

SUMMARY
Lung epithelial cells (LEC) are strategically positioned in the airway mucosa to provide barrier defense. LEC also express pattern recognition receptors and a myriad of immune genes, but their role in immunity is often concealed by the activities of “professional” immune cells, particularly in the context of fungal infection. Here, we demonstrate that NFκB signaling in LEC is essential for immunity against the pulmonary fungal pathogen Blastomyces dermatitidis. LEC orchestrate innate antifungal immunity by augmenting the numbers of IL-17A- and GM-CSF-producing innate lymphocytes, specifically “natural” Th17 (nTh17) cells. Innate lymphocyte-derived IL-17A and GM-CSF in turn enable phagocyte-driven fungal killing. LEC regulate the numbers of nTh17 cells via the production of chemokines such as CCL20, a process dependent on IL1α-interleukin-1 receptor (IL-1R) signaling on LEC. Therefore, LEC orchestrate IL-17A- and GM-CSF-mediated immunity in an IL-1R-dependent manner and represent an essential component of innate immunity to pulmonary fungal pathogens.
Keywords: Fungi, Epithelial cells, Innate Immunity, Lymphocytes
eTOC Blurb
Hernández-Santos et al. show that the lung epithelium guards against inhaled fungi by providing early signals that orchestrate innate immunity. Signaling via the lung epithelial cell receptor IL-1R is required to release soluble signals that coordinate and recruit innate lymphocytes whose soluble products arm phagocytes to kill fungi.
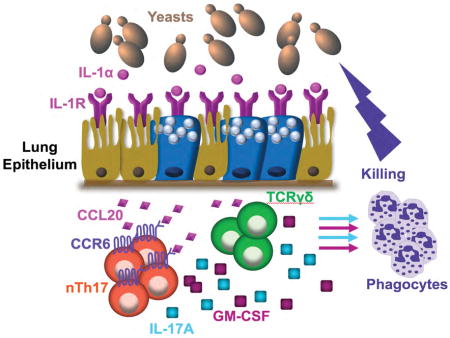
INTRODUCTION
In the air we breathe, 4–11% of the fine particle mass contains fungal spores (Frohlich-Nowoisky et al., 2009), some of which enter our airways. A fungal particles’ first point of contact in the airway is the epithelium lining the respiratory mucosa. This lining separates the environment from the lung parenchyma and enables lung epithelial cells (LEC) to guard the tissue against fungal invaders. However, the role of LEC as defenders against inhaled fungi has been overshadowed in favor of hematopoietic cells. LEC can participate in anti-microbial immunity by clearing particulates from the airway via the mucociliary escalator and by producing antimicrobial peptides and inflammatory mediators (Whitsett and Alenghat, 2015). These mediators inform the immune system of microbial assault thereby directing early decisions (Swamy et al., 2010) about innate and adaptive immunity. Despite these LEC functions, little is known about how LEC promote antifungal immunity.
IL-17 is a key component of immunity to fungi.. CD4+IL-17+ (Th17) cells were thought to be the main source of IL-17, but innate sources have now been described (Gaffen et al., 2014): TCRαβ cells, TCRγδ cells, innate lymphoid cells type 3 (ILC3), invariant natural killer T (iNKT) cells, mucosal associated invariant T cells (MAIT) cells (Rahimpour et al., 2015), neutrophils (Taylor et al., 2014), and eosinophils (Guerra et al., 2017). During oropharyngeal candidiasis (OPC), C. albicans induces natural T helper 17 (nTh17) cells, TCR γδ cells (Conti et al., 2014) and ILC3 (Gladiator et al., 2013). In keratitis due to A. fumigatus, neutrophils auto-activate via paracrine IL-17A signaling and promote fungal clearance (Taylor et al., 2014). Lastly, in pulmonary aspergillosis, eosinophils produce IL-23 and IL-17A, and associate with and kill A. fumigatus (Guerra et al., 2017). Thus, both innate and adaptive sources of IL-17 promote immunity to fungi.
Cells that produce IL-17A may make other cytokines, alone or together with IL-17A. One such cytokine is granulocyte macrophage colony stimulating factor (GM-CSF), another key component of antifungal immunity. Humans with congenital or acquired defects in GM-CSF responses (pulmonary alveolar proteinosis) are susceptible to fungal infections due to impaired macrophage and neutrophil function (Punatar et al., 2012; Uchida et al., 2007). GM-CSF−/− mice are susceptible to Pneumocystis carinii infection (Paine et al., 2000) and mice lacking the β subunit of the GM-CSF receptor (GM-CSFFRβ) show impaired reactive oxygen species (ROS) production by neutrophils and inability to kill A. fumigatus (Kasahara et al., 2016). In H. capsulatum-infected macrophages (MØ), GM-CSF causes zinc sequestration in the Golgi, prompting phagosomal zinc deprivation, induction of ROS, and fungal death (Subramanian Vignesh et al., 2013). A serine protease of Blastomyces yeast cleaves GM-CSF, promoting escape from phagocyte killing (Sterkel et al., 2016). Finally, in systemic candidiasis, IL-17 and Syk-dependent IL-23 production by dendritic cells (DC) enable NK cells to produce GM-CSF, thereby promoting candidacidal activity of neutrophils (Bar et al., 2014). These findings highlight the role of GM-CSF in potentiating phagocyte fungal killing and the link between IL-17 and GM-CSF signaling pathways during fungal infection.
The relationship of the epithelium to GM-CSF-mediated antifungal immunity is not well understood. LEC produce GM-CSF early during lung development (Guilliams et al., 2013), and alveolar epithelial cell GM-CSF orchestrates DC responses to influenza and antiviral CD8+ T cell responses (Unkel et al., 2012). To our knowledge, LEC regulation of GM-CSF immunity to fungi has not been investigated. Conversely, the epithelium and IL-17-mediated immunity has received more attention. Epithelial cells respond to IL-17. IL-17R on epithelial cells at different tissue sites regulates antimicrobial peptides and chemokines that recruit neutrophils. IL-17R on gut epithelium regulates α defensins, which maintains levels of segmented filamentous bacteria (Kumar et al., 2016). Likewise, IL-17R on oral epithelium regulates β-defensin 3, which clears C. albicans from the oral cavity (Conti et al., 2016). Similarly, IL-17R on lung club cells controls CXCL5, neutrophil recruitment, and Klebsiella pneumonia (Chen et al., 2016). Although LEC respond to IL-17, the mode by which the epithelium regulates the function of IL-17-producing cells remains ill defined.
Herein, we investigated how LEC regulate innate defense against pathogenic fungi. We addressed three questions: (i) Are LEC essential in host defense against inhaled fungi?; (ii) How do LEC sense inhaled fungi e.g. what are the signaling pathways and upstream receptor(s)?; (iii) How do LEC orchestrate innate antifungal immunity – e.g. what are the effector cells, how do they kill fungi, and how do LEC regulate these effector mechanisms. To address these questions, we exploited a murine model involving the inhaled pathogenic fungus Blastomyces dermatitidis, the causative agent of fungal pneumonia and one of the major endemic mycoses of North America. We show that the fungus rapidly activates NFκB signaling in LEC, which is essential in orchestrating innate anti-fungal immunity. LEC regulate antifungal activity by coordinating the function of innate lymphocytes, including nTh17 cells and γδ T cells. Innate lymphocyte-derived IL-17A and GM-CSF arm phagocytes to kill the fungus. This circuit is amplified by IL-1α-IL-1R signaling on LEC. These findings provide fresh insight into the earliest stages of lung host defense and highlight an unappreciated role for LEC in orchestrating innate antifungal immunity.
RESULTS
LEC mount a robust, NFκB-dependent antifungal response
To delineate the possible contribution of LEC to antifungal immunity, we first ascertained whether the fungus interacts with LEC and whether such interactions result in NFκB activation; this signaling event lies downstream of many pattern recognition pathways and mediates transcription of various immune mediators (Lawrence, 2009). Following infection, histological examination of infected lung tissue revealed yeast in close proximity to upper LEC (Fig. 1A, arrowhead 1). Yeasts also were observed in the lower airway, but inflammation obstructing the alveoli obscured the location of yeasts relative to alveolar epithelial cells (Fig. 1A, arrowhead 2). LEC response to yeast resulted in NFκB activation, as shown by GFP+ LEC in histological sections of infected NFκB reporter mice (Fig. 1B), where GFP expression is under the transcriptional control of NFκB cis-acting elements (Magness et al., 2004). Flow cytometric quantification revealed an increased proportion of GFP+ LEC upon fungal infection in these mice (Fig. 1C). We also observed translocation of p65 (RelA) into the nucleus as early as 1 hour post infection (h.p.i.) and peaking at 4 h.p.i. (Fig. S1A–B).
Figure 1. LEC mount a robust, NFκB-dependent antifungal response early upon infection (see also Fig. S1).

(A) WT mice were infected i.t. with 2×105 yeasts and 24 h.p.i. lungs were inflated and fixed in formalin. Tissue sections are stained with gomori methanimine silver. (B) NFκB-GFP reporter mice were infected with 5×105 yeast labeled with calcofluor and cryosections of lung tissue were analyzed by confocal microscopy ~3.5 h.p.i. Blue denotes yeast, green GFP (NFκB), and red CD326+ cells (LEC). Left photo = 20X mag. The right photo is the area denoted in white box at 60X mag. (C) NFκB-GFP reporter mice were infected as in panel B and analyzed for GFP expression in Ep-CAM+ (CD326+) cells from lungs at 24 h.p.i. Gates depict mean±SD % GFP+ cells (uninfected group n=5, infected group n=4). (D–F) DNTA or IKK2ΔLEC mice and littermate controls were infected i.t. with 1×105 spores or 2×104 yeast and lung CFU quantified at indicated times. Each symbol denotes a mouse. One-Way ANOVA with Bonferroni’s correction (D) and Mann-Whitney test (E–F). (G) IKK2fl/fl and IKK2ΔLEC were infected with yeast and monitored for survival over 20 days. Log-rank test. (H) IKK2fl/fl and IKK2ΔLEC were infected and analyzed for CFU as indicated in (D–F). Mann-Whitney test.
If LEC rapidly sense inhaled fungi, NFκB signaling in LEC may help restrain fungal invasion of the lungs. To test this hypothesis, we employed two murine models of infection in which NFκB is deleted either in a select subset of LEC or throughout the airway epithelium. In DNTA mice, the club cell-specific promoter CC10 drives expression of a dominant negative form of IκB upon doxycycline administration, preventing NFκB translocation to the nucleus (Cheng et al., 2007). DNTA mice treated with doxycycline exhibited a 2- to 3-log higher fungal burden than DNTA mice not treated with doxycycline or corresponding littermate controls (rtTA) (Fig. 1D). Thus, club cells may regulate early antifungal immunity. However, perturbations in the intestinal microbiota as a result of antibiotic treatment may confound interpretation of these results. Therefore, we also used IKK2ΔLEC mice (Perez-Nazario et al., 2013), in which the IKK2 (IKKβ) component of IκB kinase is deleted in most of the airway epithelium, sequestering NFκB in the cytoplasm. In this model, lung-specific surfactant protein C drives expression of cre recombinase, which recognizes loxP sites flanking exons 5 and 6 of IKK2 with a predicted efficiency of 60–100% (Fig. S1C–D). IKK2fl/fl mice phenotypically resemble wild-type (WT) and are used as littermate controls. Infection with fungal spores or yeast yielded a respective 1 and 3 log increase in fungal burden in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 1D–F). Survival post-infection was also significantly shortened in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 1G). Thus, NFκB signaling in LEC promotes antifungal immunity and prolongs survival during fungal infection.
Due to early activation of NFκB signaling upon infection (Fig. 1 and S1), we postulated that this pathway in LEC may restrain fungal invasion early during the innate phase of the host immune response. Upon kinetic analysis, the fungal burden rose significantly in IKK2ΔLEC vs. IKK2fl/fl mice by as early as 48 h.p.i. (Fig. 1H), indicating that LEC orchestrate early resistance in this model.
LEC-mediated antifungal immunity is IL-17A- and GM-CSF-dependent
Immunity to several respiratory fungal infections requires IL-17A and GM-CSF. IL-17A−/− or IL-17RA−/− mice show impaired secondary immunity (Wuthrich et al., 2011) to B. dermatitidis, as well as H. capsulatum and C. posadasii. Moreover, inactivation of GM-CSF greatly enhances virulence of B. dermatitidis yeast (Sterkel et al., 2016). Here, we analyzed the requirement for IL-17A and GM-CSF within 48 h.p.i. (day 2) to see if these products are required during the same timeframe in which LEC orchestrate innate immunity. We neutralized IL-17A and GM-CSF in WT mice during 48 h.p.i., and assayed fungal burden at the end of this interval. Neutralization of IL-17A and GM-CSF significantly increased the burden at 2 d.p.i. (Fig. 2A), suggesting a temporal association between the requirements for LEC NFκB signaling, IL-17A and GM-CSF. To assess a link, we analyzed these products in IKK2ΔLEC and IKK2fl/fl mice. IKK2ΔLEC mice had reduced IL-17A in the broncheoalveolar lavage fluid (BALF) (Fig. 2B) and also reduced numbers of IL-17A- and GM-CSF-producing innate lymphocytes compared to IKK2fl/fl mice (Fig. 2C). Thus, LEC may orchestrate innate antifungal immunity via the actions of IL-17A and GM-CSF, perhaps through regulating the function or number of innate lymphocyte populations.
Figure 2. LEC-mediated antifungal immunity is IL-17A- and GM-CSF-dependent (see also Fig. S2).

(A) WT mice were infected i.t. with yeast. IL-17A and GM-CSF were neutralized as noted in Methods. At 48 h.p.i., lung CFU was enumerated. Each panel represents a separate experiment (2 pooled experiments depicted in left panel); each symbol denotes one mouse. Mann-Whitney test (48 h.p.i.). (B) IKK2fl/fl and IKK2ΔLEC mice were infected i.t. with yeast and IL-17A content was analyzed by ELISA in BALF at 48 h.p.i. A representative of 4 experiments is depicted. Mann-Whitney test. (C) IKK2fl/fl and IKK2ΔLEC mice were infected i.t. with yeast. At 48 h.p.i., lung cell suspensions were analyzed by flow cytometry for intracellular IL-17A and GM-CSF in lymphocytes (CD90.2+ CD44hi). Two pooled experiments depicted. Mann-Whitney test. (D) WT, IL-17A−/− and GM-CSF−/− were infected with 5×105 yeasts; at 48 h.p.i., lungs were analyzed by confocal imaging. Club cells depicted in green (CC10), nuclei in blue (DAPI) and GM-CSF in red. (E) Procedure was done as in A with one further injection of antibody at 48 h.p.i. At 96 h.p.i., lung CFU were quantified. Data representative of 3 experiments. Kruskal-Wallis with Dunn’s multiple comparison.
NFκB signaling in LEC regulates IL-17A- and GM-CSF producing innate lymphocytes
We sought to delineate the cell sources of IL-17A and GM-CSF. Several innate sources of IL-17A have been described in the oral cavity; for example, nTh17, TCRγδ, and ILC3 produce IL-17A during C. albicans infection in OPC (Conti et al., 2014; Gladiator et al., 2013). We used IL-17creRosa 26ReYFP fate reporter mice (IL-17A producers are permanently labeled eYFP+) to identify cell sources in the lung (Hirota et al., 2011). In naïve lung tissue, TCRβ+CD4− cells constituted the largest proportion of IL-17A-producing cells (46%), followed by TCRβ+CD4+ (nTh17) (26%) and TCRγδ (26%), and a minor population of ILC3 (2%). Following infection, TCRβ+CD4+ cells (nTh17) increased in proportion to 41%, TCRγδ remained unchanged (25%), TCRβ+CD4− decreased to 30%, and ILC3 underwent a minor increase to 4% (Fig. S3A and Fig. 3A). We detected a minor population of IL-17A+CD8+ T cells, but no IL-17A+ neutrophils (Fig. S3B). Quantification of IL-17A-producing cells revealed an infection-dependent increase in the absolute number of all these cell populations (Fig. 3B). Upon further characterization, we found that TCRβ+CD4+ cells were CD49a−, CD49d+ (bimodal distribution in infected animals) and CD29+ (Fig. S3C), resembling the phenotype of nTh17 cells (Conti et al., 2014) and henceforth referred to as such. TCRγδ cells were CD27−, Vγ1−Vγ4+ or Vγ1−Vγ4− (Heilig and Tonegawa, 1986) (Fig. S3D), similar to reports that identified IL-17A+TCRγδ cells as CD27− (Ribot et al., 2009). The TCRβ+CD4− cells are a heterogeneous population composed of MAIT cells and a yet-undefined subset (Fig. S3E). Intracellular cytokine staining in WT animals revealed that each of these lymphocyte subsets also produce GM-CSF (Fig. 3C–D).
Figure 3. Ablation of NFκB in LEC results in decreased numbers of IL-17A- and GM-CSF-producing innate lymphocytes (see also Fig. S3).

(A–B) IL-17creRosa26ReYFP mice were infected i.t. with yeast. Lung cell suspensions were analyzed by flow cytometry at 48 h.p.i. to identify IL-17A+ innate lymphocytes. Cells were gated as CD90.2+IL-17+ and the proportion (A) and number (B) of nTh17, TCRβ+ CD4−, TCRγδ, and ILCs quantified. Total lymphocytes (B) means IL-17+ cells within the total CD90.2+ population. A representative of 3 experiments depicted; n=5/group. Mann-Whitney test (B). (C–D) IKK2fl/fl and IKK2ΔLEC mice were infected i.t. with yeast and the proportion (C) and number (D) of indicated lymphocyte populations quantified by flow cytometry at 48 h.p.i. The TCRβ+CD4− gate includes CD8+ T cells. Concatenated plots depicted in C (n=11 in IKK2fl/fl, n=9 in IKK2ΔLEC). Two pooled experiments depicted in D; each symbol is one mouse. Mann-Whitney test.
Importantly, further analysis of innate lymphocyte populations revealed that the number of cytokine-producing cells during infection was severely impaired in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 3C–D), although the total number of innate lymphocytes in the lung at baseline was unaffected by ablation of NFκB in LEC (Fig. S3F). Specifically, nTh17 and TCRγδ cells exhibited decreased numbers of IL-17A+ cells, and TCRβ+CD4− and TCRγδ showed decreased numbers of GM-CSF+ cells (Fig. 3D). Thus, NFκB signaling in LEC regulates the numbers of IL-17A- and GM-CSF-producing innate lymphocytes during fungal infection.
LEC are not a source of GM-CSF
LEC can produce GM-CSF (Unkel et al., 2012). To determine if LEC are a source of GM-CSF during fungal infection, we co-stained infected lung tissue with antibodies against GM-CSF and club cells. In WT mice, GM-CSF production was detected in cells infiltrating the airway (likely leukocytes), but not in club cells (Fig. 2D). In IL-17−/− mice, GM-CSF staining was absent, indicating that IL-17A regulates GM-CSF production in these infiltrating cells. The GM-CSF defect was not due to defective cell recruitment since the absolute numbers of lymphoid and myeloid cells were similar in IL-17A−/− and WT mice (Fig. S2). While in vivo neutralization of either IL-17A or GM-CSF impaired resistance to infection, neutralization of both together was not additive, suggesting that the two products act in the same pathway e.g. GM-CSF is a target of IL-17 signaling (Fig. 2E).
nTh17 cells are indispensable for innate antifungal immunity
We investigated the roles of IL-17A- and GM-CSF-producing innate lymphocytes in innate anti-fungal defense. Given the reduced proportion and number of IL-17A+ and GM-CSF+ TCRγδ cells upon ablation of NFκB in LEC, we first tested if TCRγδ is required for antifungal immunity at 48 h.p.i. Surprisingly, the fungal burden was similar in TCRδ−/− vs. WT mice (Fig 4A). On the other hand, the fungal load was significantly higher in TCRα−/− vs. WT mice, and similar to that in Rag−/−γc−/−, which lack all lymphocyte populations (Fig 4A). Thus, TCRαβ lymphocytes are indispensable for innate antifungal defense, whereas TCRγδ cells participate but are dispensable. TCRαβ lymphocytes are comprised of two subsets: CD4+ and CD4−. After antibody treatment in vivo, fungal load rose significantly in CD4+ cell-depleted mice vs. isotype control-treated mice and was similar to levels in TCRα−/− (Fig. 4B). Thus, TCRβ+CD4+ cells are required for antifungal immunity.
Figure 4. nTh17 cells are indispensable for innate antifungal immunity (see also Fig. S4).

(A) WT, TCRδ−/− TCRα−/− and Rag−/−γc−/− mice were infected i.t. with yeast and at 48 h.p.i. lung CFU was enumerated. Two pooled experiments depicted; each symbol denotes one mouse. One-Way ANOVA with Bonferroni’s correction. (B) WT (isotype), CD4-neutralized, and TCRα−/− mice were infected i.t. with yeast and CFU counted at 48 h.p.i. α-CD4 given i.v. at the time of infection. Two pooled experiments depicted; each symbol denotes a mouse. One-way ANOVA with Bonferroni’s correction. (C) WT, TCRδ−/− TCRα−/− mice were infected i.t. with yeast. At 48 h.p.i., the number of CCR6+ cells among TCRβ+ and TCRγδ+ cells was quantified by flow cytometry. Experiment is representative of two performed. Mann-Whitney test. (D) Summary of blastomycosis patient features. (E) Frequency of Vγ9+Vδ2+ among total CD3+ cells in peripheral blood of healthy donors and patients with active or resolved blastomycosis was quantified by flow cytometry. Each dot plot depicts a donor. (F) Proportion of total CD3+ cells in normal donors (ND) and patients with blastomycosis.
Although LEC regulate innate antifungal immunity through the requisite action of nTh17 cells, we asked if the dispensability of TCRγδ cells in this model is due to compensation (Fig. 4C). Indeed, the number of CCR6+ nTh17 cells increased greatly in TCRδ−/− vs. WT mice, whereas the number of CCR6+TCRγδ cells in TCRα−/− mice was similar to that in WT mice (Fig. 4C). Thus, nTh17 cells appear to compensate for the loss of TCRγδ+ cells.
We explored the role of these innate lymphocytes in human patients. nTh17 cells cannot be reliably detected at the time patients present with illness, but TCRγδ cells could be analyzed. We considered that whereas TCRγδ cells are dispensable for antifungal immunity in mice, this might not be the case in humans. In healthy individuals, 1–5% of the circulating CD3+ cells are TCRγδ+ (Carding and Egan, 2002) and the majority of this subset is Vγ9+Vδ2+. In psoriasis, Vγ9+Vδ2+ cells infiltrate skin lesions, and the increase of these cells in the skin corresponds to a decreased proportion of circulating Vγ9+Vδ2+ cells, which is partly restored after treatment (Laggner et al., 2011). Likewise, Vγ9+Vδ2+ exit the circulation and enter the pleural fluid of tuberculosis patients (Balbi et al., 1993). We hypothesized that, in pulmonary fungal infection, Vγ9+Vδ2+ cells leave the circulation and infiltrate the tissue in response to inflammatory signals. Thus, we analyzed the blood of patients with active or resolved blastomycosis. At the time of presentation with active infection, patients had a much lower percentage of Vγ9+Vδ2+ cells in peripheral blood compared to normal donors (Fig. 4D–E). This was not due to expansion of a different T cell subset because the proportion of total CD3+ cells was similar between normal donors and patients (Fig. 4F). Furthermore, in patients treated or cured with antifungal drugs, the frequency of Vγ9+Vδ2+ cells recovered to the levels in healthy subjects (Fig. 4D–E). Thus, during human infection, a proportion of Vγ9+Vδ2+ cells may leave the circulation and infiltrate infected tissue to promote antifungal immunity. Taken together, our data reveal an indispensible role for the LEC-nTh17 axis in innate antifungal defense in mice, and show that TCRγδ cells also participate in the response in both mice and humans.
Collaborative killing of yeast by alveolar MØ, DCs and neutrophils is dependent on LEC NFκB, IL-17A and GM-CSF
To determine how LEC promote fungal killing via the action of innate lymphocytes, we studied the recruitment and function of myeloid cells in IKK2ΔLEC and control mice. Although the levels of CXCL1 were decreased in the lungs of IKK2ΔLEC relative to IKK2fl/fl mice at 24 h.p.i. (Fig. S4A), the numbers of alveolar MØ, DCs, monocytes and neutrophils at 12, 24 and 48 h.p.i. were unaltered in the absence of NFκB signaling in LEC (Fig. S4B–C). To assess phagocyte function in IKK2ΔLEC mice, we tracked leukocyte-yeast interactions and killing in vivo using DsRed reporter yeast (Sterkel et al., 2016). Loss of DsRed fluorescence in uvitex-stained yeast denotes loss of viability e.g. uvitex+DsRed+ and Uvitex+DsRed− represent live and dead yeasts, respectively (Fig. 5A). Similar to the elevated fungal load in IKK2ΔLEC mice at 48 h.p.i (Fig. 1H), the number of DsRed+ (live) yeasts was significantly higher in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 5B, left) and paralleled the decreased proportion of dead yeasts in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 5B, right). The proportion of yeasts killed by alveolar MØ, DCs, and neutrophils was significantly reduced in IKK2ΔLEC vs. IKK2fl/fl mice (Fig. 5C), indicating that optimal killing by these phagocytes requires NFκB signaling in LEC. Neutralization of IL-17A and GM-CSF in vivo in WT mice also decreased the proportion of dead yeasts associated with lung leukocytes at 48 h.p.i. (Fig. 5D), indicating that fungal killing by leukocytes is partially dependent on IL-17A and GM-CSF. Accordingly, in vivo administration of IL-17A and GM-CSF to IKK2ΔLEC significantly enhanced killing toward the levels observed in IKK2fl/fl mice via action on leukocyte populations (Fig. 5E). Taken together, these findings support a model in which NFκB activity in LEC regulates the number of IL-17A- and GM-CSF-producing innate lymphocytes, which in turn arm phagocytes to kill the yeast.
Figure 5. Collaborative killing of yeast by alveolar MØ, DCs, and neutrophils is dependent on LEC NFκB, IL-17A and GM-CSF.

(A–B) IKK2fl/fl and IKK2ΔLEC mice were infected i.t. with DsRed yeast stained with Uvitex. The proportion and number of live (red) and dead (blue) yeast among total yeasts (red+blue) were quantified by flow cytometry. Two pooled experiments depicted. (C) Proportion of yeast-associated (uvitex+) alveolar MØ (CD11c+Siglec F+), DCs (Siglec F− CD11c+ MHCII+) and neutrophils (Siglec F− CD11b+Ly6G+) that were DsRed- (associated with dead yeast). Two pooled experiments depicted. (D) WT mice were infected i.t. with DsRed yeast and the proportion of dead yeasts was calculated by flow cytometry. IL-17A and GM-CSF were neutralized as noted in Methods. Data representative of 3 experiments. One-way ANOVA with Tukey’s multiple comparison. (E) IKK2fl/fl and IKK2ΔLEC were infected with DsRed yeast. rIL-17A and rGM-CSF were given i.t. together with the inoculum. 48 h.p.i lung cell suspensions were analyzed by flow cytometry to quantify the proportion of dead yeast (DeRed−Uvitex+) in total lung homogenate, and proportoin of phagocytes associated with dead yeast. One-Way ANOVA with Tukey’s multiple comparison.
IL-1α/IL-1R signaling pathway in LEC is essential for antifungal immunity
To uncover the LEC-intrinsic signaling pathways of antifungal immunity, we analyzed pattern recognition receptors and adaptor molecules that promote antifungal immunity. Here, CARD9−/− and MyD88−/− mice had significantly higher fungal burdens than WT mice at 48 h.p.i (Fig. S5A). We next analyzed receptors upstream of CARD9 and MyD88. Dectin-1−/−, Dectin-2−/−, Dectin-3−/−, Mincle−/−, and TLR23479−/− mice had fungal loads similar to WT mice. However, the burden in IL-1R−/− mice was significantly higher than that in WT mice and similar to MyD88−/− mice (Fig. S5A). Moreover, fungal burden was inversely correlated with IL-17A level in lung homogenate (Fig. S5B). Thus, IL-1R signaling via MyD88 and CARD9 are each required for innate antifungal immunity.
We next analyzed the tissue-specific roles of adaptors and receptors implicated above. Bone marrow chimeric mice showed that the IL-1R was required for antifungal immunity in nonhematopoietic cells. The role of CARD9, on the other hand, was restricted to the hematopoietic compartment (Fig. 6A). Furthermore, using in vivo neutralization of the ligands for IL-1R, we found that IL-1α but not IL-1β is required for IL-1R signaling in LEC, as evidenced by the significantly increased fungal load in IL-1α-neutralized mice (Fig. 6B). We also observed that IL-1α is induced in the lung by 24 h.p.i (Fig. S6A), and that LEC themselves are a source of this cytokine (Fig. S6B).
Figure 6. LEC-mediated antifungal immunity is dependent on the IL-1α/IL-1R signaling axis (see also Fig. S5 and S6).

(A) Bone marrow chimeric mice were infected i.t. with yeast and lung CFU counted at 48 h.p.i. A representative of 2 experiments is shown. One-way ANOVA with Bonferroni’s correction. (B) WT mice were infected i.t. with yeast and lung CFU counted at 48 h.p.i. IL-1α and IL-1β were neutralized at the time of infection. Two pooled experiments depicted. One-way ANOVA with Bonferroni’s correction. (C–D) IL-17A and GM-CSF production by innate lymphocytes in WT and IL-1R−/− mice were analyzed by intracellular cytokine staining. Proportion (C) and number (D) of cytokine-producing cells are depicted. Concatenated plots depicted in C (n=5 WT and 5 IL-1R−/−). A representative experiment of two is shown. Mann Whitney test. (E) IL-1Rfl/fl and IL-1RΔLEC mice were infected with yeast and CFU counted at 48 h.p.i. Mann Whitney test.
Our results imply that the IL-1α-IL-1R signaling axis may regulate expression of LEC products that promote increased innate lymphocyte numbers, thereby fostering antifungal immunity. If this hypothesis is correct, in the absence of the IL-1R, innate lymphocytes should be curtailed. Indeed, we found that both the proportion and number of nTh17 cells are reduced in IL-1R−/− vs. WT mice (Fig. 6C–D), further supporting the requirement we observed for nTh17 cells in antifungal immunity (Fig. 4A–B). IL-17A-producing TCRγδ cells were also impaired in IL-1R−/− mice (Fig. 6C–D), in accordance with prior studies that demonstrated the requisite role of IL-1R for proper TCRγδ function (Duan et al., 2010).
To specifically target the IL-1R in LEC, we generated mice in which the IL-1R is deleted only in LEC (IL-1RΔLEC). Targeted deletion of IL-1R in LEC resulted in a sharp increase in lung fungal burden compared to littermate controls (IL-1Rfl/fl) (Fig. 6E). Collectively, our data establish the indispensible role for IL-1α-IL-1R signaling in LEC for innate antifungal immunity and suggest that such signaling ultimately regulates innate lymphocyte number and function.
IL-IR regulates CCL20 production by LEC, and CCL20 partners with other chemokines to regulate nTh17 numbers
Given the requisite role of IL-1α-IL-1R signaling in LEC, and the importance of nTh17 cells in antifungal immunity, we investigated IL-1R-dependent, LEC-derived signals regulating nTh17 function during fungal infection. C-C motif chemokine ligand 20 (CCL20) is produced by epithelial cells (Reibman et al., 2003), and recruits IL-17-producing cells expressing C-C chemokine receptor 6 (CCR6) to inflammatory sites (Hirota et al., 2007). Moreover, CCL20 production is induced by IL-1 in lung epithelial cells (Starner et al., 2003). We hypothesized that B. dermatitidis induces IL-1α-IL-1R-dependent production of CCL20 in LEC, which in turn regulates nTh17 numbers. We found that CCL20 is induced in infected lung tissue at 24h.p.i (Fig. 7A). Likewise, the proportion and number of nTh17 (CCR6+IL-17A+) cells increased by 48 h.p.i. (Fig. 7B). Lung CCL20 levels were also lower in the BALF of infected IKK2ΔLEC (or IL-1R−/− mice) vs. IKK2fl/fl mice, indicating CCL20 production is contingent on LEC NFκB activity and IL-1R signaling (Fig. 7C).
Figure 7. IL-1R signaling regulates CCL20 production by LEC and CCL20 partners with other chemokines to regulate nTh17 numbers (see also Fig. S7).

(A) CCL20 levels in lung homogenate were quantified by ELISA at various times after infection with yeast. One-way ANOVA with Bonferroni’s correction. Dotted line is upper limit of detection (B) Proportion (left) and number (right) of CCR6+IL-17A+ nTh17 cells in IL-17creRosa 26ReYFP mice after infection. Mann-Whitney test. (C) CCL20 levels in BALF of IKK2fl/fl, IKK2ΔLEC and IL-1R−/− quantified by ELISA 48 h.p.i. Four pooled experiments depicted. One-way ANOVA with Bonferroni’s correction. (D) LEC (CD31−CD45−CD326+) were purified from infected mice (48 h.p.i.) and levels of CCL20 in cell lysates quantified by ELISA. Two pooled experiments depicted. One-way ANOVA. (E) Yeasts were given i.t. alone or together with rCCL20. At 48 h.p.i., lung CFU was quantified. One-way ANOVA. (F) IL-17creRosa26ReYFP were infected with yeast i.t. alone or together with PTx. At 48h.p.i., lungs were harvested and the number of IL-17A+ nTh17 and TCRγδ+ cells quantified by flow cytometry. Mann-Whitney test. (G) Yeasts were given i.t. alone or together with PTx. At 48 h.p.i, lung CFU was quantified. Mann-Whitney test. (H) Mice were infected with yeast i.t. and at 48 h.p.i. lung CFU was quantified. One-tailed, unpaired t test. (I–J) WT mice were infected with yeast and after 48h the proportion (H) and number (I) of CCR6+Ki67+ and IL-17A+ Ki67+ cells were quantified by flow cytometry. Ki67+IL-17A+ and Ki67+CCR6+ cells in the TCRβ+CD4+ gate (H) denote proliferating nTh17 cells (enumerated in I). Mann-Whitney test (I).
Since cell sources other than LEC may account for CCL20 in the BALF, we investigated LEC as a source. We purified LEC (CD31− CD45− CD326+) (Fig. S7) from infected IKK2fl/fl, IKK2ΔLEC and IL-1R−/− mice and measured CCL20 in LEC. Consistent with our findings in BALF, CCL20 levels in IKK2ΔLEC and IL-1R−/− LEC were significantly decreased relative to IKK2fl/fl (Fig. 7D). In vivo administration of recombinant CCL20 to IKK2ΔLEC mice only partially restored the fungal burden to levels in IKK2fl/fl mice (Fig. 7E), suggesting that CCL20 may collaborate with other factors to recruit or expand the numbers of CCR6+nTh17 cells in the lung upon infection.
To test the role of CCR6 signaling, we blocked this G-protein coupled receptor with pertussis toxin (PTx). The absolute number of nTh17 cells decreased sharply in mice treated with PTx, whereas the number of IL-17A+TCRγδ+ cells remained unchanged (Fig. 7F), suggesting that chemokine receptor signaling regulates the numbers of nTh17 cells in the lung. Both PTx-treated and CCR6-deficient mice exhibited significantly higher lung fungal burden (Fig. 7G–H), supporting a role for chemokine signaling, particulatly the CCL20-CCR6 axis, in antifungal immunity.
We considered that differences in innate lymphocyte numbers may arise from proliferation in situ. We quantified the number of proliferating, IL-17-producing innate lymphocytes during infection. We found that the frequency and absolute number of CCR6+Ki67+ and CCR6+IL-17A+ nTh17 and TCRγδ+ cells increased in infected mice (Fig. 7I and J). These results, together with the decreased numbers of nTh17 cells upon PTx treatment, suggest that while TCRγδ+ proliferate in situ, nTh17 are both recruited to the lung and proliferate during fungal infection. Thus, IL-1 induces CCL20 in LEC, which synergizes with other soluble factors to augment nTh17 numbers and foster fungal killing.
DISCUSSION
We report a requisite role for NFκB signaling in the lung epithelium in regulating innate immunity to an inhaled fungal pathogen. A primary and indispensible role for the lung epithelium has been neglected in favor of the general perception that hematopoietic cells such as alveolar MØ serve as the initial reservoir and sentinel of pulmonary fungal infection. For example, inhaled A. fumigatus spores are thought to initially enter resident alveolar MØ where they are either recognized and killed, or germinate and proliferate in the setting of impaired immunity (Aimanianda et al., 2009). Likewise, H. capsulatum is generally thought to parasitize and multiply in alveolar MØ. Finally, we recently reported that entry of alveolar MØ by B. dermatitidis spores early in infection accelerates conversion to yeast and escape from host immunity (Sterkel et al., 2015). These perceptions notwithstanding, MyD88 signaling in epithelium is known to regulate early recruitment of neutrophils into the lung in murine aspergillosis (Jhingran et al., 2015) and NFκB signaling in LEC promote adaptive immunity to P. carinii infection (Perez-Nazario et al., 2013).
In our study, NFκB signaling in LEC was required to contain early fungal growth, but the critical epithelial cell subset(s) remains to be determined. Although IKKΔLEC mice have impaired NFκB signaling throughout most of the lung epithelium (Fig. S1), in DNTA mice the defect is restricted to club cells (Cheng et al., 2007), indicating that club cells may regulate innate antifungal responses. The lung has seven subsets of epithelial cells, conferring distinct functions. In mouse lung, club cells comprise much of the lining of the bronchi and bronchioles (Iwasaki et al., 2016). Although our data pointing to a central role for club cells may be confounded by antibiotic (doxycycline) treatment effects on the microbiota, club cells have been implicated in host defense against Klebsiella pneumonia (Chen et al., 2016).
Our data show that NFκB signaling in airway epithelial cells orchestrate innate antifungal immunity via innate lymphocytes, particularly nTh17 cells. Innate lymphocyte-derived IL-17 and GM-CSF enabled fungal killing by alveolar MØ, DCs, and neutrophils. Airway epithelial cell and innate lymphocyte function were connected by chemokines such as CCL20, which was induced in epithelial cells by IL-1. Thus our results uncovered an unappreciated network in which LEC foster antifungal immunity, not only by responding to IL-17, but by directing the function of IL-17- and GM-CSF-producing innate lymphocytes, in part via CCL20.
The early infection-dependent translocation of NFκB to the nucleus of LEC raises the question of whether yeast induce such signaling directly through physical interactions with LEC, as in P. carinii infection (Millard et al., 1990), or indirectly via pattern recognition of fungal molecules by hematopoietic cells. Our finding of NFκB signaling in lung epithelium as early as one hour post-infection may be consistent with initial interaction of yeast with LEC. However, it remains unclear how LEC would directly sense the yeast. We excluded CLR on LEC since known CLR signal via CARD9, which we showed in bone marrow-chimeric mice exerts its effect exclusively through hematopoietic cells. It is possible that LEC signaling via MyD88, through an as yet unidentified non-CLR receptor, promotes initial recognition of yeast. A third possibility is that hematopoietic cells such as CD103+DC in apposition with airway epithelium initially sense yeast and respond by producing IL-1 that rapidly triggers engagement of LEC via the IL-1R-MyD88 signaling axis, which is amplified by autocrine production of IL-1α in a feedback loop.
We found that NFκB signaling in LEC is crucial in establishing adequate IL-17A- and GM-CSF production, which in turn regulate phagocyte killing. Traditionally, IL-17 is functionally linked to the recruitment of neutrophils to peripheral inflammatory sites via induction of C-X-C chemokines at epithelial surfaces. This is evident in K. pneumonia infection where IL-17R deficiency in club cells results in decreased expression of CXCL5 and impaired bacterial clearance from the lung (Chen et al., 2016). However, in OPC, the number of neutrophils recruited to the tongue in mice with IL-17R-deficient oral epithelium (IL-17RAΔK13) was indistinguishable from that observed in littermate controls (Conti et al., 2016). The increase in the fungal burden of IL-17RΔK13 mice was attributed to decreased production of β-defensin 3 by oral epithelium (Conti et al., 2016). Our data showed that NFκB deficiency in LEC had no effect on the recruitment of neutrophils, but it did severely affect their function. These disparities in the mechanisms of IL-17-mediated immunity suggest that the effects of IL-17 signaling in epithelial cells depend on the microbe and anatomical location of infection.
We identified several sources of IL-17 and GM-CSF upon pulmonary fungal infection, including nTh17 and TCRγδ cells, which were previously documented as IL-17 sources in the gut (Conti et al., 2014). In addition, we identified MAIT cells as a source of IL-17A during fungal infection. Our experiments in TCRδ−/− and TCRα−/− mice showed that TCRγδ cells are dispensable whereas TCRαβ, specifically CD4+ cells, are indispensable for innate antifungal immunity. This result was surprising given the preferential location of TCRγδ cells at epithelial surfaces (Chien et al., 2014) and the fact that NFκB deficiency in LEC led to severely impaired TCRγδ cell responses. Our result may be explained by compensatory TCRαβ cell responses in TCRδ−/− mice. Intestinal TCRαβ cells increase in TCRδ−/− mice (Komano et al., 1995), and dendritic epidermal T cells, which are TCRγδ+ in the skin of WT mice, are temporarily replaced by TCRαβ+ cells in TCRδ−/− mice (Jameson et al., 2004). Furthermore, in a model of hypersensitivity pneumonitis, where the main source of IL-17 is TCRγδ cells, CD4+ TCRαβ cells compensate and produce IL-17 in TCRδ−/− mice (Simonian et al., 2009). We found that the number of CCR6+ nTh17 cells increased in TCRδ−/− vs. WT mice, whereas the number of CCR6+ TCRγδ+ cells was similar in TCRα−/− and WT mice. Thus, TCRαβ cells may compensate for loss of TCRγδ cells to conceal their function in mice. However, in humans presenting with active blastomycosis, we found that the frequency of circulating Vγ9+Vδ2+ cells was decreased compared to healthy donors, suggesting that Vγ9+Vδ2+ cells leave the bloodstream to infiltrate inflamed tissue, as in psoriasis (Laggner et al., 2011). Remarkably, effective antifungal treatment restored the frequency of circulating Vγ9+Vδ2+ cells to levels found in healthy donors. TCRγδ cells infiltrate infected tissue in other granulomatous diseases such as tuberculosis (Balbi et al., 1993), but their role in such disorders remains ill defined.
The LEC cytokine and chemokine network that instructs type 17 responses is poorly understood. We show that CCL20, a product of epithelial cells at other anatomical locations (Reibman et al., 2003; Sierro et al., 2001), and a signal recruiting CCR6+ type 17 cells to sites of inflammation (Hirota et al., 2007), is made by LEC and, in conjuction with other factors, regulates the number of nTh17 cells upon fungal infection. CCL20 production by LEC required IL-1α/IL-1R signaling, itself indispensable in restraing infection. The chemokine response of alveolar epithelial cells infected with P. carini in vitro depends on IL-1R signaling (Bello-Irizarry et al., 2012), and the first wave of neutrophils recruited following A. fumigatus infection also requires IL-1R signaling in LEC (Jhingran et al., 2015). The key role of the IL-1α/IL-1R axis in LEC-mediated antifungal immunity prompts the question: “What IL-1R-dependent LEC products, in addition to CCL20, comprise the network of early signals that restrain fungal infection?” Although the answer to this question remains to be investigated, our findings highlight the role of LEC as key orchestrators of antifungal immunity and potential targets of therapeutic approaches to enhance resistance against inhaled fungal pathogens, which represent a growing public health problem worldwide.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce S. Klein (bsklein@wisc.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Peripheral blood mononuclear cells (PBMC)
Peripheral blood from healthy donors and blastomycosis patients was overlaid on Ficoll and centrifuged at 400 rcf for 20 min at RT without acceleration and without break. The PBMC layer was subsequently collected and the residual ficoll washed. Approximately 1–2 million PBMC were stained and analyzed by flow cytometry. Work with human donors was performed with informed consent from the participants and approved by the appropriate institutional IRB at University of Wisconsin-Madison.
Mice
All strains were in the C57BL/6 background, except Dectin 3−/− which are a combination of multiple genetic backgrounds. Experiments conducted with Dectin 3−/− mice included the appropriate littermate controls. Mice were bred in-house under specific pathogen free (SPF) conditions. WT animals were purchased as needed from the NCI repository. NFκB-eGFP (Magness et al., 2004), IL-17creRosa26ReYFP (Hirota et al., 2011), Dectin-1−/− (Taylor et al., 2007), Dectin-2−/− (Saijo et al., 2010), Dectin-3−/−(Wang et al., 2015), Mincle−/− (Wells et al., 2008), CARD9−/− (Hsu et al., 2007), TLR 23479−/− (Conrad et al., 2009), IL-17A−/− (Nakae et al., 2002), DNTA and rtTA mice (Cheng et al., 2007) (the latter two strains were a kind gift from Drs. Tim Blackwell and Fiona Yull at Vanderbilt University, TN) have been previously described. IL-1R−/−, IL-1Rfl/fl, MyD88−/−, TCRδ−/− and RAG−/−γc−/− were purchased from the Jackson Laboratories. SPC-CRE mice were obtained by backcrossing IKK2ΔLEC mice with WT to eliminate the IKK2fl/fl allele. IKK2ΔLEC mice were a kind gift from Dr. Terry Wright at the University of Rochester Medical Center (Bello-Irrizarry, 2013 JI). In these animals, the IKK2 component of the NFκB signaling pathway is floxed (fl) by loxP sites, and cre recombinase expression is driven by the lung-specific surfactant protein C, resulting in deletion of IKK2 exclusively in airway epithelial cells. In all experiments, the CRE-IKK2fl/fl littermates were used as controls. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC).
Growth of fungi
B. dermatitidis strain ATCC 26199 was grown in 7H10 media slants, in a humidified incubator at 37°C – 39°C, at which temperature the organism maintains its yeast morphology. In order to generate spores, B. dermatitidis strain 14081 was grown to log phase at 37°C and plated onto potato dextrose agar (PDA) to promote spore formation. Plates were incubated at 25°C for 2 weeks in BSL3 conditions and spores were subsequently harvested in PBS. For enumeration of CFU, lungs were plated onto Brain Heart Infusion (BHI) (strain 26199) or Histoplasma Macrophage Medium (HMM) (strain 14081) agar and incubated for 7 days at 37°C.
Experimental Model of Infection
Seven to twelve week old mice were anesthetized with isofluorane, intubated using the BioLITE system (Braintree Scientific, Inc.), and inoculated i.t. with 2 × 104 B. dermatitidis yeasts or 1 ×105 spores, unless stated otherwise. At various timepoints post-inoculation (indicated in each Fig.), the bronchioalveolar lavage fluid (BALF) and the lungs were collected and subsequently processed for colony forming unit (CFU) analysis, flow cytometry, or enzyme-linked immunoabsorbent assay (ELISA).
METHOD DETAILS
Flow Cytometry
The lungs were perfused by injecting 10ml of PBS into the heart’s left ventricle, mechanically disrupted with a syringe plunger, and incubated in 2 mg/ml Collagenase D and 10 μg/ml DNAse at 37°C for 20 minutes. Collagenase was inhibited with 50mM EDTA in PBS. The resulting single-cell suspension was filtered through a 40μm nylon mesh and the remaining red blood cells were lysed in ACK lysis buffer. In some experimets circulating leukocytes were excluded from analysis by injecting 2μg/mouse i.v. of fluorescently labeled anti-CD45 and allowing it to circulate for 3 minutes prior to harvesting the lungs. Cell suspensions were incubated with 10μg/ml of Brefeldin A for 4 hours at 37°C and surface and intracellular antigens were subsequently stained with fluorochrome-conjugated antibodies. Dead cells were excluded from data analysis with Live/Dead fixable dead cell stains (Molecular Probes, Life Technologies). Non-specific binding of antibodies was blocked with anti CD16/32 (Fc Block) and surface antigens were stained on ice for 20–60 minutes. Cells were then permeabilized with BD Cytofix/Cytoperm at RT for 20 minutes or at 4°C overnight and intracellular cytokines were stained on ice for 20–60 minutes. For Ki67 staining the cells were fixed and permeabilized with Foxp3 transcription factor staining buffer set (eBiosciences). Fluorochrome-conjugated antibodies were purchased from BioLegend, BD Biosciences or e-Biosciences and are listed in the key resources table. Mouse MR1 tetramer (5-OP), and the corresponding control (6-FP), for detection of MAIT cells was developed by Dr. Dale I. Godfrey (Rahimpour et al., 2015) and purchased from the NIH tetramer facility. Data were acquired in five-laser BD Fortesa or BD LSRII and analyzed with Flowjo Software v10.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse | ||
| B220 (RA3-6B2) | BD Biosciences | 553092 |
| CC10 | Seven Hills | WRAB-3950 |
| CCR6 (29-2L17) or CCR6 (140706) | BioLegend or BD Biosciences | 129819 or 557976 |
| CD103 (2E7) | BioLegend | 121426 |
| CD11b (M1/70) | BD Biosciences or BioLegend | 564443 or 101212 |
| CD11c (N418) | BioLegend or eBiosciences | 117339 or 17-0114-82 |
| CD11c-biotin (N418) LEC enrichment | eBiosciences | 13-0114-82 |
| CD27 (LG.3A10) | BioLegend | 124215 |
| CD29 (HMβ1-1) | BioLegend | 102221 |
| CD31 (390) | BioLegend | 102420 |
| CD31-biotin (390) LEC enrichment | BioLegend | 102404 |
| CD326 (G8.8) LEC enrichment | eBiosciences | 12-5791-83 |
| CD4 (GK1.5) in vivo neutralization | BioXcell | BE0003-1 |
| CD4 (RMA4-5) | BD Biosciences | 564933 or 553051 |
| CD44 (IM7) | BD Biosciences | 553133 |
| CD45-biotin (30-F11) LEC enrichment | eBiosciences | 13-0451-85 |
| CD45.2 (104) | BD Biosciences | 560697 |
| CD49a (Ha31/8) | BD Biosciences | 740375 |
| CD49d (R1-2) | BioLegend | 103607 |
| CD64 (X54-5/7.1) | BioLegend | 139304 |
| CD8α (53-6.7) | BioLegend | 100712 |
| CD90.2 (30-H12) or CD90.2 (53.2) | BioLegend or BD Biosciences | 105316 or 565257 |
| Donkey anti-chicken | Jackson Immuno Research | 703-545-155 |
| F4/80-biotin (BM8) LEC enrichment | eBiosciences | 13-4801-85 |
| GFP | Abcam | ab 13970 |
| GM-CSF (MP1-22E9) | BioLegend | 505404 |
| GM-CSF in vivo neutralization | Sterkel et al., 2016 | |
| Goat anti-rat | Invitrogen | A21434 |
| IL-17A (17F3) in vivo neutralization | BioXcell | BE0173 |
| IL-17A (TC11-18H10) | BD Biosciences | 559502 |
| IL-1α (ALF 161) | BioXcell | BE0243 |
| IL-1β (B122) | BioXcell | BE0246 |
| Ly6C (HK1.4) | BioLegend | 128014 |
| Ly6G (1A8) | BD Biosciences | 563978 |
| Ly6G-biotin (1A8) LEC enrichment | BioLegend | 127604 |
| MHC Class II (M5/114.15.2) | BioLegend | 107605 |
| MR1 tetramer (5-OP) |
Rahimpour et al., 2015 NIH Tetramer Facility |
|
| MR1 tetramer (6-FP) - control |
Rahimpour et al., 2015 NIH Tetramer Facility |
|
| NK1.1 (PK136) | BioLegend | 108710 |
| p65 (D14E12) | Cell Signaling | 8242S |
| Siglec F (E50-2440) | BD Biosciences | 565526 |
| TCRβ (H57-597) | BioLegend or BD Biosciences | 109222 or 560706 |
| TCRγδ (GL3) | BioLegend or BD Biosciences | 118120 or 563993 |
| Vγ1 (2.11) | BioLegend | 141105 |
| Vγ4 (UC3-10A6) | BioLegend | 137707 |
| Anti-human | ||
| CD3 (SK7) | BD Biosciences | 565466 |
| Ki67 (B56) | BD Biosciences | 565929 |
| TCRγδ (B1) | BD Biosciences | 564156 |
| Vγ9 (B3) | BioLegend | 331305 |
| Vδ2 (B6) | BioLegend | 331407 |
| Fungal Strains | ||
| Blastomyces dermatitidis | ATCC | 26199 |
| Blastomyces dermatitidis (spore-forming) | Clinical isolate Wisconsin State Laboratory of Hygiene | 14081 |
| Blastomyces dermatitidis DsRed | Sterkel et al., 2016 | |
| Biological Samples | ||
| Peripheral Blood Mononuclear Cells (PBMC) | Infected patients (University of Wisconsin Hospital and Clinics) and healthy controls | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant IL-17 | BioLegend | 576004 |
| Recombinant GM-CSF | PeproTech | 315-03 |
| Recombinant CCL20 | BioLegend | 582302 |
| Pertussis Toxin (PTx) | Calbiochem | 516560 |
| Critical Commercial Assays | ||
| IL-17 ELISA | R&D Systems | DY421 |
| CCL20 ELISA | R&D Systems | MCC200 |
| Experimental Models: Organisms/Strains | ||
| NFκB-eGFP | Dr. Christian Jobin Magness et al., 2004 |
|
| IL-17creRosa26ReYFP | Hirota et al., 2011 | |
| Dectin-1−/− | Dr. Gordon Brown Taylor et al., 2007 |
|
| Dectin-2−/− | Dr. Yoichiro Iwakura Saijo et al., 2010 |
|
| Dectin-3−/− | Consortium for Functional Glycomics and Mutant Mouse Resource and Research Centers (MMRRC) | 031935-UCD |
| CARD9−/− | Dr. Xin Lin Hsu et al., 2007 |
|
| Mincle−/− | Consortium for Functional Glycomics and Mutant Mouse Resource and Research Centers (MMRRC) | 031936-UCD |
| TLR 23479−/− | Dr. Carsten Kirschning Conrad et al., 2009 |
|
| DNTA and rtTA | Dr. Timothy S. Blackwell Cheng et al., 2007 |
|
| IL-1R−/− | The Jackson Laboratory | 003245 |
| IL-1RloxP/loxP (IL-1Rfl/fl) | The Jackson Laboratory | 028398 |
| MyD88−/− | The Jackson Laboratory | 009088 |
| TCRδ−/− | The Jackson Laboratory | 002120 |
| RAG−/−γc−/− | Taconic | 4111 |
| IKK2ΔLEC | Dr. Terry Wright Bello-Irrizarry, 2013 JI |
|
| IL-17A−/− | Dr. Yoichiro Iwakura Nakae et al., 2002 |
|
| Cas9-GFP | The Jackson Laboratory | 026175 |
| SPC-Cre | Backcrossed IKK2ΔLEC to WT to eliminate IKK2fl/fl allele | |
| Software and Algorithms | ||
| Prism | GraphPad Software | |
| FlowJo v10 | FLOWJO, LLC | |
In vivo Antibody Neutralization and Depletion
Two hundred and fifty micrograms of anti-IL-1α (BioXcell ALF 161), anti-IL-1β (BioXcell B122), anti-IL-17A (BioXcell 17F3), anti-GM-CSF (BioXcell MP1-22E9) and anti-CD4 (BioXcell GK1.5) were injected i.v retroorbitally at the time of inoculation with B. dermatitidis.
In vivo Administration of CCL20 and Pertussis Toxin (PTx)
100 ng of rCCL20 (BioLegend) and 400 ng of PTx were given i.t at the time of infection together with the inoculum (2 × 104 yeasts) in 20μl.
IL-17A and CCL20 ELISA
Mice were bled retroorbitaly and lungs were perfused with 10ml of PBS. BAL was collected by instilling 1ml of ice-cold PBS containing protease inhibitors (Completen Mini EDTA-free protease inhibitor tablets, Roche) via the trachea and aspirating it back with a syringe. BALF was obtained by centrifuging at maximum speed at 4°C to pellet the cells. The supernatant was collected and stored at −70°C. The lungs were collected in PBS containing protease inhibitors and homogenized. 1% Triton-X was added to the homogenate and debris was eliminated by centrifuging at maximum speed at 4°C and the supernatant was stored at −70°C. IL-17A and CCL20 content in BALF and lung homogenate was analyzed by ELISA according to the manufacturer’s specifications (R&D Systems).
In vivo Killing Assay
Mice were inoculated with DsRed yeast stained with 20μg/ml Uvitex and at 48 h.p.i. the lungs were harvested and processed for analysis by flow cytometry as described (Sterkel et al., 2016). When cytokine neutralization was required, antibodies were given i.v. at the time of inoculation.
Epithelial Cell Purification
The protocol for purification of airway epithelial cells was adapted from Messier, E.M. et. al. (Messier et al., 2012). Mice were bled retroorbitally and the lungs were perfused with 10ml of PBS. The lungs were then instilled with 2–3ml of undiluted 37°C Dispase via the trachea. The backflow of enzyme was prevented by injecting 0.5ml of 1% low-melt agarose subsequently solidified by icing the trachea. The lungs were then incubated in 2ml of Dispase at 37°C for 6 minutes and homogenized in pre-warmed (37°C) complete High Glucose DMEM containing DNAse (10 μg/ml) using a Gentle MACS dissociator (Miltenyi Biotec). The resulting single-cell suspensions were filtered sequentially through 70μm and 40μm nylon meshes. Non-specific binding of antibodies was blocked with anti-CD16/32, leukocytes (anti-CD45, anti-CD11c, anti-Ly6G, anti F4/80) and endothelial cells (anti-CD31) were labeled with biotinylated antibodies and subsequently depleted by positive selection with Streptavidin Microbeads (Miltenyi Biotec) in LS columns (Miltenyi Biotec). The resulting leukocyte- and endothelial cell-depleted fraction was labeled with anti-CD326 PE and epithelial cells were positively selected with anti-PE magnetic Microbeads and LS Columns. The purity of the epithelial cell fraction was verified by flow cytometry (Fig S7). Purified epithelial cells were lysed in NP40 lysis buffer (1% NP40, 50mM NaCl, 50mM Tris, 5mM EDTA) and lysates were analyzed for cytokine content by ELISA.
Bone Marrow Chimeras
Recipient mice received lethal irradiation in two sequential doses of 550Gy each (1,100Gy total), with a 3-hour resting period in between doses. Twenty-four hours post-irradiation, mice received 1 × 106 donor bone marrow cells i.v. Eight weeks after transplant, chimeric animals were infected with B. dermatitidis yeasts and 48-hours later the lungs were plated onto BHI agar for quantification of fungal burden.
NFκB Immunofluorescence
Mice were infected with 5 × 105 B. dermatitidis yeasts. In some experiments, yeast were labeled with 100 μg/ml calcofluor white. Lung tissue was fixed in 10% buffered formalin for 48 hours, embedded in paraffin and sectioned. In order to assess nuclear localization of NFκB by immunofluorescence, sections were deparaffinized and antigen retrieval was performed in citrate buffer pH 6.0 (10mM citric acid, 0.05% tween 20) for 3 minutes in a Biocare decloaker (Biocare Medical, Concord, CA)). After cooling and serum blocking, sections were incubated with 1:800 rabbit anti-p65 antibody (Cell Signaling, 8242S) in PBS with 1% goat serum overnight at 4C. After washing, sections were subsequently treated with 1:500 Alexa Fluor 488 goat antirabbit immunoglobulin (Invitrogen) for half an hour at room temperature, at which timepoint sections were washed and counter-stained with Prolong Gold antifade with DAPI (Invitrogen). All images displayed were taken at a total magnification of 400X, with identical microscope settings and image processing for all pictures. Individual airways from lung sections from each time point were examined, and the percent of lung epithelial cells with nuclear-localized NFκB was determined. When cryosections from NFκB-GFP reporter mice were used, the tissue was processed as described in the section Immunofluorescence to detect GM-CSF in the lung. Samples were stained with primary antibodies to GFP (1:1000) and CD326 (1:100) followed by secondary antibodies donkey anti-chicken Alexa Fluor 488 and goat antirat Alexa Fluor 555.
Immunofluorescence to detect GM-CSF in the lung
WT and GM-CSF−/− were infected with 5 × 105 B. dermatitidis yeasts and the lungs were processed for tissue sectioning 48 h.p.i. Six hours prior to harvest, mice were injected with 500μg i.p. and 42μg i.n. of Brefeldin A in order to retain cytokines in the cytoplasm. The lungs were fixed in 10% buffered formalin for 1 hour and subsequently cryoprotected by infusion with 30% sucrose for approximately 3 days. The left lobes were embedded in OCT and sectioned. Non-specific binding of antibodies was subsequently blocked for 1 hr at room temperature with vector lab animal-free blocking solution with 1% Tween. Samples were stained with 1:20,000 anti-CC10 (Seven Hills) and 1:100 anti-GM-CSF diluted in blocking solution overnight at 4°C. Antigens were visualized upon secondary staining with 1:1000 antirat Alexa Fluor 564 (Invitrogen) and 1:1000 antirabbit Alexa Fluor 488 (Jackson Immune Res) for 2 hours at room temperature in blocking solution. Confocal Images were collected on Nikkon A1R+ 20X optical with 2X digital zoom.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism and is specified in each figure legend.
Supplementary Material
Highlights.
Lung epithelial cells (LEC) mount a robust NFκB-dependent response to inhaled fungi
NFκB signaling in LEC regulates IL-17A- and GM-CSF producing innate lymphocytes
Fungal killing by innate immune cells is dependent on LEC NFκB, IL-17A and GM-CSF
IL-1α/IL-1R signaling in LEC promotes a CCL20-CCR6 axis to regulate innate lynphocytes
Acknowledgments
We thank the University of Wisconsin Carbone Cancer Center and Joe Hardin for preparation of Immunofluorescent lung sections; and Robert Gordon, Department of Pediatrics, for assistance with graphic illustrations. The work was supported by Burroughs Wellcome Fund grant 133-AAB4476 (NHS), NIAID T32AI007635 (AJM), American Heart Association 17POST32790004 (JSF), USPHS grant AI035681 (BSK), and by the Carbone Cancer Center Grant P30 CA014520.
Footnotes
AUTHOR CONTRIBUTIONS
N.H.S., D.W., J.S.F., A.M., T.W., M.W. designed and performed experiments; N.H.S. drafted the paper. B.S.K. helped design and supervise the study and write the paper.
Declaration of Interests. The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aimanianda V, Bayry J, Bozza S, Kniemeyer O, Perruccio K, Elluru SR, Clavaud C, Paris S, Brakhage AA, Kaveri SV, et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117–1121. doi: 10.1038/nature08264. [DOI] [PubMed] [Google Scholar]
- Balbi B, Valle MT, Oddera S, Giunti D, Manca F, Rossi GA, Allegra L. T-lymphocytes with gamma delta+ V delta 2+ antigen receptors are present in increased proportions in a fraction of patients with tuberculosis or with sarcoidosis. The American review of respiratory disease. 1993;148:1685–1690. doi: 10.1164/ajrccm/148.6_Pt_1.1685. [DOI] [PubMed] [Google Scholar]
- Bar E, Whitney PG, Moor K, Reis ESC, Leibundgut-Landmann S. IL-17 Regulates Systemic Fungal Immunity by Controlling the Functional Competence of NK Cells. Immunity. 2014 doi: 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Bello-Irizarry SN, Wang J, Olsen K, Gigliotti F, Wright TW. The alveolar epithelial cell chemokine response to pneumocystis requires adaptor molecule MyD88 and interleukin-1 receptor but not toll-like receptor 2 or 4. Infection and immunity. 2012;80:3912–3920. doi: 10.1128/IAI.00708-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nature reviews Immunology. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- Chen K, Eddens T, Trevejo-Nunez G, Way EE, Elsegeiny W, Ricks DM, Garg AV, Erb CJ, Bo M, Wang T, et al. IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe. 2016;20:596–605. doi: 10.1016/j.chom.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE, Blackwell TS. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. 2007;178:6504–6513. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- Chien YH, Meyer C, Bonneville M. gammadelta T cells: first line of defense and beyond. Annual review of immunology. 2014;32:121–155. doi: 10.1146/annurev-immunol-032713-120216. [DOI] [PubMed] [Google Scholar]
- Conrad ML, Ferstl R, Teich R, Brand S, Blumer N, Yildirim AO, Patrascan CC, Hanuszkiewicz A, Akira S, Wagner H, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. The Journal of experimental medicine. 2009;206:2869–2877. doi: 10.1084/jem.20090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Bruno VM, Childs EE, Daugherty S, Hunter JP, Mengesha BG, Saevig DL, Hendricks MR, Coleman BM, Brane L, et al. IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis. Cell Host Microbe. 2016;20:606–617. doi: 10.1016/j.chom.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Peterson AC, Brane L, Huppler AR, Hernandez-Santos N, Whibley N, Garg AV, Simpson-Abelson MR, Gibson GA, Mamo AJ, et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. The Journal of experimental medicine. 2014 doi: 10.1084/jem.20130877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe. 2010;7:140–150. doi: 10.1016/j.chom.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich-Nowoisky J, Pickersgill DA, Despres VR, Poschl U. High diversity of fungi in air particulate matter. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12814–12819. doi: 10.1073/pnas.0811003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nature reviews Immunology. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. 2013;190:521–525. doi: 10.4049/jimmunol.1202924. [DOI] [PubMed] [Google Scholar]
- Guerra ES, Lee CK, Specht CA, Yadav B, Huang H, Akalin A, Huh JR, Mueller C, Levitz SM. Central Role of IL-23 and IL-17 Producing Eosinophils as Immunomodulatory Effector Cells in Acute Pulmonary Aspergillosis and Allergic Asthma. PLoS pathogens. 2017;13:e1006175. doi: 10.1371/journal.ppat.1006175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig JS, Tonegawa S. Diversity of murine gamma genes and expression in fetal and adult T lymphocytes. Nature. 1986;322:836–840. doi: 10.1038/322836a0. [DOI] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nature immunology. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. The Journal of experimental medicine. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nature immunology. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Foxman EF, Molony RD. Early local immune defences in the respiratory tract. Nature reviews. Immunology. 2016 doi: 10.1038/nri.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson JM, Cauvi G, Witherden DA, Havran WL. A keratinocyte-responsive gamma delta TCR is necessary for dendritic epidermal T cell activation by damaged keratinocytes and maintenance in the epidermis. J Immunol. 2004;172:3573–3579. doi: 10.4049/jimmunol.172.6.3573. [DOI] [PubMed] [Google Scholar]
- Jhingran A, Kasahara S, Shepardson KM, Junecko BA, Heung LJ, Kumasaka DK, Knoblaugh SE, Lin X, Kazmierczak BI, Reinhart TA, et al. Compartment-Specific and Sequential Role of MyD88 and CARD9 in Chemokine Induction and Innate Defense during Respiratory Fungal Infection. PLoS pathogens. 2015;11:e1004589. doi: 10.1371/journal.ppat.1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara S, Jhingran A, Dhingra S, Salem A, Cramer RA, Hohl TM. Role of Granulocyte-Macrophage Colony-Stimulating Factor Signaling in Regulating Neutrophil Antifungal Activity and the Oxidative Burst During Respiratory Fungal Challenge. The Journal of infectious diseases. 2016;213:1289–1298. doi: 10.1093/infdis/jiw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komano H, Fujiura Y, Kawaguchi M, Matsumoto S, Hashimoto Y, Obana S, Mombaerts P, Tonegawa S, Yamamoto H, Itohara S, et al. Homeostatic regulation of intestinal epithelia by intraepithelial gamma delta T cells. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:6147–6151. doi: 10.1073/pnas.92.13.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, Vikram A, Good M, Schoenborn AA, Bibby K, et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity. 2016;44:659–671. doi: 10.1016/j.immuni.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggner U, Di Meglio P, Perera GK, Hundhausen C, Lacy KE, Ali N, Smith CH, Hayday AC, Nickoloff BJ, Nestle FO. Identification of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J Immunol. 2011;187:2783–2793. doi: 10.4049/jimmunol.1100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor perspectives in biology. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol. 2004;173:1561–1570. doi: 10.4049/jimmunol.173.3.1561. [DOI] [PubMed] [Google Scholar]
- Messier EM, Mason RJ, Kosmider B. Efficient and rapid isolation and purification of mouse alveolar type II epithelial cells. Experimental lung research. 2012;38:363–373. doi: 10.3109/01902148.2012.713077. [DOI] [PubMed] [Google Scholar]
- Millard PR, Wakefield AE, Hopkin JM. A sequential ultrastructural study of rat lungs infected with Pneumocystis carinii to investigate the appearances of the organism, its relationships and its effects on pneumocytes. Int J Exp Pathol. 1990;71:895–904. [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Paine R, 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, Reed JA, Ross G, Whitsett JA, Beck JM. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol. 2000;164:2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- Perez-Nazario N, Rangel-Moreno J, O’Reilly MA, Pasparakis M, Gigliotti F, Wright TW. Selective Ablation of Lung Epithelial IKK2 Impairs Pulmonary Th17 Responses and Delays the Clearance of Pneumocystis. J Immunol. 2013;191:4720–4730. doi: 10.4049/jimmunol.1301679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect. 2012;65:173–179. doi: 10.1016/j.jinf.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, Chen Z, Whittle B, Liu L, Fairlie DP, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. The Journal of experimental medicine. 2015;212:1095–1108. doi: 10.1084/jem.20142110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reibman J, Hsu Y, Chen LC, Bleck B, Gordon T. Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. American journal of respiratory cell and molecular biology. 2003;28:648–654. doi: 10.1165/rcmb.2002-0095OC. [DOI] [PubMed] [Google Scholar]
- Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, Silva-Santos B. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nature immunology. 2009;10:427–436. doi: 10.1038/ni.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Sierro F, Dubois B, Coste A, Kaiserlian D, Kraehenbuhl JP, Sirard JC. Flagellin stimulation of intestinal epithelial cells triggers CCL20-mediated migration of dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13722–13727. doi: 10.1073/pnas.241308598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonian PL, Roark CL, Wehrmann F, Lanham AM, Born WK, O’Brien RL, Fontenot AP. IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. J Immunol. 2009;182:6540–6549. doi: 10.4049/jimmunol.0900013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starner TD, Barker CK, Jia HP, Kang Y, McCray PB., Jr CCL20 is an inducible product of human airway epithelia with innate immune properties. American journal of respiratory cell and molecular biology. 2003;29:627–633. doi: 10.1165/rcmb.2002-0272OC. [DOI] [PubMed] [Google Scholar]
- Sterkel AK, Lorenzini JL, Fites JS, Subramanian Vignesh K, Sullivan TD, Wuthrich M, Brandhorst T, Hernandez-Santos N, Deepe GS, Jr, Klein BS. Fungal Mimicry of a Mammalian Aminopeptidase Disables Innate Immunity and Promotes Pathogenicity. Cell Host Microbe. 2016;19:361–374. doi: 10.1016/j.chom.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkel AK, Mettelman R, Wuthrich M, Klein BS. The unappreciated intracellular lifestyle of Blastomyces dermatitidis. J Immunol. 2015;194:1796–1805. doi: 10.4049/jimmunol.1303089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS., Jr Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39:697–710. doi: 10.1016/j.immuni.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy M, Jamora C, Havran W, Hayday A. Epithelial decision makers: in search of the ‘epimmunome’. Nature immunology. 2010;11:656–665. doi: 10.1038/ni.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Roy S, Leal SM, Jr, Sun Y, Howell SJ, Cobb BA, Li X, Pearlman E. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nature immunology. 2014;15:143–151. doi: 10.1038/ni.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nature immunology. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, Carey BC, Filippi MD, Wert SE, Denson LA, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356:567–579. doi: 10.1056/NEJMoa062505. [DOI] [PubMed] [Google Scholar]
- Unkel B, Hoegner K, Clausen BE, Lewe-Schlosser P, Bodner J, Gattenloehner S, Janssen H, Seeger W, Lohmeyer J, Herold S. Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. The Journal of clinical investigation. 2012;122:3652–3664. doi: 10.1172/JCI62139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li M, Lerksuthirat T, Klein B, Wuthrich M. The C-Type Lectin Receptor MCL Mediates Vaccine-Induced Immunity against Infection with Blastomyces dermatitidis. Infection and immunity. 2015;84:635–642. doi: 10.1128/IAI.01263-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CA, Salvage-Jones JA, Li X, Hitchens K, Butcher S, Murray RZ, Beckhouse AG, Lo YL, Manzanero S, Cobbold C, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008;180:7404–7413. doi: 10.4049/jimmunol.180.11.7404. [DOI] [PubMed] [Google Scholar]
- Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nature immunology. 2015;16:27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthrich M, Gern B, Hung CY, Ersland K, Rocco N, Pick-Jacobs J, Galles K, Filutowicz H, Warner T, Evans M, et al. Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. The Journal of clinical investigation. 2011;121:554–568. doi: 10.1172/JCI43984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.