

By the age of 3 years, his mother noted that his lower extremities grew in volume. Her attention was first drawn to this enlargement of his calves which entered his stockings with difficulty.
—Duchenne’s description of his first patient, Joseph Sarrazin, as cited by Tyler1
Mutations in the human dystrophin gene cause 2 clinical phenotypes, Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), distinguished principally based on the age at which patients lose the ability to ambulate.2,3 Boys with DMD become wheelchair users before 14 years of age, whereas those with BMD walk beyond age 16 years.4 The basis for this clinical distinction can largely be traced to the fact that DMD mutations result in the loss of the mRNA reading frame, virtually eliminating dystrophin protein production, and BMD mutations preserve the reading frame, allowing production of a partially functional protein.5 Muscles of patients with both forms express variable pathologic changes that generally lead to profound muscle atrophy. In contrast, some muscles, most notably the gastrocnemius, enlarge. Although hypertrophy hastypically been attributed to deposition of fat and connective tissue, so-called pseudohypertrophy,6,7 imaging studies have shown true hypertrophy in some individuals.
There are 3 mammalian models in which spontaneous dystrophin gene mutations lead to distinct phenotypes of muscular dystrophy: the mdx mouse,8,9 golden retriever muscular dystrophy (GRMD) dog,10,11 and feline hypertrophic muscular dystrophy (FHMD) cat.12,13 The GRMD model most closely mirrors DMD at multiple levels, including progressive disease that leads primarily to muscle atrophy. The mdx mouse progresses through an initial phase of muscle hypertrophy followed in old age by atrophy. In contrast, the FHMD cat has persistent muscle hypertrophy. The role of true hypertrophy has been more broadly accepted in these models, with less attention paid to contributions made by fat and connective tissue in muscle enlargement.
In principle, relative muscle sparing or hypertrophy could be either beneficial or detrimental. The benefits are obvious because preservation of strength should enhance motor function in activities ranging from ambulation to breathing. Harmful effects are less clear and relate to the potential for muscle hypertrophy to exacerbate contractures and postural instability. Additional deleterious consequences occur due to difficulties in eating because of glossal hypertrophy and regurgitation resulting from either esophageal hypomotility or obstruction at the level of the hypertrophied diaphragm. Whether the hypertrophy results from an actual increase in muscle mass or fat and connective tissue, studies directed at defining underlying mechanisms could provide insight into the pathogenesis of dystrophin deficiency and inform treatment development.
VARIABLE MUSCLE INVOLVEMENT AND HYPERTROPHY
Effects of dystrophin deficiency vary among species, individuals, and muscles. Reasons for phenotypic variation are poorly understood and raise questions about primary versus secondary effects of dystrophin deficiency.14
DMD and BMD
Extensor muscles that undergo eccentric muscle contraction, such as the quadriceps femoris, are particularly vulnerable in DMD.15 In contrast, the extraocular muscles16 are largely spared, and other muscles undergo striking paradoxical hypertrophy. Although attention has focused on gastrocnemius (calf) enlargement (Fig. 1), hypertrophy occurs in many other muscles. Presumptive cases of DMD characterized by hypertrophy of the calves, deltoid, and infraspinatus muscles were seen in Italy and England as early as the 1830s well before Duchenne’s classic account.1 In a 1995 review of 84 patients with DMD from India, 94% had calf enlargement, followed by the infraspinatus (88%), deltoid (52%), and tibialis anterior (40%).17 The selective muscular involvement extended to different heads of the deltoid and quadriceps, which showed concomitant atrophy and hypertrophy. Calf hypertrophy was evident on physical examination in 20 of 26 (77%) BMD cases in another study.18 These patients were further studied with computed tomography to determine the pattern and course of muscle involvement. Hypertrophy was seen in the calves (42%), sartorius (42%), gracilis (42%), adductor longus (38%), semitendinosus (19%), and rectus femoris (11%). Patients with DMD and/or BMD have also been shown to have hypertrophy of the tongue (macroglossia),19 diaphragm,20 and hypothenar (palm)21 muscles, among others.
Fig. 1.
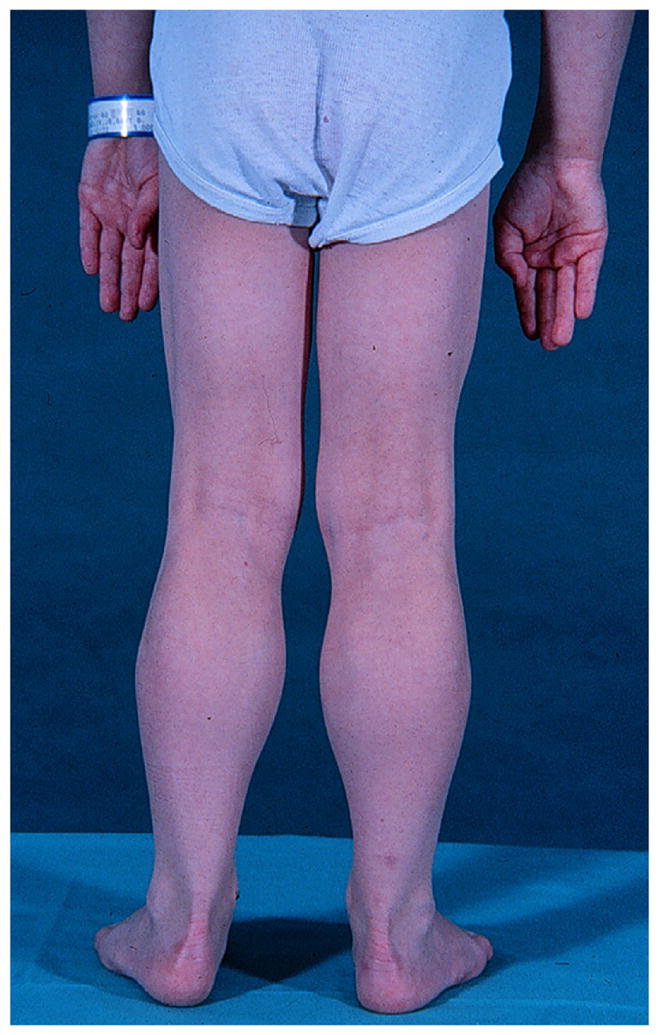
Nine-year-old boy with DMD demonstrating characteristic calf hypertrophy. (Courtesy of James F. Howard Jr.)
Dating to Duchenne’s monograph, muscle hypertrophy in DMD and BMD has been attributed to deposition of fat and connective tissue, giving rise to the term pseudohypertrophic muscular paralysis.1 Indeed, histopathologic studies have documented fibrosis and fatty change in the calves and other hypertrophied muscles.6,7 Walton22 speculated that true hypertrophy also contributes to muscle enlargement, perhaps occurring early in the disease course, followed by pseudohypertrophy. However, because of inherent limitations of sequential muscle sampling in human patients, the time course and relative roles of true hypertrophy and pseudohypertrophy have remained unsettled. Specialized imaging techniques have complemented and, in some cases, essentially replaced histopathologic evaluation of muscle biopsy samples. Increasingly, use of these techniques has documented features consistent with a true increase in contractile mass. In one such study, 10 of 16 patients with BMD had calf enlargement on ultrasonography, and 9 of them were judged to have true hypertrophy.23 Another study, cited earlier, found that enlarged muscles visualized with computed tomography were often rounded and had normal densities, suggesting true hypertrophy.18 A further paper, in which magnetic resonance imaging (MRI) was evaluated, showed that the gracilis and sartorius muscles were relatively spared and/or hypertrophied in 10 patients with DMD.24 We are particularly intrigued by the relative sparing of the sartorius muscle (Fig. 2) because of our own studies of cranial sartorius hypertrophy in GRMD (see later).25
Fig. 2.
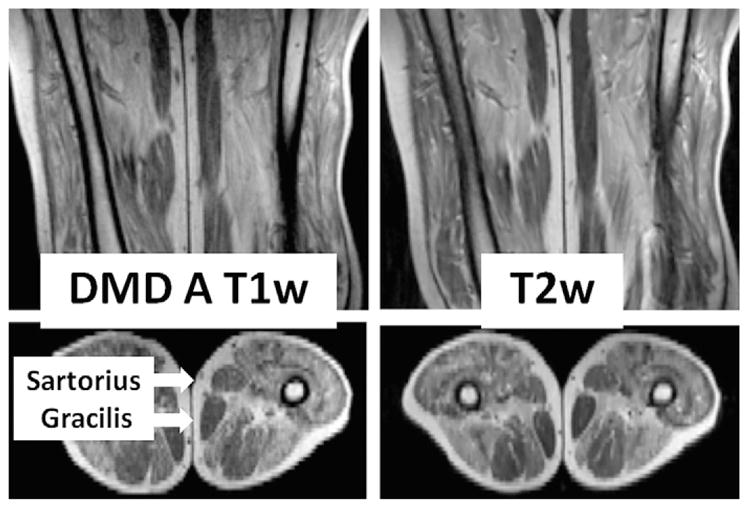
T1- and T2-weighted magnetic resonance images of the proximal leg of a boy with DMD in the anteroposterior (top) and transverse (bottom) planes. The transverse image is at the level of the midfemur. Most muscles have been partially to near totally replaced with signal-intense material compatible with fat. Note that the subcutaneous fat has comparable signal intensity in both T1- and T2-weighted images. In contrast to the properties of most muscles seen, the sartorius and gracilis muscles (arrows) are largely unaffected.
Ideally, imaging and histopathologic results should be correlated to define the relative contributions that fat, fibrous connective tissue, and myofiber hyperplasia or hypertrophy make to muscle enlargement. Few such studies have been completed in DMD. In one paper that correlated computed tomography and histochemistry findings, apparent true hypertrophy of the gastrocnemius in a 7-year-old boy with DMD was judged to be caused by myofiber hyperplasia rather than hypertrophy.6 With this said, hypertrophied myofibers are also seen early in the disease and persist throughout life.7
The mdx Mouse
More extensive pathologic studies can be done with animal models, potentially allowing better definition of mechanisms contributing to differential muscle involvement. Just as in DMD, muscles are variably affected in the mdx mouse, ranging from the unaffected extraocular muscles26 to the severely involved diaphragm.27 Limb muscles undergo dramatic necrosis at 3 to 4 weeks of age followed by robust regeneration and muscle hypertrophy.28–31 Changes vary among muscles, with the predominantly slow twitch soleus being more involved at 3 to 4 weeks29,32,33 and the fast twitch extensor digitorum longus at 32 weeks and beyond.29,33 Muscle hypertrophy causes mdx mice to be larger than normal between about 10 and 40 weeks of age, after which they lose weight in concert with a loss of muscle mass.28,29,33 Reflecting the early strong regenerative response, mdx mice have increased numbers of small regenerating fibers with central nuclei at 3 weeks of age.28,32,34 The percentage of larger regenerated myofibers increases with age, approaching 70% and 90% in the soleus and extensor digitorum longus, respectively, by 26 weeks.32 Because populations of both small and large fibers occur concomitantly, mean fiber diameter of mdx mice typically is normal.28,35 The concomitant occurrence of large and small fibers suggests that both hyperplasia and hypertrophy contribute to muscle hypertrophy.31
Unlike DMD, mdx mice have minimal fatty change and fibrosis,33 indirectly implicating true hypertrophy as the cause for muscle enlargement.31 The time course of pathologic lesions in the mdx mouse leads to corresponding functional changes, with affected mice being weaker at 2 to 4 weeks and then recovering.36–38 Absolute tension generated by mdx muscles is typically higher by 8 to 16 weeks of age.31,36,39 When corrected for cross-sectional area, isomeric soleus muscle force of younger (≤100 days) mdx mice was lower than normal, whereas values for older (≥100 days) mdx mice were higher.39 Coulton and colleagues39 commented that, “when mdx mice lose muscle fibers by necrosis, they do not just replace them with equally efficient new muscle, but with heavier, stronger, muscles than the wild-type.” Typical of this pattern and consistent with our own findings in the GRMD dog (see later), isometric tension generated by the tibialis anterior is considerably lower in mdx mice at 3 to 4 weeks36 but exceeds normal values by 8 to 16 weeks.31,36 Pathologic changes in the tibialis anterior also occur earlier than those in the soleus and gastrocnemius muscles.36 Taken together, these mdx mouse data show clear evidence that early necrosis leads to a dramatic regenerative response that can lead to functional hypertrophy.
Contraction kinetics in mdx mice also differ from normal. Relaxation times were increased in the soleus, independent of age,40 and in the tibialis anterior at 3 to 4 weeks36 but not at later ages.36,37 Although twitch-tetany ratios were decreased compared with normal in the soleus of variably aged mdx mice,40 a reverse relationship was seen in the tibialis anterior at 7 to 8 weeks.37 Relaxation times of the mdx tibialis anterior were also increased at 3 to 4 weeks,36 but values normalized in older mice.36,37
FHMD
Male cats with weakness, histologic features typical of muscular dystrophy, and gross and histologic evidence of muscle hypertrophy were characterized before the discovery of dystrophin.41 Subsequently, an analogous syndrome was reported in cats with dystrophin deficiency.12,13,42 One of these FHMD cats was shown to have a dystrophin gene deletion that included the skeletal muscle and neuronal Purkinje cell promoters and first exons.42 The most striking clinical feature in all these cats was gross hypertrophy of their axial and appendicular muscles, as well as the tongue, diaphragm, and esophageal muscularis. In one study, 4 muscles (biceps brachii, cranial tibialis, gastrocnemius, and diaphragm) from 2 FHMD cats weighed twice as much as those from comparably sized normal cats.12 Physical evidence of hypertrophy has been noted as early as 3 months13 and seems to become more pronounced with age. As an example, the circumference of the neck in one cat increased from 28 to 33 cm between 14 and 25 months of age.12 On microscopic examination, FHMD cats have myofiber size variation, with increased populations of both small regenerating and hypertrophied myofibers and an overall mean myofiber diameter in the normal range.43 In keeping with progressive hypertrophy, the mean myofiber diameter of FHMD cats increased more so than that of normal cats between 3 to 4 and 6 to 9 months of age. In addition, there was myofiber necrosis and splitting and both gross and individual myofiber mineralization. The relative absence of fibrosis in FHMD cats up to 2 years of age suggests that the muscle enlargement is most likely because of true hypertrophy.12
GRMD Dog
As with DMD and the mdx mouse, the extraocular muscles are largely spared in GRMD.44,45 Other muscles become involved as a function of age and usage. Muscles that are used heavily in utero and early in life, such as the tongue, diaphragm, and limb flexors, are acutely necrotic during the neonatal period.44,45 Extensor muscles demonstrate a more delayed pattern of involvement, reflecting their greater use in weight bearing. As with the mdx mouse, muscles that undergo early necrosis may then regenerate and even hypertrophy. In one of the original GRMD dogs studied by our group, the hamstrings, tongue, diaphragmatic crura, and esophageal muscularis were enlarged.10
A further study in our laboratory showed that most GRMD pelvic limb muscles atrophy, whereas the caudal and cranial sartorius and popliteus hypertrophy.25 Cranial sartorius muscle weights were corrected for body weight and endomysial space to determine true muscle weights (g/kg) in 3 GRMD age groups (4–10, 13–26, and 33–66 months) and grouped normal dogs (6–20 months) (Table 1). Corrected GRMD weights in the younger dogs were greater than those of normal dogs, indicating that the cranial sartorius undergoes initial true muscle hypertrophy. Values of both older groups were less than those of the younger dogs, suggesting that the cranial sartorius muscle atrophies over time, with an associated increase in the endomysial space because of deposition of fat and connective tissue.
Table 1.
Morphometric GRMD vs. normal canine cranial sartorius histopathologic data (mean ± SD)
| Pathologic Lesion | Normal (6–20 mo; n = 12) | GRMD | ||
|---|---|---|---|---|
| Group 1 | Group 2 | Group 3 | ||
| 4–10 mo; n = 15 | 13–26 mo; n = 4 | 33–66 mo; n = 4 | ||
| Corrected muscle weight (g/kg)a | 1.3075 ± 0.2079 | 3.0573 ± 0.7635 | 2.4725 ± 0.7556 | 1.4650 ± 0.6575 |
| Percentage of endomysial spaceb | 2.8083 ± 1.3468 | 27.1857 ± 15.3869 | 44.1275 ± 14.6462 | 58.9100 ± 14.2060 |
| True muscle weight (g/kg)c | 1.2699 ± 0.1966 | 2.2063 ± 0.6884 | 1.3758 ± 0.5078 | 0.5720 ± 0.2423 |
| Mean fiber diameter (μm)d | 42.1658 ± 4.3542 | 57.4980 ± 11.7419 | 63.1295 ± 13.3033 | 47.0850 ± 17.7029 |
Normal<GRMD, Group 1 (P<.05); GRMD Group 1>GRMD Group 3 (P<.05).
Normal<GRMD Groups 1–3 (all P<.05).
Normal<GRMD, Group 1 (P<.001); GRMD Group 1>GRMD Groups 2 (P<.05) and 3 (P<.001).
Normal<GRMD Groups 1 and 2; P<.01 for Group 1 and P<.05 for Group 2.
Data from Kornegay JN, Cundiff DD, Bogan DJ, et al. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord 2003;13:493–500.
Our studies of force/torque generated by individual and grouped GRMD muscles are in keeping with these pathologic data and findings from mdx mice. We initially evaluated tension generated by the peroneus longus muscle.46 Absolute twitch tension and both muscle- and body-weight-corrected twitch tension in GRMD dogs were low compared with normal littermates at 3 months of age. Tetanic tension was affected similarly. However, although absolute values were still reduced at 6 months, twitch and tetanic tension corrected for either muscle or body weight was not statistically different (Fig. 3), suggesting that the peroneus longus recovers from an initial period of necrosis. Moving forward, we have primarily evaluated force/torque generated by tarsal joint flexors (including the cranial tibialis and peroneus longus) and extensors (including the gastrocnemius and superficial digital flexor that is analogous to the soleus).47 For these measurements, the peroneal and tibial nerves are stimulated percutaneously so that the paw pulls (peroneal nerve, flexion) or pushes against (tibial nerve, extension) a lever interfaced with a force transducer. In our initial study, force values were measured at 3, 4.5, 6, and 12 months of age. While absolute and body-weight-corrected GRMD twitch and tetanic force values were lower than normal at all ages, tarsal flexion and extension were differentially affected (Fig. 4). Flexion values were especially low at 3 months, whereas extension was affected more at later ages.
Fig. 3.
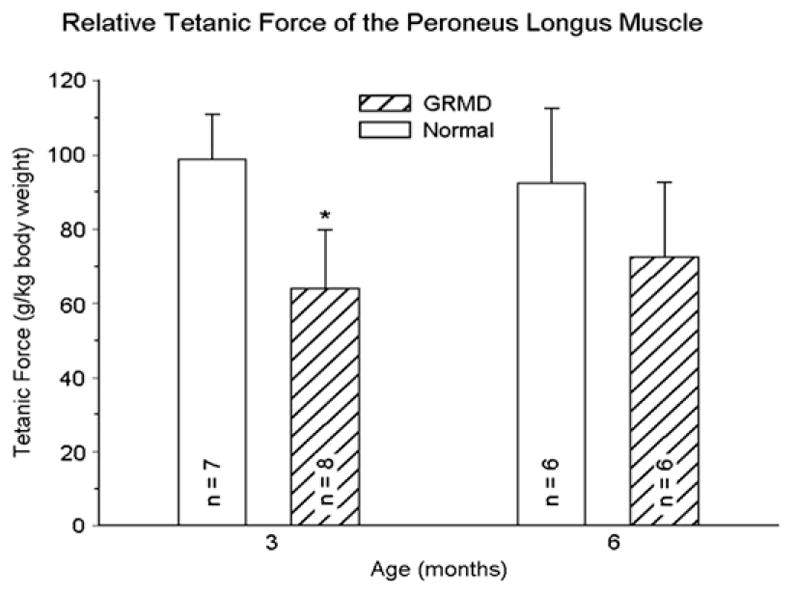
Tetanic force corrected for body weight (g/kg) generated by the peroneus longus muscle of normal and GRMD dogs at 3 and 6 months of age. Values for the GRMD dogs are lower (P<.05)* at 3 months. However, although body-weight-corrected force is proportionally lower in normal dogs at 6 months, it has increased in GRMD dogs, such that values for the 2 groups are no longer significantly different. (Data from Kornegay JN, Sharp NJ, Bogan DJ, et al. Contraction tension and kinetics of the peroneus longus muscle in golden retriever muscular dystrophy. J Neurol Sci 1994;123:100–7.)
Fig. 4.

Tetanic force, corrected for body weight (N/kg), generated by tarsal joint flexors (left) and extensors (right) from normal dogs and GRMD dogs at 3, 4.5, 6, and 12 months of age. Values for GRMD dogs are lower (P<.01 for all) than those of normal dogs at all ages. However, the differential between GRMD and normal dogs differs. Flexion values are especially low at 3 months, whereas extension is affected more at later ages. (From Kornegay JN, Bogan DJ, Bogan JR, et al. Contraction force generated by tibiotarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci 1999;166:119; with permission.)
We have used tetanic tarsal joint force measurements to evaluate effects of prednisone (2 mg/kg) given to GRMD dogs for a 4-month period beginning at 2 months of age.48 Tarsal extension force increased in treated versus control GRMD dogs, whereas flexion paradoxically decreased. In light of our force studies, we assumed that the paradoxical decline in flexion occurred because prednisone attenuated early necrosis in muscles such as the peroneus longus and cranial tibialis that would have otherwise led to functional hypertrophy.
Contraction kinetics in GRMD dogs for both the peroneus longus46 and grouped tarsal joint flexors and extensors47 also differed from normal. Post-tetanic potentiation for the peroneus longus was more pronounced in GRMD versus normal dogs at both 3 and 6 months. Twitch contraction and relaxation times were dramatically prolonged, and there was concomitant sustained electrical activity at or before 6 months of age in some severely affected dogs. For the grouped muscles, the twitch-tetany ratio was generally lower, post-tetanic potentiation for flexion values was less marked, and extension relaxation and contraction times were longer. As discussed further later, prolonged relaxation times with underlying sustained electrical activity and more generalized complex repetitive discharges could play a role in muscle hypertrophy.
POSTURAL INSTABILITY AND CONTRACTURES
Comparative aspects of gait abnormalities, postural changes, and joint contractures in humans and animal models must be interpreted in light of fundamental differences in conformation, some of which arise because of quadrupedal versus bipedal locomotion. As an example, the line of the pelvis from the tuber ischium to the wing of the ilium extends in a vertical plane, perpendicular to the walking surface, in humans but is oriented more horizontally, largely parallel to the ground, in quadrupeds. Thus, changes in pelvic orientation innately differ among patients with DMD, mdx mice, and GRMD dogs. In some cases, muscle weakness in DMD causes the posture to shift toward one normally assumed by quadrupeds and vice versa. The pelvis tilts anteriorly toward a more horizontal position to maintain postural stability in DMD49 but shifts toward a vertical plane in GRMD dogs50 (see later). Another factor relates to whether the leg/limb stance is plantigrade (humans and mice) or digitigrade (dogs).51,52 Humans and mice walk on their phalanges, carpal, and tarsal bones, whereas dogs normally walk only on their distal and intermediate phalanges (digits). With neuromuscular diseases, in general, dogs become more plantigrade in the pelvic limbs,53,54 adopting a stance more in keeping with that normally used by humans. A third stance, termed unguligrade, involves walking only on the tip of the distal digit.52,55 The fact that horses have this posture explains use of the term equinus in DMD patients who toe walk.
DMD and BMD
Most DMD natural history studies include measurements of muscle strength, joint contractures, and timed function tests. Results from these tests are used to track disease progression and offer insight on clinical milestones, such as the loss of ambulation and the need for ventilatory support. Contracture and muscle strength scores generally correlate, deteriorate synchronously over time, and contribute mutually to postural instability.56,57 As discussed later, unequal muscle weakness in DMD precipitates a vicious cycle that can lead to debilitating contractures and loss of ambulation (Fig. 5).58
Fig. 5.
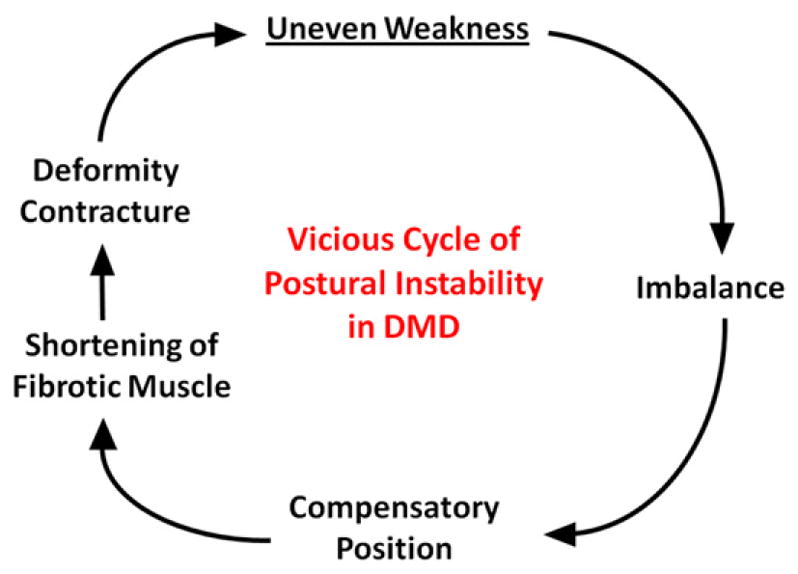
Vicious cycle of postural instability that leads to loss of ambulation in patients with DMD. Uneven weakness leads to imbalance and compensatory postural changes that ultimately result in shortening of muscles and contractures. The process is self-perpetuating. (Modified from Roy L, Gibson DA. Pseudohypertrophic muscular dystrophy and its surgical management: review of 30 patients. Can J Surg 1970;13:14; with permission.)
Generally, contractures are caused by inactivity and restricted motion of the affected joint,59,60 with a subsequent increase of collagen cross-links in periarticular connective tissue.61 Major causes include forced joint immobilization to stabilize fractures,60 spasticity associated with upper motor neuron lesions,62 and primary neuromuscular diseases.63 Joint contractures occur more commonly in DMD than other neuromuscular diseases63 and have long been recognized as a major factor in disease morbidity. In one review that included 43 patients with DMD, the ankle (34/43), knee (29/43), hip (29/43), and elbow (28/43) were affected most frequently.63 The ankle was also most commonly affected in BMD, occurring in 4 of 7 patients. Despite the prominent role that joint contractures play in these dystrophies, causative mechanisms are not fully understood. Underlying abnormal positioning presumably occurs because of an imbalance of forces acting on the joint, because diseased muscles are either disproportionately weakened or shortened by fibrosis. Relevant to this review, several studies have assessed the proportional strength of agonist and antagonist muscles operating at joints of patients with DMD. Some studies have concluded that muscular imbalance contributes to contractures,57,64–66 whereas another found no such association.67 Those finding a relationship noted a strong negative correlation between extensor muscle weakness and flexor contracture severity in DMD. As opposing extensor muscles weakened, flexor contractures worsened. Disproportionate flexor muscle hypertrophy would logically exaggerate this process. In a somewhat similar vein, contractures occurring because of spasticity also are magnified by enhanced flexor muscle activity.62
Postural instability and leg contractures are of special interest in DMD because of their role in the loss of ambulation. The relative sequence and proportional involvement of flexor and extensor muscles is critical. Early weakness of the hip (gluteus maximus) and knee (quadriceps femoris) extensors necessitates postural changes to maintain ambulation. Increased anterior pelvic tilt and lumbar lordosis are adopted to shift the center of gravity forward of the knee and behind the hip, respectively.49 Johnson68 suggested that this posture allows passive ligamentous stabilization of these joints. Of comparative interest, a somewhat analogous system of ligamentous support (stay apparatus) allows horses to remain standing while sleeping.69 Relative preservation of hip (including the sartorius) and knee (hamstrings) flexors in DMD creates destabilizing torque forces and also contributes to contractures at both levels.70 Toe walking is adopted to stabilize the knee and later plays a role in the development of ankle equinus.71 Plantar flexor contractures associated with equinus are aggravated by unbalanced muscle activity at the ankle, with selective weakening of the tibialis anterior and peroneus longus muscles and relative sparing of the triceps surae (collectively, the 2 heads of the gastrocnemius and soleus).57,71 One investigator characterized this as a return to the infantile digitigrade pattern of walking, perhaps in an effort to put less stress on the weakened tibialis anterior muscle.72 These contractures may initially have beneficial effects because tension in the gastrocnemius muscles pulls on the femoral condyles, extending the knee.73 Iliotibial band (hip) contractures also extend the knee, providing additional stability. However, in advanced stages, heel cord and iliotibial band tightening destabilize gait, prompting the development of various corrective surgical procedures.65,74 Scoliosis is typically a late complication of DMD, occurring in boys after they have gone into wheelchairs, with the side of convexity almost always toward the dominant hand.75 The developing spine is thought to become deformed by excessive unbalanced forces from the dominant extremity. Scoliosis restricts expansion of the chest and tracks closely with respiratory disease.76 Importantly, this biomechanical interplay in scoliosis shows that application of disproportionate (asymmetric) muscle force to the developing skeleton can cause bony maldevelopment with clinical consequences.
As discussed earlier, mechanisms contributing to preferential calf and other flexor muscle sparing or hypertrophy, starting with whether there is an actual increase in contractile tissue, are especially intriguing. If one accepts that these muscles undergo true hypertrophy in at least some patients, the timing and functionality of the muscle enlargement and whether it is playing a beneficial or detrimental role become critical. This would be particularly relevant with treatments, such as myostatin inhibition, that are intended to increase muscle mass (see later).
The mdx Mouse
Kyphosis analogous to scoliosis in DMD has been characterized in the mdx mouse. Just as with DMD, there is an association between spinal deformity and respiratory compromise. Mechanisms to account for kyphosis have not been defined, although Laws and Hoey77 speculated that muscle hypertrophy could contribute. In a separate proof-of-concept study, this same group evaluated the potential for intramuscular injection of antisense oligonucleotides into paraspinal muscles to ameliorate kyphosis.78 The treated group had less thoracic deformity than controls, but other outcome parameters did not differ between the groups. Weights of the latissimus dorsi, diaphragm, and intercostals muscles were increased in mdx mice in both treated and control mice. Force generation by these muscles was not increased, and histologic features were more in keeping with muscle degeneration and fibrosis than true hypertrophy. Thus, although not the central focus of this study, there was not a definite link between muscle hypertrophy and kyphosis.
Contractures of the tarsal (talocrural, ankle) joint have recently been characterized in the mdx mouse.79 As discussed earlier, both mice and humans have a plantigrade stance, so there could be analogous operative forces. In keeping with findings from DMD, mdx mice had plantar flexor contractures, evidenced by decreased dorsiflexion and range of motion. There was an associated increase in gastrocnemius wet muscle weight, and torque generated by the dorsiflexors was proportionally lower than that of the gastrocnemius muscles.
FHMD
Because of their muscle hypertrophy, FHMD cats have a stiff stilted gait with their hocks adducted.12,13 They adopt a “falling down technique” to move into lateral recumbency and are unable to turn their heads and groom.12 FHMD cats also do not close their mouths completely, presumably because of glossal hypertrophy,12,13 which interferes with eating and swallowing, ultimately causing dehydration and azotemia.13 The esophagus may have decreased contractility13 and can be constricted by the hypertrophied diaphragm,12,13 resulting in regurgitation. A syndrome similar to malignant hyperthermia has been characterized in several cats that died during stress or anesthesia (see discussion linking calcium homeostasis in this condition and muscle hypertrophy).42,80
GRMD Dog
Gait and postural changes in GRMD must be considered in the context of marked phenotypic variation among affected dogs.81,82 Severely affected dogs may die within the first 10 days of life,81 whereas others live well into adulthood.83 By 6 weeks of age, GRMD dogs advance their pelvic limbs simultaneously (bunny hopping) and subsequently exhibit a progressively more stilted gait.81 Those with a severe phenotype develop a characteristic plantigrade stance between 3 and 6 months, as evidenced by hyperextension of the carpus and hyperflexion of the tarsus.81,84 Over this same period, the elbows become abducted and the hocks are adducted. Concomitantly, the pelvic limbs shift forward, as the tuber ischium of the pelvis moves ventrally and cranially. In severe cases, the line of the pelvis may be oriented in an essentially vertical plane, perpendicular to the walking surface (Fig. 6).50 Some of these dogs lose the ability to walk and must be euthanized.
Fig. 6.

Lateral (A) and ventrodorsal (B) radiographs of the pelvis of a 4-year-old GRMD dog and a normal dog (C and D) for comparison. Note the marked vertical tilt of the pelvis in the GRMD dog (A), so that the angle formed by the wing of the ilium and lumbar spine is much more acute at approximately 90° (angle marked by lines within the red circle). The wings of the ilia flare laterally in the GRMD dog (red circle in B). (From Brumitt JW, Essman SC, Kornegay JN, et al. Radiographic features of Golden Retriever muscular dystrophy. Vet Radiol Ultrasound 2006;47:578; with permission.)
Beyond the initial 6 months of life, clinical signs in GRMD dogs tend to stabilize and further adaptive changes help to maintain postural stability. With both video gait analysis85 and accelerometry,86 GRMD dogs walk more slowly and their stride length is decreased. Accelerometry demonstrated a redistribution of power at gait, with a decrease in the craniocaudal plane and a compensatory increase mediolaterally to maintain balance.86 On video gait analysis, older, generally mildly affected dogs had a more upright stance, with relatively greater extension of the stifle and lesser flexion of the tarsus.85 This posture presumably is adopted in an effort to stabilize their stance in the face of quadriceps weakness. In our experience, GRMD dogs rarely become nonambulatory after the initial critical period of destabilization at 3 to 6 months of age. Some have a normal life span; we currently have a 12-year-old GRMD dog with a remarkably mild phenotype in our colony.
One of our earlier studies showed that 6-month-old GRMD dogs positioned in dorsal recumbency for force measurements have abnormally acute (contracted) tarsal joint angles.46,84 Other investigators have subsequently described methods to measure joint angles at maximal flexion and extension in normal dogs.87,88 We now use the method suggested by Jaegger and colleagues87 to measure pelvic limb joint angles and range of motion. By 6 months of age, GRMD dogs tend to have more restricted maximal flexion of the hip joint, increased maximal stifle extension, and more acute maximal tarsal flexion. To objectively characterize the cranioventral shift of the pelvis, we also measure the angle formed by 2 lines extending cranially from the tuber ischium, one drawn parallel to the lumbar spine and the other extending to the midpoint of the tuber coxae. This angle is larger in dogs with GRMD than in normal dogs at 6 months of age.
Mechanisms contributing to postural and joint angle changes in GRMD dogs have not been defined. The cranioventral pelvic shift may be an adaptive response, as affected dogs move their pelvic limbs under the torso to maintain balance. The resultant posture is similar to that achieved by boys with DMD when they shift their pelvis forward.49 Alternatively, unbalanced hip flexor and extensor strength might play a role. Relative preservation of the hamstring muscles in GRMD dogs could pull the tuber ischium ventrally and also contribute to decreased maximal hip flexion values. As discussed earlier, considering the role that the sartorius and iliotibial band play in hip flexor contractures in DMD,74 true hypertrophy of the cranial sartorius could be playing an analogous role in GRMD. In support of a relationship, we have previously shown that cranial sartorius circumference correlates negatively with tarsal joint angle in affected dogs.25 Still, it is unclear whether there is truly a cause-and-effect relationship. The hypertrophied cranial sartorius could actively pull the stifle joint forward, with the tarsus passively following to assume a plantigrade position. On the other hand, cranial sartorius hypertrophy and plantigrade stance might have common root causes but no direct functional relationship. In any case, cranial sartorius hypertrophy or contracture does seem to affect the developing pelvis in young GRMD dogs, as the ilial wings from which it originates flare laterally (see Fig. 6),50 presumably in response to unopposed torque. This is somewhat analogous to scoliosis resulting from unbalanced force applied by the dominant arm in boys with DMD75 and emphasizes the potential for disproportionate muscle size and strength to cause skeletal deformity.
Local imbalance of agonist and antagonist muscles could also be playing a role in postural changes at the tarsal and carpal joints of GRMD dogs. Consistent with findings in DMD, we have shown that GRMD extensor and flexor muscles operating at the tarsal joint are differentially affected. Flexion values are especially low at 3 months, whereas extension is affected more at later ages (see Fig. 4).47 At 6 months of age, the tarsal extension-flexion force ratio correlates positively with tarsal joint angle, which is to say that dogs with stronger extensors have larger joint angles and a less severe phenotype.89 We have not systematically studied joint angles in the thoracic limb and can only speculate on mechanisms involved in carpal hyperextension. It seems unlikely that a reverse pattern of muscle involvement, whereby extensor muscles are relatively preserved, is responsible. Importantly, plantigrade stance is a nonspecific clinical sign in dogs and cats with neuromuscular disease,53,54 independent of underlying cranial sartorius hypertrophy. This suggests that the pelvic limb plantigrade posture, coupled with hyperextension of the carpus (so-called palmigrade stance),53 may simply reflect distal muscle weakness. The pivot point would logically differ between the carpus (hyperextension) and tarsus (hyperflexion) to keep the footpads in contact with the walking surface.
Spinal curvature is not a major feature of GRMD. Lumbar kyphosis may occur in tandem with the cranioventral shift of the pelvis; lordosis can be seen more chronically.81 The lordotic posture could occur because of unequal application of force on the developing spine, with relative restriction of vertebral growth dorsally (posteriorly) or exaggerated growth ventrally (anteriorly).90,91 Relative preservation of the psoas major muscle in GRMD dogs25 might exert disproportionate force on the ventral lumbar spine, contributing to both hip flexor contractures and lordosis.
MECHANISMS CONTRIBUTING TO MUSCLE HYPERTROPHY
Differences in the severity, distribution, and nature of pathologic lesions leading to muscle enlargement in DMD and the 3 animal models should offer clues on mechanisms contributing to muscle hypertrophy in dystrophin deficiency. In the mdx mouse and GRMD cranial sartorius, muscle hypertrophy follows early necrosis. We believe that a convergence of factors driving muscle regeneration and differentiation leads to a disordered or overly robust proliferative response in GRMD. Although it is tempting to speculate on a similar mechanism in DMD and FHMD, no direct evidence links early necrosis and hypertrophy in these diseases. The relative lack of fatty infiltration in muscles of the mammalian models raises additional mechanistic questions relating to the efficiency of muscle regeneration among species and their propensity for fatty change secondary to muscle injury. These questions are generally beyond the scope of this article. Here, we review central concepts and suggest potential drivers for muscle hypertrophy in light of findings in affected animals.
Muscle mass can expand through either increased cell number (hyperplasia) or size (hypertrophy), with proliferation playing a greater role in developing muscle92 and hypertrophy being more important in maturity.93 Both myofiber hyperplasia and hypertrophy are thought to contribute to increased muscle size in DMD and the animal models.6,7,25,31,43 Muscle size is regulated by a complex set of genes that establish a balance between forces that would otherwise lead to either atrophy or hypertrophy.94 With regard to muscle mass in DMD, much attention has focused on insulin-like growth factor 1, which is known to promote muscle hypertrophy by activating the phosphatidylinositol 3 kinase/Akt pathway and, in turn, mTOR and further downstream targets.95 Activation of the Akt-mTOR pathway is associated with muscle hypertrophy in the mdx mouse, and Akt expression is increased in DMD muscles.96 These findings led the investigators to conclude that “vigorous activation of hypertrophic processes during prenecrotic stages of disease might reduce the severity of pathology or extend the prenecrotic stage of disease.”96
Many other factors could be driving or supporting muscle hypertrophy. Anderson and colleagues97 found increased binding activity of basic fibroblast growth factor (bFGF) in mdx mouse versus DMD and GRMD muscle, suggesting that its expression might augment the regenerative response by recruiting more muscle precursor cells. However, in a subsequent study, administration of bFGF did not enhance muscle regeneration in mdx mice.98 Transforming growth factor β2 (TGF-β2) levels are increased in both newly formed myotubes of regenerating muscle99 and in muscle samples from patients with DMD.100 The glycoprotein osteopontin (OPN; secreted phosphoprotein 1) functions as a proinflammatory/fibrotic cytokine in mdx mice101 and serves as a genetic modifier in DMD.102 Importantly, OPN also promotes myogenesis103 and so could play a dual role in transitioning muscle from an inflamed to regenerative state. We have found increased levels of both TGF-β2 and OPN in GRMD muscles; moreover, TGF-β2 tends to track positively with the degree of cranial sartorius hypertrophy (Nghiem and colleagues, unpublished data, 2011).
The complex repetitive discharges seen on electromyography in the 3 dystrophin-deficient animal models10,13,104 and, to a lesser extent, in DMD105 offer an additional mechanism to account for muscle hypertrophy. Sustained electrical activity (myotonia) and muscle contractions associated with chloride and sodium channelopathies can lead to dramatic muscle hypertrophy in the nondystrophic myotonias.106,107 Spontaneous discharges in dystrophin-deficient muscle may occur because of altered ion currents across the damaged muscle cell membrane, with an associated shift in the resting membrane potential. Sustained muscle contraction would presumably augment muscle size, just as it does in myotonia. A positive correlation was observed between Akt pathway activity and muscle hypertrophy in a group of patients with myotonic dystrophy.108
While correlative data are largely circumstantial, abnormal calcium homeostasis could predispose to both muscle hypertrophy and susceptibility to malignant hyperthermia–like syndromes. Myofiber calcium levels are increased in DMD109 and each of the 3 mammalian models.13,110,111 Increased cytosolic levels of calcium also occur in malignant hyperthermia caused by mutations in the ryanodine receptor gene.112 Myofiber hypertrophy is a predictor of susceptibility to malignant hyperthermia in humans113 and occurs in susceptible dogs.114 Although patients with DMD are not at increased risk for malignant hyperthermia itself, they do occasionally develop a similar syndrome when anesthetized with volatile gases.115 Among the dystrophin-deficient animal models, cats are particularly prone to a malignant hyperthermia–like syndrome80 and demonstrate the most striking muscle hypertrophy.12,13 We have seen a similar metabolic syndrome in GRMD dogs anesthetized with isoflurane and sevoflurane. In addition, changes in contraction kinetics of the GRMD peroneus longus muscle occur as it is increasing in size and are compatible with those seen with a delay in calcium sequestration.46
MYOSTATIN INHIBITION IN MUSCLE DISEASE
Beyond the incalculable emotional trauma for patients with DMD and their families, society pays a huge price. Yearly health care costs exceed $30,000 per patient with DMD, 10 to 20 times those of other hospitalized groups.116 In Australia, overall annual costs for each patient, including personal care and lost productivity, have been estimated to be $126,000 and yearly costs due to muscular dystrophy have been placed at approximately $1.5 billion.117 These figures offer a window into the broader societal impact of more common degenerative muscle disorders such as sarcopenia and cancer cachexia. Sarcopenia incidence increases over life, with approximately 15% of women and 30% of men affected at age 80 years.118 Direct health care costs attributable to sarcopenia in the United States approached $20 billion in 2000.119
The collective incidence and cost of health care for the muscular dystrophies and other muscle wasting disorders is a major driver for the pharmaceutical industry.120,121 One strategy for promoting muscle regeneration involves inhibiting myostatin (Mstn; growth/differentiation factor 8), a negative regulator of muscle growth. Humans,122 cattle,123 sheep,124 and dogs125 with myostatin mutations have dramatic muscle hypertrophy. Dystrophin-deficient mdx mice in which myostatin is knocked out (Mstn−/−)126 or inhibited postnatally127 also have a less severe phenotype. Conversely, results from other dystrophic murine models in which myostatin was knocked out have varied,128,129 with some mice showing increased morbidity.130 There are questions about potential senescence of myostatin-deficient cells that have undergone multiple divisions.129 Moreover, abnormalities have been identified in muscle tendons from Mstn−/− mice.131
Although genetically engineered mice have provided an extremely powerful tool to study the molecular pathogenesis of disease, results do not necessarily extrapolate to humans, presumably because of differences between murine and human size and physiology.132 These shortcomings are partially obviated with canine models, which have been used extensively to study disease pathogenesis and treatment efficacy.133,134 This trend toward the use of canine genetic models such as GRMD to study human disease will likely accelerate with the recent sequencing of the canine genome.135 Thus, ideally, results from myostatin-deficient mdx mice should be corroborated in dystrophic dogs in which the gene has been knocked out. Unfortunately, transgenic technology in the dog is very cumbersome and inefficient, essentially precluding use of this approach.136 With this in mind, we have developed a canine (GRippet) model by crossbreeding dystrophin-deficient GRMD dogs with myostatin-heterozygous (Mstn+/−) whippets.
A total of 4 dystrophic Mstn+/− GRippets and 3 dystrophic Mstn wild-type (Mstn+/+) dogs from 2 litters have been evaluated using various phenotypic tests at 6 to 8 months of age. Rather than showing improvement, the dystrophic Mstn+/− GRippets were more clinically affected than their dystrophic Mstn+/+ littermates, apparently because of disproportionate enlargement of certain muscles. In particular, unequal hypertrophy of proximal pelvic limb muscles, as determined by MRI (Fig. 7 and Table 2) and necropsy, seemed to exaggerate postural abnormalities. Gross cranial sartorius muscle hypertrophy was particularly prominent. In general, atrophy or hypertrophy of individual muscles in dystrophic Mstn+/+ dogs was exaggerated in the dystrophic Mstn+/− GRippets. Some dystrophic Mstn+/− GRippets had commensurate, more pronounced atrophy/hypoplasia of the quadriceps femoris muscle.
Fig. 7.

T2-weighted magnetic resonance images with fat signal suppression (FS) of pelvic limb muscles at midthigh from 3 crossbred GRMD and myostatin-heterozygous (Mstn+/−) dogs from the first litter. Note the proportional enlargement of the sartorius and hamstring muscles and the associated atrophy/hypoplasia of the quadriceps of the dystrophic Mstn+/+ dog, Flash, relative to the normal dog, Racer, and the even more dramatic differential size of these muscles in the dystrophic Mstn+/− dog, Dash (also see volumetric measurements from these sections in Table 2). Segmentation was done using ITK-SNAP (http://www.itksnap.org/pmwiki/pmwiki.php).137
Table 2.
MRI volumetric measurements of proximal pelvic limb muscles in crossbred GRMD and Mstn+/− dogsa
| Muscle | Racer (Normal/Mstn+/+)
|
Flash (Dys/Mstn+/+)
|
Dash (Dys/Mstn+/−)
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Volume (mm3) | Volume (mm3/kg) | Percent of Section | Volume (mm3) | Volume (mm3/kg) | Percent of Section | Volume (mm3) | Volume (mm3/kg) | Percent of Section | |
| Cranial sartorius | 101 | 5.87 | 1.77 | 168 | 16.15 | 4.25 | 309 | 30.00 | 8.01 |
|
| |||||||||
| Caudal sartorius | 29 | 1.69 | 0.51 | 52 | 5.0 | 1.32 | 109 | 10.78 | 2.83 |
|
| |||||||||
| Sartorius total | 130 | 7.56 | 2.28 | 220 | 21.15 | 5.57 | 418 | 40.78 | 10.84 |
|
| |||||||||
| Rectus femoris | 279 | 16.22 | 4.89 | 181 | 17.40 | 4.58 | 69 | 6.70 | 1.79 |
|
| |||||||||
| Vastus lateralis | 635 | 36.91 | 11.13 | 419 | 40.29 | 10.60 | 232 | 22.52 | 6.01 |
|
| |||||||||
| Vastus medialis | 351 | 20.41 | 6.16 | 127 | 12.21 | 3.21 | 132 | 12.82 | 3.42 |
|
| |||||||||
| Vastus intermedius | 480 | 27.91 | 8.42 | 207 | 19.90 | 5.23 | 130 | 12.62 | 3.37 |
|
| |||||||||
| Quadriceps total | 1745 | 101.45 | 30.60 | 934 | 89.80 | 23.62 | 563 | 54.66 | 14.59 |
|
| |||||||||
| Semimembranosus | 807 | 46.92 | 14.16 | 578 | 55.58 | 14.62 | 807 | 78.35 | 20.92 |
|
| |||||||||
| Semitendinosus | 284 | 16.51 | 4.98 | 509 | 48.94 | 12.88 | 680 | 66.02 | 17.63 |
|
| |||||||||
| Hamstring total | 1091 | 63.43 | 19.14 | 1087 | 104.52 | 27.50 | 1487 | 144.37 | 38.55 |
|
| |||||||||
| Biceps femoris | 1156 | 67.21 | 20.28 | 643 | 61.83 | 16.27 | 612 | 59.42 | 15.86 |
|
| |||||||||
| Gracilis | 634 | 36.86 | 11.12 | 214 | 20.58 | 5.41 | 176 | 17.09 | 4.56 |
|
| |||||||||
| Adductor | 945 | 54.94 | 16.57 | 855 | 82.11 | 21.63 | 602 | 58.45 | 15.60 |
|
| |||||||||
| Total | 5701 | 331.45 | 99.99 | 3953 | 380.10 | 100 | 3858 | 374.77 | 100 |
Data from these GRippet dogs indicate that mechanisms contributing to selective muscle hypertrophy or atrophy/hypoplasia in dystrophin-deficient muscle may be exaggerated by partial loss of myostatin. Clearly, findings from a genetic model in which myostatin is inhibited from conception will not necessarily predict the outcome of staged postnatal treatments. However, at the very least, our results imply that differential effects of myostatin inhibition on muscle could have deleterious consequences, in keeping with various other findings reviewed in this article.
SUMMARY
Mutations in the dystrophin gene cause Duchenne and Becker muscular dystrophy in humans and syndromes in mice (mdx mouse), dogs (golden retriever muscular dystrophy and other canine mutations), and cats (feline hypertrophic muscular dystrophy). While affected humans and dogs have progressive disease that leads primarily to muscle atrophy, certain muscles undergo paradoxical hypertrophy. Mdx mice progress through an initial phase of muscle hypertrophy followed by atrophy. Cats have persistent muscle hypertrophy. In both humans and animals, muscles are not uniformly affected, with some atrophying as others enlarge. Disproportionate involvement of flexor and extensor muscles acting at joints can exaggerate contractures. Muscle hypertrophy in humans has generally been attributed to deposition of fat and connective tissue (pseudohypertrophy), but recent imaging studies suggest that increased muscle mass (true hypertrophy) also occurs. True hypertrophy plays a more prominent role than fibrosis and fatty deposition in the animal models. Various mechanisms could potentially account for increased muscle mass and, thereby, be manipulated therapeutically. However, for maximal benefit, treatments should uniformly increase mass or preferentially improve strength of clinically-important muscles. Treatments that result in disproportionate muscle enlargement may exaggerate postural instability and joint contractures. These deleterious consequences of muscle hypertrophy should be considered when developing treatments for muscular dystrophy and other muscle wasting disorders.
References
- 1.Tyler KL. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve. 2003;28:402–22. doi: 10.1002/mus.10435. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1998;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Prior TW, Bridgeman SJ. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn. 2005;7:317–26. doi: 10.1016/S1525-1578(10)60560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malhotra S, Hart K, Klamut H, et al. Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science. 1988;242:755–9. doi: 10.1126/science.3055295. [DOI] [PubMed] [Google Scholar]
- 5.Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 6.Jones DA, Round JM, Edwards RH, et al. Size and composition of the calf and quadriceps muscles in Duchenne muscular dystrophy. A tomographic and histochemical study. J Neurol Sci. 1983;60:307–22. doi: 10.1016/0022-510x(83)90071-0. [DOI] [PubMed] [Google Scholar]
- 7.Cros D, Harnden P, Pellissier JF, et al. Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study. J Neurol. 1989;236:43–7. doi: 10.1007/BF00314217. [DOI] [PubMed] [Google Scholar]
- 8.Bulfield G, Siller WG, Wight PA, et al. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–92. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil. 1999;20:605–25. doi: 10.1023/a:1005545325254. [DOI] [PubMed] [Google Scholar]
- 10.Kornegay JN, Tuler SM, Miller DM, et al. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11:1056–64. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- 11.Cooper BJ, Winand WN, Stedman H, et al. The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature. 1988;334:154–6. doi: 10.1038/334154a0. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter JL, Hoffman EP, Romanul FC, et al. Feline muscular dystrophy with dystrophin deficiency. Am J Pathol. 1989;135:909–19. [PMC free article] [PubMed] [Google Scholar]
- 13.Gaschen FP, Hoffman EP, Gorospe JR, et al. Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci. 1992;110:149–59. doi: 10.1016/0022-510x(92)90022-d. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman EP, Gorospe RM. The animal models of Duchenne muscular dystrophy: windows on the pathophysiological consequences of dystrophin deficiency. Curr Top Membr. 1991;38:113–55. [Google Scholar]
- 15.Edwards RH, Jones DA, Newham DJ, et al. Role of mechanical damage in the pathogenesis of proximal myopathy in man. Lancet. 1984;1:548–52. doi: 10.1016/s0140-6736(84)90941-3. [DOI] [PubMed] [Google Scholar]
- 16.Karpati G, Carpenter S. Small-caliber skeletal muscle fibers do not suffer deleterious consequences of dystrophic gene expression. Am J Med Genet. 1986;25:653–8. doi: 10.1002/ajmg.1320250407. [DOI] [PubMed] [Google Scholar]
- 17.Pradhan S, Mittal B. Infraspinatus muscle hypertrophy and wasting of axillary folds important signs in Duchenne muscular dystrophy. Clin Neurol Neurosurg. 1995;97:134–8. doi: 10.1016/0303-8467(95)00013-a. [DOI] [PubMed] [Google Scholar]
- 18.de Visser M, Verbeeten B., Jr Computed tomography of the skeletal musculature in Becker-type muscular dystrophy and benign infantile spinal muscular atrophy. Muscle Nerve. 1985;8:435–44. doi: 10.1002/mus.880080514. [DOI] [PubMed] [Google Scholar]
- 19.Ardran GM, Hamilton A, Kemp FH. Enlargement of the tongue and changes in the jaws with muscular dystrophy. Clin Radiol. 1973;24:359–64. doi: 10.1016/s0009-9260(73)80058-3. [DOI] [PubMed] [Google Scholar]
- 20.De Bruin PF, Ueki J, Bush A, et al. Diaphragm thickness and inspiratory strength in patients with Duchenne muscular dystrophy. Thorax. 1997;52:472–5. doi: 10.1136/thx.52.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renard D, Labauge P. Thenar and hypothenar muscle hypertrophy in Becker muscular dystrophy. Neuromuscul Disord. 2010;20:281. doi: 10.1016/j.nmd.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Walton JN. Clinical examination of the neuromuscular system. In: Walton JN, editor. Disorders of voluntary muscle. London: Churchill Livingstone; 1981. pp. 448–80. [Google Scholar]
- 23.Reimers CD, Schlotter B, Eicke BM, et al. Calf enlargement in neuromuscular diseases: a quantitative ultrasound study in 350 patients and review of the literature. J Neurol Sci. 1996;143:46–56. doi: 10.1016/s0022-510x(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 24.Marden FA, Connolly AM, Siegel MJ, et al. Compositional analysis of muscle in boys with Duchenne muscular dystrophy using MR imaging. Skeletal Radiol. 2005;34:140–8. doi: 10.1007/s00256-004-0825-3. [DOI] [PubMed] [Google Scholar]
- 25.Kornegay JN, Cundiff DD, Bogan DJ, et al. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2003;13:493–500. doi: 10.1016/s0960-8966(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 26.Karpati G, Carpenter S, Prescott S. Small-caliber skeletal muscle fibers do not suffer necrosis in mdx mouse dystrophy. Muscle Nerve. 1988;11:795–803. doi: 10.1002/mus.880110802. [DOI] [PubMed] [Google Scholar]
- 27.Stedman HH, Sweeney HL, Shrager JB, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–9. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 28.Coulton GR, Morgan JE, Partridge TA, et al. The mdx mouse skeletal muscle myopathy: 1. A histological, morphometric, and biochemical investigation. Neuropathol Appl Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 29.Pastoret C, Sebille A. Mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129:97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 30.DiMario JX, Uzman A, Strohman RC. Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev Biol. 1991;148:314–21. doi: 10.1016/0012-1606(91)90340-9. [DOI] [PubMed] [Google Scholar]
- 31.Sacco P, Jones DA, Dick JR, et al. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin Sci. 1992;82:227–36. doi: 10.1042/cs0820227. [DOI] [PubMed] [Google Scholar]
- 32.Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J Neurol Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- 33.Anderson JE, Bressler BH, Ovalle WK. Functional regeneration in the hindlimb skeletal muscle of the mdx mouse. J Muscle Res Cell Motil. 1988;9:499–515. doi: 10.1007/BF01738755. [DOI] [PubMed] [Google Scholar]
- 34.Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110:269–99. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- 35.Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69:91–5. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- 36.Dangain J, Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve. 1984;7:700–4. doi: 10.1002/mus.880070903. [DOI] [PubMed] [Google Scholar]
- 37.Quinlan JG, Johnson SR, McKee MK, et al. Twitch and tetanus in mdx mouse muscle. Muscle Nerve. 1992;15:837–42. doi: 10.1002/mus.880150713. [DOI] [PubMed] [Google Scholar]
- 38.Muntoni F, Mateddu A, Marchei F, et al. Muscular weakness in the mdx mouse. J Neurol Sci. 1993;120:71–7. doi: 10.1016/0022-510x(93)90027-v. [DOI] [PubMed] [Google Scholar]
- 39.Coulton GR, Curtin NA, Morgan JE, et al. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol Appl Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 40.Dupont-Versteegden EE, McCarter RJ. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve. 1992;15:1105–10. doi: 10.1002/mus.880151008. [DOI] [PubMed] [Google Scholar]
- 41.Vos JH, van der Linde-Sipman JS, Goedegebuure SA. Dystrophy-like myopathy in the cat. J Comp Pathol. 1986;96:335–41. doi: 10.1016/0021-9975(86)90053-8. [DOI] [PubMed] [Google Scholar]
- 42.Winand NJ, Edwards M, Pradhan D, et al. Deletion of the dystrophin muscle promoter in feline muscular dystrophy. Neuromuscul Disord. 1984;4:433–45. doi: 10.1016/0960-8966(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 43.Gaschen F, Burgunder JM. Changes of skeletal muscle in young dystrophin-deficient cats: a morphological and morphometric study. Acta Neuropathol. 2001;101:591–600. doi: 10.1007/s004010000299. [DOI] [PubMed] [Google Scholar]
- 44.Valentine BA, Cooper BJ, Cummings JF, et al. Canine X-linked muscular dystrophy: morphologic lesions. J Neurol Sci. 1990;97:1–23. doi: 10.1016/0022-510x(90)90095-5. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen F, Cherel Y, Guigand L, et al. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Pathol. 2002;126:100–8. doi: 10.1053/jcpa.2001.0526. [DOI] [PubMed] [Google Scholar]
- 46.Kornegay JN, Sharp NJ, Bogan DJ, et al. Contraction tension and kinetics of the peroneus longus muscle in golden retriever muscular dystrophy. J Neurol Sci. 1994;123:100–7. doi: 10.1016/0022-510x(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 47.Kornegay JN, Bogan DJ, Bogan JR, et al. Contraction force generated by tibio-tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. 1999;166:115–21. doi: 10.1016/s0022-510x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- 48.Liu JM, Okamura CS, Bogan DJ, et al. Effects of prednisone in canine muscular dystrophy. Muscle Nerve. 2004;30:767–73. doi: 10.1002/mus.20154. [DOI] [PubMed] [Google Scholar]
- 49.Sutherland DH, Olshen R, Cooper L, et al. The pathomechanics of gait in Duchenne muscular dystrophy. Dev Med Child Neurol. 1991;23:3–22. doi: 10.1111/j.1469-8749.1981.tb08442.x. [DOI] [PubMed] [Google Scholar]
- 50.Brumitt JW, Essman SC, Kornegay JN, et al. Radiographic features of Golden Retriever muscular dystrophy. Vet Radiol Ultrasound. 2006;47:574–80. doi: 10.1111/j.1740-8261.2006.00188.x. [DOI] [PubMed] [Google Scholar]
- 51.Charteris J, Leach D, Taves C. Comparative kinematic analysis of bipedal and quadrupedal locomotion: a cyclographic technique. J Anat. 1979;128:803–19. [PMC free article] [PubMed] [Google Scholar]
- 52.Polly PD. Limbs in mammalian evolution. In: Hall BK, editor. Fins into limbs: evolution, development, and transformation. Chicago: University of Chicago Press; 2007. pp. 245–68. [Google Scholar]
- 53.Hanson SM, Smith MO, Walker TL, et al. Juvenile-onset distal myopathy in Rottweiler dogs. J Vet Intern Med. 1998;12:103–8. doi: 10.1111/j.1939-1676.1998.tb02103.x. [DOI] [PubMed] [Google Scholar]
- 54.Bensfield AC, Evans J, Pesayco JP, et al. Recurrent demyelination and remyelination in 37 young Bengal cats with polyneuropathy. J Vet Intern Med. 2011;25:882–9. doi: 10.1111/j.1939-1676.2011.0740.x. [DOI] [PubMed] [Google Scholar]
- 55.Wentink GH. Dynamics of the hind limb at walk in horse and dog. Anat Embryol. 1979;155:179–90. doi: 10.1007/BF00305750. [DOI] [PubMed] [Google Scholar]
- 56.Vignos PJ, Jr, Spencer GE, Jr, Archibald JC. Management of muscular dystrophy of childhood. JAMA. 1963;184:89–96. doi: 10.1001/jama.1963.03700150043007. [DOI] [PubMed] [Google Scholar]
- 57.Brooke MH, Fenichel GM, Griggs RC, et al. Clinical investigation in Duchenne dystrophy: 2. Determination of the “power” of therapeutic trials based on the natural history. Muscle Nerve. 1983;6:91–103. doi: 10.1002/mus.880060204. [DOI] [PubMed] [Google Scholar]
- 58.Roy L, Gibson DA. Pseudohypertrophic muscular dystrophy and its surgical management: review of 30 patients. Can J Surg. 1970;13:13–21. [PubMed] [Google Scholar]
- 59.Kottke FJ. The effects of limitation of activity upon the human body. JAMA. 1966;196:825–30. [PubMed] [Google Scholar]
- 60.Akeson WH, Amiel D, Abel MF, et al. Effects of immobilization on joints. Clin Orthop Relat Res. 1987;219:28–37. [PubMed] [Google Scholar]
- 61.Akeson WH, Amiel D, Mechanic GL, et al. Collagen cross-linking alterations in joint contractures: changes in the reducible cross-links in periarticular connective tissue collagen after nine weeks of immobilization. Connect Tissue Res. 1977;5:15–9. doi: 10.3109/03008207709152607. [DOI] [PubMed] [Google Scholar]
- 62.Young RR, Wiegner AW. Spasticity. Clin Orthop Relat Res. 1987;219:50–62. [PubMed] [Google Scholar]
- 63.Johnson ER, Fowler WM, Jr, Lieberman JS. Contractures in neuromuscular disease. Arch Phys Med Rehabil. 1992;73:807–10. [PubMed] [Google Scholar]
- 64.Archibald KC, Vignos PJ., Jr A study of contractures in muscular dystrophy. Arch Phys Med Rehabil. 1959;40:150–7. [PubMed] [Google Scholar]
- 65.Siegel IM. Equinocavovarus in muscular dystrophy. Its treatment by percutaneous tarsal medullostomy and soft tissue release. Arch Surg. 1972;104:644–6. doi: 10.1001/archsurg.1972.04180050020005. [DOI] [PubMed] [Google Scholar]
- 66.Wagner MB, Vignos PJ, Jr, Carlozzi C. Duchenne muscular dystrophy: a study of wrist and hand function. Muscle Nerve. 1989;12:236–44. doi: 10.1002/mus.880120313. [DOI] [PubMed] [Google Scholar]
- 67.McDonald CM, Abresch RT, Carter GT, et al. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74:S70–92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 68.Johnson EW. Walter J. Zeiter Lecture: pathokinesiology of Duchenne muscular dystrophy: implications for management. Arch Phys Med Rehabil. 1977;58:4–7. [PubMed] [Google Scholar]
- 69.Schuurman SO, Kersten W, Weijs WA. The equine hind limb is actively stabilized during standing. J Anat. 2003;202:355–62. doi: 10.1046/j.1469-7580.2003.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsu JD. Orthopedic approaches for the treatment of lower extremity contractures in the Duchenne muscular dystrophy patient in the United States and Canada. Semin Neurol. 1995;15:6–8. doi: 10.1055/s-2008-1041000. [DOI] [PubMed] [Google Scholar]
- 71.Hsu JD, Furumasu J. Gait and posture changes in the Duchenne muscular dystrophy child. Clin Orthop Relat Res. 1993;288:122–5. [PubMed] [Google Scholar]
- 72.Trias D, Gioux M, Cid M, et al. Gait analysis of myopathic children in relation to impairment level and energy cost. J Electromyogr Kinesiol. 1994;4:67–81. doi: 10.1016/1050-6411(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 73.Mukherjee AK, Mokashi MG. Epidemiology. The incidence and management of joint contracture in India. Clin Orthop Relat Res. 1987;219:87–92. [PubMed] [Google Scholar]
- 74.Rideau Y. Treatment of orthopedic deformity during the ambulatory stage of Duchenne muscular dystrophy. In: Serratrice G, Cros D, Desnuelle C, editors. Neuromuscular disease. New York: Raven; 1984. pp. 557–64. [Google Scholar]
- 75.Johnson EW, Yarnell SK. Hand dominance and scoliosis in Duchenne muscular dystrophy. Arch Phys Med Rehabil. 1976;57:462–4. [PubMed] [Google Scholar]
- 76.Rideau Y, Jankowski LW, Grellet J. Respiratory function in the muscular dystrophies. Muscle Nerve. 1981;4:155–64. doi: 10.1002/mus.880040213. [DOI] [PubMed] [Google Scholar]
- 77.Laws N, Hoey A. Progression of kyphosis in mdx mice. J Appl Physiol. 2004;97:1970–7. doi: 10.1152/japplphysiol.01357.2003. [DOI] [PubMed] [Google Scholar]
- 78.Laws N, Cornford-Nairn RA, Irwin N, et al. Long-term administration of antisense oligonucleotides into the paraspinal muscles of mdx mice reduces kyphosis. J Appl Physiol. 2008;105:662–8. doi: 10.1152/japplphysiol.00068.2008. [DOI] [PubMed] [Google Scholar]
- 79.Garlich MW, Baltgalvis KA, Call JA, et al. Plantarflexion contracture in the mdx mouse. Am J Phys Med Rehabil. 2010;89:976–85. doi: 10.1097/PHM.0b013e3181fc7c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaschen F, Gaschen L, Seiler G, et al. Lethal peracute rhabdomyolysis associated with stress and general anesthesia in three dystrophin-deficient cats. Vet Pathol. 1998;35:117–23. doi: 10.1177/030098589803500205. [DOI] [PubMed] [Google Scholar]
- 81.Valentine BA, Cooper BJ, de Lahunta A, et al. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88:69–81. doi: 10.1016/0022-510x(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 82.Ambrósio CE, Fadel L, Gaiad TP, et al. Identification of three distinguishable phenotypes in golden retriever muscular dystrophy. Genet Mol Res. 2009;8:389–96. doi: 10.4238/vol8-2gmr581. [DOI] [PubMed] [Google Scholar]
- 83.Ambrósio CE, Valadares MC, Zucconi E, et al. Ringo, a Golden Retriever Muscular Dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscul Disord. 2008;18:892–3. doi: 10.1016/j.nmd.2008.06.385. [DOI] [PubMed] [Google Scholar]
- 84.Kornegay JN, Sharp NJ, Schueler RO, et al. Tibiotarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab Anim Sci. 1994;44:331–3. [PubMed] [Google Scholar]
- 85.Marsh AP, Eggebeen JD, Kornegay JN, et al. Kinematics of gait in golden retriever muscular dystrophy. Neuromuscul Disord. 2010;20:16–20. doi: 10.1016/j.nmd.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barthélémy I, Barrey E, Thibaud JL, et al. Gait analysis using accelerometry in dystrophin-deficient dogs. Neuromuscul Disord. 2009;19:788–96. doi: 10.1016/j.nmd.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 87.Jaegger G, Marcellin-Little DJ, Levine D. Reliability of goniometry in Labrador retrievers. Am J Vet Res. 2002;63:979–86. doi: 10.2460/ajvr.2002.63.979. [DOI] [PubMed] [Google Scholar]
- 88.Nicholson HL, Osmotherly PG, Smith BA, et al. Determinants of passive hip range of motion in adult Greyhounds. Aust Vet J. 2007;85:217–21. doi: 10.1111/j.1751-0813.2007.00145.x. [DOI] [PubMed] [Google Scholar]
- 89.Kornegay JN, Bogan JR, Bogan DJ, et al. Golden retriever muscular dystrophy (GRMD): developing and maintaining a colony and physiological functional measurements. In: Duan D, editor. Muscle gene therapy: methods and protocols, Methods in molecular biology. Vol. 709. New York: Humana Press; 2011. pp. 105–23. [DOI] [PubMed] [Google Scholar]
- 90.Coleman SS. The effect of posterior spine fusion on vertebral growth in dogs. J Bone Joint Surg Am. 1968;50:879–96. [PubMed] [Google Scholar]
- 91.Kioschos HC, Asher MA, Lark RG, et al. Overpowering the crankshaft mechanism. The effect of posterior spinal fusion with and without stiff transpedicular fixation on anterior spinal column growth in immature canines. Spine. 1996;21:1168–73. doi: 10.1097/00007632-199605150-00008. [DOI] [PubMed] [Google Scholar]
- 92.Moss FP, Leblond CP. Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec. 1971;170:421–35. doi: 10.1002/ar.1091700405. [DOI] [PubMed] [Google Scholar]
- 93.McCarthy JJ, Mula J, Miyazaki M, et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011;138:3657–66. doi: 10.1242/dev.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology. 2008;23:160–70. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 95.Shavlakadze T, Chai J, Maley K, et al. A growth stimulus is needed for IGF-1 to induce skeletal muscle hypertrophy in vivo. J Cell Sci. 2010;123:960–71. doi: 10.1242/jcs.061119. [DOI] [PubMed] [Google Scholar]
- 96.Peter AK, Crosbie RH. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. 2006;312:2580–91. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 97.Anderson JE, Kakulas BA, Jacobsen PF, et al. Comparison of basic fibroblast growth factor in X-linked dystrophin-deficient myopathies of human, dog and mouse. Growth Factors. 1993;9:107–21. [PubMed] [Google Scholar]
- 98.Mitchell CA, McGeachie JK, Grounds MD. The exogenous administration of basic fibroblast growth factor to regenerating skeletal muscle in mice does not enhance the process of regeneration. Growth Factors. 1996;13:37–55. doi: 10.3109/08977199609034565. [DOI] [PubMed] [Google Scholar]
- 99.McLennan IS, Koishi K. Cellular localisation of transforming growth factor-beta 2 and -beta 3 (TGF-beta2, TGF-beta3) in damaged and regenerating skeletal muscles. Dev Dyn. 1997;208:278–89. doi: 10.1002/(SICI)1097-0177(199702)208:2<278::AID-AJA14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 100.Murakami N, McLennan IS, Nonaka I, et al. Transforming growth factor-beta2 is elevated in skeletal muscle disorders. Muscle Nerve. 1999;22:889–98. doi: 10.1002/(sici)1097-4598(199907)22:7<889::aid-mus12>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 101.Vetrone SV, Montecino-Rodriguez E, Kudryashova E, et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β. J Clin Invest. 2009;119:1583–94. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pegoraro E, Hoffman EP, Piva L, et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76:219–26. doi: 10.1212/WNL.0b013e318207afeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Uaesoontrachoon K, Yoo HJ, Tudor EM, et al. Osteopontin and skeletal muscle myoblasts: association with muscle regeneration and regulation of myoblast function in vitro. Int J Biochem Cell Biol. 2008;40:2303–14. doi: 10.1016/j.biocel.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 104.Kurihara T, Kishi M, Saito N, et al. Electrical myotonia and cataract in X-linked muscular dystrophy (mdx) mouse. J Neurol Sci. 1990;99:83–92. doi: 10.1016/0022-510x(90)90202-x. [DOI] [PubMed] [Google Scholar]
- 105.Hausmanowa-Petrusewicz I, Jedrzejowska H. Correlation between electromyographic findings and muscle biopsy in cases of neuromuscular disease. J Neurol Sci. 1971;13:85–106. doi: 10.1016/0022-510x(71)90209-7. [DOI] [PubMed] [Google Scholar]
- 106.Trip J, Pillen S, Faber CG, et al. Muscle ultrasound measurements and functional muscle parameters in non-dystrophic myotonias suggest structural muscle changes. Neuromuscul Disord. 2009;19:462–7. doi: 10.1016/j.nmd.2009.06.369. [DOI] [PubMed] [Google Scholar]
- 107.Sinha MK, Chaurasia RN, Verma R. A family with autosomal recessive generalised myotonia with Herculean appearance. J Assoc Physicians India. 2011;59:120–2. [PubMed] [Google Scholar]
- 108.Li X, Zhang W, Lv H, et al. Activities of Akt pathway and their correlation with pathological changes in myotonic dystrophy. Beijing Da Xue Xue Bao. 2010;42:526–9. [in Chinese] [PubMed] [Google Scholar]
- 109.Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–46. doi: 10.1212/wnl.28.5.439. [DOI] [PubMed] [Google Scholar]
- 110.Ruegg UT, Nicolas-Métral V, Challet C, et al. Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul Disord. 2002;12(Suppl 1):S155–61. doi: 10.1016/s0960-8966(02)00095-0. [DOI] [PubMed] [Google Scholar]
- 111.Valentine BA, Cooper BJ, Gallagher EA. Intracellular calcium in canine muscle biopsies. J Comp Pathol. 1989;100:223–30. doi: 10.1016/0021-9975(89)90099-6. [DOI] [PubMed] [Google Scholar]
- 112.Stowell KM. Malignant hyperthermia: a pharmacogenetic disorder. Pharmacogenomics. 2008;9:1657–72. doi: 10.2217/14622416.9.11.1657. [DOI] [PubMed] [Google Scholar]
- 113.Mezin P, Payen JF, Bosson JL, et al. Histological support for the difference between malignant hyperthermia susceptible (MHS), equivocal (MHE) and negative (MHN) muscle biopsies. Br J Anaesth. 1997;79:327–31. doi: 10.1093/bja/79.3.327. [DOI] [PubMed] [Google Scholar]
- 114.O’Brien PJ, Cribb PH, White RJ, et al. Canine malignant hyperthermia: diagnosis of susceptibility in a breeding colony. Can Vet J. 1983;24:172–7. [PMC free article] [PubMed] [Google Scholar]
- 115.Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109:1043–8. doi: 10.1213/ane.0b013e3181aa5cf6. [DOI] [PubMed] [Google Scholar]
- 116.Ouyang L. Health care utilization and expenditures for children and young adults with muscular dystrophy in a privately insured population. J Child Neurol. 2008;23:883–8. doi: 10.1177/0883073808314962. [DOI] [PubMed] [Google Scholar]
- 117.Report. Access Economics Pty Limited for the Muscular Dystrophy Association; Canberra, Melbourne, and Sydney (Australia): Oct 17, 2007. The cost of muscular dystrophy. [Google Scholar]
- 118.Morley JE, Baumgartner RN, Roubenoff R, et al. Sarcopenia. J Lab Clin Med. 2001;137:231–43. doi: 10.1067/mlc.2001.113504. [DOI] [PubMed] [Google Scholar]
- 119.Janssen I, Shepard DS, Katzmarzyk R, et al. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–5. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- 120.Bradley L, Yaworsky PJ, Walsh FS. Myostatin as a therapeutic target for musculoskeletal disease. Cell Mol Life Sci. 2008;65:2119–24. doi: 10.1007/s00018-008-8077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patel K, Amthor H. The function of myostatin and strategies of myostatin blockade—new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscul Disord. 2005;15:117–26. doi: 10.1016/j.nmd.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 122.Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–8. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 123.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94(12):457–61. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Clop A, Marcq F, Takeda H, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–8. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 125.Mosher DS, Quignon P, Bustamante CD, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:779–86. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wagner KR, McPherron AC, Winik N, et al. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–6. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 127.Wagner KR, Liu X, Chang X, et al. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102:2519–24. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mendias CL, Marcin JE, Calerdon DR, et al. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol. 2006;101:898–905. doi: 10.1152/japplphysiol.00126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parsons SA, Millay DP, Sargent MA, et al. Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am J Pathol. 2006;168:1975–85. doi: 10.2353/ajpath.2006.051316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li ZF, Shelton GD, Engvall E. Elimination of myostatin does not combat muscular dystrophy in dy mice but increases postnatal lethality. Am J Pathol. 2005;166:491–7. doi: 10.1016/S0002-9440(10)62271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mendias CL, Bakhurin KI, Faulkner JA. Tendons of myostatin-deficient mice are small, brittle, and hypocellular. Proc Natl Acad Sci USA. 2008;105:388–93. doi: 10.1073/pnas.0707069105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lin JH. Applications and limitations of genetically modified mouse models in drug discovery and development. Curr Drug Metab. 2008;9:419–38. doi: 10.2174/138920008784746355. [DOI] [PubMed] [Google Scholar]
- 133.Schneider MR, Wolf E, Braun J, et al. Canine embryo-derived stem cells and models for human diseases. Hum Mol Genet. 2007;17(R1):R42–7. doi: 10.1093/hmg/ddn078. [DOI] [PubMed] [Google Scholar]
- 134.Tsai KL, Clark LA, Murphy KE. Understanding hereditary diseases using the dog and human as companion model systems. Mamm Genome. 2007;18:444–51. doi: 10.1007/s00335-007-9037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lindblad-Toh K, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–19. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 136.Hong SG, Kim MK, Jang G, et al. Generation of red fluorescent protein transgenic dogs. Genesis. 2009;47:314–22. doi: 10.1002/dvg.20504. [DOI] [PubMed] [Google Scholar]
- 137.Yushkevich PA, Piven J, Hazlett HC, et al. User-guided 3D active contour segmentation of anatomic structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–28. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]