Renzo F, Devoisselle J-M, Li YY, Sailor MJ. Confinement of Thermoresponsive Hydrogels in Nanostructured Porous Silicon Dioxide Templates. Adv Funct Mater. 2007;17:1153–1162. [Google Scholar]
- 29.Petrova-Koch V, Muschik T, Kux A, Meyer BK, Koch F, Lehmann V. Rapid thermal oxidized porous Si- the superior photoluminescent Si. Appl Phys Lett. 1992;61(8):943–945. [Google Scholar]
- 30.Cullis AG, Canham LT, Calcott PDJ. The structural and luminescence properties of porous silicon. J Appl Phys. 1997;82(3):909–965. [Google Scholar]
- 31.Gelloz B, Koshida N. Highly enhanced photoluminescence of as-anodized and electrochemically oxidized nanocrystalline p-type porous silicon treated by high-pressure water vapor annealing. Thin Solid Films. 2006;508(1–2):406–409. [Google Scholar]
- 32.Ray M, Sarkar S, Bandyopadhyay NR, Hossain SM, Pramanick AK. Silicon and silicon oxide core-shell nanoparticles: Structural and photoluminescence characteristics. J Appl Phys. 2009;105(7):074301. [Google Scholar]
- 33.Joo J, Cruz JF, Vijayakumar S, Grondek J, Sailor MJ. Photoluminescent Porous Si/SiO2 Core/Shell Nanoparticles Prepared by Borate Oxidation. Adv Funct Mater. 2014;24:5688–5694. [Google Scholar]
- 34.Wolkin MV, Jorne J, Fauchet PM, Allan G, Delerue C. Electronic states and luminescence in porous silicon quantum dots: The role of oxygen. Phys Rev Lett. 1999;82(1):197–200. [Google Scholar]
- 35.Saar A. Photoluminescence from silicon nanostructures: The mutual role of quantum confinement and surface chemistry. J Nanophotonics. 2009;3:032501. [Google Scholar]
- 36.Wilson WL, Szajowski PF, Brus LE. Quantum Confinement in Size selected, Surface-Oxidized Si Nanocrystals. Science. 1993;262:1242–1244. doi: 10.1126/science.262.5137.1242. [DOI] [PubMed] [Google Scholar]
- 37.Zacharias M, Heitmann J, Scholz R, Kahler U, Schmidt M, Blasing J. Size-controlled highly luminescent silicon nanocrystals: A SiO/SiO2 superlattice approach. Appl Phys Lett. 2002;80(4):661–663. [Google Scholar]
- 38.Ledoux G, Guillois O, Porterat D, Reynaud C, Huisken F, Kohn B, Paillard V. Photoluminescence properties of silicon nanocrystals as a function of their size. Phys Rev B: Condens Matter Mater Phys. 2000;62(23):15942–15951. [Google Scholar]
- 39.Delerue C, Allan G, Lannoo M. Theoretical aspects of the luminescence of porous silicon. Phys Rev B: Condens Matter Mater Phys. 1993;48(15):11024–11036. doi: 10.1103/physrevb.48.11024. [DOI] [PubMed] [Google Scholar]
- 40.Campbell IH, Fauchet PM. The effects of microcrystal size and shape on the one phonon Raman spectra of crystalline semiconductors. Solid State Commun. 1986;58(10):739–741. [Google Scholar]
- 41.Kanemitsu Y, Uto H, Masumoto Y, Matsumoto T, Futagi T, Mimura H. Microstructure and optical properties of free-standing porous silicon films: Size dependence of absorption spectra in Si nanometer-sized crystallites. Phys Rev B: Condens Matter Mater Phys. 1993;48(4):2827–2830. doi: 10.1103/physrevb.48.2827. [DOI] [PubMed] [Google Scholar]
- 42.Li BB, Yu DP, Zhang SL. Raman spectral study of silicon nanowires. Phys Rev B: Condens Matter Mater Phys. 1999;59(3):1645–1648. [Google Scholar]
- 43.Su TJ, Lu JR, Thomas RK, Cui ZF. Effect of pH on the adsorption of bovine serum albumin at the silica/water interface studied by neutron reflection. J Phys Chem B. 1999;103:3727–3736. [Google Scholar]
- 44.Perelman LA, Pacholski C, Li YY, VanNieuwenhze MS, Sailor MJ. pH-Triggered Release of Vancomycin from Protein-Capped Porous Silicon Films. Nanomedicine. 2008;3(1):31–43. doi: 10.2217/17435889.3.1.31. [DOI] [PubMed] [Google Scholar]
- 45.Kang J, Joo J, Kwon EJ, Skalak M, Hussain S, She Z-G, Ruoslahti E, Bhatia SN, Sailor MJ. Self-Sealing Porous Silicon-Calcium Silicate Core–Shell Nanoparticles for Targeted siRNA Delivery to the Injured Brain. Adv Mater. 2016;28:7962–7969. doi: 10.1002/adma.201600634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson SHC, Elliott H, Wallis DJ, Canham LT, Powell JJ. Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Phys Status Solidi A-Appl Res. 2003;197(2):331–335. [Google Scholar]
- 47.Wang Y, Wang G, Wang H, Liang C, Cai W, Zhang L. Chemical-Template Synthesis of Micro/Nanoscale Magnesium Silicate Hollow Spheres for Waste-Water Treatment. Chem - Eur J. 2010;16(11):3497–3503. doi: 10.1002/chem.200902799. [DOI] [PubMed] [Google Scholar]
- 48.Mullin JB, Riley JP. The colorimetric determination of silicate with reference to sea and natural waters. Anal Chim Acta. 1955;12:162–176. [Google Scholar]
- 49.Low SP, Voelcker NH, Canham LT, Williams KA. The biocompatibility of porous silicon in tissues of the eye. Biomaterials. 2009;30(15):2873–2880. doi: 10.1016/j.biomaterials.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Chen MY, Sailor MJ. Charge-Gated Transport of Proteins in Nanostructured Optical Films of Mesoporous Silica. Anal Chem. 2011;83:7186–7193. doi: 10.1021/ac201636n. [DOI] [PubMed] [Google Scholar]
- 51.Sailor MJ. Porous Silicon in Practice: Preparation, Characterization, and Applications. Wiley-VCH; Weinheim, Germany: 2012. p. 249. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.