

Abstract
Objective
Duchenne muscular dystrophy (DMD) is caused by the inability to produce dystrophin protein at the myofiber membrane. A method to rescue dystrophin production by antisense oligonucleotides, termed `exon-skipping', has been reported for the mdx mouse and in four DMD patients by local intramuscular injection. We sought to test efficacy and toxicity of intravenous oligonucleotide (morpholino) induced exon skipping in the DMD dog model.
Methods
We tested a series of antisense drugs singly and as cocktails, both in primary cell culture, and two in vivo delivery methods (intramuscular injection, and systemic intravenous injection). The efficiency and efficacy of multi-exon skipping (exons 6-9) was tested at the mRNA, protein, histological, and clinical levels.
Results
Weekly or biweekly systemic intravenous injections with a three morpholino cocktail over the course of 5–22 weeks induced therapeutic levels of dystrophin expression throughout the body, with an average of about 26% normal levels. This was accompanied by reduced inflammatory signals examined by MRI and histology, improved or stabilized timed running tests and clinical symptoms. Blood tests indicated no evidence of toxicity.
Interpretation
This is the first report of widespread rescue of dystrophin expression to therapeutic levels in the dog model of DMD. This study also provides a proof of concept for systemic multi-exon skipping therapy. Use of cocktails of morpholinos, as shown here, allows broader application of this approach to a greater proportion of DMD patients (90%), and also offers the prospect of selecting deletions that optimize the functionality of the dystrophin protein.
Introduction
Duchenne Muscular Dystrophy (DMD) and its milder form, Becker Muscular Dystrophy (BMD), are caused by mutations in the DMD gene1. DMD is a progressive and fatal X-linked myopathy arising from the absence of functional dystrophin at the myofiber plasma membrane2. Most DMD mutations are caused by out-of frame (frame shift) or non-sense gene mutations, while the majority of BMD mutations are in-frame and thus compatible with production of an mRNA transcript that can be translated into a partly-functional quasi-dystrophin (reading frame rule)3. Some BMD patients with deletions as large as 33 exons (46% of the gene) can show little or no clinical symptoms, with only elevated serum creatine kinase4. This raises the possibility of using anti-sense-mediated removal of exons carrying nonsense mutations, or whose presence disrupts the open reading-frame at the site of the mutation so as to restore the translational reading frame and thus to convert DMD to a milder BMD phenotype5. Recently, intramuscular injection of 2'O-methylated phosphorothioate (2'O-MePs) has been shown to induce limited dystrophin expression in four DMD boys6. These studies, and extensive mdx mouse model systemic intravenous delivery reports, have rescued dystrophin expression by targeting a single exon. However, many DMD patients would require skipping of two or more exons to restore the reading frame. The ability to use cocktails of anti-sense oligonucleotides targeting multiple exons would permit designing resulting dystrophin proteins that retain more functionality7, Finally, the use of cocktails could lead to FDA-approved mixtures that would successfully treat a large group of DMD patients with distinct but overlapping deletions. This might alleviate the problem of performing toxicology tests and FDA approvals for each individual antisense sequence; a formidable barrier to clinical application. Skipping of more than one exon could, theoretically, increase the applicability to some 90% of DMD patients7, 8 while aiming to produce the most functionally favorable dystrophin variants7.
The Canine X-linked muscular dystrophy (CXMD) harbors a point mutation within the acceptor splice site of exon 7, leads to exclusion of exon 7 from the mRNA transcript9. We employed the Beagle model here, the mutation of which originates from the Golden Retriever model, but which is less severely affected. To restore the open reading frame, at least two further exons (exons 6 and 8; Fig.1) must be skipped (multi-exon skipping), therefore, it is more challenging to rescue dystrophic dogs with exon-skipping strategy. Previously, McClorey et al showed transfection with antisense oligo targeting exon 6 and 8 restored reading frame of mRNA in cultured myotubes from dystrophic dogs in vitro10. Here we identified a PMO cocktail that, using either intramuscular injection or systemic intravenous delivery, was not toxic and resulted in extensive dystrophin expression to therapeutic levels and associated with significant functional stabilization in dystrophic dogs in vivo.
Figure 1. Mutation in CXMD and strategy for exon skipping treatment.

The dystrophic dog harbors a point mutation at splice site in intron 6, which leads to lack of exon 7 in mRNA. Single exon skipping of exon 6 or exon 8 leads to out of frame products. Exclusion of at least two further exons (exon 6 and exon 8) is required to restore reading frame. 1) Vertical line means that the exons begin by the first nucleotide of a codon, 2) Arrows toward left mean that the exons begin with the second nucleotide of a codon, 3) Arrows toward right mean that the exon begin with the third nucleotide of a codon.
Subjects and Methods
Animals
CXMD affected dogs and wild type littermates from 2 months – 5 years old were used in this study11 (SupplementaryTable 1). Institutional animal care and use committee of National Center of Neurology and Psychiatry (NCNP) Japan approved all experiments using CXMD.
Antisense sequences and chemistries
We designed four anti-sense sequences to target exons 6 and 8 of the dog dystrophin mRNA as follows:
Ex6A (.GTTGATTGTCGGACCCAGCTCAGG),
Ex6B (ACCTATGACTGTGGATGAGAGCGTT),
Ex8A (CTTCCTGGATGGCTTCAATGCTCAC),
Ex8B (ACCTGTTGAGAATAGTGCATTTGAT).
Sequences were designed to target exonic sites of exon 6 (Ex6A) and exon 8 (Ex8A), or exon/intron boundary between exon 6 and intron 6 (Ex6B), or exon 8 and intron 8 (Ex8B) as illustrated (Fig.1). Two donor site sequences (Ex6B, and Ex8B) were designed to target 23 bp of exon and 2 bp of intron. Sequences were synthesized using two different backbone chemistries: 2'O-MePs (Eurogentec, Inc), and morpholino (Gene-Tools, LLC)12. We determined these sites based on the ESE motifs, GC contents, and secondary structures. We also avoided self/hetero dimers. U (uracil) was used instead of T (thymidine) for the synthesis of 2'O-MePs oligos. A discussion and figure showing the alternative chemistries can be found in Yokota et al. (in press).13
In vitro cell transfections
Primary myoblast cells from neonatal CXMD dogs were obtained by standard methods using pre-plating method14. Normal control (Wild-type; WT) or dystrophic (CXMD) myoblasts (1.5×105 cells) were cultured in growth medium containing F-10 growth media containing 20% fetal calf serum, bFGF (2.5 ng/ml), penicillin (200 U/ml), and streptomycin (200 μg/ml) for 72 hrs, followed by antisense oligonucleotides (2'O-MePs) administration (0.25–5 μg/ml, 30–600 nM) for 3 hrs with Lipofectin (Invitrogen) following manufacturer's instruction (AOs vs lipofectin =1:2). The cells were cultured in differentiation medium containing DMEM with HS (2%), penicillin (200 U/ml), and streptomycin (200 μg/ml), for 4–5 days before analyses for RNA and protein. Morpholino antisense oligonucleotides carry no charge, and can not be transfected into cells efficiently, so only 2'OMePs chemistry was utilized for in vitro studies.
Intramuscular injections
Animals were anesthetized with thiopental sodium induction and maintained by isoflurane for all intramuscular injections and muscle biopsies. Skin was excised over the site of injection, muscle exposed, and the injection site marked with a suture in the muscle. Antisense oligonucleotides were delivered as intramuscular injection using 1 ml saline bolus into the tibialis anterior (TA) or extensor carpi ulnaris (ECU) muscles using a 27G needle. Antisense oligonucleotides were delivered either singly or in mixtures. Both 2'OMePs and morpholino chemistries were tested. Muscle biopsies were obtained at 2 weeks after antisense injection.
Intravenous systemic delivery
Three dogs were treated and all were given an equimolar mixture of morpholinos Ex6A, Ex6B, and Ex8A at 32 mg/ml each. Between 26 ml to 62 ml was injected into the right saphenous vein using a 22G indwelling catheter, leading to a cumulative (combined) dose of 120–200 mg/kg per injection. Morpholinos were injected 5–11 times at weekly or biweekly intervals (Supplementary Table 1). Tissues were examined at 2 weeks after the last injection.
RT-PCR and cDNA sequencing
Total RNA was extracted from myoblasts or frozen tissue sections using Trizol (Invitrogen). Then RT-PCR was performed on 200 ng of total RNA for 35 cycles of amplification using One-Step RT-PCR kit (Qiagen) following manufacturer's instruction with 0.6 mM of either an exon 5 (CTGACTCTTGGTTTGATTTGGA), or an exon 3/4 junction (GGCAAAAACTGCCAAAAGAA) forward primer. Reverse primers were either exon 10 (TGCTTCGGTCTCTGTCAATG), or exon 13 (TTCATCGACTACCACCACCA). The resulting PCR bands were extracted by using Gel extraction kit (Qiagen). BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) was used for cDNA sequencing with the same primers.
HE and immunostaining
8-μm cryosections were cut from flash-frozen muscle biopsies at an interval of every 200 μm, then placed on poly-L-lysine–coated slides, and air-dried. Anti-Dystrophin rod (DYS-1) or C-terminal monoclonal Ab (DYS-2) (Novocastra) were used as primary antibodies. Rabbit anti-nNOS (Zymed) was used for nNOS staining. Alexa 468 or 594 (Invitrogen) was used as secondary antibody. DAPI containing mounting agent (Invitrogen) was used for nuclear counter staining (Blue). The number of positive fibers for DYS1 were counted and compared in sections where their biggest number of the positive fibers were as previously described15. Hematoxylin and Eosin (HE) staining was performed with Harris hematoxylin and eosin solutions. Images were analyzed and quantified by using ImageJ software16.
Western blotting analysis
Muscle proteins from cryo-sections were extracted with lysis buffer containing 75 mM Tris-HCl (pH 6.8), 10% SDS, 10 mM EDTA, and 5% 2-mercaptoethanol. 4–40 μg of proteins were loaded onto pre-case 5% resolving SDS-PAGE gels following manufacturer's instructions. The gels were transferred by semi-dry blotting (Bio-Rad) at 240 mA for 2 hours. DYS-2 (Novocastra) antibody against dystrophin, rabbit polyclonal antibody against α-sarcoglycan, and rabbit polyclonal antibody desmin were used as primary antibodies17. HRP conjugated anti-mouse or anti-rabbit goat immunoglobuline (Cederlane) was used as secondary antibodies. ECL kit (GE) was used for the detection. Blots were analyzed by ImageJ software.
Blood tests
Creatine phosphokinase activity, blood counts, serum biochemistry, and toxicology test in canine serum was assayed with the Fuji Drychem system and Sysmex F-820 according to manufacturer's instruction.
MRI
For imaging studies, animals were anesthetized with thiopental sodium and maintained by isoflurane. We employed a superconducting 3.0 Tesla MRI device (MAGNETOM Trio, Siemens-Asahi Medical Technologies, Japan) with an 18-cm diameter / 18-cm length human extremity coil. The acquisition parameters for T2WI were; TR / TE = 4000 / 85 ms, slice thickness = 6 mm, slice gap = 0 mm, field of view = 18×18 cm, matrix size = 256×256, and number of acquisitions = 3 during fast spin echo.
Functional testing
Clinical evaluation of dogs was performed as described in our previous report18. Grading of clinical signs in CXMD dogs were as follows:
Gait disturbance, Grade 1: None, Grade 2: Sitting with hind legs extended. Grade 3: Bunny hops with hind legs. Grade 4: Shuffling walk. Grade 5: Unable to walk.
Mobility disturbance; Grade 1: None. Grade 2: lying down more than normal. Grade 3: Cannot jump on hind legs. Grade 4: Increasing difficulty moving around. Grade 5: Unable to get up and move around.
Limb or temporal muscle atrophy. Grade 1: None. Grade 2: Suspect hardness. Grade 3: Can feel hardness or apparently thin. Grade 4: Between Grade 3 and 5, Grade 5: Extremely thin or hard.
Drooling; Grade 1: None. Grade 2: ccasionally dribbles saliva when sitting. Grade 3: Some drool when eating and drinking. Grade 4: Strings of drool when eating or drinking. Grade 5: Continuous drool.
Macroglossia; Grade 1: None. Grade 2: Slightly enlarged, Grade 3: Extended outside dentition. Grade 4: Enlarged and slightly thickened. Grade 5: Enlarged and thickened.
Dysphagia; Grade 1: None. Grade 2: Takes time and effort in taking food. Grade 3: Difficulty in taking food from plate. Grade 4: Difficulty in chewing, swallowing or drinking. Grade 5: Unable to eat.
For timed running tests, each dog was encouraged to run down a hallway (15 meters), and elapsed time was recorded. Single tests were done due to easy tiring of dystrophic dogs.
Results
In vitro screening of antisense oligonucleotides in dog primary myoblasts
The CXMD dog harbors a splice site mutation of exon 7, leading to an out-of-frame mRNA transcript fusing exons 6 to 8 (Fig. 1). To restore the reading frame, both exons 6 and 8 must be excluded (skipped) from the mRNA. Four antisense oligonucleotides were designed against exons 6 and 8. Ex6A and Ex8A were designed to bind exonic splicing enhancers (ESE), and Ex6B and Ex8B were directed against the 5' splice boundaries of each exon (Fig.1). The four AOs were transfected as 2`O-MePs either singly or in mixture (cocktails; 5 μg/ml each or 600 nM each) into cultures of primary myoblasts isolated from the skeletal muscle of neonatal CXMD dogs. Four days after differentiation into myotubes, RNA was isolated and tested for specific exon skipping by RT-PCR. A cocktail of all 4 AOs produced a single 101 bp band with 100% of RT-PCR product corresponding to a desired in-frame splice product of exon 5-10 (Fig. 2A). It is of interest that exon 9, known to be an alternatively spliced exon in the dystrophin mRNA, was consistently skipped although no antisense oligonucleotide was used against this exon (Fig. 2, Supplemental fig 1)10. Each of the four sequences were also transfected individually, and either Ex6A or Ex6B successfully induced skipping as a single AO (100% in-frame product) (Fig. 2A). The precise skipping of exons was confirmed by cDNA sequencing (Fig. 2B). We found a dose/response relationship where use of 30 nM and 60 nM of either Ex6A or Ex6B induced multi-exon skipping of exons 5-10, although with less efficiency and more intermediate out-of-frame products (Supplemental Fig. 1A). In contrast, the Ex8A AO alone induced skipping of mainly exon 8 and 9, an out of frame transcript while the Ex8B AO induced no detectable exon skipping (Fig. 2A). Dystrophin protein production from in-frame mRNA was confirmed by immunocytochemistry using either the four sequence cocktail, or Ex6A alone (Fig. 2C). We tested the same sequences in normal (wild-type) control beagle cells (Supplemental Fig. 1B). When transfected into wild-type beagle myoblasts, exon 9 was again routinely removed from the transcripts. The anti-exon 8 AO pair excised exon 8, similar to dystrophic cells, whereas the exon 6-specific AO-pair excised exon 6 as well as 9, but leaving exons 7 and 8 in place.
Figure 2. In vitro screening of antisense oligonucleotides and recovery of dystrophin expression by single antisense oligos in dog primary myoblasts.

(A) Detection of exon 6-9 skipped in-frame products (101 bp) using RT-PCR at 4 days after the transduction of 5 μg (600 nM) each AOs of single (Ex6A, or Ex6B) or cocktail AOs (Ex6A, Ex6B, Ex8A, and Ex8B) as indicated. The faint 585 bp out-of-frame band is detected in Ex8B treated myotubes. Non-treated myotubes (NT) show little RT-PCR product, likely due to nonsense-mediated decay. (B) cDNA sequencing after antisense oligonucleotide treatment at 4 days after the transduction of Ex6A alone, showing the desired in-frame exon 5-10 skip. (C) Immunocytochemistry with dystrophin C-terminal antibody (Dys-2; Red) and nuclear counter staining (Blue) for primary myotubes from CXMD cells at 4 days after transfection with cocktail or single antisense 2'O-MePs targeting exon 6 and 8 (5 μg each/ml, or 600 nM), non-treated wild-type (WT) and CXMD cells (CXMD). Bar; 50 μm.
Efficient skipping in vivo requires an three-AO cocktail
Given that Ex8B seemed ineffective by all in vitro assays, we did not continue with this sequence. Intramuscular injections were done using Ex6A alone, and equimolar mixtures of Ex6A, Ex6B, and Ex8A. Intramuscular injections into the tibialis anterior (TA) or Extensor carpi ulnaris (ECU) muscles of 0.5–5 year old CXMD dogs were done for both 2'OMePs and morpholino chemistries, at a dose of 0.12 mg to 1.2 mg of each sequence (Fig. 3, Supplemental figure 2). Injection sites, marked by suture threads, were biopsied 2 weeks later. Dystrophin positive fibers were concentrated around the injection site, and the absolute number of dystrophin-positive fibers was counted in cross-section.
Figure 3. Recovery of dystrophin expression by in vivo intramuscular injection of a three-AO cocktail, but not Ex6A alone.

(A) Restoration of dystrophin expression in TA at 14 days after single injection with 1.2 mg of Ex6A only, or cocktail containing 0.12 mg each, 0.6 mg each, or 1.2 mg each of antisense morpholino Ex6A, Ex6B, and Ex8A are shown. Age-matched non-treated CXMD (NT) and wild-type (WT) dogs are shown as controls. HE staining at 14 days after 1.2 mg each of the cocktail injection and age-matched non-treated control (NT) with consecutive cryosection stained with DYS-1 and DAPI show histological correction of the dystrophy. Bar; 100 μm.
(B) and (C) show the number of dystrophin (DYS1) positive fibers in TA or extensor carpi ulnaris at 14 days after a single injection with cocktail (Ex6A+Ex6B+Ex8A), or indicated combinations, at 1.2 mg each (morpholino; B), or 120 μg each 2'O-MePs (C). Values are mean ± s. e. m. (D) RT-PCR analysis at 2 weeks after intramuscular injection of cocktail or Ex6A morpholino at 1.2 mg each. The percentage of the in-frame exons 5-10 skip is shown under the gel image for treated muscle; normal control (WT) muscle shows the normal full-length in-frame transcript at the expected 100%.
In contrast to the skipping patterns observed with in vitro cell transfections, injection of Ex6A alone induced skipping of only exon 6 in experiments using either morpholinos or 2'O-MePs chemistries (Fig. 3, Supplemental figure 2). By contrast, the cocktail of Ex6A, Ex6B, and Ex8A induced robust dystrophin expression in a highly dose dependent manner), with 1.2 mg/each morpholino showing areas of complete dystrophin rescue, and high levels of dystrophin by immunohistochemical analysis and immunoblot (Fig.3A, 3B, 3D; Supplemental fig. 2). Intramuscular injections using the same cocktail with 2'O-MePs chemistry showed similar results, with greater dystrophin rescue using the three AO cocktail compared to Ex6A alone (Fig.3C). Two pair-wise combinations of Ex8A with either Ex6A or Ex6B were tested with morpholino chemistry, and neither combination proved as efficient as the three-sequence cocktail (Fig. 3B).
RT-PCR analyses of injected muscles showed the Ex6A/Ex6B/Ex8A morpholino cocktail to drive efficient skipping of exons 6-10 skipped products, with between 61% to 83% of RT-PCR products showing the desired in-frame product in the three muscles tested (Fig. 3D). Additional out-of-frame products were observed with Ex6A alone, and as a minority of products in the cocktail-treated muscles (Fig. 3D). Histological analyses of the muscle injected with the 3 morpholino cocktail (1.2 mg/each) showed significant histological improvement of the dystrophy, relative to uninjected muscle, using (Fig. 3A; lower panels).
By immunoblotting, intramuscular injection of the optimal cocktail dystrophin induced dystrophin to 50% normal levels in a 2-year-old dog but only to 25% in a more clinically-severe 5-year-old dog (Supplemental Fig. 2A). This result implies that the muscle quality influences the amount of dystrophin that can be produced.
Intravenous systemic delivery of a morpholino cocktail induces body-wide dystrophin expression
To be of therapeutic value AOs must be deliverable systemically. Accordingly we undertook intravenous infusion of the three morpholino cocktail showing the most success in the intramuscular experiments (Ex6A, Ex6B, and Ex8A). Three CXMD dogs were studied using intravenous doses similar to that used in mdx mouse studies (30–40 mg/kg/injection), with weekly or biweekly dosing. The first dog received 120 mg/kg morpholinos (40 mg/kg/each sequence) in weekly intravenous injections, with five doses/kg over five weeks. The second dog was given the same dose 11 times at 2-week intervals over the course of 5.5 months. The third dog received a higher dose, 200 mg/kg (66 mg/kg of each morpholino.) 7 times at weekly intervals (Supplementary Table 1). All dogs were euthanized 2 weeks after the last injection, and multiple muscles examined.
All skeletal muscles of each treated dog showed evidence of de novo dystrophin expression by immunofluorescence of cryosections, although the degree of rescue was variable (Fig. 4A). Histopathology was markedly improved in regions showing high dystrophin expression (Fig. 4A). Immunoblotting confirmed expression up to ~50 % of normal levels, but some muscles expressed only trace amounts (Fig.4C–D). Dystrophin expression was also detected in cardiac muscles but, as in the mdx mouse19, less than in skeletal muscles and concentrated in small patches (Fig.4A). Of the three dogs, the average dystrophin protein expression level was greatest in the dog given 7 × weekly doses of 200 mg/kg PMO, with an average of 26% dystrophin levels.
Figure 4. Wide-spread dystrophin expression and improved histology by intravenous systemic delivery of cocktail morpholinos in CXMD dogs.

(A) Dystrophin (DYS1) staining and histology in bilateral tibialis anterior muscles (TA), diaphragm (DIA), sternocleidomastoid (SCM) and heart at 2 weeks after final injection after 5 × weekly iv injection of 120 mg/kg of cocktail morpholinos containing Ex6A, Ex6B, and Ex8A (2001MA). Comparisons were made with TA from normal control (wild-type; WT) and from non-treated CXMD littermate (NT) tibialis anterior (TA) and heart. Intravenous morpholino treatment resulted in extensive though variable dystrophin production in multiple muscles, but with only limited evidence of rescue in heart (isolated cardiocytes). Paired dystrophin immunostaining and histology from treated dog (TA, lower panels) showed improved histopathology relative to untreated littermate (NT TA) histology. Bars; 200 μm, except for higher magnification picture of DIA and hearts (100 μm).
(B) Quantitation of centrally nucleated fibers (CNFs) in TA, intercostal (IC), Quadriceps (QUA), diaphragm (DIA), and sternocleidomastoid (SCM) in treated dog (blue bars; 2001MA) and untreated dog (red bars; 2008MA).
(C) Western blotting analysis for detection of dystrophin at 2 weeks after final injection after 5 × weekly iv injection of 120 mg/kg of cocktail morpholinos containing Ex6A, Ex6B, and Ex8A (2001MA). Dystrophin rescue is variable with high expression in right extensor carpi ulnaris (ECU(R)) and left biceps femoris (BF(L)), and less in posterior or anterior esophagus (ESO(P), ESO(A)), and sternocleidomastoid (SCM).
(D) Immunoblot analysis of dystrophin in intravenous morpholino treated dog (2703MA; 7× weekly dosing) and controls (normal control [WT], non-treated [NT]). Desmin immunoblot is shown as a loading control. Dystrophin shows high levels (>25% control levels) in triceps brachii (TB), diaphragm (DIA) and masseter (MAS).
ESO Esophagus (posterior or anterior); ECU, Extensor carpi ulnaris; SCM, Sternocleidomastoid; BF, Biceps Femoris; TB, Triceps Brachii; BB, Biceps Brachii; DIA, Diaphragm; ADD, Adductor; EDL, Extensor digitorum longus; MAS, Masseter.
Selected muscles were studied for quantitative rescue of histopathology, and for biochemical rescue of dystrophin-interacting proteins (dystrophin-associated glycoproteins, and nNOS). A commonly utilized quantitative marker for muscle pathology is central nucleation of myofibers, where increased central nucleation is reflective of increased degeneration and regeneration. Quantitation of central nucleation in treated dogs compared to untreated littermates showed that intravenous antisense treatment reduced central nucleation in all five muscle groups examined (Fig. 4B).
Both nNOS and α-sarcoglycan are dystrophin-associated proteins that co-localize with dystrophin in normal muscle, and are reduced in DMD muscle. nNOS immunofluorescence and α-sarcoglycan immunoblotting were done on a series of muscles from treated and control dogs. By immunoblot, α-sarcoglycan was seen to be increased in all muscles examined (Fig. 5B). Likewise, nNOS was seen to re-localize to the membrane in dystrophin-positive regions in systemically treated dogs (Fig. 5A).
Figure 5. Recovery of localization and expression of dystrophin associated proteins after systemic delivery of cocktail morpholinos to CXMD dogs.
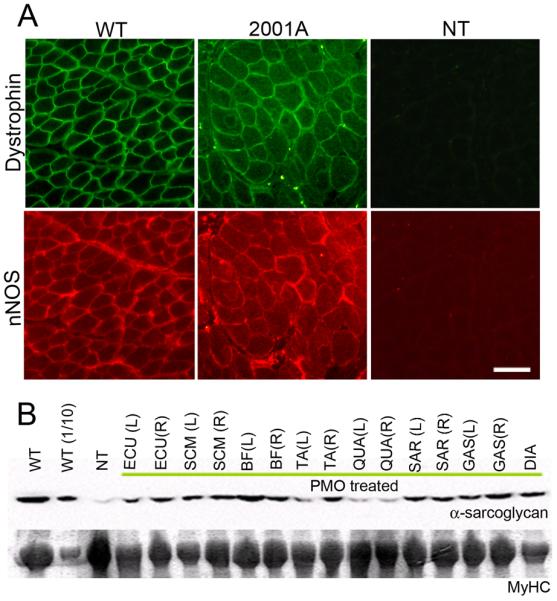
nNOS (A) and α-sarcoglycan (B) expression at 2 weeks after 5 × weekly 120 mg/kg cocktail (2001MA) or 7 × weekly 200 mg/kg cocktail morpholino injections (2703MA) CXMD dogs. Recovery of nNOS expression at sarcolemma was observed by double immuno-fluorescence against dystrophin (DYS-1) and nNOS. Bar; 50 μm. By immunoblot (B), α-sarcoglycan levels are increased in treated dog muscles, compared to untreated dystrophic controls (NT). Myosin heavy chain (MyHC) shown as a loading control. WT; wild-type normal controls, WT(1/10); wild-type (1/10 diluted samples, i.e. 4 μg loaded), NT; non-treated CXMD muscles (tibialis anterior) ECU; Extensor carpi ulnaris, SCM; Sternocleidomastoid, BF; Biceps Femoris, TA; Tibialis Anterior, QUA; Quadriceps, SAR; Sartorius, GAS; Gastrocnemius, DIA; Diaphragm. (L) and (R) stand for left side and right side respectively.
Muscle imaging and clinical grading scores are improved by systemic antisense
A global improvement in muscle pathology was further supported by the T2-weighted MRI examination (Fig. 6). The high intensity T2 signal, indicative of inflammation and increased water content, was diminished in PMO-treated dogs compared with pre-treated and un-treated control dogs in most muscles (Fig.6).
Figure 6. Amelioration of pathology and reduced inflammation signal in MRI.
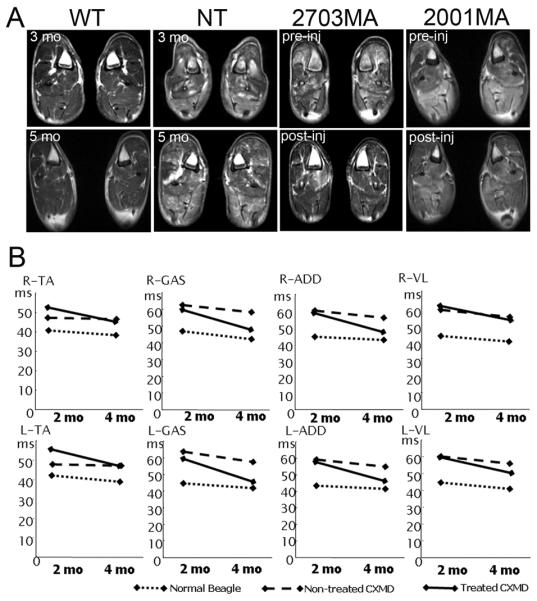
T2-weighted MRI of hind legs at 1 week before initial injection (pre-inj), and at 2 weeks after final injection (post-inj) of 7 × weekly iv injection of 200 mg/kg of cocktail morpholinos (2703MA) or 5 × weekly iv injection of 120 mg/kg of cocktail morpholinos (2001MA). Age-matched un-treated dogs (WT [normal control] and NT [non-treated dystrophic control]) are shown for comparison. (B) Changes of T2 value examined by MRI at 2 weeks after 7 × weekly 200 mg/kg of cocktail morpholino injections. Changes of T2 values in hind-legs at 1 week before initial injection and at 2 weeks after final injection are shown. Intravenous morpholino treatment resulted in decreased T2 signal in all muscles examined. TA, Tibialis Anterior; GAS, Gastrocnemius; VL, Vastus lateralis; ADD, Adductor.
Functional improvement of treated dogs was also assessed by a 15 meter timed running test, and by a combined clinical grading score, as we have previously published18. Three dogs treated with intravenous morpholinos were compared to three untreated, at the ages of both 2 or 5 months (pre-treatment) and 4 or 7 months (post-treatment) (Fig. 7B–C). The untreated littermates became slower over the treatment time, while all treated dogs ran faster after treatment. The single dog treated at an older age with more advanced symptoms showed greater improvement relative to untreated littermates (Fig. 7B).
Figure 7. Stabilization of clinical symptoms by systemic morpholino treatment.

(A) A combined clinical grading scores before (black lines) and after starting treatment (red lines) of the three treated dogs. Clinical grades of gait disturbance, mobility disturbance, limb or temporal muscle atrophy, drooling, macroglossia, Dysphagia are scored as described in methods. A series of untreated dogs (n=6-13) were studied for comparison (dashed line, standard error bars).
(B–C) Fifteen meter timed running tests in treated dogs and untreated littermates. A CXMD dog treated from 5–7 months (2001MA) of age showed decreased timed 15 m run after treatment, whereas untreated littermates showed slowed running ability (B). Similarly, two littermate dogs treated at 2–4 months of age (2703MA; 2702FA) showed quicker 15 m times following treatment compared to non-treated littermate (C).
The combined clinical grading score similarly showed improvement or stabilization of disease progression following antisense treatment, relative to natural history controls (Fig. 7A). Videos documenting running ability of treated dogs and untreated littermates are available as supplemental data (Supplemental movie 1–5).
Serum creatine kinase (CK) was assessed before and after intravenous treatment, and compared to natural history controls (Supplemental Fig. 3). While serum CK was variable, post-treatment CK levels were consistently below natural history controls.
Intravenous high-dose morpholino cocktail shows no evidence of toxicity
No local inflammatory reactions or organ dysfunctions were recorded in the morpholino-treated dogs. Twice-weekly serum toxicology screens of the three systemically treated dogs showed no evidence of liver or kidney dysfunction (Supplemental Fig. 4). Levels of urea nitrogen (BUN), gamma glutamyl transpeptidase (GGT, or gamma-GTP) and creatinine, all remained within the normal ranges In addition, no significant changes were observed in amylase, total protein, total bilirubin, c-reactive protein (CRP), sodium, potassium, or chloride. Growth of body weight was also within normal range in all treated dogs (data not shown).
Discussion
This is the first report of widespread induction of dystrophin expression to therapeutic levels in the dog model of DMD. Overall, our findings provide a promising message for DMD patients. Specifically, we show that intravenous morpholino antisense (PMOs) can generate body-wide production of functional dystrophin in a model clinically more severe than DMD, resulting in stabilization or improvement of the clinical disease. Beneficial effects were documented by histology, MRI, and functional tests (running and combined clinical grading scores).
We encountered some unexpected findings that raise important questions as to how to pursue this promising approach into human clinical trials. Clearly, the choice of specific antisense sequences is a crucial determinant of the ultimate success of targeted exon skipping. To date, specific AO sequences have been assessed for efficiency of exon skipping using cell-based experimental (in vitro) systems, with the optimal sequences then used for in vivo experiments. In studies presented here, antisense oligonucleotides directed against exon 6 were able to efficiently induce the desired exon 5 to exon 10 splicing in vitro, but not in vivo. Our observations of discrepant outcomes for ex-vivo and in vivo in the dystrophic dog tell us that we do not currently possess a reliable means of screening for sequences that induce efficient skipping of a particular exon in a particular mutational context. Data obtained from application of sequences as 2'O-MePs in primary myogenic cells or as PMOs incubated ex vivo with muscle fragments failed to predict the effects when PMO sequences were tested in vivo. The very high percentage of in-frame products here might be related to nonsense-mediated decay of out-of-frame products, or quality of RNA from cell culture, however, the in vitro experiments were consistent using three different concentrations (600 nM, 60 nM, and 30 nM), with two different sequences (Ex6A, and Ex6B). The results were confirmed by RT-PCR, IHC, and cDNA sequences (Fig. 2, Supplemental figure 1). The in vitro effect of the exon 6-specific sequence was, in addition, context-dependent. For, when transfected into wild-type beagle myoblasts the exon 8 AO pair again excised exons 8 and 9 whereas the exon 6-specific AO-pair excised exons 6 and 9, leaving exons 7 and 8 in place (Supplemental fig.1). Thus, excision of exon 8 by the exon 6-specific sequences occurs only in the context of the mutant exon 7 splice site. Together, the differences between patterns of skipping in vivo versus in vitro and between wild-type versus mutant genotypes tell us that efficiency of skipping during transcription is dominated by variables other than the availability or otherwise of specific local sequence. Thus it is prudent to consider testing of selected sequences in multiple systems with human dystrophin mRNA as the target before committing to a specific sequence for clinic trials.
We observed efficient skipping of exon 9, even though no anti-sense sequence targeted its removal in both wild-type and CXMD (Fig. 2–3), which is known as alternative splice site20. AOs targeting exon 8 have been reported to induce skipping of both exon 8 and exon 9 in both human and dog studies (Fig. 2–3)10, 21. It seems likely that the small size of intron 8 compared with intron 7 (1.1 versus 110 Kb) predisposes to splicing of exon 8 to exon 9 prior to splicing its splicing to exon 7.
In the systemically treated dogs, we found widespread expression of dystrophin in all muscles analyzed but with considerable variation (Fig. 4). No difference in dystrophin expression between fiber types was evident (data not shown). Even contralateral muscles differed from one another, suggesting that variation in efficacy of dystrophin production is a reflection of transient sporadic events such as myopathic episodes or changes in vascularization or circulation rather than any intrinsic muscle specific properties. Pathological stages of degeneration/regeneration might be also involved. Overall most studies to-date suggest that 10–20% normal dystrophin levels are needed to improve muscle function22, 23, and the data on systemic morpholino-induced exon skipping presented here implies that some, but not all, muscle groups reached this therapeutic level.
Systemic delivery of morpholinos in CXMD dogs as in mdx mice induced only modest dystrophin production in the heart (Fig. 4)19. The reason is not clear, but it has been suggested that dystrophic skeletal muscle fibers may give greater access to AOs because they have more “leaky” membranes than the smaller cardiac cells and because the syncytial structure of myofibers may permit wider diffusion of PMO molecules from each site of entry24. Cardiac ischemia, as indicated by an abnormal Q-waves in CXMD25, may also limit access of AOs to cardiomyocytes. Some improvement in delivery has been reported with cell-penetrating peptide tagged morpholinos, or use of microbubbles and ultrasound which may enhance uptake efficiency in the heart by facilitating penetration of cell membranes, although toxicity of these strategies are not clear26,27.
Considerable evidence for functional and histological improvement was seen in the three systemically treated dogs (Fig. 4–7). All were stabilized compared with their untreated littermates in motor function tests, general clinical condition, and serum creatine kinase levels. MRI images also showed reduction of T2-weighted signal, interpreted as a sign of diminished inflammation, following morpholino delivery (Fig.6). However, longer-term experiments are required to investigate whether AOs can reduce infiltration by fibro-fatty tissue, and to what extent functional loss can be recovered. Many DMD patients require two or more exons to be skipped to restore the reading frame. The data shown here are the first demonstrations of efficient skipping of multiple exons systemically through intravenous delivery. The dog model required skipping of 2–3 exons to restore the reading frame, and we were able to show efficient skipping of three exons by both intramuscular and intravenous delivery methods. Multi-exon skipping has also been shown in vitro cell cultures, and in mdx mice by intramuscular injections8, 28–30. Multi-exon skipping increase the range of potentially treatable DMD patients, and also raises the prospect of selecting the most functionally favorable in-frame dystrophins7, although skipping larger stretches of exons has a yet not been achieved, and may currently not be feasible, Specific morpholino cocktails able to treat a large proportion of DMD patients with optimized quasi-dystrophin production might be submitted for regulatory approval as a single `drug'. For example, a cocktail of AOs targeting exon 45-55 would be applicable up to 63% of patients with dystrophin deletions, and this specific deletion is associated with asymptomatic or very mild BMD clinical phenotypes31, 32
Supplementary Material
(A) Western blotting analysis with dystrophin DYS-2 antibody 2 weeks after the intramuscular injection of the cocktail morpholinos as indicated. (B) RT-PCR with left (forward) primer at exon 3/4 junction and right (reverse) primer in exon 13 reveals larger, exons 6-10 skipped in-frame transcripts as well as exon 6-9 skipped one in some samples from 1.2 mg each of cocktail PMO treated dogs. Non-treated muscle (NT) showed almost no detectable band, probably due to nonsense-mediated decay. (C) Dystrophin expression at 2 weeks after intramuscular injection of 120 μg each cocktail or 120 μg single AOs in 1 ml saline into left or right extensor carpi ulnaris muscles. Bar; 50 μm.
Running test of 11 × biweekly 120 mg/kg morpholino-treated dog (2702FA) at 7 months of age
(A) 5 × weekly 120 mg/kg of (2001MA), (B) 7 × weekly 200 mg/kg (2703MA), and (C) 11 × bi-weekly 120 mg/kg (2702FA) of PMO injected dogs were tested twice-weekly. Broken lines indicate mean of age-matched untreated CXMD dogs (n=6-13). Values are mean ± s. e. m.
The change of BUN (A–B), GGT (C–D), creatinine (E–F) in serum before and during the course of 11 × bi-weekly 120 mg/kg (2702FA; A, C, E) or 7 × weekly 200 mg/kg (2703MA; B, D, F) of cocktail morpholino injections. Maximum reference ranges for parameters in normal dogs are indicated by dashed lines.
(A) RT-PCR analysis to detect exons 5-10 skipped in-frame products 4 days after 0.25–0.5 μg/ml (30–60 nM) of single transfection with Ex6A or Ex6B or lipofectin only (60 μg/ml) in CXMD dog myotubes. (B) RT-PCR analysis after AO-treatment in wild-type myotubes (600 nM for each oligo). Non-treated myotubes (NT) show 584 bp splice variant band, which skips exon 9, as well as 704 bp full-length band. By transfection targeting exon 8 (Ex8A +Ex8B), 402 bp of exon 8 and 9 skipped products were observed, while by transfection targeting exon 6 (Ex6A +Ex6B), 411 bp of exon 6 and 9 skipped products were observed. By transfection targeting both exon 6 and exon 8 (cocktail), 230 bp of exon 6, 8 and 9 skipped products were observed. The pattern of exon skipped product was further confirmed by cDNA sequencing (C).
Running test of non-treated littermate dog (2008MA) at 7 months of age
Running test of non-treated littermate dog (2003MA) at 7 months of age
Running test of 5 × weekly 120 mg/kg morpholino-treated dog (2001MA) at 7 months of age
Running test of 7 × weekly 200 mg/kg morpholino-treated dog (2703MA) at 5 months of age
Acknowledgements
Authors thank Drs Stephanie Duguez, Javad Nazarian, Heather Gordish-Dressman (Children's National Medical Center, Washington DC), Yoshitsugu Aoki, Takashi Saito, Katsutoshi Yuasa, Naoko Yugeta, Sachiko Ohshima, Zin-Hong Shin, Michiko Wada, Kazuhiro Fukushima, Satoru Masuda, Kazue Kinoshita, Hideki Kita, Shin-ichi Ichikawa, Yumiko Yahata, Takayuki Nakayama (National Institute of Neuroscience, Tokyo, Japan), Adam Rabinowitz, and Jonathan Beauchamp (Imperial College, London, UK) for discussions and technical assistance. Supported by the Foundation to Eradicate Duchenne (FED; www.DuchenneMD.org), the Department of Defense CDMRP program, the Jain Foundation, the Crystal Ball of Virginia Beach (Muscular Dystrophy Association USA), the National Center for Medical Rehabilitation Research (www.ncmrr.org), a collaborative grant from the NIH Wellstone Muscular Dystrophy Research Centers (http://www.wellstone-dc.org) (USA), Grants-in-Aid from the Research on Nervous and Mental Disorders (16B-2, 19A-7), Health and Labor Sciences Research Grants for Translation Research (H19-translational research-003) and Health Sciences Research Grants for Research on Psychiatry and Neurological Disease and Mental Health (H18-kokoro-019) from the Ministry of Health, Labor, and Welfare of Japan.
Footnotes
Authors claim no conflict of interest.
References
- 1.Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Matsumura K, Campbell KP. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- 3.Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 4.England SB, Nicholson LV, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- 5.Dunckley MGMM, Villiet P, Eperon IC, Dickson G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet. 1998;7:1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- 6.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 7.Yokota T, Duddy W, Partridge T. Optimizing exon skipping therapies for DMD. Acta Myol. In Press. [PMC free article] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, Janson AA, Kaman WE, et al. Antisense-induced multiexon skipping for Duchenne muscular dystrophy makes more sense. Am J Hum Genet. 2004;74:83–92. doi: 10.1086/381039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharp NJ, Kornegay JN, Van Camp SD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13:115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- 10.McClorey G, Moulton HM, Iversen PL, et al. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006 doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 11.Shimatsu Y, Katagiri K, Furuta T, et al. Canine X-linked muscular dystrophy in Japan (CXMDJ) Exp Anim. 2003;52:93–97. doi: 10.1538/expanim.52.93. [DOI] [PubMed] [Google Scholar]
- 12.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 13.Yokota T, Takeda S, Lu QL, Partridge TA, Nakamura A, Hoffman EP. A renaissance for anti-sense oligonucleotide drugs in neurology: Exon-skipping breaks new ground. Arch. Neurol. doi: 10.1001/archneurol.2008.540. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jankowski RJ, Haluszczak C, Trucco M, Huard J. Flow cytometric characterization of myogenic cell populations obtained via the preplate technique: potential for rapid isolation of muscle-derived stem cells. Hum Gene Ther. 2001;12:619–628. doi: 10.1089/104303401300057306. [DOI] [PubMed] [Google Scholar]
- 15.Yokota T, Lu QL, Morgan JE, et al. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J Cell Sci. 2006;119:2679–2687. doi: 10.1242/jcs.03000. [DOI] [PubMed] [Google Scholar]
- 16.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 17.Araishi K, Sasaoka T, Imamura M, et al. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum Mol Genet. 1999;8:1589–1598. doi: 10.1093/hmg/8.9.1589. [DOI] [PubMed] [Google Scholar]
- 18.Shimatsu Y, Yoshimura M, Yuasa K, et al. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ. Acta Myol. 2005;24:145–154. [PubMed] [Google Scholar]
- 19.Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 20.Reiss J, Rininsland F. An explanation for the constitutive exon 9 cassette splicing of the DMD gene. Hum Mol Genet. 1994;3:295–298. doi: 10.1093/hmg/3.2.295. [DOI] [PubMed] [Google Scholar]
- 21.Aartsma-Rus A, De Winter CL, Janson AA, et al. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: indication for steric hindrance of SR protein binding sites. Oligonucleotides. 2005;15:284–297. doi: 10.1089/oli.2005.15.284. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura M, Sakamoto M, Ikemoto M, et al. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther. 2004;10:821–828. doi: 10.1016/j.ymthe.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 23.Liang KW, Nishikawa M, Liu F, et al. Restoration of dystrophin expression in mdx mice by intravascular injection of naked DNA containing full-length dystrophin cDNA. Gene Ther. 2004;11:901–908. doi: 10.1038/sj.gt.3302239. [DOI] [PubMed] [Google Scholar]
- 24.Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert opinion on biological therapy. 2007;7:831–842. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- 25.Yugeta NUN, Fujii Y, Yoshimura M, Yuasa K, Wada MR, Nakura M, Shimatsu Y, Tomohiro M, Takahashi A, Machida N, Wakao Y, Nakamura A, Takeda S. Cardiac involvement in Beagle-based canine X-linked muscular dystrophy in Japan (CXMDJ): electrocardiographic, echocardiographic, and morphologic studies. BMC Cardiovasc Disord. 2006;6:47. doi: 10.1186/1471-2261-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jearawiriyapaisarn N, Moulton HM, Buckley B, et al. Sustained Dystrophin Expression Induced by Peptide-conjugated Morpholino Oligomers in the Muscles of mdx Mice. Mol Ther. 2008 doi: 10.1038/mt.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vannan M, McCreery T, Li P, et al. Ultrasound-mediated transfection of canine myocardium by intravenous administration of cationic microbubble-linked plasmid DNA. J Am Soc Echocardiogr. 2002;15:214–218. doi: 10.1067/mje.2002.119913. [DOI] [PubMed] [Google Scholar]
- 28.Fall AM, Johnsen R, Honeyman K, et al. Induction of revertant fibres in the mdx mouse using antisense oligonucleotides. Genet Vaccines Ther. 2006;4:3. doi: 10.1186/1479-0556-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aartsma-Rus A, Janson AA, van Ommen GJ, van Deutekom JC. Antisense-induced exon skipping for duplications in Duchenne muscular dystrophy. BMC Med Genet. 2007;8:43. doi: 10.1186/1471-2350-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aartsma-Rus A, Kaman WE, Weij R, et al. Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exons. Mol Ther. 2006;14:401–407. doi: 10.1016/j.ymthe.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura A, Yoshida K, Fukushima K, et al. Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci. 2008;15:757–763. doi: 10.1016/j.jocn.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Beroud C, Tuffery-Giraud S, Matsuo M, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat. 2007;28:196–202. doi: 10.1002/humu.20428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Western blotting analysis with dystrophin DYS-2 antibody 2 weeks after the intramuscular injection of the cocktail morpholinos as indicated. (B) RT-PCR with left (forward) primer at exon 3/4 junction and right (reverse) primer in exon 13 reveals larger, exons 6-10 skipped in-frame transcripts as well as exon 6-9 skipped one in some samples from 1.2 mg each of cocktail PMO treated dogs. Non-treated muscle (NT) showed almost no detectable band, probably due to nonsense-mediated decay. (C) Dystrophin expression at 2 weeks after intramuscular injection of 120 μg each cocktail or 120 μg single AOs in 1 ml saline into left or right extensor carpi ulnaris muscles. Bar; 50 μm.
Running test of 11 × biweekly 120 mg/kg morpholino-treated dog (2702FA) at 7 months of age
(A) 5 × weekly 120 mg/kg of (2001MA), (B) 7 × weekly 200 mg/kg (2703MA), and (C) 11 × bi-weekly 120 mg/kg (2702FA) of PMO injected dogs were tested twice-weekly. Broken lines indicate mean of age-matched untreated CXMD dogs (n=6-13). Values are mean ± s. e. m.
The change of BUN (A–B), GGT (C–D), creatinine (E–F) in serum before and during the course of 11 × bi-weekly 120 mg/kg (2702FA; A, C, E) or 7 × weekly 200 mg/kg (2703MA; B, D, F) of cocktail morpholino injections. Maximum reference ranges for parameters in normal dogs are indicated by dashed lines.
(A) RT-PCR analysis to detect exons 5-10 skipped in-frame products 4 days after 0.25–0.5 μg/ml (30–60 nM) of single transfection with Ex6A or Ex6B or lipofectin only (60 μg/ml) in CXMD dog myotubes. (B) RT-PCR analysis after AO-treatment in wild-type myotubes (600 nM for each oligo). Non-treated myotubes (NT) show 584 bp splice variant band, which skips exon 9, as well as 704 bp full-length band. By transfection targeting exon 8 (Ex8A +Ex8B), 402 bp of exon 8 and 9 skipped products were observed, while by transfection targeting exon 6 (Ex6A +Ex6B), 411 bp of exon 6 and 9 skipped products were observed. By transfection targeting both exon 6 and exon 8 (cocktail), 230 bp of exon 6, 8 and 9 skipped products were observed. The pattern of exon skipped product was further confirmed by cDNA sequencing (C).
Running test of non-treated littermate dog (2008MA) at 7 months of age
Running test of non-treated littermate dog (2003MA) at 7 months of age
Running test of 5 × weekly 120 mg/kg morpholino-treated dog (2001MA) at 7 months of age
Running test of 7 × weekly 200 mg/kg morpholino-treated dog (2703MA) at 5 months of age