

Abstract
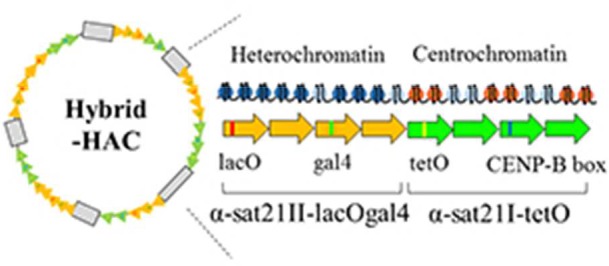
It is generally accepted that chromatin containing the histone H3 variant CENP-A is an epigenetic mark maintaining centromere identity. However, the pathways leading to the formation and maintenance of centromere chromatin remain poorly characterized due to difficulties of analysis of centromeric repeats in native chromosomes. To address this problem, in our previous studies we generated a human artificial chromosome (HAC) whose centromere contains a synthetic alpha-satellite (alphoid) DNA array containing the tetracycline operator, the alphoidtetO-HAC. The presence of tetO sequences allows the specific targeting of the centromeric region in the HAC with different chromatin modifiers fused to the tetracycline repressor. The alphoidtetO-HAC has been extensively used to investigate protein interactions within the kinetochore and to define the epigenetic signature of centromeric chromatin to maintain a functional kinetochore. In this study, we developed a novel synthetic HAC containing two alphoid DNA arrays with different targeting sequences, tetO, lacO and gal4, the alphoidhybrid-HAC. This new HAC can be used for detailed epigenetic engineering studies because its kinetochore can be simultaneously or independently targeted by different chromatin modifiers and other fusion proteins.
Keywords: centromere, chromosome segregation, human artificial chromosome, kinetochore, mitosis
Centromeres define the site of the assembly of the kinetochore, a multiprotein complex that directs chromosome segregation during cell division.1 In humans, endogenous centromeres typically form on chromosome-specific higher-order alphoid DNA arrays, which are composed of 171 bp alpha-satellite monomer units that are tandemly arranged in a directional head-to-tail fashion.2 Independent of this sequence preference, specific deposition of the centromere-specific histone H3 variant CENP-A forms the basis for an epigenetic maintenance of centromere identity.3,4 The epigenetic control of centromere activity is illustrated by the inactivation of centromeres on dicentric chromosomes5 and by the formation of rare neocentromeres that recruit CENP-A and assemble fully functional kinetochore structures on nonalphoid DNA.6
Microscopic investigation of stretched kinetochore fibers revealed that blocks of CENP-A nucleosomes are interspersed with canonical histone H3 nucleosomes that contain transcription-associated modifications, such as H3K4me2 and H3K36me2.7−9 This special chromatin was termed “centrochromatin”7 and suggests a functional link between the local chromatin environment and kinetochore function.1 These observations raise several questions about the exact nature of the chromatin that specifies kinetochore assembly and propagation. (1) What combination of histone modifications defines the elusive epigenetic state that is centrochromatin? (2) Can histone modifications be manipulated to turn normal chromatin into centrochromatin or, inversely, to inactivate established centromeres? (3) What barriers prevent heterochromatin spreading into centromeres? Answering these questions with native human chromosomes is extremely challenging.
Knowledge of the structure and function of human centromeres has dramatically increased since the first reconstitution of functional human centromeres, when DNA satellite repeats were transfected into human cells, forming the first human artificial chromosomes (HACs).10,11 Subsequently, many HACs have been constructed using different strategies of satellite DNA cloning and delivery into human cells12−23
The development of RCA-TAR cloning by our group provided the first method for constructing synthetic alphoid DNA arrays with precisely defined DNA sequence variation and the possibility of manipulating alphoid DNA arrays.24 This approach involves rolling circle amplification (RCA) of alphoid DNA oligomers as small as a dimer followed by assembly of the amplified fragments by transformation-associated recombination (TAR) in yeast.25,26 Using the RCA-TAR method, synthetic alphoid DNA arrays up to 140 kb have been generated and used for de novo HAC formation.24 The method also permitted generation of a synthetic HAC with a conditional centromere, the alphoidtetO-HAC,27 which has been instrumental in resolving a role for chromatin structure in kinetochore function.23 The alphoidtetO-HAC is based on a dimeric alphoid DNA array that contains alternating monomers with either CENP-B boxes or tetracycline operator (tetO) sequences. The latter sequences allow specific engineering of the alphoidtetO-HAC using chromatin modifiers fused to the tetracycline repressor (tetR). The alphoidtetO-HAC has been extensively used to investigate protein interactions within the kinetochore and to define the epigenetic signature of centromeric chromatin to maintain a functional kinetochore.9,23,28−33 Studies with this HAC revealed that nucleating heterochromatin or high levels of active transcription within centrochromatin disrupts kinetochore function27−29 and that a low level of transcription is needed to maintain an active kinetochore.9,33 Recent studies using the alphoidtetO-HAC showed that histone modifications and centromeric transcription block heterochromatin spreading into centrochromatin, thus preventing kinetochore inactivation.33 Histone modifications linked to transcription might act as an epigenetic barrier between centrochromatin and pericentromeric heterochromatin.
Although heterochromatin occurs in close proximity to centromeres, its role in chromosome segregation varies among species. S. pombe may provide an extreme example with its dependence on heterochromatin for de novo deposition of CENP-A and for proper cohesion dynamics.34 In contrast, the role of pericentromeric heterochromatin on kinetochore function in humans remains poorly understood. Results from different groups showed that disruption of pericentromeric heterochromatin is associated with chromosome mis-segregation and tumorigenesis.35−37
In this work, we have constructed a new alphoidhybrid-HAC containing two distinct synthetic alphoid arrays to investigate the role of heterochromatin domains on kinetochore maintenance and function. This HAC is based on a synthetic hybrid array consisting of an alphoidtetO array linked to a novel alphoid DNA array derived from monomeric alpha-satellite DNA lacking CENP-B boxes and containing lac operators (lacO) and yeast Gal4 binding sites. Transfection of the hybrid array into human HT1080 cells resulted in HAC formation. The alphoidhybrid-HAC was used to examine whether heterochromatin or centrochromatin can be induced to spread laterally by targeting chromatin modifiers.
Results and Discussion
Construction of the Hybrid tetO/lacOgal4 Synthetic Arrays by RCA-TAR Cloning
We wished to construct a human artificial chromosome (HAC) containing centrochromatin and pericentromeric heterochromatin domains (alphoidhybrid-HAC) to manipulate the epigenetic status of both domains independently. As the basis for the kinetochore domain, we designed a 343 bp alphoid 21-I dimer from high-order repeats (HORs) of chromosome 21 in which sequences corresponding to the CENP-B box in one monomer were replaced by a 42 bp tetracycline operator sequence (tetO), the binding site for E. coli tetracycline repressor (tetR) (Figure 1). As the basis for the pericentromeric heterochromatin domain, we synthesized a novel 2,078 bp alphoid 21-II-lacOgal4 12-mer that lacked CENP-B boxes but contained four gal4 binding sites (each 21 bp in size) and four lacO sequences (each 36 bp in size). These permit targeting the heterochromatin domain with chimeric proteins containing the yeast gal4 protein and/or E. coli lactose repressor (lacI) (Figure 2).
Figure 1.

Sequence of the alphoid dimer used for construction of the α21-I-tetO array. Both monomers are derived from a chromosome 21 alphoid type I (HOR). One monomer contains a CENP-B box (shaded blue). In the second monomer, the position corresponding to the CENP-B box sequence was replaced by a 42 bp tetO motif (shaded yellow).
Figure 2.

Sequence of the alphoid 12-mer used for construction of the α21-II-lacOgal4 array. All monomers were derived from a chromosome 21 alphoid type II array. Four gal4 sequences (two 21 bp in size and two dimers 42 bp in size; shaded green) and four lacO sequences (each 36 bp in size; shaded red) were incorporated into a 12-mer. Fifteen MseI sites are present in the 12-mer.
HAC formation requires input naked alphoid DNA of at least 30 kb in size for functional CENP-A core assembly.38 For this purpose, an artificial alphoid 21-I (α21-I) dimer and a synthetic α21-II-lacOgal4 12-mer were extended by rolling circle amplification (RCA) using phage ϕ29 DNA polymerase followed by yeast Transformation-associated recombination (TAR) cloning (Figure 3).24,25 These steps yielded two arrays, i.e. ∼ 40 kb α21-I-tetO and ∼40 kb α21-II-lacOgal4 arrays, cloned into the 10,209 bp YAC/BAC-based RCA-Sat43 vector containing the mammalian selectable marker blasticidin (Bsr)24 (Figures 3b, 4,a and 4b; see Materials and Methods for details). The tandem repeat structure of the α21-II-lacOgal4 array was confirmed by AlwN1 digestion, which revealed a 2,078 bp 12-mer (Figure 4c). The tandem repeat structure of the α21-I-tetO array was confirmed by EcoRI digestion, which revealed a 343 bp 2-mer (Figure 4d).
Figure 3.

Schematic representation of construction of synthetic tandem arrays. (a) Step one includes amplification of either a 2,078 bp 21-II-lacOgal4 12-mer or a 343 bp 21-I-tetO dimer by rolling circle amplification (RCA) reaction up to 1–3 kb fragments. (b) Step two includes construction of long alphoid arrays by transformation-associated recombination (TAR) cloning in yeast. The RCA-amplified fragments are cotransformed into yeast cells along with the MluI-linearized RCA-Sat43 vector (the MluI restriction site is located between the hooks). This vector contains a BAC cassette (a BAC replicon and a Clm marker), a YAC cassette (a selectable marker HIS3, a centromere sequence CEN6 from yeast chromosome VI, and yeast origin of replication ARSH4), and a mammalian marker Bsr (the blasticidin gene) that allows the vector to propagate in yeast, bacterial, and mammalian cells and alphoid-specific hooks of 40 bp each (Ebersole et al. 2005). Recombination of the RCA-amplified fragments accompanied by their recombination with the hooks results in the rescue of long arrays as circular YAC/BACs, and 40 kb α21-I-tetO and 40 kb α21-II-lacOgal4 arrays were chosen for further experiments. (c) Construction of the hybrid tetO-CENPB+-lacOgal4-CENPB– array. Recombination between the arrays accompanied by their recombination with the vector hooks leads to formation of the hybrid arrays. Ultimately, a molecule containing a 20 kb lacOgal4 array and 25 kb tetO array was chosen for HAC formation.
Figure 4.

Hybrid alphoid-DNA array construction. (a) CHEF analysis of 13 BACs with α21-II-lacOgal4 arrays of different size. The BAC DNAs were linearized by AvaII to release a vector part and an array. BAC #12 has an array of ∼40 kb in size. (b) CHEF analysis of the BAC with the α21-I-tetO array of ∼40 kb in size. The BAC DNA was digested by NheI/SpeI to release a vector fragment and the array. (c) Confirmation of the tandem repeat structure of 40 and 60 kb α21-II-lacOgal4 arrays by AlwN1 digestion. CHEF analysis revealed 2,461 bp 12-mer α21-II-lacOgal4 repeat units. (d) Conformation of the tandem repeat structure of a 40 kb α21-I-tetO array by EcoRI digestion. CHEF analysis revealed 343 bp 2-mer 21-I-tetO repeat units. (E) CHEF analysis of the hybrid α21-I-tetO/a21-II-lacOgal4 arrays. (Lane 1) Array consisting of 10 kb of the α21-II-lacOgal4 array and 30 kb of the a21-I-tetO array. (Lane 2) Array consisting of 25 kb α21-II-lacOgal4 array and 30 kb α21-I-tetO array. The array in lane 2 (in red) was chosen for HAC formation. (Lane 3) Array consisting of 15 kb α21-II-lacOgal4 array and 40 kb α21-I-tetO array.
To generate a hybrid array containing both α21-I-tetO and α21-II-lacOgal4, the two arrays were combined by recombination in yeast. After screening ∼190 colonies by CHEF gel electrophoresis, we obtained a molecule consisting of 25 kb α21-II-lacOgal4 and 30 kb α21-I-tetO arrays in the RCA-Sat43 vector (Figures 3c and 4e). This hybrid YAC/BAC DNA molecule of ∼65 kb in size (∼55 kb alphoid array plus ∼10 kb vector) was moved to bacterial cells and served as the input DNA for further HAC formation.
HAC Formation Using the Hybrid α-Satellite Array in Human HT1080 Cells
For HACs to be obtained, the RCA-Sat43 vector containing the hybrid array was purified from large-scale bacterial cultures (see Materials and Methods). The quality of the hybrid array DNA was checked by CHEF gel electrophoresis (data not shown). Input DNA was transfected into human HT1080 cells, and blasticidin S (BS)-resistant clones were selected for cytogenetic analysis. A total of 17 BS-resistant clones were obtained and expanded after transfection of the hybrid array DNA construct. To determine the fate of the hybrid array in HT1080 cells (expected to be either episomal HACs or chromosome integrations), these clones were processed for fluorescence in situ hybridization (FISH) using the RCA-Sat43 vector backbone as DNA probe directly labeled with Spectrum Green (Abbott Molecular) (Figure 5a). FISH analyses showed that most of the BS-resistant clones (13 out of 17; 76.5%) contain array integrations into endogenous chromosome arms (Figure 5b). Indeed, the most common fate of the input DNA construct in HAC formation experiments is integration into endogenous chromosome arms11,24,27,39 (Figure 5b). No preferential chromosomes or chromosome positions were observed for these array integrations.
Figure 5.

Hybrid HAC formation in HT1080 cells. (a) Representative FISH images of clones containing a HAC (left) and an array integration in an endogenous chromosome (right). (b, c) Screening of blasticidin-resistant clones by FISH. Diagrams represent the frequency of metaphases with HACs (black bars) and array integrations (gray bars) (N = 25) in HT1080 cells without (b) and with (c) CENP-A overexpression. (d) Frequency of HAC-containing clones with (CENP-A OE) and without (CENP-A WT) transient CENP-A overexpression during HAC formation. Only clones with a minimum of 10% metaphases containing HACs were considered as positive (10 vs 33%). (e) Representative two-color oligo-FISH images showing different hybrid HACs (clone 20.CA.07-top and 20.CA.24-bottom) containing tetO (red) and lacOgal4 (green) domains. Images were captured at optimized exposure times to clearly distinguish both signals in either clone (for signal intensity comparison between clones, see Figure S2). (f) Representative image of an HT1080 cell containing HAC clone 20.CA.24 and expressing both lacI-GFP (green) and tetR-mCherry (red) fusion proteins. Merged image (right panel) represents the overlay of GFP, mCherry, and DAPI channels. (g) Frequency of HAC-containing metaphases in the indicated clones containing HACs in the presence of blasticidin and after 30 days after blasticidin washout. The HAC loss rate is indicated in red. (h) Representative immunofluorescence images on metaphase spreads of HAC clone 20.CA.24 and stained with the indicated antibodies. Scale bars = 10 μm.
FISH analysis revealed that the hybrid array formed HACs in 2 of 17 BS-resistant cell lines (clones 20.05 and 20.07), showing 11 and 60% of HAC-containing metaphases, respectively (Figure 5b). Clone 20.05 also showed metaphases containing array integrations into endogenous chromosomes. A third clone (clone 20.22) showed a single HAC-containing metaphase plus 40% of metaphases containing array integrations (Figure 5b) and was not considered as a HAC-containing cell line. Only clones showing a minimum of 5% of metaphases with detectable HACs (at least 2 out of the 25 metaphases analyzed) were considered. The remaining BS-resistant clones showed neither HACs nor array integrations in any metaphase analyzed, suggesting that the clones may have acquired only the BSR gene. Overall, we observed HAC formation with an efficiency of 11.7% after transfection of the hybrid alphoid DNA array. This is consistent with the efficiency observed previously by others using synthetic alphoid DNA arrays for HAC formation experiments.24,27
These results showed that the hybrid array can form HACs in human HT1080 cells with an efficiency similar to those of other synthetic alphoid DNA arrays.
CENP-A Overexpression Significantly Increases the Efficiency of HAC Formation
The low efficiency of de novo HAC formation is one limitation for the wider application of HAC technology.20,23,40 We were therefore interested when it was recently reported that elevated levels of CENP-A increase the frequency of centrochromatin assembly and minichromosome formation in S. pombe.41 Moreover, we previously observed that seeding of centromeric chromatin at ectopic array integrations under different conditions is enhanced in cells overexpressing CENP-A.42
To investigate the effect of CENP-A levels on the efficiency of de novo HAC formation, we transfected the hybrid array into HT1080 cells transiently overexpressing CENP-A. As in previous HAC formation experiments, BS-resistant clones were selected and further expanded for cytogenetic analysis. FISH experiments revealed HACs in 11 of the 23 BS-resistant clones obtained after transfection with a range of 8–76% of HAC-containing metaphases (Figure 5c). Two additional clones showed HACs in a single metaphase and therefore were not considered as HAC-positive cell lines (Figure 5c). Some clones showed both array integrations and HACs as previously described for other HACs.27,43 For further analysis, we chose two clones, 20.CA.07 and 20.CA24 (Figure 5c), that contained only autonomously replicating HACs.
Thus, transient CENP-A overexpression increases the efficiency of HAC formation by 3-fold (Figure 5d), overcoming an important limitation of the HAC technology.
The Alphoidhybrid-HAC Contains Both Alphoid DNA Arrays
Previous molecular analyses have revealed that HACs are formed by amplification of the input DNA that may be accompanied by structural rearrangements, generating complex structures containing alternating blocks of α-satellite DNA and vector backbone.10,11,43,44 Because we used a hybrid α-satellite array as input DNA for HAC formation, it was possible that the resulting alphoidhybrid-HAC could have lost one of the two original arrays as a consequence of DNA reorganization in HT1080 cells. Therefore, we used oligo-FISH with labeled oligonucleotides that specifically recognize the tetO, lacO, and gal4 sequences to examine whether the HAC contains both α-satellite arrays. Oligo-FISH was first developed by Matera and Ward45 to detect repetitive sequences such as specific α-satellite DNA families. The use of small oligonucleotide probes, which show superior hybridization kinetics, has proven to be superior for faster and more sensitive FISH protocols. In our case, the repetitive nature of the hybrid HAC allows the visualization of oligonucleotide probes, as they hybridize in large numbers, thus allowing specific detection of the two different arrays in the HAC.
This experiment revealed that the HACs in both selected clones (20.CA.07 and 20.CA.24) have FISH signals for tetO and lacO + gal4 oligonucleotide probes (Figure 5e and Figure S1a). Therefore, the novel HACs contain both synthetic α-satellite DNA arrays. Importantly, a control experiment using the 1C7 cell line, which contains the alphoidtetO-HAC,28 showed a signal only for the tetO DNA array with no signal for the lacO + gal4 DNA array. This confirms the specificity of the probes used for oligo-FISH (Figure S1a).
To further confirm the presence of both synthetic arrays in the alphoidhybrid-HAC, we expressed tetR-mCherry and lacI-EGFP in these HT1080 cell lines and then analyzed the tethering of these fusion proteins to the hybrid HACs. As expected, the results showed the presence of signals for both tetR-mCherry and lacI-EGFP in interphase nuclei (Figure 5f). The signals were also detected on the HACs in metaphase chromosome spreads, where the HACs can be identified by DAPI staining (Figure S1b).
These data demonstrate the successful construction of novel synthetic HACs containing both α21-I-tetO and α21-II-lacOgal4 synthetic alphoid-DNA arrays. Such alphoidhybrid-HACs represent a novel targeting system for future epigenetic engineering studies of the centromere.
The Alphoidhybrid-HAC Segregates Accurately during Cell Division
Two independent clones containing alphoidhybrid-HACs (clones 20.CA.07 and 20.CA.24) were selected for further studies. In clone 20.CA.24, only 20% of the cells carry the alphoidhybrid-HAC; for this reason, we subcloned this cell line by limiting dilution to obtain a homogeneous population of HAC-containing cells. A total of 37 BS-resistant subclones were analyzed by FISH for the presence of the alphoidhybrid-HAC. Among them, one subclone (subclone 5B10) was shown to carry single alphoidhybrid-HACs in 100% of the metaphases analyzed (N = 25). The other subclones showed neither HACs nor integration in chromosome arms, suggesting that only the bsr gene was integrated.
To analyze the mitotic stability of these HACs, we grew the cells in the presence and absence of BS selection for 30 days (∼30 cell divisions) and monitored the presence of HACs by FISH. As a positive control, we included the 1C7 cell line, which contains the previously constructed alphoidtetO-HAC.27,28 The daily loss rate of the HAC (R) was calculated using the formula Nn= N0 × (1 – R)n, where N0 is the number of metaphase chromosome spreads showing HAC in the cells cultured under selection, Nn is the number of HAC-containing metaphase chromosome spreads after n days of culture in the absence of selection.
In 1C7 cells, the alphoidtetO-HAC was present in 22 out of 25 metaphases analyzed (88%) when cells were grown in the presence of BS. In the absence of BS selection for 30 days, the alphoidtetO-HAC was present in 20 out of 25 metaphases (80%). Thus, the daily loss rate of the alphoidtetO-HAC was established at 0.0032 (Figure 5g). This is consistent with the values previously observed (0.0021–0.0054).27
The alphoidhybrid-HAC in clone 20.CA.07 showed a decrease of HAC-containing metaphases from 76 to 72% when BS selection was removed for 30 days, and the frequency of metaphases with the HAC in clone 20.CA.24–5B10 decreased from 96 to 80% after BS washout (Figure 5g). Thus, the daily rates of loss observed in clones 20.CA.07 and 20.CA.24–5B10 were 0.0018 and 0.0054, respectively, showing a mitotic behavior similar to that of the alphoidtetO-HAC. These results indicate that the alphoidhybrid-HACs have a high mitotic stability in the absence of selection, indicating that these HACs replicate and segregate accurately during cell division.
The high mitotic stability observed for the alphoidhybrid-HACs suggests that these synthetic chromosomes contain the basic elements to maintain chromosome function. To confirm this, we performed immunofluorescence (IF) on unfixed metaphase spreads with antibodies that recognize different complexes that are important for chromosome segregation (Figure 5h). IF showed the presence of the outer kinetochore protein HEC1 on the alphoidhybrid-HAC (Figure 5h). Other protein complexes important for centromere function were also detected on the HAC at levels similar to endogenous chromosomes: specifically, the chromosome passenger complex shown by the presence of survivin staining of the HAC, the chromosome scaffold compartment shown by SMC2 staining of the HAC, and pericentromeric heterochromatin shown by the presence of HP1α in the HAC (Figure 5h). We thus conclude that the alphoidhybrid-HAC contains a functional kinetochore resembling that of endogenous chromosomes.
The alphoidhybrid-HAC has a high mitotic stability comparable to the synthetic alphoidtetO-HAC described previously.27 Furthermore, the alphoidhybrid-HAC resembles natural chromosomes, containing the basic protein complexes necessary for chromosome structure, function, and stability.
Structural Characterization of the Alphoidhybrid-HAC Shows Contiguous Tandem Copies of the Input DNA
Because the alphoidhybrid array forms mitotically stable HACs that resemble natural chromosomes, we went on to perform cytogenetic and molecular biology experiments to characterize the molecular organization of both alphoidhybrid-HAC clones. Oligo-FISH experiments on metaphase spreads clearly showed that the HAC in clone 20.CA.24–5B10 is larger than the HAC in clone 20.CA.07, as shown by DAPI and FISH signal intensities for both the tetO and lacOgal4 domains (Figure S2). Importantly, these results were confirmed by expressing tetR-EYFP and lacI-mCherry fusion proteins in HT1080 cells containing the alphoidhybrid-HACs. Whereas the alphoidhybrid-HAC in clone 20.CA.24–5B10 showed clear signals for both fusion proteins tethered to tetO and lacO sites, those signals were hardly visible in clone 20.CA.07 (data not shown). For this reason, we used the 20.CA.24–5B10 alphoidhybrid-HAC for further characterization. Interestingly, although alphoidhybrid-HAC 20.CA.07 is significantly smaller, its mitotic stability is higher compared to those of other synthetic HACs. Therefore, HAC size does not correlate in a simple manner with mitotic stability.
As shown above, FISH analysis of interphase and metaphase cells revealed the presence of lacOgal4 and tetO arrays in the alphoidhybrid-HAC in clone 20.CA.24 (Figure 5e). We performed high-resolution fiber-FISH analysis to see how the arrays are organized in this HAC. DNA fibers were prepared from HT1080 cells containing the HAC and hybridized with either tetO-specific or a mixture of the lacO plus gal4-specific oligonucleotide probes46 (see Materials and Methods). Representative images are shown in Figure 6a. This fiber-FISH analysis revealed alternating blocks of α21-I-tetO and α21-II-lacOgal4 arrays. The results are consistent with those obtained by interphase FISH analysis of the 20.CA.07 alphoidhybrid-HAC (Figure S3a–c), which show α21-I-tetO and α21-II-lacOgal4 blocks that are repeated in tandem.
Figure 6.

Structural analysis of the hybrid HAC propagated in human HT1080 cells. (a) Representative fiber-FISH images of clone 20.CA.24 HAC using oligonucleotide probes for tetO (red) and lacO + gal4 sequences (green). Different degrees of fiber stretching are shown (compare upper and lower panels). (b) Genomic DNA possessing the original HAC clone 20.CA.24 (left panel) and its subclone (5B10; right panel) were digested with SpeI endonuclease and separated by CHEF gel electrophoresis (range 10–100 kb). The SpeI recognition site is present once in the RCA-SAT43 vector at position 812 but not in the hybrid array. The transferred membrane was hybridized with radioactively labeled tetO-specific or lacO + gal4-specific probes. The 5B10 subclone has a HAC with a remarkably conserved array. Arrows indicate fragments of 95, 65, 40, and 30 kb in size that are specific to both probes. (c) Diagram illustrating multimerization of input DNA during de novo HAC formation in human HT1080 cells. Input DNA consists of 65 kb hybrid array and 10,209 bp RCA-Sat43 vector sequence.
Structural analysis of the alphoidhybrid-HACs in both clones was also carried out by Southern blot hybridization. Genomic DNA containing the alphoidhybrid-HACs was digested by SpeI endonuclease. This endonuclease cuts the vector RCA-Sat43 sequence once but does not have recognition sites in the alphoid α21-I-tetO or α21-II-lacOgal4 DNA arrays. SpeI-digested genomic DNA was separated by CHEF gel electrophoresis and hybridized with two different probes. One probe was specific to the tetO-alphoid sequence. The other was specific for both the lacO and gal4 sequences (see Materials and Methods for details). If the HACs were formed by simple concatenation involving rolling-circle amplification of the input DNA and had not undergone structural rearrangements, only one band of 65 kb in size (∼55 kb alphoid array plus ∼10 kb vector part) would be observed on the Southern blot after SpeI digestion. For the original clone 20.CA.24 (containing ∼25% of HAC-containing metaphases; see Figure 5), four major bands of 95, 65, 40, and 30 kb were detected with the tetO probe, and a similar profile with two additional minor bands of 60 and 8 kb was observed with the lacOgal4 probe (Figure 6b). For clone 20.CA.07, two major bands of 95 and 65 kb were detected with the tetO and lacOgal4 probes (Figure S3d).
Unexpectedly, for 20.CA.24 subclone 5B10, only one major band of 65 kb in size was observed (∼55 kb array plus ∼10 kb vector). These results indicate that the original clones 20.CA.24 and 20.CA.07 are likely a mixture of the cells with different-sized HACs. In contrast, the 5B10 subclone contains only one HAC with a regular structure containing alternating tetO and lacOgal4. This could suggest a rolling circle amplification mechanism of formation of this HAC. These results are in agreement with the alternating organization of α21-I-tetO and α21-II-lacOgal4 DNA blocks observed by fiber-FISH analysis (Figures 6a). Taking these data together, a proposed diagram of the hybrid HAC in subclone 5B10 is presented in Figure 6c.
Interestingly, this novel alphoidhybrid-HAC (subclone 5B10) had much more regular structure than the original synthetic alphoidtetO-HAC (subclone AB2.218.21).27 Southern blot hybridization of the SpeI-digested genomic DNA harboring the alphoidtetO-HAC revealed 15 fragments of different size, indicating complex rearrangements of 50 kb input alphoidtetO-DNA array during HAC formation.43 It also contained a large region from the long arm of chromosome 13, suggesting that at one point during its formation it may have integrated into that chromosome. It is possible that the more regular structure of the alphoidhybrid-HAC may be due to a more efficient process of de novo HAC formation, possibly as a result of CENP-A overexpression.
These results show that de novo alphoidhybrid-HAC formation was accompanied, as expected, by amplification of the input DNA molecule but that amplification can occur without gross structural rearrangements. Future experiments will determine whether this is due to HAC formation in the presence of overexpressed CENP-A.
Epigenetic Engineering Showed a Functional Two-Domain Kinetochore in Alphoidhybrid-HAC That Can Be Manipulated with Different Targeting Systems
As shown above, the alphoidhybrid-HAC in 20.CA24 subclone 5B10 is composed of regular alternating tandem blocks of α21-I-tetO and α21-II-lacOgal4 repeats.
We next asked where the kinetochore is localized in this HAC using chromatin immunoprecipitation with CENP-A antibodies followed by qPCR (ChIP-qPCR). Surprisingly, the results showed equal amounts of CENP-A on both the α21-I-tetO and α21-II-lacOgal4 arrays (Figure 7a). It was previously demonstrated that CENP-B box sequences are a requisite for de novo kinetochore nucleation.11,24 This suggested that either (i) seeding of CENP-A chromatin on the alphoidhybrid-HAC was initiated on both consecutive α21-I-tetO and α21-II-lacOgal4 blocks or (ii) CENP-A chromatin assembled first on the α21-I-tetO domains containing CENP-B boxes and subsequently spread to the flanking α21-II-lacOgal4 domains that lack CENP-B.
Figure 7.

Epigenetic engineering shows the presence of a two-domain centromere in the alphoidhybrid HAC. (a) ChIP-qPCR analysis of CENP-A levels in HT1080 clone 5B10 containing the alphoidhybrid HAC. The α21-I-tetO (tetO), α21-II-lacOgal4 (lacOgal4) hybridHAC domains, satellite D17Z1 (Chr17), and degenerate satellite type-II (Sat2) repeats were assessed. (b) Representative images of HT1080–5B10 cells expressing the indicated tetR and lacI-fusion proteins and stained with H3K9me3 (second panel) and CENP-A (third panel) antibodies. Merge images represent the overlay of GFP (green), H3K9me3 (blue), and CENP-A (red). (c) Quantification of HAC-associated CENP-A staining in individual cells transfected with the indicated fusion proteins and plotted as A.F.U. Solid bars indicate the medians, and error bars represent the s.e.m. n = two independent experiments for each time point and staining. Asterisks indicate a significant difference (*P < 0.05; **P < 0.01; Mann–Whitney test). Scale bars = 10 μm.
To look at the ability of proteins to spread across the two arrays (array “cross-talk”), we transiently expressed KAP1 fused to either tetR-EYFP or lacI-GFP in 5B10 cells. KAP1 is a chromatin modifier that disrupts the kinetochore by seeding heterochromatin marks, including H3K9me3.28 Expression of tetR-EYFP-KAP1 caused a significant decrease of CENP-A levels on the alphoidhybrid-HAC compared to that of control experiments tethering tetR-EYFP (Figure 7b and c). The decrease of CENP-A levels observed after tethering tetR-EYFP-KAP1 was accompanied by an increase of HAC mis-segregation, as revealed by a significantly increased frequency of cells showing an abnormal number of HACs compared to controls (Figure 8c). On the other hand, tethering lacI-GFP-KAP1 to the alphoidhybrid-HAC did not result in significant differences in CENP-A levels compared to control tethering of either lacI-GFP or tetR-EYFP (Figures 7b and c). LacI-GFP-KAP1 tethering also did not cause an increase in HAC segregation defects, as the cells maintained a stable HAC number (Figure 8c). These results are consistent with the previous results using the alphoidtetO-HAC.28
Figure 8.

Alphoidhybrid HAC shows epigenetically distinct centromeric domains. (a) Representative images of HT1080–5B10 cells expressing the indicated tetR (first panel) and lacI-fusion proteins (second panel) and stained with antibodies recognizing H3K9me3 (third panel) and CENP-A (fourth panel). Merged images represent the overlay of TMR-SNAP, GFP, and H3K9me3 (MERGE 1; fifth panel) and GFP, H3K9me3 and CENP-A (MERGE 2; sixth panel). (b) Quantification of HAC-associated CENP-A staining in individual cells transfected with the indicated fusion proteins and plotted as A.F.U. Solid bars indicate the medians, and error bars represent the s.e.m. n = two independent experiments for each time point and staining. Asterisks indicate a significant difference (**P < 0.01; Mann–Whitney test). (c) Quantification of alphoidhybrid HAC copy-numbers as determined by counting the GFP and/or TMR-SNAP spot in interphase nuclei of cells transfected with the indicated fusion proteins. Data represent the mean (and s.e.m.) of three independent assays of each time point after doxycycline washout (n = 1,000 nuclei per condition; *P < 0.05, **P < 0.0001; χ2-test). (d) ChIP-qPCR analysis in HT1080–5B10 cells using the indicated antibodies. The α21-I-tetO (tetO), α21-II-lacOgal4 (lacOgal4) hybridHAC domains, the satellite D17Z1 (Chr17), and the degenerate satellite type-II (Sat2) repeats were assessed. Scale bars = 10 μm.
This suggests that heterochromatin nucleated on the α21-II-lacOgal4 array did not spread efficiently into the core centromere domain on the α21-I-tetO array.
Recent models suggest that chromatin modifications present at centrochromatin allow CENP-A and kinetochore assembly and maintenance.23 In particular, chromatin modifications associated with transcriptionally active chromatin, such as H3K4me2 and H3K9 acetylation, increase the CENP-A level and prevent heterochromatin spreading to centrochromatin that can ultimately inactivate the kinetochore.9,33 To examine the functional interactions between centrochromatin containing CENP-A and flanking heterochromatin, the alphoidhybrid-HAC was targeted with combinations of chromatin modifiers binding to the two different HAC domains. As a proof of principle, we simultaneously targeted the C-terminal transactivation domain of NF-ΚB p65 to the α21-I-tetO domains and KAP1 to the α21-II-lacOgal4 domains as tetR-SNAP and lacI-GFP fusion proteins, respectively. Tethering of tetR-SNAP-p65 together with lacI-GFP significantly increased CENP-A levels on the alphoidhybrid-HAC compared to that of a control tethering of tetR-SNAP with lacI-GFP (Figures 8a and b). As previously reported for the alphoidtetO-HAC,29 tethering tetR-SNAP-p65 to the alphoidhybrid-HAC increases levels of H3K9ac (Figure S4b). In parallel experiments, tethering lacI-GFP-KAP1 to the alphoidhybrid-HAC increased the H3K9me3 level (Figure S4a). Simultaneous tethering of lacI-GFP-KAP1 together with tetR-SNAP maintained the CENP-A level similar to that observed in controls (Figure 8a and b).
Strikingly, simultaneous tethering of tetR-SNAP-p65 with lacI-GFP-KAP1 seems to balance the levels of H3K9ac and H3K9me3 (Figure S4), and the levels of CENP-A are maintained similar to those observed in controls (Figure 8a and b). Thus, although there does appear to be cross-talk between the two domains of the HAC, kinetochore function is preserved as demonstrated by the proper segregation of the alphoidhybrid-HAC when these fusion proteins were tethered together (Figure 8c).
The epigenetic environment in the centromere is important for kinetochore assembly and maintenance.23 To study the epigenetic differences between the different centromeric domains in the alphoidhybrid-HAC, we performed ChIP-qPCR experiments pulling down different histone H3 modifications typically associated with centromeric chromatin, such as H3K4me2, H3K36me2, and H3K9me3 (Figure 8d). The results showed that there is no difference in the amount of the heterochromatin marker H3K9me3 between α21-I-tetO and α21-II-lacOgal4 domains or between the alphoidhybrid-HAC and the centromere of the endogenous chromosome 17 (Figure 8d). Thus, the level of heterochromatin is similar between the alphoidhybrid-HAC centromere and endogenous centromeres. The lower level of H3K9me3 in the Sat2 region could be explained by the lower level of heterochromatin in HT1080 cells (Figure 8d).36,47
We also observed different levels of chromatin marks associated with actively transcribed chromatin on the α21-I-tetO and α21-II-lacOgal4 arrays. Whereas H3K4me2 is preferentially enriched on the α21-II-lacOgal4 arrays, H3K36me2 was preferentially enriched on the α21-I-tetO arrays (Figure 8d). The different localizations of H3K4me2 and H3K36me2 on centrochromatin suggest that these histone modifications may play different roles in kinetochore assembly and/or maintenance. Previous results showed that removing H3K4me2 from the alphoidtetO-HAC centromere disrupts kinetochore maintenance and function.33 The present results suggest that it will be important in the future to explore the role of H3K36me2 in centrochromatin in greater detail.
Our proof of principle experiments reveal that the alphoidhybrid-HAC containing two synthetic domains can be manipulated for epigenetic engineering studies. Our results with this novel HAC reinforce the model that centromeres are assembled and maintained by a specific chromatin environment. Moreover, our experiments combining the opposing chromatin modifiers p65 and KAP1 revealed that, although there is cross-talk between the kinetochore and heterochromatin domains, induced heterochromatin disrupts the centromere only when it is nucleated within centrochromatin. Inducing heterochromatin in the pericentromeric domains did not disrupt the kinetochore, possibly due to the presence of specific histone modifications and/or active RNAP II transcription at centrochromatin that protects it from heterochromatin spreading.
Conclusions
We developed and characterized a novel synthetic alphoidhybrid-HAC (human artificial chromosome) containing two distinct alphoid DNA arrays. The two synthetic arrays can remain remarkably preserved during HAC formation in contrast to the gross rearrangements observed with the previous generation of alphoidtetO HACs. The alphoidhybrid-HAC allows simultaneous independent targeting of chromatin modifiers to different centromeric compartments using independent tetO and lacO/gal4 targeting systems, and our preliminary experiments reveal that there is some cross-talk between the chromatin in two arrays. The alphoidhybrid-HAC may be used in future studies to clarify whether the endogenous CENP-A chromatin is continuous or interrupted with blocks of heterochromatin in natural human chromosomes.
Materials and Methods
Construction of α21-I-tetO and α21-II-lacOgal4 Alphoid Arrays by Rolling-Circle Amplification (RCA) Followed by Transformation-Associated Recombination (TAR) in Yeast
A method to rapidly convert any desirable DNA fragment, as small as 100 bp, into long tandem DNA arrays up to 140 kb in size is described in detail in a previous publication.48 The method includes rolling-circle phi29 amplification (RCA) of the sequence in vitro and assembly of the RCA products in vivo by homologous recombination in the yeast Saccharomyces cerevisiae. In our case, for the RCA reaction we used a 343 bp 21-I alphoid dimer from high-order repeats (HORs) of chromosome 21 containing CENP-B boxes and a 2,078 bp synthesized 21-II-lacOgal4/CENPB minus-12-mer. In one of the monomers of the 21-I alphoid dimer, a CENP-B box was replaced by a 42 bp tetO motif (see Figure 1). In the 21-II alphoid 12-mer, four gal4 sequences (each 21 bp in size) and four lacO sequences (each 36 bp in size) were incorporated (see Figure 2). RCA reactions were performed using an Amersham TempliPhi kit according to the manufacturer’s instructions except that reactions were scaled up to 100 μL and were spiked with a template-specific primer mix to a final concentration of 2 pmol/μL (Figure 3a). Reaction products were phenol/chloroform extracted and ethanol precipitated prior to recombinational cloning. As a second step, the length of the resulting RCA products was extended by transformation-associated homologous recombination (TAR) in yeast25,26,49 using a targeting RCA-Sat43 vector24 (Figure 3B). The RCA-Sat43 vector (10,209 bp in size) contains YAC (HIS3, CEN6, ARSH4) and BAC (Cm, ori F) cassettes as well as a mammalian selectable marker (BSR). Also, the RCA-Sat43 vector contains the appropriate alphoid satellite hooks of ∼40 bp each. Before yeast transformation, the vector was linearized by MluI to release targeting hooks. The highly transformable Saccharomyces cerevisiae strain VL6–48N (MATalpha, his3-Δ200, trp1-Δ1, ura3-Δ1, lys2, ade2–101, met14), which has HIS3 and URA3 deletions, was used for transformation. Conditions for spheroplast transformation were described previously.50 Typically, each transformation used 2–3 μg of RCA product and 0.02 μg of the linearized vector. Under such conditions, 200–1000 His+ transformants were usually obtained. As a third step, yeast transformants were combined into pools, and then purified genomic DNA from the yeast clones was electroporated into Escherichia coli cells (DH10B, Invitrogen). The insert size of α21-II-lacOgal4 arrays was determined by CHEF (Bio-Rad) after AvaII digestion, which releases an ∼6.5 kb vector fragment and an array plus ∼3.5 kb vector fragment. Representative CHEF analysis of 13 BACs is shown (Figure 4a). Digestion of the α21-II-lacOgal4 arrays by AlwNI reveals 12-mers of 2,078 bp in size (Figure 4b). The insert size of α21-I-tetO arrays was determined by CHEF (Bio-Rad) after NotI digestion that releases an ∼2 kb fragment of the RCA-Sat43 vector and an array plus ∼7 kb vector fragment. Representative CHEF analysis of one BAC is shown (Figure 4c). Digestion of the α21-I-tetO arrays by EcoRI reveals 2-mers of 343 bp in size (Figure 4d).
Construction of the Hybrid tetO-CENPB+/lacOgal4-CENPB– Array by Recombinational Cloning in Yeast
A general scheme of construction of the synthetic hybrid tetO-CENPB+/lacOgal4-CENPB- array is presented in Figure 3c. The ∼40 kb α21-II-lacOgal4 array (Figure 4a; lane 12) and ∼40 kb α21-I-tetO array (Figure 4c) were chosen for construction of the hybrid tetO-CENPB+/lacOgal4-CENPB– array. Restriction of BAC DNAs was done by endonucleases that cleave the molecule at insert/vector junctions and by an endonuclease that cuts the vector part completely. The 40 kb α21-II-lacOgal4 array was released by digestion with MspI/NsiI endonucleases (33 MspI recognition sites are present in the RCA-Sat43 vector). The 40 kb α21-I-tetO array was released by digestion with MseI/SpeI/NheI endonucleases (57 MseI recognition sites are present in the RCA-Sat43 vector). The α21-II-lacOgal4 and α21-I-tetO arrays were gel-purified after CHEF separation, mixed with MluI-digested RCA-Sat43 vector (the MluI site is present between the hooks and not present in the arrays) and transformed into Saccharomyces cerevisiae strain VL6–48N. Typically, 0.5 μg of each array and 0.02 μg of the linearized RCA-Sat43 vector were used for one transformation. Under such conditions, 100–300 His+ transformants were usually obtained. Next, genomic DNA from yeast clones washed from plates was isolated and electroporated into Escherichia coli cells (DH10B, Invitrogen). Because homology between α21-II-lacOgal4 and α21-I-tetO arrays is ∼70–74%, the percentage of the hybrid arrays was very low, i.e., ∼0.1%. The insert size of the entire hybrid arrays in the BACs was checked by MspI digestion. The size of the α21-I-tetO part in the hybrid arrays was checked by MseI digestion (this endonuclease completely digests a vector part and a α21-II-lacOgal4 array). The size of the α21-II-lacOgal4 array in the hybrid arrays was checked by MslI or AlwI (these endonucleases digest a vector part and the α21-I-tetO array). A BAC containing a hybrid array consisting of ∼25 kb α21-II-lacOgal4 array and ∼30 kb α21-I-tetO array was chosen for further HAC development experiments (Figure 4e).
Southern Blot Hybridization Analysis
Southern blot hybridization was performed with a 32P-labeled probe as described previously51 with minor changes. Genomic DNA was prepared in agarose plugs and restriction-digested by SpeI in the buffer recommended by the manufacturer. The digested DNA was CHEF (CHEF Mapper, Bio-Rad) separated (autoprogram, 5–150 kb range, 16 h transfer), transferred to membrane (Amersham Hybond-N+), and blot-hybridized with an 82 bp probe for tetO and a 74 bp probe for lacO and gal4. DNA sequences for the probes were amplified by PCR using the primers and synthetic DNA fragment as a template (tetO_south_21_M: 5′-TTTGTGGAAGTGGACATTTACTAGCAGCAGAGCTCTCCCTATCAGTGATAGAGACTAGCCCATAAAAATAGACAGAAGCATT-3′, tetO_south_21_F1: 5′-TTTGTGGAAGTGGACATTTC-3′, tetO_south_21_R1: 5′-AATGCTTCTGTCTATTTTTA-3′; lacO_south3_M: 5′-TGTGGAAGTGGACATTTCGACCACATGTGGAATTGTGAGCGGATAACAATTTGTGGCCCATAAAAATAGACAGA-3′, lacO_south3_F1: 5′-TGTGGAAGTGGACATTTCGA-3′, lacO_south3_R1: 5′-TCTGTCTATTTTTATGGGCC-3′; gal4_south1_M: 5′-AATGGACATTTCGACGGAGGACAGTCCTCCGTCGACGGAGGACAGTCCTCCGCATAAAATCTA-3′, Gal4_south1_F1: 5′-AATGGACATTTCGACG-3′, gal4_south1_R1: 5′-TAGATTTTATGCGGAG-3′). The blot was incubated for 2 h at 65 °C for prehybridization in Church’s buffer (0.5 M Na-phosphate buffer containing 7% SDS and 100 μg/mL of unlabeled salmon sperm carrier DNA). The labeled probe was heat denatured in a boiling water bath for 5 min and snap-cooled on ice. The probe was added to the hybridization Church’s buffer and allowed to hybridize for 48 h at 65 °C. Blots were washed twice in 2× SSC (300 mM NaCl, 30 mM sodium citrate, pH 7.0), 0.05% SDS for 10 min at room temperature, then twice in 2× SSC, 0.05% SDS for 5 min at 60 °C. Blots were exposed to X-ray film for 5 days at −80 °C.
Cell Culture, Transfections, and HAC Formation
Human HT1080 cells were maintained in DMEM supplemented with 10% FBS (Invitrogen) plus 100 U/ml penicillin G and 100 μg/mL of streptomycin sulfate (Invitrogen). Cells were grown at 37 °C in 5% CO2 in a humidified atmosphere. Transfections were performed using Xtremegene-9 (Roche) following the manufacturer’s instructions. In brief, for transfections of cells growing in 10 cm dishes, transfection complexes containing 15 μL of Xtremegene-9 reagent and 5 μg of plasmid DNA were prepared in 500 μL of OptiMEM (Invitrogen). After 20 min of incubation at room temperature, 500 μL of transfection complexes was added dropwise in 10 mL of media. After 24 h, transfected cells were selected adding 4 μg/mL of blasticidin S (Sigma) and grown for 2–3 weeks until separate resistant colonies were present. Resistant colonies were isolated by trypsinization in cloning cylinders (Thermo Scientific). Isolated clones were expanded in the presence of 4 μg/mL of blasticidin S. For cotransfection experiments with the CENP-A-SNAP plasmid, 5 μg of plasmid DNA was added to the transfection reaction.
Fluorescent in Situ Hybridization (FISH)
Samples were processed as previously described by us.46 In brief, metaphase chromosomes were obtained following standard procedures: 3 h before harvesting, cells were treated with Colcemid (Invitrogen) at a final concentration of 0.2 μg/mL. They were then resuspended in warm hypotonic solution (0.075 M KCl) for 10 min at 37 °C and fixed in methanol:acetic acid (3:1). To obtain stretched chromatin fibers, 2 mL of a cell culture was centrifuged, and the pellets were washed in 1× PBS. Pellets were resuspended in 1× PBS to reach a final concentration of 2 × 106 cells/mL and spread on slides. Once the slides were mounted on the Shandon Sequenza cover plates (Thermo Scientific), DNA fibers were released applying a lysis solution (0.07 M NaOH in ethanol) and fixed in methanol. Slides were kept at −20 °C until they were processed for FISH.
The RCA-Sat43 vector backbone was used as DNA probe. BAC DNA extraction was performed using the EndoFree Plasmid Maxi kit (Qiagen GmBh; Hilden, Germany) following the manufacturer’s instructions. BAC DNA was labeled by Nick translation using Spectrum Green dUTPs (Abbott Molecular) following the manufacturer’s instructions. Probes were mixed with 10 μg of Cot 1 DNA (Invitrogen; Carlsbad, USA), ethanol precipitated, and resuspended in 1× hybridization buffer (50% formamide, 1× SSC, and 10% dextran sulfate) to a final concentration of 40 ng/μL. For oligo-FISH experiments, oligonucleotides recognizing the tetO sequence (5′- ACTAGCAGCAGAGCTCTCCCTATCAGTGATAGAGACTAG-3′) labeled with Digoxigenin, and oligonucleotides recognizing both lacO (5′- CATGTGGAATTGTGAGCGGATAACAATTTGTGG-3′) and Gal4 (5′- TCGACGGAGGACAGTCCTCCG-3′) sequences labeled with Biotin were synthesized (Sigma). Oligonucleotides were mixed at 100 ng/ μL and resuspended in 2× hybridization buffer (Cellay Inc.), 10× blocking reagent (Cellay Inc.), and 50 μg/mL of salmon sperm DNA (Sigma).
FISH was carried out following standard procedures.46 Briefly, the DNA probe was denatured at 80 °C for 5 min and preannealed at 37 °C for 30 min. Slides were denatured in 70% formamide/2× SSC at 70 °C for 1 min and hybridized in a humid chamber at 37 °C overnight. Slides were washed in 0.7×SSC/0.03%NP40 for 1 min at 70 °C and in 2×SSC/0.03%NP40 for 2 min at room temperature. For Oligo-FISH experiments, slides were denatured in 70% formamide/2×SSC at 70 °C for 1 min and hybridized in a humid chamber at 37 °C for 2 h. Slides were thereafter washed in 20%formamide/2×SSC for 5 min and in 2×SSC/0.1%Tween-20 for 5 min at 37 °C. Oligonucleotide probes were detected with rhodamine-conjugated antidigoxigenin (Roche) and fluorescein-conjugated streptavidin (Vector Laboratories). Slides were mounted with Vectashield (Vector Laboratories) containing 4′,6-diamidino-2-phenylindole (DAPI) for chromosome counterstaining.
Indirect Immunofluorescence Staining and Microscopy
Indirect immunofluorescence staining of cells fixed in 4% formaldehyde/1×PBS was performed following standard procedures. Immunofluorescence on unfixed metaphase spreads was performed as described previously.9 The following antibodies were used: mouse anti-CENP-A (clone A1, 1:500), mouse anti-HP1α (Millipore MAB3584, 1:1000), mouse anti-HEC1 (abcam AC3612, 1:1000), rabbit anti-SMC2 (A. Losada, 1:1000), rabbit anti-Survivin (Cell Signaling, 1:400), rabbit anti-H3K9me3 (abcam 8898; 1:200), and rabbit anti-H3K9ac (B. Turner, 1:200).
Microscope images were acquired on a DeltaVision Core system (Applied Precision) using an Olympus IX-71 inverted microscope stand with an Olympus UPlanSApo 100× oil immersion objective (numerical aperture (NA) 1.4) and an LED light source. Camera (Photometrics Cool Snap HQ), shutter, and stage were controlled through SoftWorx (Applied Precision). Z-series were collected with a spacing of 0.2 μm, and image stacks were subsequently deconvolved in SoftWorx. For CENP-A signal quantification, a custom-made macro in ImageJ (National Institutes of Health, Bethesda, MD) modified from Bodor et al.52 was used.
Chromatin Immunoprecipitation and Quantitative PCR (ChIP-qPCR)
Exponentially growing cells were washed in D-PBS (Gibco) and subsequently harvested with TrypLE Express (Gibco). Cells were resuspended in D-PBS up to a concentration of 1 × 106 cells/ml and cross-linked in a final 1% formaldehide solution (Sigma) for 5 min at room temperature, followed by quenching with 2.5 M glycine for 5 min at room temperature. Cells at a concentration of 5 × 106 cells/ml were lysed in lysis buffer (10 mM Tris pH 8.0; 10 mM NaCl; 0.5% NP-40) containing protease inhibitors (1 μg/mL of CLAP; 0.5 μg/mL of Aprotinin; 1 mM PMSF) for 10 min on ice. Nuclei were briefly washed in lysis buffer with protease inhibitors in 300 μL of dilution buffer 1 (50 mM Tris pH 8.0; 2 mM EDTA; 0.2% SDS; 134 mM NaCl; 0.88% Triton X-100; 0.088% Na-deoxycholate). Chromatin was sheared by sonication in a Bioruptor sonicator (Diagenode) for 14 cycles (30s ON/30S OFF) at high setting and 4 °C. Supernatant products of sonication were diluted with 300 μL of dilution buffer 1, 500 μL of dilution buffer 2 (50 mM Tris pH 8.0; 167 mM NaCl; 1.1% Triton X-100; 0.11% Na-deoxycholate), and 500 μL of RIPA buffer containing 150 μL of NaCl (RIPA-150) and protease inhibitors. Antimouse IgG Dynabeads (Invitrogen) were coated with the relevant antibodies for 6 h with RIPA-150/0.5% BSA at 4 °C and washed twice with RIPA-150/0.5% BSA, and 500 μL of sheared chromatin was incubated with the beads at 4 °C overnight. Beads were afterward washed twice with RIPA-150 and RIPA buffer containing 500 mM NaCl (RIPA-500) and a final wash with TE pH 8.0. Antibody/chromatin complexes were de-cross-linked with 10% Chelex-100 resin (BioRad) in water at 93 °C and treated with RNase A and Proteinase K. DNA was subsequently recovered by aliquoting 60 μL of the supernatant in a new eppendorf tube. ChIPed DNA was subjected to RT-PCR using a SYBR Green Master Mix (Roche) using the following oligonucleotides: tetO-Fw (5′-CCACTCCCTATCAGTGATAGAGAA-3′), tetO-Rv (5′-TCGACTTCTGTTTAGTTCTGTGCG-3′) for the α21-I-tetO domain of the hybrid HAC, lacOgal4-Fw (5′-TATGGTGTCGACGGAGGACA-3′), and lacOgal4-Rv (5′-CCGCTCACAATTCCACATGTG-3′) for the α21-II-lacOgal4 domain of the hybrid HAC, chr17-Fw (5′-TTGTGGTTTGTGGTGGAAAA-3′) and chr17-Rv (5′-CTCAAAGCGCTCCAAATCTC-3′) for the alphoidchr17 array, bsr-Fw (5′-CAGGAGAAATCATTTCGGCAGTAC-3′) and bsr-Rv (5′-TCCATTCGAAACTGCACTACCA-3′) for the blasticidin resistance gene, sat2-Fw (5′-TCGCATAGAATCGAATGGAA-3′) and sat2-Rv (5′-GCATTCGAGTCCGTGGA-3′) for the pericentromeric alphoidchr1, act-Fw (5′-GCCGGGACCTGACTGACTAC-3′) and act-Rv (5′-AGGCTGGAAGAGTGCCTCAG-3′) for actin.
Acknowledgments
This work was funded by the UK Research Councils’ Synthetic Biology for Growth programme core grant BB/M018040/1 to the BBSRC/EPSRC/MRC Synthetic Biological Research Centre. Additional funding of the Earnshaw lab is provided by Wellcome, of which W.C.E. is a Principal Research Fellow (grant number 073915). O.M. was funded in part by the European Molecular Biology Organization (long-term EMBO fellowship; ALTF-453-2012). Additional funding was provided by the Intramural Research Program of the NIH, NCI Center for Cancer Research (V.L. and N.K.) and MEXT KAKENHI (grant numbers 23247030 and 23114008) and the Kazusa DNA Research Institute Foundation (H.M.).
Supporting Information Available
Supplementary Figures: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.8b00018.
The alphoidhybrid HACs containing both alphoid DNA arrays, the alphoidhybrid HAC in clone 20.CA.24 being larger than in clone 20.CA.07, structural analysis of clone HT1080–20.CA.07, and the alphoidhybrid HAC showing epigenetically distinct centromeric domains (PDF)
Author Contributions
E.P.: data acquisition, analysis and interpretation, manuscript revision; N.K.: data acquisition, analysis and interpretation, wrote the manuscript, and made all arrays; N.K. and M.L.: performed Southern blot experiments; J.A.C.: contributed to the optimization of oligo-FISH experiments; V.L. and H.M.: contributed new experimental and analytical tools and revised the article critically for important intellectual content; W.C.E.: experimental design, data analysis and interpretation, manuscript drafting and revision; O.M.: experimental design, data acquisition, analysis and interpretation, wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fukagawa T.; Earnshaw W. C. (2014) The centromere: chromatin foundation for the kinetochore machinery,. Dev. Cell 30, 496–508. 10.1016/j.devcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler M. G.; Sullivan B. A. (2006) Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 7, 301–313. 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- Earnshaw W. C.; Migeon B. R. (1985) Three related centromere proteins are absent from the inactive centromere of a stable isodicentric chromosome. Chromosoma 92, 290–296. 10.1007/BF00329812. [DOI] [PubMed] [Google Scholar]
- Sekulic N.; Black B. E. (2012) Molecular underpinnings of centromere identity and maintenance. Trends Biochem. Sci. 37, 220–229. 10.1016/j.tibs.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan B. A.; Schwartz S. (1995) Identification of centromeric antigens in dicentric Robertsonian translocations: CENP-C and CENP-E are necessary components of functional centromeres. Hum. Mol. Genet. 4, 2189–2197. 10.1093/hmg/4.12.2189. [DOI] [PubMed] [Google Scholar]
- du Sart D.; Cancilla M. R.; Earle E.; Mao J. I.; Saffery R.; Tainton K. M.; Kalitsis P.; Martyn J.; Barry A. E.; Choo K. H. (1997) A functional neo-centromere formed through activation of a latent human centromere and consisting of non-alpha-satellite DNA,. Nat. Genet. 16, 144–153. 10.1038/ng0697-144. [DOI] [PubMed] [Google Scholar]
- Sullivan B. A.; Karpen G. H. (2004) Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 11, 1076–1083. 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro S. A.; Vagnarelli P.; Dong Y.; Hori T.; McEwen B. F.; Fukagawa T.; Flors C.; Earnshaw W. C. (2010) A super-resolution map of the vertebrate kinetochore,. Proc. Natl. Acad. Sci. U. S. A. 107, 10484–10489. 10.1073/pnas.1002325107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann J. H.; Rodriguez M. G.; Martins N. M.; Kimura H.; Kelly D. A.; Masumoto H.; Larionov V.; Jansen L. E.; Earnshaw W. C. (2011) Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 30, 328–340. 10.1038/emboj.2010.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington J. J.; Van Bokkelen G.; Mays R. W.; Gustashaw K.; Willard H. F. (1997) Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 15, 345–355. 10.1038/ng0497-345. [DOI] [PubMed] [Google Scholar]
- Ikeno M.; Grimes B.; Okazaki T.; Nakano M.; Saitoh K.; Hoshino H.; McGill N. I.; Cooke H.; Masumoto H. (1998) Construction of YAC-based mammalian artificial chromosomes. Nat. Biotechnol. 16, 431–439. 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- Ebersole T. A.; Ross A.; Clark E.; McGill N.; Schindelhauer D.; Cooke H.; Grimes B. (2000) Mammalian artificial chromosome formation from circular alphoid input DNA does not require telomere repeats. Human molecular genetics 9, 1623–1631. 10.1093/hmg/9.11.1623. [DOI] [PubMed] [Google Scholar]
- Grimes B. R.; Schindelhauer D.; McGill N. I.; Ross A.; Ebersole T. A.; Cooke H. J. (2001) Stable gene expression from a mammalian artificial chromosome. EMBO Rep. 2, 910–914. 10.1093/embo-reports/kve187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N.; Ebersole T.; Koriabine M.; Pak E.; Rogozin I. B.; Katoh M.; Oshimura M.; Ogi K.; Peredelchuk M.; Solomon G.; Brown W.; Barrett J. C.; Larionov V. (2003) Cloning of human centromeres by transformation-associated recombination in yeast and generation of functional human artificial chromosomes. Nucleic acids research 31, 922–934. 10.1093/nar/gkg182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J.; Stromberg G.; Compitello G.; Willard H. F.; Van Bokkelen G. (2005) Rapid creation of BAC-based human artificial chromosome vectors by transposition with synthetic alpha-satellite arrays. Nucleic acids research 33, 587–596. 10.1093/nar/gki207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco Z. L.; Moralli D. (2006) Progress in artificial chromosome technology. Biochem. Soc. Trans. 34, 324–327. 10.1042/BST0340324. [DOI] [PubMed] [Google Scholar]
- Basu J.; Willard H. F. (2006) Human artificial chromosomes: potential applications and clinical considerations. Pediatr. Clin. North Am. 53, 843–853. 10.1016/j.pcl.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Ikeno M.; Suzuki N. (2011) Construction and use of a bottom-up HAC vector for transgene expression. Methods Mol. Biol. 738, 101–110. 10.1007/978-1-61779-099-7_7. [DOI] [PubMed] [Google Scholar]
- Kouprina N.; Earnshaw W. C.; Masumoto H.; Larionov V. (2013) A new generation of human artificial chromosomes for functional genomics and gene therapy,. Cell. Mol. Life Sci. 70, 1135–1148. 10.1007/s00018-012-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N.; Tomilin A. N.; Masumoto H.; Earnshaw W. C.; Larionov V. (2014) Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin. Drug Delivery 11, 517–535. 10.1517/17425247.2014.882314. [DOI] [PubMed] [Google Scholar]
- Moralli D.; Monaco Z. L. (2015) Developing de novo human artificial chromosomes in embryonic stem cells using HSV-1 amplicon technology, Chromosome research: an international journal on the molecular. Chromosome Res. 23, 105–110. 10.1007/s10577-014-9456-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimura M.; Uno N.; Kazuki Y.; Katoh M.; Inoue T. (2015) A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges,. Chromosome Res. 23, 111–133. 10.1007/s10577-014-9459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina O.; Kouprina N.; Masumoto H.; Larionov V.; Earnshaw W. C. (2017) Using human artificial chromosomes to study centromere assembly and function. Chromosoma 126, 559–575. 10.1007/s00412-017-0633-x. [DOI] [PubMed] [Google Scholar]
- Ebersole T.; Okamoto Y.; Noskov V. N.; Kouprina N.; Kim J. H.; Leem S. H.; Barrett J. C.; Masumoto H.; Larionov V. (2005) Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Res. 33, e130. 10.1093/nar/gni129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N.; Larionov V. (2006) TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet. 7, 805–812. 10.1038/nrg1943. [DOI] [PubMed] [Google Scholar]
- Kouprina N.; Larionov V. (2016) Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 125, 621–632. 10.1007/s00412-016-0588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M.; Cardinale S.; Noskov V. N.; Gassmann R.; Vagnarelli P.; Kandels-Lewis S.; Larionov V.; Earnshaw W. C.; Masumoto H. (2008) Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell 14, 507–522. 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale S.; Bergmann J. H.; Kelly D.; Nakano M.; Valdivia M. M.; Kimura H.; Masumoto H.; Larionov V.; Earnshaw W. C. (2009) Hierarchical inactivation of a synthetic human kinetochore by a chromatin modifier. Molecular biology of the cell 20, 4194–4204. 10.1091/mbc.E09-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann J. H.; Jakubsche J. N.; Martins N. M.; Kagansky A.; Nakano M.; Kimura H.; Kelly D. A.; Turner B. M.; Masumoto H.; Larionov V.; Earnshaw W. C. (2012) Epigenetic engineering: histone H3K9 acetylation is compatible with kinetochore structure and function. J. Cell Sci. 125, 411–421. 10.1242/jcs.090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shono N.; Ohzeki J.; Otake K.; Martins N. M.; Nagase T.; Kimura H.; Larionov V.; Earnshaw W. C.; Masumoto H. (2015) CENP-C and CENP-I are key connecting factors for kinetochore and CENP-A assembly. J. Cell Sci. 128, 4572–4587. 10.1242/jcs.180786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins N. M.; Bergmann J. H.; Shono N.; Kimura H.; Larionov V.; Masumoto H.; Earnshaw W. C. (2016) Epigenetic engineering shows that a human centromere resists silencing mediated by H3K27me3/K9me3. Molecular biology of the cell 27, 177–196. 10.1091/mbc.E15-08-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J.; Shono N.; Otake K.; Martins N. M.; Kugou K.; Kimura H.; Nagase T.; Larionov V.; Earnshaw W. C.; Masumoto H. (2016) KAT7/HBO1/MYST2 Regulates CENP-A Chromatin Assembly by Antagonizing Suv39h1-Mediated Centromere Inactivation. Dev. Cell 37, 413–427. 10.1016/j.devcel.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina O.; Vargiu G.; Abad M. A.; Zhiteneva A.; Jeyaprakash A. A.; Masumoto H.; Kouprina N.; Larionov V.; Earnshaw W. C. (2016) Epigenetic engineering reveals a balance between histone modifications and transcription in kinetochore maintenance. Nat. Commun. 7, 13334. 10.1038/ncomms13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten J. O.; Szilagyi Z.; Liu B.; Lopez M. D.; Szaszi E.; Djupedal I.; Nystrom T.; Ekwall K.; Gustafsson C. M.; Zhu X. (2012) Mediator promotes CENP-a incorporation at fission yeast centromeres. Molecular and cellular biology 32, 4035–4043. 10.1128/MCB.00374-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgo R. J.; Siddiqui H.; Fox S.; Solomon D.; Sansam C. G.; Yaniv M.; Muchardt C.; Metzger D.; Chambon P.; Roberts C. W.; Knudsen E. S. (2009) SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Molecular biology of the cell 20, 3192–3199. 10.1091/mbc.E08-12-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee R. B.; Steiner C. M.; Herbert B. S.; Vance G. H.; Hickey R. J.; Schwarz T.; Christan S.; Radovich M.; Schneider B. P.; Schindelhauer D.; Grimes B. R. (2012) Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene 31, 3244–3253. 10.1038/onc.2011.502. [DOI] [PubMed] [Google Scholar]
- Molina O.; Carmena M.; Maudlin I. E.; Earnshaw W. C. (2016) PREditOR: a synthetic biology approach to removing heterochromatin from cells, Chromosome research: an international journal on the molecular. Chromosome Res. 24, 495–509. 10.1007/s10577-016-9539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y.; Nakano M.; Ohzeki J.; Larionov V.; Masumoto H. (2007) A minimal CENP-A core is required for nucleation and maintenance of a functional human centromere,. EMBO J. 26, 1279–1291. 10.1038/sj.emboj.7601584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H.; Ikeno M.; Nakano M.; Okazaki T.; Grimes B.; Cooke H.; Suzuki N. (1998) Assay of centromere function using a human artificial chromosome. Chromosoma 107, 406–416. 10.1007/s004120050324. [DOI] [PubMed] [Google Scholar]
- Larin Z.; Mejia J. E. (2002) Advances in human artificial chromosome technology. Trends Genet. 18, 313–319. 10.1016/S0168-9525(02)02679-3. [DOI] [PubMed] [Google Scholar]
- Catania S.; Pidoux A. L.; Allshire R. C. (2015) Sequence features and transcriptional stalling within centromere DNA promote establishment of CENP-A chromatin. PLoS Genet. 11, e1004986. 10.1371/journal.pgen.1004986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T.; Shang W. H.; Toyoda A.; Misu S.; Monma N.; Ikeo K.; Molina O.; Vargiu G.; Fujiyama A.; Kimura H.; Earnshaw W. C.; Fukagawa T. (2014) Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Dev. Cell 29, 740–749. 10.1016/j.devcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N.; Samoshkin A.; Erliandri I.; Nakano M.; Lee H. S.; Fu H.; Iida Y.; Aladjem M.; Oshimura M.; Masumoto H.; Earnshaw W. C.; Larionov V. (2012) Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synth. Biol. 1, 590–601. 10.1021/sb3000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moralli D.; Jefferson A.; Valeria Volpi E.; Larin Monaco Z. (2013) Comparative study of artificial chromosome centromeres in human and murine cells. Eur. J. Hum. Genet. 21, 948–956. 10.1038/ejhg.2012.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera A. G.; Ward D. C. (1992) Oligonucleotide probes for the analysis of specific repetitive DNA sequences by fluorescence in situ hybridization. Hum. Mol. Genet. 1, 535–539. 10.1093/hmg/1.7.535. [DOI] [PubMed] [Google Scholar]
- Molina O.; Blanco J.; Anton E.; Vidal F.; Volpi E. V. (2012) High-resolution fish on DNA fibers for low-copy repeats genome architecture studies. Genomics 100, 380–386. 10.1016/j.ygeno.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J.; Bergmann J. H.; Kouprina N.; Noskov V. N.; Nakano M.; Kimura H.; Earnshaw W. C.; Larionov V.; Masumoto H. (2012) Breaking the HAC Barrier: histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 31, 2391–2402. 10.1038/emboj.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov V. N.; Lee N. C.; Larionov V.; Kouprina N. (2011) Rapid generation of long tandem DNA repeat arrays by homologous recombination in yeast to study their function in mammalian genomes. Biol. Proced. Online 13, 8. 10.1186/1480-9222-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larionov V.; Kouprina N.; Graves J.; Chen X. N.; Korenberg J. R.; Resnick M. A. (1996) Specific cloning of human DNA as yeast artificial chromosomes by transformation-associated recombination. Proc. Natl. Acad. Sci. U. S. A. 93, 491–496. 10.1073/pnas.93.1.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N.; Larionov V. (2008) Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 3, 371–377. 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- Liskovykh M.; Lee N. C.; Larionov V.; Kouprina N. (2016) Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther.--Methods Clin. Dev. 3, 16043. 10.1038/mtm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor D. L.; Mata J. F.; Sergeev M.; David A. F.; Salimian K. J.; Panchenko T.; Cleveland D. W.; Black B. E.; Shah J. V.; Jansen L. E. (2014) The quantitative architecture of centromeric chromatin. eLife 3, e02137. 10.7554/eLife.02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.