

Abstract
Background
Electrical conduction from the cardiac sinoatrial node to the ventricles is critical for normal heart function. Genome-wide association studies (GWAS) have identified more than a dozen common genetic loci that are associated with PR interval. However, it is unclear whether rare and low-frequency variants also contribute to PR interval heritability.
Methods and Results
We performed large-scale meta-analysis of the PR interval that included 83,367 participants of European ancestry and 9,436 of African ancestry. The Illumina HumanExome BeadChip examined both common and rare variants. We identified 31 genetic loci that were significantly associated with PR interval after Bonferroni correction (P<1.2×10−6), including 11 novel loci that have not been reported previously. Many of these loci are involved in heart morphogenesis. In gene-based analysis, we found that multiple rare variants at MYH6 (P = 5.9×10−11) and SCN5A (P=1.1×10−7) were associated with PR interval. SCN5A locus also was implicated in the common variant analysis, whereas MYH6 was a novel locus.
Conclusion
We identified common variants at 11 novel loci and rare variants within two gene regions that were significantly associated with PR interval. Our findings provide novel insights to the current understanding of atrioventricular conduction, which is critical for cardiac activity and an important determinant of health.
Keywords: ECG, genetics, association studies, epidemiology, genetics, PR interval, exome chip
Journal Subject Terms: Electrophysiology, Epidemiology, Genetic Association Studies
Introduction
Electrical conduction from the cardiac sinoatrial node to the ventricles is critical for normal heart function. Abnormalities of atrioventricular conduction can cause significant morbidity, and have been associated with atrial fibrillation (AF),1,2 need for pacemaker implantation,2 cardiac malformations, and sudden death.3,4 Conduction from the sinus node through the atria, atrioventricular node, and His-Purkinje fibers is readily evaluated from surface electrocardiogram (ECG), by measurement of the duration of PR interval. Despite the critical role that the cardiac conduction system plays in cardiac physiology and disease, the formation and regulation of the conduction system remains incompletely understood.
Recent data indicate that cardiac conduction measurements are heritable5–7 and have a genetic basis.8–11 To date, genetic studies of PR interval have been relatively modest-sized largely European-ancestry samples, and have implicated cardiac expressed ion channels, cardiac developmental transcription factors, signaling molecules, as well as novel pathways not previously known to be involved in cardiac conduction processes. Nevertheless, existing studies have focused on the role of common and predominantly noncoding genetic variants, which account for only a modest proportion of trait heritability.6
To better understand the biological and potential clinical implications of genetic variation underlying cardiac conduction, there is a need to examine both common and rare variation underlying atrioventricular conduction in large, well-powered, multiethnic studies. Moreover, assessment of genetic variation that alters protein coding has the potential to more directly implicate genes involved in processes critical to cardiac conduction. We therefore sought to examine PR interval duration in relation to predominantly coding genetic variants, in large, multi-ethnic analyses using the exome chip.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results, subject to Data Use/Sharing Agreements adopted by individual participating cohorts. The summary results from the current manuscript are available at the Broad Cardiovascular Disease Knowledge Portal (www.broadcvdi.org).
Study participants
The current project included participants of European ancestry (EA) from 22 studies: Age, Gene/Environment Susceptibility Study (AGES); Atherosclerosis Risk in Communities study (ARIC); British Genetics of Hypertension (BRIGHT); Massachusetts General Hospital Cardiology and Metabolic Patient cohort (CAMP); Cardiovascular Health Study (CHS); Erasmus Rucphen Family Study (ERF); Framingham Heart Study (FHS); Genes for Cerebral Hemorrhage on Anticoagulation (GOCHA); Genetic Regulation of Arterial Pressure In Humans in the Community (GRAPHIC); INTER99; Cooperative Health Research in the Region Augsburg (KORA); CROATIA-Korcula (KORCULA); LifeLines Cohort Study (LifeLines); Multi-Ethnic Study of Atherosclerosis (MESA); The Netherlands Epidemiology of Obesity (NEO); Rotterdam Study (RS); Generation Scotland: Scottish Family Health Study (GS:SFHS); Study of Health in Pomerania (SHIP); TwinsUK; Utrecht Health Project (UHP); Women’s Health Initiative (WHI); and Young Finns Study (YFS).
In addition, we included participants of African ancestry (AA) from five studies. These studies included ARIC, CHS, Jackson Heart Study (JHS), MESA and WHI.
Institutional Review Boards or Ethics Committees approved study procedures at each contributing site. All participants provided written informed consent to participate in genetic research.
Measurement of PR interval
PR interval duration, in milliseconds, was measured from the onset of the P wave to the onset of the QRS interval for each cohort. The following exclusions were applied: extreme PR values (≤ 80 ms or ≥ 320 ms); second or third degree heart block; atrial fibrillation on baseline ECG; history of myocardial infarction, heart failure, or Wolff–Parkinson–White syndrome; pacemaker placement; use of class I or III blocking medications (ATC code prefix C01B); digoxin use (ATC code C01AA05) or pregnancy.
Genotyping
Genotyping was performed independently in each study using the Illumina Human Exome BeadChip (v1.0, 1.1, or 1.2). Data were called and cleaned according to CHARGE ExomeChip best practices.12 Detailed information for each study regarding genotyping platforms, variant calling, and quality control metrics is shown in Supplementary Table 1. All studies used the same set of reference alleles to recode variants to ensure consistency.
Statistical analyses
Prior to association analysis, PR interval was first adjusted for covariates by taking residuals from a linear regression of PR on age, sex, height, body mass index, and RR interval. Each cohort additionally adjusted as necessary for cohort-specific variables, such as clinic sites, family structure, and population structure. To reduce sensitivity to extreme PR values, the residuals were inverse-normal transformed and used as the outcome for association testing.
Because single-marker based analyses typically have low power to identify associations between rare variants and traits, we separated the analysis for common and rare variants based on minor allele frequency (MAF). Common variants were defined as those with MAF≥1%, and the remaining variants were defined as rare variants (MAF<1%). For each of the common variants, we evaluated its association with the transformed PR interval, and accounted for multiple testing by Bonferroni correction (P < 0.05/42075=1.2×10−6). For the rare variants, we restricted analyses to nonsynonymous or splicing variants with MAF <1%, because such variants are more likely to be functional than synonymous or more common variants. As we expect some rare variants may act in the same or opposite directions even in the same gene region,13 we used a modified version of the Sequence Kernel Association Test (SKAT),14 which avoids problems of signals cancelling out each other in burden test results. Many gene regions had few or no rare nonsynonymous or splicing variants. Monomorphic variants from each study also were reported in the cohort level results as they were used for the cumulative MAF computations in gene-based tests. Gene regions with a cumulative MAF of rare variants <1% were excluded, resulting in 5,761 gene regions that were tested (see results below). Therefore, Bonferroni-corrected significance threshold for our gene-based tests was P<0.05/5,761=8.7×10−6. In secondary analyses, we limited the analysis to damaging variants, defined as nonsense variants or variants predicted to be damaging by PolyPhen-215 or SIFT.16
Analyses were performed using the “prepScores” function of the “seqMeta” R package. Family-based studies implemented the “kins” option in “prepScores” to specify kinship matrices. Each study provided single variant z-statistics from score tests, as well as genotype covariance matrices, which were then combined by fixed effects meta-analysis. The heterogeneity across studies was assessed by the Cochran’s Q, which is a non-parametric statistical test defined as the weighted sum of squared differences between individual study effects and the pooled effect. We performed both race stratified and race combined meta-analyses, and the race combined results were used for the remaining sections unless stated otherwise.
Comparison with genetic loci associated with AF and P-wave indices (PWI)
We also compared genetic loci associated with PR interval with those associated with AF and PWI to see if there are any shared genetic mechanisms. “AF loci” were identified by a recent exome chip analysis that included 22,806 AF cases and 132,612 referents.17 “PWI loci” were identified from a meta-analysis of P-wave duration and P-wave terminal force that included 44,456 participants.18 In addition, for each of the top variants associated with PR, we also examined its association with AF and PWI.
Examine potential function of PR-related variants for gene expression, regulation and biological pathways
Pathway analysis was performed by MAGENTA19 with default settings. The summary result for the common variants was used as the input, and significant pathways were defined as those with a false discovery rate (FDR)20 <0.05. The implication of genetic variants on cardiac gene expression (eQTL analysis) was performed by querying the GTEx database.21 At each PR-related locus, we identified the top variant and its neighboring variants that were within 500kb and in linkage disequilibrium with the top variant (r2 ≥ 0.5). Four heart and vascular tissues were queried, including artery aorta, artery coronary, atrial appendage and heart left ventricle. Significant eQTLs were defined as those with FDR<0.05. Regulatory regions were downloaded from the ENCODE Project22 and the NIH Roadmap Epigenomics Program.23 Four tracks were created: 1) included all 98 cell types from Roadmap epigenomics H3K27ac sites; 2) included only four heart tissues (aorta, right atrium, left ventricle, right ventricle) from Roadmap epigenomics H3K27ac sites; 3) included all 125 cell lines from ENCODE DNaseHS sites; 4) included only three heart-derived cell lines (cardiac fibroblasts, atrial fibroblasts, cardiac myocytes). The enrichment of PR-related loci in regulatory regions was examined by the “VSE” R package.24 For comparison, we randomly created 1,000 variant sets with MAF values and LD structures similar to those seen for PR-related loci.
Results
The current analyses included a total of 92,803 individuals from 27 cohorts, with 83,367 individuals from 22 studies of European ancestry and 9,436 individuals from 5 studies of African ancestry. Clinical characteristics of the study participants are in Table 1.
Table 1.
Clinical characteristics of the participating studies
| Ancestry | Study | Total N | Men, N (%) | Age, yrs, mean |
PR interval, ms, mean± SD |
RR interval, ms, mean± SD |
BMI, kg/m2, mean ± SD |
Height, cm, mean ± SD |
SBP, mmHg, mean ± SD |
Beta blockers (%) |
Diuretics (%) |
Calcium antagonists* (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| European ancestry | AGES | 2052 | 742 (36.2) | 75.9±5.4 | 170.5±26.8 | 895±129 | 27.0±4.4 | 166±9 | 143±20 | 635 (31.0) | ND | 108 (5.3) |
| ARIC | 9828 | 4528 (46.1) | 54.1±5.7 | 160.3±23.3 | 928±136 | 26.9±4.7 | 169±9 | 118±17 | 789 (8.0) | 1085 (11.0) | 176 (1.8) | |
| BRIGHT | 841 | 324 (38.9) | 57.6±10.7 | 161.1±19.9 | 960±169 | 27.5±3.8 | 166±9 | 153±24 | 248 (29.5) | 260 (30.9) | 18 (2.1) | |
| CAMP | 2493 | 1394 (55.9) | 60.7±11.6 | 163.0±26.8 | 926±166 | 28.5±5.8 | 171±10 | ND | Excluded | ND | Excluded | |
| CHS | 3247 | 1313 (40.4) | 72.4±5.4 | 167.8±28.2 | 956±151 | 26.4± 4.4 | 165±9 | 136±21 | 366 (11.3) | 750 (23.1) | 206 (6.3) | |
| ERF | 982 | 447 (45.5) | 48.2±14.3 | 152.9±23.2 | 982±159 | 26.9±4.6 | 168±10 | 140±20 | 3 (0.3) | 133 (13.5) | 25 (2.5) | |
| FHS | 7580 | 3428 (45.2) | 39.3±9.8 | 152.0±22.1 | 910±175 | 26.0±5.0 | 169±10 | 119±15 | Excluded | ND | Excluded | |
| GOCHA | 355 | 161 (45.4) | 73.2±8.2 | 167.6±27.7 | 913±173 | 26.1±4.6 | 169±10 | N/A | Excluded | ND | ND | |
| GRAPHIC | 1755 | 893 (50.9) | 39.1±14.5 | 153.0±24.0 | 934±145 | 26.1±4.6 | 171±9 | 128±19 | 39 (2.2) | ND | ND | |
| INTER99 | 5836 | 2843 (48.7) | 46.1±7.9 | 158.2±22.4 | 921±150 | 26.3±4.6 | 172±9 | 130±18 | ND | ND | ND | |
| KORA | 2617 | 1247 (47.6) | 48.3±13.0 | 162.1±22.2 | 944±149 | 26.9±4.4 | 168±9 | 127±19 | 199 (7.6) | 152 (5.8) | 13 (0.5) | |
| KORCULA | 293 | 106 (36.2) | 55.0±13.4 | 159.8± 24.0 | 929±127 | 28.0± 4.3 | 168± 9 | 139± 14 | 8 (2.7) | 3 (1.0) | 6 (2.0) | |
| LifeLines | 1934 | 781 (40.3) | 45.2±13.1 | 156.7±24.7 | 896±145 | 25.9±4.5 | 175±9 | 122±16 | 64 (3.3) | 39 (2.0) | 23 (1.2) | |
| MESA | 2455 | 1171 (47.7) | 62.8±10.2 | 164.7±25.2 | 1047±158 | 27.8±5.1 | 169±10 | 123±21 | Excluded | ND | Excluded | |
| NEO | 5782 | 2717 (47.0) | 55.9±5.9 | 164.5±23.4 | 940±151 | 30.0±4.8 | 174±10 | 133±17 | Excluded | ND | ND | |
| RS | 2358 | 1086 (46.1) | 68.6±8.1 | 168.2±24.7 | 871±144 | 26.3±3.6 | 168± 9 | ND | 293 (12.4) | ND | ND | |
| GS:SFHS | 9168 | 3786 (41.3) | 52.0±13.6 | 164.1± 24.9 | 886±146 | 26.9± 5.1 | 168± 10 | 134± 18 | 192 (2.1) | ND | ND | |
| SHIP | 6493 | 2608 (40.2) | 49.2±15.3 | 158.5±23.3 | 897±146 | 27.5±5.0 | 170±9 | 131±20 | ND | ND | ND | |
| TwinsUK | 465 | 32 (6.9) | 52.3±11.7 | 159.6±22.6 | 923±148 | 26.8±5.4 | 163±7 | 119±16 | ND | ND | ND | |
| UHP | 1735 | 779 (44.9) | 39.1±13.0 | 155.9±22.5 | 950±151 | 24.9±3.9 | 175±10 | 125±17 | 69 (4.6) | 32 (1.8) | 18 (1.0) | |
| WHI | 13252 | 0 (0) | 66.0±6.5 | 161.4±24.0 | 921±138 | 28.7±5.6 | 162±6 | 130±18 | 735 (5.5) | 1715 (13.3) | 1230 (9.3) | |
| YFS | 1846 | 824 (44.6) | 41.9±5.0 | 156.2±22.6 | 1028±165 | 26.4±4.9 | 172±9 | 119±14 | 38 (2.1) | 24 (1.3) | 1 (0.1) | |
| African ancestry | ARIC | 3366 | 1291 (38.4) | 53.3±5.8 | 171.2±26.8 | 929±151 | 29.4±6.1 | 168±9 | 128±22 | 315 (9.4) | 717 (21.3) | 222 (6.6) |
| CHS | 627 | 232 (37.0) | 72.4±5.5 | 170.2±28.1 | 918±161 | 28.4± 5.5 | 165±9 | 142± 22 | 54 (8.6) | 217 (34.6) | 60 (9.6) | |
| JHS | 2220 | 833 (37.5) | 52.7±12.5 | 172.7±27.3 | 956±150 | 31.4±6.4 | 169±9 | 126±18 | Excluded | ND | Excluded | |
| MESA | 1565 | 718 (45.9) | 62.3±10.0 | 170.9±26.3 | 1050±172 | 30.2±5.9 | 168±10 | 132±21 | Excluded | ND | Excluded | |
| WHI | 1658 | 0 (0) | 64.6±6.4 | 167.1± 24.8 | 921±148 | 31.1±5.8 | 162±7 | 134±17 | 87 (5.2) | 393 (23.7) | 341 (20.6) |
Exclusion criteria are given in Supplementary Table 1. SBP, systolic blood pressure; BMI, body mass index; ND, not determined; SD, standard deviation;
Non-dihydropyridine calcium antagonists.
Identification of 31 loci associated with PR interval
A total of 42,075 common variants were analyzed (MAF ≥ 1%). As shown in Figure 1 and Table 2, 31 loci were significantly associated with PR interval after Bonferroni correction (P < 1.2×10−6), including 22 loci that reached the conventional genome-wide significance threshold (P < 5×10−8). The results of the random effects meta-analysis were similar to those of the fixed effects analysis (Supplementary Table 2). The most significant locus was tagged by rs6795970 (P= 4.0×10−240), a missense variant in SCN10A, which encodes a sodium channel that has been associated previously with the PR interval (r2=0.97 with the top SNP rs6599250 reported previously).8 Highly associated variants clustered in the linker region between the second and third domains of SCN10A (Figure 2). The top variants at 12 loci are missense variants. In addition, the top variants at 4 loci (including 3 novel loci) are low-frequency variants (1% < MAF < 5%), illustrating the power of exome chip analyses to identify low-frequency coding associations. Detailed information of the nearest gene to each genome wide significant locus is given in Supplementary Table 3.
Figure 1. Manhattan plot showing the association between common variants and PR interval from combined ancestry analysis.
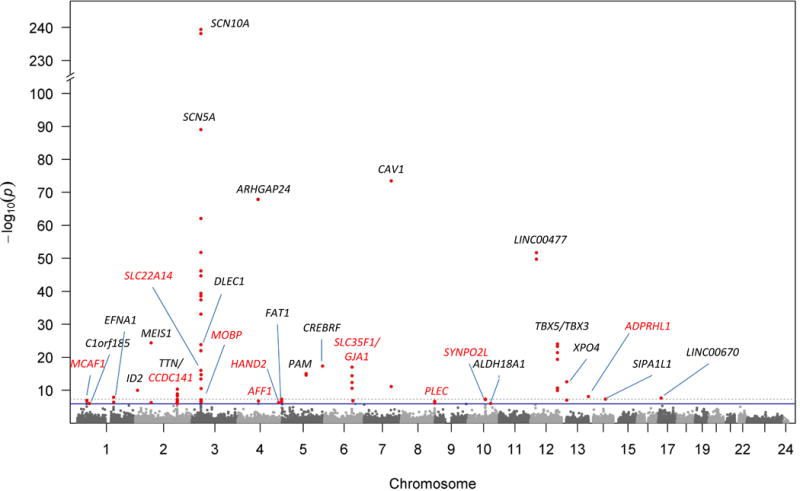
The x-axis represents the chromosomal position for each SNP, and the y-axis represents the –log10(p-value) of the association with PR interval. The dashed line represents the genome-wide significance cutoff of 5×10−8, and the blue line represents the Bonferroni P-value cutoff of 1.3×10−6. Black color represents known loci, whereas red color represents novel loci.
Table 2.
Common variants significantly associated with PR interval from meta-analysis of all studies
| SNP | Locus | Closest gene | Function | Coding allele | CAF* | Beta | SE | P value | Number of studies‡ | Prolong or shorten PR interval | Novel locus |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs6795970 | 3p22.2 | SCN10A | Missense | A | 0.37 | 0.1705 | 0.0052 | 4.0×10−240 | 27 | Prolong | |
| rs3922844 | 3p22.2 | SCN5A | Intronic | A | 0.34 | −0.1069 | 0.0053 | 9.3×10−90 | 26 | Shorten | |
| rs3807989 | 7q31.2 | CAV1 | Intronic | A | 0.43 | 0.0908 | 0.0050 | 3.0×10−74 | 27 | Prolong | |
| rs7660702 | 4q21.23 | ARHGAP24 | Intronic | C | 0.33 | −0.0921 | 0.0053 | 1.2×10−68 | 27 | Shorten | |
| rs17287293 | 12p12.1 | LINC00477 | Intergenic | G | 0.14 | −0.1084 | 0.0071 | 1.9×10−52 | 27 | Shorten | |
| rs11897119 | 2p14 | MEIS1 | Intronic | C | 0.39 | 0.0566 | 0.0055 | 4.2×10−25 | 25 | Prolong | |
| rs1896312 | 12q24.21 | TBX3 | Intergenic | G | 0.28 | 0.0564 | 0.0055 | 8.7×10−25 | 26 | Prolong | |
| rs883079 | 12q24.21 | TBX5 | 3′UTR | G | 0.29 | 0.0550 | 0.0054 | 4.5×10−24 | 26 | Prolong | |
| rs116202356 | 3p22.2 | DLEC1 | Missense | A | 0.02 | −0.1953 | 0.0199 | 1.0×10−22 | 27 | Shorten | |
| rs251253 | 5q35.1 | CREBRF | Intergenic | G | 0.42 | −0.0439 | 0.0051 | 4.7×10−18 | 26 | Shorten | |
| rs11153730 | 6q22.31 | SLC35F1 | Intergenic | C | 0.47 | −0.0420 | 0.0049 | 9.5×10−18 | 27 | Shorten | Novel |
| rs35658696 | 5q21.1 | PAM | Missense | G | 0.04 | 0.0956 | 0.0119 | 8.5×10−16 | 27 | Prolong | |
| rs2070492 | 3p22.2 | SLC22A14 | Missense | T | 0.10 | 0.0624 | 0.0083 | 4.0×10−14 | 27 | Prolong | Novel |
| rs2585897 | 13q12.11 | XPO4 | Intronic | A | 0.17 | 0.0471 | 0.0064 | 2.8×10−13 | 27 | Prolong | |
| rs2042995 | 2q31.2 | TTN | Missense | C | 0.26 | 0.0375 | 0.0057 | 4.3×10−11 | 27 | Prolong | |
| rs4399693 | 2p25.1 | ID2 | Intergenic | A | 0.34 | 0.0374 | 0.0058 | 9.1×10−11 | 25 | Prolong | |
| rs41306688 | 13q34 | ADPRHL1 | Missense | C | 0.03 | 0.1002 | 0.0173 | 7.4×10−9 | 22 | Prolong | Novel |
| rs4745 | 1q22 | EFNA1 | Missense | T | 0.49 | 0.0299 | 0.0053 | 1.2×10−8 | 26 | Prolong | |
| rs11078078 | 17p12 | LINC00670 | Intronic | A | 0.40 | 0.0281 | 0.0050 | 2.2×10−8 | 27 | Prolong | |
| rs60632610 | 10q22.2 | SYNPO2L | Missense | T | 0.15 | −0.0371 | 0.0068 | 4.5×10−8 | 27 | Shorten | Novel |
| rs11848785 | 14q24.2 | SIPA1L1 | Intronic | G | 0.24 | 0.0317 | 0.0058 | 4.6×10−8 | 27 | Prolong | |
| rs3733414 | 4q35.2 | FAT1 | Missense | A | 0.38 | 0.0280 | 0.0051 | 4.8×10−8 | 27 | Prolong | |
| rs17362588 | 2q31.2 | CCDC141 | Missense | A | 0.08 | −0.0491 | 0.0090 | 5.5×10−8 | 27 | Shorten | Novel |
| rs2296172 | 1p34.3 | MACF1 | Missense | G | 0.20 | 0.0326 | 0.0061 | 1.1×10−7 | 27 | Prolong | Novel |
| rs9398652 | 6q22.31 | GJA1 | Intergenic | A | 0.14 | 0.0390 | 0.0074 | 1.3×10−7 | 26 | Prolong | Novel |
| rs442177 | 4q22.1 | AFF1 | Intronic | C | 0.42 | −0.0262 | 0.0050 | 1.8×10−7 | 26 | Shorten | Novel |
| rs7002002 | 8q24.3 | PLEC | Missense | A | 0.38 | −0.0272 | 0.0052 | 2.1×10−7 | 25 | Shorten | Novel |
| rs1768208 | 3p22.1 | MOBP | Intron | T | 0.25 | 0.0288 | 0.0057 | 3.6×10−7 | 27 | Prolong | Novel |
| rs2119788 | 4q34.1 | HAND2 | Intergenic | C | 0.52 | −0.0246 | 0.0049 | 5.6×10−7 | 27 | Shorten | Novel |
| rs17391905† | 1p32.3 | C1orf185 | Intergenic | G | 0.03 | −0.0694 | 0.0142 | 9.6×10−7 | 27 | Shorten | |
| rs524295 | 10q24.1 | ALDH18A1 | Intergenic | A | 0.40 | −0.0261 | 0.0053 | 9.7×10−7 | 26 | Shorten |
Coding allele frequency
SNP was not significant if African participants were excluded.
Some variants did not reach pass the quality filtering in respective studies and thus were excluded.
Figure 2. Diagram of sodium voltage-gated channel alpha subunit 10 (SCN10A).
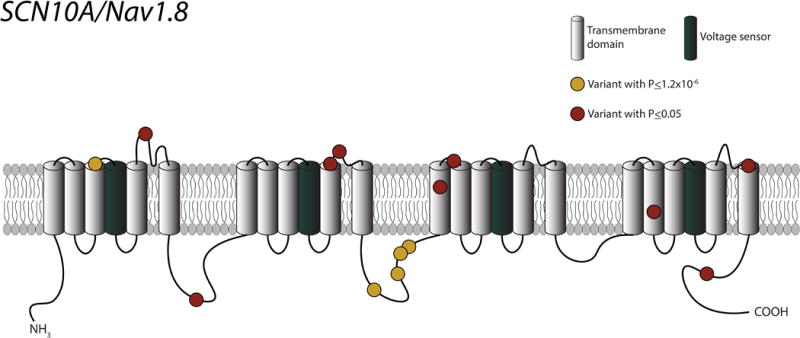
Each yellow circle represents a genetic variant with a P-value less than the significance cutoff (1.2 × 10−6). Each red circle represents a genetic variant with a P-value greater than the significance cutoff, but less than 0.05.
We then examined the associations between these top PR variants with AF and electrocardiographic PWI. Eight out of 31 PR loci identified in our analysis were associated with AF after Bonferroni correction (P<0.05/31=1.6×10−3), consistent with some shared mechanisms between the regulation of PR interval and AF. Variants in SCN10A most significantly associated with PR interval were also significantly associated with AF (Supplementary Table 4). Among PR-related SNPs, rs60632610 at the SYNPO2L locus was most significantly associated with AF (Odds ratio: 1.90 (0.87-0.93), P=1.5×10−10). Supplementary Figure 1 shows the overlap among loci associated with PR interval, AF, and PWI.
We also performed a sensitivity analysis that separated samples of European and African ancestry. As shown in Supplementary Table 5 and Supplementary Figure 2, all of the 31 loci except rs17391905 at the 1p32.3 locus (P = 2.6×10−6) were also significant in the analysis of European-only samples. Supplementary Table 6 and Supplementary Figure 3 show the result for the analysis of African ancestry-only samples. Three loci were significant: SCN5A (rs3922844), SCN10A (rs6795970), and TBX5 (rs883079) after Bonferroni correction; P < 1.3 × 10−6. All three loci were also significant in the analysis of European-only samples. The result from each individual study is shown in Supplementary Table 7.
Rare variations in MYH6 and SCN5A are associated with PR interval
We next examined the association between PR interval and rare variants (MAF<1%) in gene regions. Variation in two gene regions, MYH6 (P = 5.9×10−11) and SCN5A (P = 1.1×10−7), was associated with PR interval (Table 3). Supplementary Tables 8 and 9 show the association of each rare variant within MYH6 and SCN5A with PR interval, respectively. MYH6 encodes a cardiac myosin heavy chain subunit, and SCN5A encodes the major cardiac sodium channel and was previously found to be associated with PR interval.8 MYH6 was also recently found to associate with PWI.18 We also performed an ancestry-stratified analysis in the same way as the combined analysis. The same two gene regions were significant using data from European samples alone (P = 4.1×10−12 and 8.3×10−7 for MYH6 and SCN5A, respectively). These two genes did not reach the significance cutoff in African samples (P = 0.03 and 0.01 for MYH6 and SCN5A, respectively). Two other genes, HEATR2 (P = 2.2×10−−6) and THRAP3 (P = 4.2×10−6), were significantly associated in African samples alone. However, in the combined analysis, these two genes were not significant (P=0.02 and 0.06 for HEATR2 and THRAP3, respectively), probably due to a low cumulative allele frequency.
Table 3.
Top 10 gene regions associated with PR interval by the SKAT test*
| Gene | P value | Qmeta† | CMAF‡ | #Variants | Number of studies with at leat one rare variant | Average number of variants in each study |
|---|---|---|---|---|---|---|
| MYH6 | 5.9×10−11 | 23537340 | 0.0215 | 32 | 27 | 12 |
| SCN5A | 1.1×10−7 | 16604843 | 0.0289 | 35 | 27 | 13 |
| GORASP1 | 1.3×10−5 | 14361252 | 0.0308 | 16 | 27 | 6 |
| NEBL | 1.9×10−5 | 11787699 | 0.0309 | 36 | 27 | 11 |
| TRIML2 | 1.2×10−4 | 10173978 | 0.0223 | 23 | 27 | 10 |
| SLC22A11 | 1.5×10−4 | 6539656 | 0.0136 | 11 | 27 | 6 |
| MTRF1 | 2.8×10−4 | 9073098 | 0.0235 | 10 | 26 | 3 |
| CD36 | 3.5×10−4 | 8001777 | 0.0156 | 28 | 27 | 9 |
| CAPRIN2 | 3.7×10−4 | 6886375 | 0.0169 | 15 | 27 | 7 |
| PIK3R6 | 6.0×10−4 | 9763336 | 0.0316 | 23 | 26 | 8 |
The analysis included only nonsynonymous and splice site rare variants (MAF<1%) within the gene regions
Qmeta: The SKAT Q-statistic, defined as , where is the weight, and is the squared score.
CMAF: Cumulative minor allele frequency; SKAT: Sequence Kernel Association Test
The significance level for gene-based tests after Bonferroni correction was P<0.05/5759=8.7×10−6; the two genes that reached this significant cutoff are highlighted in bold font.
In our secondary analysis of pooled samples, we analyzed only damaging variants, defined as nonsense mutations or alternations predicted to be damaging by PolyPhen-215 or SIFT.16 Three genes reached the signifiance cutoff (P<0.05/2030=2.5×10−5), including GORASP1 (P=1.1×10−5), NEBL (P=1.9×10−5), and SCN5A (P=2.2×10−5) (Supplementary Table 10).
Expression quantitative trait loci (eQTL) analysis
We also performed eQTL analysis to determine if any of the novel PR-related variants were associated with cardiac gene expression using data from GTEx.21 Eight loci were associated with expression of at least one gene in the atrial appendage, left ventricle, coronary artery, or aorta, suggesting the importance of these loci in the regulation of gene expression in heart or vascular tissues (Supplementary Table 11).
Enrichment of PR-related variants in regulatory regions
We examined involvement of PR-related variants in regulatory function. As shown in Supplementary Figure 4, PR-related variants were significantly enriched in regulatory regions in both primary heart tissues (Padj=3.7×10−9) and heart-derived cell lines (Padj=0.002), but not in all tissues (Padj>0.05). The observed enrichment suggested involvement of these loci in tissue-specific regulatory functions. In addition, the variants also tended to locate within evolutionarily conserved regions (Padj=2.8×10−5 for primates and 6.4×10−5 for mammals).
Enrichment of PR-related variants in biological pathways
We examined the enrichment of PR-related variants in biological pathways by MAGENTA.19 Supplementary Table 12 shows the top pathways identified. The most significant pathway was heart morphogenesis (P=3.6×10−5, FDR=0.049), suggesting that many PR-related genes might be involved in cardiac development. The pathway was only the significant pathway after correction for multiple testing (FDR<0.05).
Discussion
We conducted a large-scale analysis of the genetic determinants of atrioventricular conduction in 92 803 individuals by studying the electrocardiographic PR interval. In total, we observed 31 genetic loci that were associated with atrioventricular conduction, 11 of which are novel. In aggregate, the results implicate loci containing genes encoding ion channels in the heart, sarcomeric proteins, cardiac transcription factors, and other proteins with unknown cardiac function. Our findings provide new insights to the current understanding of atrioventricular conduction, which is critical for cardiac function.
Interestingly, rare variants in SCN5A and MYH6 were associated with PR interval. A missense mutation (D1275N) in SCN5A has previously been reported in a large family with multiple members affected by dilated cardiomyopathy, conduction disorder, and arrhythmia.25 The mutation, together several other mutations within the same gene, has also been associated with dilated cardiomyopathy,26 atrial fibrillation,27 and long-QT syndrome.28–31 Rare mutations within MYH6 were associated with sick sinus syndrome,28 congenital heart defects,32 and atrial septal defects.33
Our observations support and extend prior analyses of cardiac conduction. Most previous genome-wide association studies involved the study of common genetic variation in smaller samples of up to 28,517 individuals.8,10,11 In keeping with those prior studies, we again observed that SCN10A is the most prominent gene involved in atrioventricular conduction. Our recent GWAS based on 105K samples corroborates many of our current findings.34 However, our current study had greater power than those earlier analyses for assessment of rare coding variation.
Our study has two major implications. First, our results underscore the utility of assessing coding variation as an efficient way to identify functional molecular domains. In particular, our findings provide insights into the functional topology of SCN10A. The SCN10A sodium channel gene is widely expressed in the nervous system and heart,21 but it has only recently been implicated in cardiac conduction8,34–36 and arrhythmias such as AF35 and Brugada syndrome.37 SCN10A encodes an alpha subunit (with six transmembrane spanning regions), which forms tetrameric, voltage gated sodium channels responsible for the Nav 1.8 late sodium channel current.38,39 We found a collection of amino acid substitutions in the linker region between the second and third domains of SCN10A that were associated with PR duration (Figure 2). Variants in this linker region that were associated with the PR interval also were associated with AF, suggesting that function of this domain may have important clinical implications.
Prior work on the homologous SCN5A cardiac sodium channel gene – which is also a cardiac conduction locus – indicates that this linker region is critical for sodium channel inactivation. Sodium influx is predominantly responsible for cardiomyocyte depolarization. Moreover, channel inactivation is essential for restoration of the hyperpolarized state needed for cyclic cardiomyocyte depolarization and contraction. Therefore, variations in this linker region might be involved in Nav 1.8 inactivation. Other data are necessary to identify relationships among variation in the linker region, the late sodium channel current, and channel inactivation in both healthy and diseased states.
Together with previously discovered susceptibility genes, our findings implicate genes in different functional classes that regulate atrioventricular conduction such as ion channels and cardiac transcription factors. In many cases, anomalies in these genes have been found to cause human cardiac diseases, such as congenital heart defects, primary cardiac conduction abnormalities, and syndromes predisposing to sudden cardiac death (Supplementary Table 3). Interestingly, some of the genes are not expressed (in high abundance) in the right atrial appendage or the left ventricle, according to existing data sets – although most are active in the heart (Supplementary Table 13). Atrioventricular nodal conduction also can be influenced by external tone from the autonomic nervous system. Therefore, further work is necessary to determine the mechanisms by which identified genes that are not expressed in the heart influence the PR interval.
We acknowledge several limitations of our study. Because PR interval was measured across many cohorts, it is possible that there is some heterogeneity that would diminish our power to detect modest associations. We excluded individuals with extreme values of PR interval, which might have been gleaned from large variations in cardiac conduction. We also performed inverse normal transformation on the raw PR interval to reduce the heterogeneity, which on the other hand might reduce the interpretability. Although we performed single-variant and gene-based tests, we did not examine the association of haplotype patterns with PR interval, so it is unclear if there are any haplotypes that might be associated with PR interval. Most of the genetic variants analyzed were in exons. Therefore the effects of variants within regulatory regions were not investigated. We note that the variants identified may not be causally related to the studied phenotypes (PR interval, AF, and PWI), but may be in LD with causal variants. We anticipate that future increases in sample size with additional replications and more comprehensive genotyping platforms, such as denser SNP arrays or genome sequencing, will help address these limitations.
In conclusion, we studied genetic variants associated with PR interval duration and identified 31 common loci – including 11 that were novel – and two rare variant regions. Our findings greatly expand our knowledge of the genes that underlie atrioventricular conduction in the heart.
Supplementary Material
Clinical Perspective.
The duration of PR interval is an important biomarker of the cardiac conduction system. Increasing evidences suggest that cardiac conduction measurements including PR interval are heritable. It is thus interesting to understand the biological and potential clinical implications of genetic variation underlying cardiac conduction. We performed a large-scale meta-analysis of PR interval that included 83,367 participants of European ancestry and 9,436 of African ancestry using the Illumina exome chip. Thirty-one genetic loci were significantly associated with PR interval after Bonferroni correction, including 11 loci that have not been previously reported. Our findings provide new insights to the current understanding of atrioventricular conduction, which is critical for cardiac activity and an important determinant of health.
Acknowledgments
Sources of Funding: This work was partly supported by grants from the National Institutes of Health to Drs. Ellinor, Benjamin, and Lunetta (2RO1HL092577), Ellinor and Benjamin (R01HL128914), Ellinor (K24HL105780), and Arking and Sotoodehnia (R01HL116747). Dr. Ellinor is also supported by an Established Investigator Award from the American Heart Association (13EIA14220013) and by the Fondation Leducq (14CVD01). Dr. Lin was partly supported by Boston University Digital Health Initiative, and the National Center for Advancing Translational Sciences, National Institutes of Health, through BU-CTSI Grant Number 1UL1TR001430. Niek Verweij is supported by ICIN-NHI and Marie Sklodowska-Curie GF (call: H2020-MSCA-IF-2014, Project ID: 661395). Dr. Lubitz is supported by NIH grants K23HL114724 and a Doris Duke Charitable Foundation Clinical Scientist Development Award 2014105. Dr. Sotoodehnia is supported by NIH grants HL116747 and HL111089. Folkert W. Asselbergs is supported by a Dekker scholarship-Junior Staff Member 2014T001 – Netherlands Heart Foundation and UCL Hospitals NIHR Biomedical Research Centre. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Disclosures: Dr. Patrick Ellinor is PI of a grant from Bayer HealthCare to the Broad Institute focused on the genetics and therapeutics of AF. Dr. Bruce Psaty serves on the DSMB of a clinical trial funded by the manufacturer (Zoll LifeCor) and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Dr. Lubitz receives sponsored research support from Bayer HealthCare, Biotronik, and Boehringer Ingelheim, and has consulted for St. Jude Medical and Quest Diagnostics.
References
- 1.Soliman EZ, Prineas RJ, Case LD, Zhang ZM, Goff DC., Jr Ethnic distribution of ecg predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the atherosclerosis risk in communities (aric) study. Stroke; a journal of cerebral circulation. 2009;40:1204–1211. doi: 10.1161/STROKEAHA.108.534735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton-Cheh C, Levy D, et al. Long-term outcomes in individuals with prolonged pr interval or first-degree atrioventricular block. JAMA. 2009;301:2571–2577. doi: 10.1001/jama.2009.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao HB, Roy C, Fujimoto S, Gibson DG. Natural history of abnormal conduction and its relation to prognosis in patients with dilated cardiomyopathy. Int J Cardiol. 1996;53:163–170. doi: 10.1016/0167-5273(95)02502-2. [DOI] [PubMed] [Google Scholar]
- 4.Thiene G, Pennelli N, Rossi L. Cardiac conduction system abnormalities as a possible cause of sudden death in young athletes. Human pathology. 1983;14:704–709. doi: 10.1016/s0046-8177(83)80143-9. [DOI] [PubMed] [Google Scholar]
- 5.Pilia G, Chen WM, Scuteri A, Orru M, Albai G, Dei M, et al. Heritability of cardiovascular and personality traits in 6,148 sardinians. PLoS Genet. 2006;2:e132. doi: 10.1371/journal.pgen.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva CT, Kors JA, Amin N, Dehghan A, Witteman JC, Willemsen R, et al. Heritabilities, proportions of heritabilities explained by gwas findings, and implications of cross-phenotype effects on pr interval. Hum Genet. 2015;134:1211–1219. doi: 10.1007/s00439-015-1595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton-Cheh C, Guo CY, Wang TJ, O’Donnell CJ, Levy D, Larson MG. Genome-wide association study of electrocardiographic and heart rate variability traits: The framingham heart study. BMC Med Genet. 2007;8(Suppl 1):S7. doi: 10.1186/1471-2350-8-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, et al. Genome-wide association study of pr interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, et al. Several common variants modulate heart rate, pr interval and qrs duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 10.Butler AM, Yin X, Evans DS, Nalls MA, Smith EN, Tanaka T, et al. Novel loci associated with pr interval in a genome-wide association study of 10 african american cohorts. Circ Cardiovasc Genet. 2012;5:639–646. doi: 10.1161/CIRCGENETICS.112.963991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JG, Magnani JW, Palmer C, Meng YA, Soliman EZ, Musani SK, et al. Genome-wide association studies of the pr interval in african americans. PLoS Genet. 2011;7:e1001304. doi: 10.1371/journal.pgen.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, et al. Best practices and joint calling of the humanexome beadchip: The charge consortium. PLoS ONE. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. American journal of human genetics. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. American journal of human genetics. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using polyphen-2. Current protocols in human genetics/editorial board, Jonathan L. Haines … [et al.] 2013 doi: 10.1002/0471142905.hg0720s76. Chapter 7: Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. doi: 10.1038/ng.3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christophersen IE, Magnani JW, Yin X, Barnard J, Weng LC, Arking DE, et al. Fifteen genetic loci associated with the electrocardiographic p wave. Circ Cardiovasc Genet. 2017;10 doi: 10.1161/CIRCGENETICS.116.001667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segre AV, Consortium D, investigators M. Groop L, Mootha VK, Daly MJ, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 21.Consortium GT. Human genomics. The genotype-tissue expression (gtex) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.ENCODE Project Consortium. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chadwick LH. The nih roadmap epigenomics program data resource. Epigenomics. 2012;4:317–324. doi: 10.2217/epi.12.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed M, Sallari RC, Guo H, Moore JH, He HH, Lupien M. Variant set enrichment: An r package to identify disease-associated functional genomic regions. BioData mining. 2017;10:9. doi: 10.1186/s13040-017-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, et al. Scn5a mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 26.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Zhang W, Fang C, Jiang S, Shu C, Cheng H, et al. Polymorphism h558r in the human cardiac sodium channel scn5a gene is associated with atrial fibrillation. The Journal of international medical research. 2011;39:1908–1916. doi: 10.1177/147323001103900535. [DOI] [PubMed] [Google Scholar]
- 28.Qureshi SF, Ali A, John P, Jadhav AP, Venkateshwari A, Rao H, et al. Mutational analysis of scn5a gene in long qt syndrome. Meta gene. 2015;6:26–35. doi: 10.1016/j.mgene.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwasa H, Itoh T, Nagai R, Nakamura Y, Tanaka T. Twenty single nucleotide polymorphisms (snps) and their allelic frequencies in four genes that are responsible for familial long qt syndrome in the japanese population. Journal of human genetics. 2000;45:182–183. doi: 10.1007/s100380050207. [DOI] [PubMed] [Google Scholar]
- 30.Modell SM, Lehmann MH. The long qt syndrome family of cardiac ion channelopathies: A huge review. Genet Med. 2006;8:143–155. doi: 10.1097/01.gim.0000204468.85308.86. [DOI] [PubMed] [Google Scholar]
- 31.Ter Bekke RMA, Isaacs A, Barysenka A, Hoos MB, Jongbloed JDH, Hoorntje JCA, et al. Heritability in a scn5a-mutation founder population with increased female susceptibility to non-nocturnal ventricular tachyarrhythmia and sudden cardiac death. Heart Rhythm. 2017;14:1873–1881. doi: 10.1016/j.hrthm.2017.07.036. [DOI] [PubMed] [Google Scholar]
- 32.Granados-Riveron JT, Ghosh TK, Pope M, Bu’Lock F, Thornborough C, Eason J, et al. Alpha-cardiac myosin heavy chain (myh6) mutations affecting myofibril formation are associated with congenital heart defects. Hum Mol Genet. 2010;19:4007–4016. doi: 10.1093/hmg/ddq315. [DOI] [PubMed] [Google Scholar]
- 33.Posch MG, Waldmuller S, Muller M, Scheffold T, Fournier D, Andrade-Navarro MA, et al. Cardiac alpha-myosin (myh6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS ONE. 2011;6:e28872. doi: 10.1371/journal.pone.0028872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Setten J, Brody JA, Jamshidi Y, Swenson BR, Butler AM, Campbell H, et al. Genome-wide association meta-analysis of pr interval identifies 47 novel loci associated with atrial and atrioventricular electrical activity. bioRxiv. doi: 10.1038/s41467-018-04766-9. DOI: https://doi.org/10.1101/241489. [DOI] [PMC free article] [PubMed]
- 35.Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, et al. Genetic variation in scn10a influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 36.Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with qrs duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, et al. Common variants at scn5a-scn10a and hey2 are associated with brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. doi: 10.1038/ng.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, et al. Functional nav1.8 channels in intracardiac neurons: The link between scn10a and cardiac electrophysiology. Circ Res. 2012;111:333–343. doi: 10.1161/CIRCRESAHA.112.274035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.