

Abstract
Tumor necrosis factor-α (TNF-α) is a polypeptide cytokine that has been associated with muscle wasting and weakness in inflammatory disease. Despite its potential importance in muscle pathology, the direct effects of TNF-α on skeletal muscle have remained undefined until recently. Studies of cultured muscle cells indicate that TNF-α disrupts the differentiation process and can promote catabolism in mature cells. The latter response appears to be mediated by reactive oxygen species and nuclear factor-κB which upregulate ubiquitin/proteasome activity. This commentary outlines our current understanding of TNF-α effects on skeletal muscle and the mechanism of TNF-α action.
Keywords: antioxidants, cachexia, cytokines, free radicals, skeletal muscle
Introduction
TNF-α is a polypeptide cytokine that promotes antitumor and immune responses [1]. TNF-α has long been associated with muscle pathology and was originally designated 'cachectin' in recognition of its catabolic action. Experimental animals lose muscle mass when treated with TNF-α [2,3] or exposed to interventions that elevate endogenous TNF-α (e.g. sepsis or tumor implantation). In humans, muscle catabolism has been attributed to TNF-α in inflammatory diseases that include cancer [4], congestive heart failure [5], AIDS [6], and chronic obstructive pulmonary disease (COPD) [7]. In the latter case, malnourished individuals with COPD have elevated serum levels of TNF-α [8] which may reflect exaggerated production by peripheral blood monocytes [9]. Loss of muscle mass contributes to weakness, fatigue, and loss of mobility for individuals with COPD and other inflammatory diseases. Despite its potential importance, the effects of TNF-α on skeletal muscle and the mechanisms of TNF-α action have remained largely undefined until recently.
Cellular mechanism of TNF-alpha action: a working model
This commentary outlines the current perspective of the authors regarding TNF-α effects on differentiated muscle. Our concepts are summarized in an experimental model depicted in Figure 1.
Figure 1.
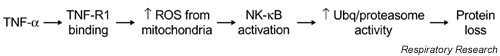
Proposed events regulating TNF-α-induced muscle catabolism. TNF-α binding to the type 1 TNF-α receptor (TNFR1) stimulates increased production of reactive oxygen species (ROS) by mitochondrial electron transport, thereby activating nuclear factor-κB (NF-κB). Subsequently, NF-κB increases activity of the ubiquitin (Ubq)/proteasome pathway, accelerating protein degradation.
In brief, we propose that TNF-α can act directly on muscle cells to stimulate protein loss, an action mediated by nuclear factor-κB (NF-κB) which is a transcription factor. Intermediate steps in TNF-α/NF-κB signaling include stimulation of the type 1 TNF-α receptor (TNFR1) and an increase in reactive oxygen species (ROS) production via mitochondrial electron transport. NF-κB appears to increase activity of the ubiquitin/proteasome pathway, which accelerates the regulated degradation of muscle proteins and promotes muscle weakness. Subsequent sections address major components of the proposed process and provide a brief overview of the underlying evidence.
TNF-alpha and protein loss
The mechanism of TNF-α effects in vivo remains largely enigmatic, although it has long been recognized that TNF-α may stimulate catabolism via indirect mechanisms. TNF-α alters circulating levels of hormones that regulate muscle growth and affects tissue sensitivity to such factors. TNF-α also stimulates production of catabolic cytokines and induces anorexia. Any of these effects could indirectly promote muscle wasting. Mechanisms by which TNF-α might directly stimulate catabolism are less clear. One potential mechanism is by inhibiting myoblast differentiation [10,11], an action of TNF-α that could limit the regenerative response of satellite cells to muscle injury [12]. A second mechanism, apoptosis, appears less important [13,14]. The third mechanism, a direct catabolic effect on differentiated muscle, is the focus of this commentary. Early experiments investigated the catabolic action of TNF-α using excised rodent muscles in vitro. Incubation with supraphysiologic TNF-α concentrations for up to 3 hours yielded no detectable change in protein breakdown, leading previous investigators to conclude that TNF-α does not directly stimulate protein loss [3,15,16].
More recently, cell culture techniques have enabled the use of longer-term protocols. In the absence of exogenous anabolic stimuli, TNF-α directly stimulates a time-dependent and concentration-dependent decrement in total muscle protein content and loss of muscle-specific proteins, including adult fast-type myosin heavy chain (MHCf) [13,14]. MHCf losses are not accompanied by a change in synthesis rate [13], suggesting TNF-α stimulates degradation of myofibrillar proteins. Accelerated protein loss can be induced using TNF-α levels that do not stimulate cell death, by either apoptosis or necrosis, and are within the range measured clinically [13,14]. The catabolic program activated under these conditions thus closely mimics the changes observed in muscles of cachectic humans (i.e. fiber atrophy without overt cell death).
Available data suggest that the catabolic response to TNF-α in cell culture can be overridden by the superimposition of anabolic stimuli. Guttridge and co-workers [10] reported that TNF-α did not stimulate MHCf loss in myotubes that were simultaneously exposed to pharmacologic levels of insulin. More recently, Langen and colleagues [17] used a cell culture matrix of collagen IV, laminin, heparin sulfate proteoglycan, and entactin to enhance myocyte differentiation. Myotubes grown on this matrix were refractory to the catabolic effects of TNF-α. Only marginal changes were seen in total protein content, MHCf content, or creatine kinase activity.
The TNF-alpha/NF-kappaB pathway
TNF-α stimulates a complex array of postreceptor signaling events that evoke pleiotropic, cell-type-specific responses. At least three major pathways mediate the cellular response to TNF-α. One pathway stimulates apoptosis via interaction with the TNF-α-receptor complex and the Fas-associated protein with death domain. A second pathway activates Jun-N-terminal kinases and the transcription factor AP-1. The third pathway activates NF-κB, a primary mediator of transcriptional control and a major candidate for catabolic signaling. We [13,14,18,19] and others [10,17,20] have shown that TNF-α stimulates the activation and nuclear translocation of NF-κB in skeletal muscle cells. This is a rapid, dose-dependent response that involves phosphorylation and proteasomal degradation of the NF-κB-inhibitory protein, Iκ-Bα [13]. NF-κB activity in the cell nucleus peaks within 30 minutes of TNF-α exposure and then rapidly decays. This transient stimulus alters gene expression and causes prolonged changes in muscle protein levels. The contribution of NF-κB was established using a dominant negative approach by which TNF-α activation of NF-κB could be selectively inhibited [14]. Myotubes derived from this dominant negative cell line do not exhibit a catabolic response to TNF-α; neither total protein content nor muscle-specific protein levels are diminished by prolonged TNF-α stimulation [14]. These findings suggest that NF-κB signaling is essential for TNF-α-induced catabolism in differentiated muscle cells. The differentiation process also appears to be modulated by TNF-α/NF-κB signaling [10,11,17,19]. This represents a second mechanism by which this pathway could influence muscle adaptation, both during development and during activation of satellite cells following muscle injury [12].
Receptor-mediated signaling: oxidants as second messengers
The responses of muscle cells to TNF-α are mediated by two sarcolemmal receptor populations, TNFR1 (55 kDa) and type2 TNF-α receptor (TNFR2) (75 kDa) [21]. Ligand binding stimulates a complex cascade of postreceptor signaling events that are subtype-specific. While NF-κB may be activated via either TNFR1 or TNFR2, the existing data implicate TNFR1 as the receptor subtype by which TNF-α stimulates loss of muscle protein [22,23]. This pathway appears to be redox sensitive. Sen and colleagues [20] used L6 myoblasts to demonstrate that TNF-α activation of NF-κB is regulated by the glutathione cycle, a primary mechanism of antioxidant buffering. Buck and Chojkier [2] have shown that antioxidants and nitric oxide (NO) synthase blockade inhibit muscle wasting in a mouse model of TNF-α-induced cachexia. In mature myotubes, TNF-α activation of NF-κB is blunted by catalase, which enzymatically dehydrates hydrogen peroxide; conversely, exogenous hydrogen peroxide activates NF-κB in the absence of TNF-α [18]. ROS thus appear to function as second messengers for TNF-α in skeletal muscle, activating NF-κB either directly or indirectly. Data from myotube studies indicate the most likely source of TNF-α-induced ROS is the mitochondrial electron transport chain [18]. NO derivatives do not appear to be intrinsic elements of this pathway. NO synthase blockade does not affect TNF-α/NF-κB signaling, and NF-κB is insensitive to NO donors [18].
The ubiquitin/proteasome pathway as downstream effector
Existing data suggest that TNF-α stimulates muscle catabolism by activating the ubiqutin/proteasome pathway [24,25,26]. As reviewed elsewhere [27], the ubiquitin/proteasome pathway degrades the bulk of all intracellular proteins and is responsible for regulated proteolysis in signal transduction, cell-cycle progression, transcriptional regulation, and antigen presentation. Pathway activity depends upon coordinated interactions among several enzyme families. These interventions result in tagging of substrate proteins with polymeric ubiquitin chains that mark the protein for degradation. The marked protein is then degraded by the 26S-proteasome complex, by an ATP-dependent process. In catabolic states, the activity of the ubiquitin/ proteasome pathway is increased by upregulation of selected pathway components [28]. The level of circulating TNF-α is also elevated in these conditions and is a potential stimulus for pathway upregulation. Acute, intravenous injection of TNF-α causes time-dependent increases in both free and conjugated ubiquitin [24], and ubiquitin mRNA [25] in the limb muscles of intact rats. The limited data currently available suggest that TNF-α acts directly on muscle fibers to upregulate the pathway. Llovera et al [26] have shown that ubiquitin mRNA levels are elevated in excised muscle exposed to TNF-α in vitro. Preliminary studies suggest the ubiquitin/proteasome pathway is sensitive to TNF-α signaling events, including elevated ROS levels and NF-κB activation [29], but this link has yet to be established formally.
Conclusion
It appears that TNF-α can act directly on mature muscle to accelerate protein degradation. The cellular mechanisms that regulate this response are beginning to be understood. The early mediators of TNF-α action (ROS and NF-κB) are classical components of the inflammatory response and are sensitive to other ligand/receptor interactions (e.g. interleukin-1 and interleukin-6). Perhaps ROS and NF-κB represent upstream elements of a common pathway that integrates catabolic cytokine input to skeletal muscle. Such a pathway would have obvious clinical relevance, providing potential targets for therapeutic interventions to inhibit or reverse cachexia. Of course, ROS and NF-κB are ubiquitous signaling elements that mediate a variety of cellular responses. Treatment of cachexia will require the identification of one or more downstream signals that are specific to skeletal muscle. It is our hope that continued research will establish muscle-specific targets and define therapeutic approaches by which cachexia can be prevented.
Abbreviations
COPD = chronic obstructive pulmonary disease; kDa = kiloDalton; MHCf = adult fast-type myosin heavy chain; NF-κB = nuclear factor-κB; NO = nitric oxide; ROS = reactive oxygen species; TNF-α = tumor necrosis factor-α; TNFR1 = type 1 TNF-α receptor; TNFR2 = type 2 TNF-α receptor.
Acknowledgments
Acknowledgement
Our work in this area has been sponsored by National Institutes of Health grant HL-59878.
References
- Schutze S, Machleidt T, Kronke M. Mechanisms of tumor necrosis factor action. Semin Oncol. 1992;2:16–24. [PubMed] [Google Scholar]
- Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxi-dants. EMBO J. 1996;15:1753–1765. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez C, Lopez-Soriano FJ, Argiles JM. Acute treatment with tumour necrosis factor-alpha induces changes in protein metabolism in rat skeletal muscle. Mol Cell Biochem. 1993;125:11–18. doi: 10.1007/BF00926829. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Wasting in cancer. J Nutr. 1999;129:243S–246S. doi: 10.1093/jn/129.1.243S. [DOI] [PubMed] [Google Scholar]
- Anker SD, Rauchaus M. Insights into the pathogenesis of chronic heart failure: immune activation and cachexia. Curr Opin Cardiol. 1999;14:211–216. doi: 10.1097/00001573-199905000-00004. [DOI] [PubMed] [Google Scholar]
- Moldawer LL, Sattler FR. Human immunodifficency virus-associated wasting and mechanisms associated with inflammation. Semin Oncol. 1998;25:73–81. [PubMed] [Google Scholar]
- Farber MO, Mannix ET. Tissue wasting in patients with chronic obstructive pulmonary disease. Neurol Clin. 2000;18:245–262. doi: 10.1016/s0733-8619(05)70188-2. [DOI] [PubMed] [Google Scholar]
- Di Francia M, Barbier D, Mege JL, Orehek J. Tumor necrosis factor-α levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;150:1453–1455. doi: 10.1164/ajrccm.150.5.7952575. [DOI] [PubMed] [Google Scholar]
- de Godoy I, Donahoe M, Calhoun WJ, Mancino J, Rogers RM. Elevated TNF-α production by peripheral blood monocytes of weight-losing COPD patients. Am J Respir Crit Care Med. 1996;153:633–638. doi: 10.1164/ajrccm.153.2.8564110. [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS. NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- Layne MD, Farmer SR. Tumor necrosis factor-alpha and basic fibroblast growth factor differentially inhibit the insulin-like growth factor-I induced expression of myogenin in C2C12 myoblasts. Exp Cell Res. 1999;249:177–187. doi: 10.1006/excr.1999.4465. [DOI] [PubMed] [Google Scholar]
- Thaloor D, Miller KJ, Gephart J, Mitchell PO, Pavlath GK. Systemic administration of the NF-κB inhibitor curcumin stimulates muscle regenerations after traumatic injury. Am J Physiol. 1999;277:C320–C329. doi: 10.1152/ajpcell.1999.277.2.C320. [DOI] [PubMed] [Google Scholar]
- Li Y-P, Schwartz RJ, Waddell ID, Holloway BR, Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-κB activation in response to tumor necrosis factor α. FASEB J. 1998;12:871–880. doi: 10.1096/fasebj.12.10.971. [DOI] [PubMed] [Google Scholar]
- Li Y-P, Reid MB. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1165–R1170. doi: 10.1152/ajpregu.2000.279.4.R1165. [DOI] [PubMed] [Google Scholar]
- Rofe AM, Conyers RAJ, Bais R, Gamble JR, Vadas MA. The effects of recombinant tumour necrosis factor (cachectin) on metabolism in isolated rat adipocyte, hepatocyte and muscle preparations. Biochem J. 1987;247:789–792. doi: 10.1042/bj2470789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MN. Tumor necrosis factor induces skeletal muscle protein breakdown in rats. Am J Physiol. 1991;260:E727–E730. doi: 10.1152/ajpendo.1991.260.5.E727. [DOI] [PubMed] [Google Scholar]
- Langen RCJ, Schols AMWJ, Kelders MCJM, Wouters EFM, Janssen-Heininger YMW. Inflammatory cytokines inhibit myo-genic differentiation through activation of nuclear factor-κB. FASEB J. 2001;15:1169–1180. doi: 10.1096/fj.00-0463. [DOI] [PubMed] [Google Scholar]
- Li Y-P, Atkins CM, Sweatt JD, Reid MB. Mitochondria mediate tumor necrosis factor-α/NF-κB signaling in skeletal muscle myotubes. Antioxid Redox Signal. 1999;1:97–104. doi: 10.1089/ars.1999.1.1-97. [DOI] [PubMed] [Google Scholar]
- Li Y-P, Schwartz RJ. TNF-α regulates early dfferentiation of C2C12 myoblasts in an autocrine fashion [abstract]. FASEB J. 2001;15:A1080. doi: 10.1096/fj.00-0632fje. [DOI] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Resznick AZ, Roy S, Packer L. Glutathione regulation of tumor necrosis factor-α-induced NF-κB activation in skeletal muscle-derived L6 cells. Biochem Biophys Res Comm. 1997;237:645–649. doi: 10.1006/bbrc.1997.7206. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- Llovera M, Garcia-Martinez C, Lopez-Soriano J, Carbo N, Agell M, Lopez-Soriano FJ, Argiles JM. Role of TNF receptor 1 in protein turnover during cancer cachexia using gene knockout mice. Mol Cell Endocrinol. 1998;142:183–189. doi: 10.1016/S0303-7207(98)00105-1. [DOI] [PubMed] [Google Scholar]
- Llovera M, Garcia-Martinez C, Lopez-Soriano J, Agell M, Lopez-Soriano FJ, Garcia I, Argiles JM. Protein turnover in skeletal muscle of tumour-bearing mice overexpressing the soluble TNF receptor-1. Cancer Lett. 1998;130:19–27. doi: 10.1016/S0304-3835(98)00137-2. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez C, Agell N, Llovera M, Lopez-Soriano FJ, Argiles JM. Tumour necrosis factor-α increases the ubiquitinization of rat skeletal muscle proteins. FEBS Lett. 1993;323:211–214. doi: 10.1016/0014-5793(93)81341-V. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez C, Llovera M, Agell N, Lopez-Soriano FJ, Argiles JM. Ubiquitin gene expession in skeletal muscle is increased during sepsis: involvement of TNF-α but not IL-1. Biochem Biophys Res Comm. 1995;217:839–844. doi: 10.1006/bbrc.1995.2848. [DOI] [PubMed] [Google Scholar]
- Llovera M, Garcia-Martinez C, Lopez-Soriano FJ, Argiles JM. TNF can directly induce the expression of ubiquitin-dependent proteolytic system in rat soleus muscles. Biochem Biophys Res Comm. 1997;230:238–241. doi: 10.1006/bbrc.1996.5827. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- Hasselgren PO, Fischer JE. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg. 2001;233:9–17. doi: 10.1097/00000658-200101000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-P, Reid MB. TNF-α and H2O2 stimulate ubiquitin conjugation of proteins in skeletal muscle myotubes [abstract]. FASEB J. 2001;15:A1080. [Google Scholar]