

Abstract
Background
Epilepsy is a common neurological disorder that affects approximately 50 million people worldwide. Both risk of epilepsy and response to treatment partly depend on genetic factors, and gene identification is a promising approach to target new prediction, treatment, and prevention strategies. However, despite significant progress in the identification of genes causing epilepsy in families with a Mendelian inheritance pattern, there is relatively little known about the genetic factors responsible for common forms of epilepsy and so-called epileptic encephalopathies.
Study design
The Epilepsy Phenome/Genome Project (EPGP) is a multi-institutional, retrospective phenotype–genotype study designed to gather and analyze detailed phenotypic information and DNA samples on 5250 participants, including probands with specific forms of epilepsy and, in a subset, parents of probands who do not have epilepsy.
Results
EPGP is being executed in four phases: study initiation, pilot, study expansion/establishment, and close-out. This article discusses a number of key challenges and solutions encountered during the first three phases of the project, including those related to (1) study initiation and management, (2) recruitment and phenotyping, and (3) data validation. The study has now enrolled 4223 participants.
Conclusions
EPGP has demonstrated the value of organizing a large network into cores with specific roles, managed by a strong Administrative Core that utilizes frequent communication and a collaborative model with tools such as study timelines and performance-payment models. The study also highlights the critical importance of an effective informatics system, highly structured recruitment methods, and expert data review.
Introduction
This article describes lessons learned thus far from the Epilepsy Phenome/Genome Project (EPGP), a large, multi-institutional international study that is creating the world's most comprehensive database of phenotypic characteristics of participants with epilepsy paired to biobanked DNA samples (Table 1). Sponsored by the National Institute of Neurological Disorders and Stroke (NINDS), the study was launched in May 2007 and aims to enroll 1500 pairs of first-degree relatives (termed ‘family pairs’) who both have either idiopathic generalized epilepsy (IGE) or localization-related epilepsy (LRE), and 750 probands with epilepsy related to infantile spasms (IS), Lennox–Gastaut syndrome (LGS), or specific malformations of cortical development. In the second group (termed ‘triads’), both biological parents without epilepsy are enrolled. Enrollment, which can occur directly at EPGP clinical centers or via phone and mail from remote distance, involves structured interviews, collection of relevant medical records, magnetic resonance imaging (MRI) studies and electroencephalogram (EEG) findings, and sending blood samples for DNA preparation at the NINDS Repository at Coriell Institute of Medical Research.
Table 1.
EPGP Collaborative authors and contributions.
| Author | Institution | Study PI | Clinical site PI |
Clinical site Co-PI |
Referral Center PI |
Administrative Core |
Phenotyping Core |
Informatics Core |
Neurophysiology Core |
Imaging Core |
Pharmacogenomics Core |
Genomics and Data Analysis Core |
Data Review Core |
Publications Committee |
Writing of manuscript |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abou-Khalil, Bassel, MD | Vanderbilt University Medical Center | X | X | ||||||||||||
| Alldredge, Brian, PharmD | University of California, San Francisco | X | |||||||||||||
| Bautista, Jocelyn, MD | Cleveland Clinic | X | |||||||||||||
| Berkovic, Sam, MD | The University of Melbourne | X | |||||||||||||
| Bluvstein, Judith, MD | New York University School of Medicine | X | X | ||||||||||||
| Boro, Alex, MD | Albert Einstein College of Medicine | X | |||||||||||||
| Cascino, Gregory, MD | Mayo Clinic College of Medicine Rochester, Minnesota | X | X | ||||||||||||
| Consalvo, Damian, MD, PhD | Hospital General de Agudos José Maria Ramos Mejía | X | |||||||||||||
| Cristofaro, Sabrina, RN, BSN, Phenotyping Director | New York University | X | X | ||||||||||||
| Crumrine, Patricia, MD | Children's Hospital of Pittsburgh of University of Pittsburgh Medical Center | X | |||||||||||||
| Devinsky, Orrin, MD | New York University School of Medicine | X | X | X | X | X | |||||||||
| Dlugos, Dennis, MD, MCSE | The Children's Hospital of Philadelphia | X | X | X | X | ||||||||||
| Epstein, Michael, PhD | Emory University School of Medicine | X | |||||||||||||
| Fahlstrom, Robyn, MPH, Data Monitor | University of California, San Francisco | X | X | X | |||||||||||
| Fiol, Miguel, MD | University of Minnesota Medical Center | X | |||||||||||||
| Fountain, Nathan, MD | University of Virginia Health System | X | |||||||||||||
| Fox, Kristen, RN, MS, Recruitment Director | University of California, San Francisco | X | X | ||||||||||||
| French, Jacqueline, MD | New York University School of Medicine | X | |||||||||||||
| Freyer Karn, Catharine, Project Director | University of California, San Francisco | X | X | X | |||||||||||
| Friedman, Daniel, MD | New York University School of Medicine | X | |||||||||||||
| Geller, Eric, MD | St. Barnabas Health Care System | X | |||||||||||||
| Glauser, Tracy, MD | Cincinnati Children's Hospital Medical Center | X | X | ||||||||||||
| Glynn, Simon, MD | University of Michigan | X | |||||||||||||
| Haas, Kevin, MD | Vanderbilt University Medical Center | X | |||||||||||||
| Haut, Sheryl, MD, MS | Albert Einstein College of Medicine | X | |||||||||||||
| Hayward, Jean, MD | Kaiser Permanente: Oakland Medical Center | X | |||||||||||||
| Helmers, Sandra, MD | Emory University School of Medicine | X | |||||||||||||
| Joshi, Sucheta, MD | University of Michigan | X | |||||||||||||
| Kanner, Andres, MD | Rush University Medical Center | X | |||||||||||||
| Kirsch, Heidi, MD, MS | University of California, San Francisco | X | X | ||||||||||||
| Knowlton, Robert, MD | University of Alabama at Birmingham School of Medicine | X | X | ||||||||||||
| Kossoff, Eric, MD | The Johns Hopkins University School of Medicine | X | |||||||||||||
| Kuperman, Rachel, MD | Children's Hospital & Research Center Oakland | X | |||||||||||||
| Kuzniecky, Ruben, MD | New York University School of Medicine | X | X | X | X | X | X | ||||||||
| Lowenstein, Daniel, MD | University of California, San Francisco | X | X | X | X | X | X | ||||||||
| McGuire, Shannon, MD | Louisiana State University Health Sciences Center | X | |||||||||||||
| Motika, Paul, MD | Rush University Medical Center | X | |||||||||||||
| Nesbitt, Gerard, MBA, Director of Informatics | University of California, San Francisco | X | X | X | |||||||||||
| Novotny, Edward, MD | Seattle Children's Hospital | X | |||||||||||||
| Ottman, Ruth, PhD | Columbia University | X | X | X | X | ||||||||||
| Paolicchi, Juliann, MD | Vanderbilt University Medical Center | X | X | X | |||||||||||
| Parent, Jack, MD | University of Michigan | X | |||||||||||||
| Park, Kristen, MD | Children's Hospital Colorado | X | |||||||||||||
| Poduri, Annapurna, MD | Children's Hospital Boston | X | X | X | |||||||||||
| Risch, Neil, PhD | University of California, San Francisco | X | |||||||||||||
| Sadleir, Lynette, MBChB, FRACP, MD | Wellington School of Medicine and Health Sciences, University of Otago | X | |||||||||||||
| Scheffer, Ingrid, MBBS, PhD | The University of Melbourne | X | |||||||||||||
| Shellhaas, Renee, MD | University of Michigan | X | |||||||||||||
| Sherr, Elliott, MD, PhD | University of California, San Francisco | X | X | ||||||||||||
| Shih, Jerry J., MD | Mayo Clinic College of Medicine Jacksonville, Florida | X | X | X | |||||||||||
| Shinnar, Shlomo, MD, PhD | Albert Einstein College of Medicine | X | |||||||||||||
| Singh, Rani, MD | University of Michigan | X | |||||||||||||
| Sirven, Joseph, MD | Mayo Clinic College of Medicine Scottsdale, Arizona | X | |||||||||||||
| Smith, Michael, MD | Rush University Medical Center | X | |||||||||||||
| Sullivan, Joe, MD | University of California, San Francisco | X | |||||||||||||
| Thio, Liu Lin, MD, PhD | Washington University in St. Louis | X | X | ||||||||||||
| Venkat, Anu, MD | The Children's Hospital of Philadelphia | X | |||||||||||||
| Vining, Eileen, MD | The Johns Hopkins University School of Medicine | X | X | ||||||||||||
| Von Allmen, Cretchen, MD | University of Texas Health Science Center at Houston | X | |||||||||||||
| Weisenberg, Judith, MD | Washington University in St. Louis | X | X | ||||||||||||
| Widdess-Walsh, Peter, MB, FRCPI | St. Barnabas Health Care System | X | X | ||||||||||||
| Winawer, Melodie, MD, MS | Columbia University | X | X |
EPCP: Epilepsy Phenome/Cenome Project; PI: principal investigator.
Background and study design
Epilepsy affects approximately 50 million people worldwide and has a lifetime risk of about 3% [1–3]. More than 1,750,000 people develop epilepsy every year [4]. The World Health Organization estimated that epilepsy accounts for 1% of the global burden of disease, as measured by disability-adjusted life years (DALYs: number of years lost due to disability or premature death). Among people with epilepsy who have access to treatment (a minority worldwide), 30%–40% have seizures that are refractory to medication [5]. People with refractory epilepsy have poorer employment status, significantly limited activities, impaired quality of life, and increased mortality rates compared to the general population. Even people with well-controlled seizures or those whose seizures completely remit have relatively poor educational and social outcomes years after their seizures have resolved [6].
In the search for new strategies to reduce the burden of epilepsy, the discovery of specific genes that influence risk offers a novel opportunity to clarify pathogenic mechanisms, to identify susceptible individuals prior to seizure onset, and to treat and prevent seizures in people at risk. Converging data support a pathogenic role of genetics in epilepsy: epidemiologic studies show a two- to four-fold increased risk in the first-degree relatives of people with epilepsy of unknown cause (either IGE or non-acquired LRE) [7–10]; twin studies show higher concordance in monozygotic than dizygotic pairs, with heritability estimates of 70%–80% [11–14]; and more than 20 genes with a major effect on susceptibility have been identified [15–17]. However, the known genes affect risk in a very small proportion of individuals – primarily those with extensive family histories consistent with Mendelian inheritance. Most epilepsies occur without a significant family history, and identifying and characterizing the genetic mechanisms in these ‘complex epilepsies’ are major challenges in the coming years [18].
The genetic architecture of complex epilepsies could involve common variants with small effects on risk, consistent with the ‘common disease, common variant hypothesis’ [19,20] or multiple rare variants with larger risk-raising effects [21]. Although the distribution of these types of variants in the epilepsies is uncertain, evidence from epilepsy [22,23] and other disorders [24,25] suggests that common variants contribute little to epilepsy heritability. In contrast, accumulating evidence for association of complex epilepsies with rare microdeletions suggests that rare variants are important [26–30]. In addition, de novo genetic events can cause some ‘catastrophic epilepsies’, such as IS and LGS [31–33], and certain malformations of cortical development that include intractable epilepsy as a principal feature [34–40].
The EPGP was designed to address the hypothesis that genetic variation contributes to risk of developing epilepsy and pharmacological intractability and that the variation may (1) include common polymorphisms, rare variants, or private mutations; (2) range in scale from single nucleotide alterations to large genomic rearrangements; (3) be inherited or arise de novo; and (4) occur in the germ line or somatically. EPGP is a multi-institutional, collaborative network of 27 academic epilepsy centers throughout the United States, Australia, New Zealand, and Argentina, which is carrying out detailed phenotyping and banking DNA samples from 1500 pairs of first-degree relatives (3000 individuals) with either IGE or LRE, and 750 triads (2250 individuals) with one child with either IS, LGS, or specific malformations of cortical development (polymicrogyria or periventricular nodular heterotopia), plus both biological parents. Completion of this work will set the stage for large-scale genome analyses of these individuals. The overall design of the study is as follows.
Participants
The inclusion criteria for participants and epilepsy types are listed in Table 2.
Table 2.
Epilepsy Phenome/Genome Project (EPGP) inclusion and exclusion criteria.
| All participants with epilepsy |
|---|
|
Idiopathic generalized epilepsy (IGE)
|
Localization-related epilepsy (LRE)
|
Infantile spasms (IS) or Lennox–Gastaut syndrome (LGS) – cryptogenic
|
IS or LGS – FCD
|
Polymicrogyria (PMG) or bilateral periventricular nodular heterotopia (PVNH)
|
Participant recruitment
Participants are identified primarily through the EPGP Clinical Centers by prospective screening of clinic patients, retrospective medical record reviews, and ongoing education of colleagues within the institution and from neighboring hospitals and practices. There is also an EPGP National Recruitment Campaign to recruit eligible families outside of the Clinical Centers through community advocacy groups, the Internet, news articles, advertising, and other forms of communications with patients and healthcare professionals.
Phenotype data collection
Figure 1 and Table 3 provide an overview of the EPGP clinical data collection protocol. After giving informed consent, participants undergo a series of detailed, semistructured web-based interviews (including Subject Demographics, Eligibility Screening, and a Diagnostic Interview) administered by study coordinators. Additional information is extracted from the participant's medical records to support the epilepsy diagnosis, categorize response to antiepileptic drugs (AEDs), and to provide additional phenotypic data. Informatics tools were developed for the recording and reviewing of responses to AEDs. The tools include (1) a decision tree that leads the study coordinator through a series of decision points that will ultimately result in the phenotypic classification of individual AED trials as either uninformative, success, or failure, (2) a web-based data collection form to collect the subject's AED trials and drug response history, and (3) a web-based AED review process that allows the AED Core to review all AED data.
Figure 1.

EPGP enrollment, phenotyping, and data review process.
IGE: idiopathic generalized epilepsy; LRE: localization-related epilepsy; EEG: electroencephalogram; MRI: magnetic resonance imaging; EPGP: Epilepsy Phenome/Genome Project; AED: antiepileptic drug.
Table 3.
Activities included in data collection protocol, by participant type.
| Activity name | IGE participant | LRE participant | IS/LGS proband | PMG/PVNH proband | Parent controls |
|---|---|---|---|---|---|
| Eligibility checklist | Yes | Yes | Yes | Yes | Yes |
| Consent | Yes | Yes | Yes | Yes | Yes |
| Screening interview | Yes | Yes | Yes | Yes | Yes |
| Specimen collection | Yes | Yes | Yes | Yes | Yes |
| Subject demographics | Yes | Yes | Yes | Yes | Yes |
| Diagnostic interview | Yes | Yes | |||
| Medical record abstraction | Yes | Yes | Yes | Yes | |
| Pharmacogenomics form | Yes | Yes | |||
| AED web form | Yes | Yes | |||
| Electroencephalography review | Yes | Yes | Yes | ||
| MRI review | Optional | Yes | Yes | Yes | |
| Final diagnosis form | Yes | Yes | Yes | Yes |
IGE: idiopathic generalized epilepsy; LRE: localization-related epilepsy; IS: infantile spasms; LGS: Lennox–Gastaut syndrome; PMG: polymicrogyria; PVNH: periventricular nodular heterotopia; AED: antiepileptic drug; MRI: magnetic resonance imaging.
The source medical records are de-identified, scanned, and stored at the Clinical Center and uploaded to the EPGP Data Repository. EEG and MRI studies are also submitted by the Clinical Center and uploaded to the EPGP Data Repository, reviewed by the EEG and MRI Cores, and used to confirm the diagnoses. Finally, at each EPGP Clinical Center, the site principal investigator (PI) examines all collected clinical data (interviews, medical record abstraction, MRI, and EEG) and completes the web-based Final Diagnosis Form, where details regarding seizure semiology and epilepsy classification are documented.
Blood, cell line and DNA logistics
For each enrolled participant, two tubes of blood are submitted to the NINDS Human Genetics Repository at the Coriell Institute for Medical Research, where DNA is extracted and cell lines are generated to serve as a perpetual resource to the research community. EPGP uses an advanced web-based system, designed by the Informatics Core, for tracking specimen shipments from the clinical site to the Coriell Institute for Medical Research and for recording when a specimen has been accessioned at the Coriell Institute for Medical Research.
Data surveillance and quality control
Systematic quality reviews are conducted to identify and correct errors in phenotypic data. The following activities are conducted on an ongoing basis: (1) qualitative and quantitative data monitoring activities by the EPGP statistician, (2) automated error checks programmed by the Informatics Core, (3) in-person data review meetings to examine forms and medical records for a subset of participants, and (4) expert reviews by EPGP scientific cores, including EEG, MRI, Pharmacogenomic, Phenotyping, and Data Review Cores.
Informatics infrastructure
EPGP developed a suite of custom web-based informatics solutions for participant tracking, electronic data collection, and data review. Details of the platform design, along with an interim assessment of its effectiveness by end users, are provided in Ref. [41]. The core application is called Participant Activity Tracker, which allows new patients to be registered and data collection forms to be completed. The forms and activities completed for a study participant vary depending upon the participant's study arm and participant type. Therefore, it was necessary to define these rules in the database, so that the execution and completion status of these activities could be tracked in real time and reported accurately.
Collecting the data electronically using web-based forms enabled EPGP to streamline the data collection process and reduce data errors. It has decreased the time taken to collect data and increased the data accuracy. Methods used to realize these benefits include specifying allowable values and data ranges in each form field, automatically hiding questions that do not apply to the study participant, and branching to only the relevant sections in the form based on specified conditions. Input fields that do not satisfy the data validation criteria prompt the end user to rectify the data error, preventing the end user from proceeding to the next page in the form until all data errors are corrected. Another advantage of using web-based forms is that the research data are available immediately, enabling monitoring, reporting, and review in real time.
EPGP also collects source files belonging to study participants. These include medical records, EEGs, and MRIs. On receipt of these files, any residual protected health information (PHI) that could potentially identify a patient is removed from the source files, and the files are converted to an accessible format. Medical records are converted to portable document format (PDF), EEGs are converted to Persyst Insight format, and MRIs are converted to digital imaging and communications in medicine (DICOM) format. All source files are stored on a secure, password-protected File Transfer Protocol (FTP) file server.
A number of electronic workflows were designed and implemented to facilitate peer reviews performed by the EEG, MRI, and Data Review Cores. Web-based tools were developed for this purpose and enable the EEGs and MRIs to be characterized accurately using structured data collection forms and deemed acceptable or not for the study. Once the entire study participant's data are collected and their source EEG, MRI, and medical records are available (where applicable), the Data Review Core is alerted automatically, at which point the study participant's complete data and final epilepsy classification are reviewed and confirmed, or corrected if necessary.
The entire informatics platform was designed and developed in approximately an 8-month timeframe. There was a lack of any suitable off-the-shelf solution in the marketplace when we commenced the study, and a custom-developed solution was the only option available at that time. We continued to develop new enhancements and changes to the platform over the duration of the EPGP study, and it has served its purpose well. EPGP's data warehouse contains over 3.5 million completed data points, 1775 MRIs, and 2,245 EEGs. More than 33,000 web-based activities have been completed thus far.
Results
EPGP is organized into four phases of execution: study initiation, pilot, study expansion/establishment, and close-out. Each of these phases is presented here for the purpose of emphasizing the challenges and lessons learned.
Study initiation and management
Study initiation, which occurred during the first 6 months, involved hiring of staff, approval of Institutional Review Board (IRB) applications, development of phenotyping tools and training materials, and programming of informatics systems. Key decisions were made during this period that contributed to the overall success of the project: the network was organized into cores and managed by an Administrative Core, study timelines were generated and followed closely, communications were frequent and standardized, and a pay-for-performance financial model was established.
Core-based organizational structure
In addition to the Clinical Centers staffed by site investigators and study coordinators, the decision was made at the start to rely on an organizational structure that included cores serving specific administrative and scientific functions (Figure 2); the design of this structure was generated empirically based on the complex organizational needs of the project.
Figure 2.
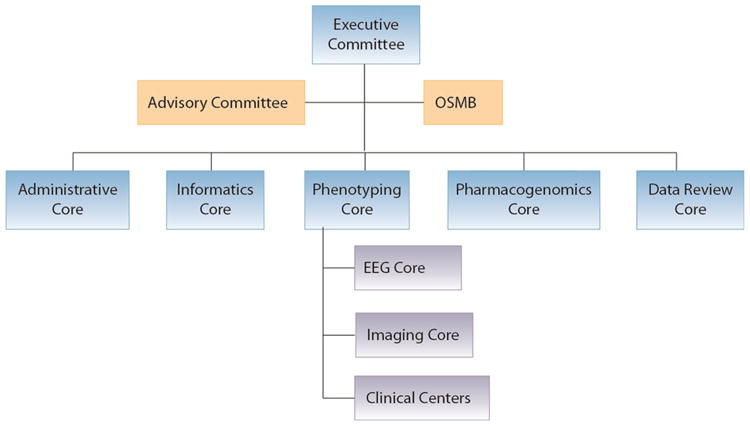
The EPGP organizational structure.
EPGP: Epilepsy Phenome/Genome Project; OSMB: Observational Study Monitoring Board; EEG: electroencephalogram.
Overall leadership of EPGP is provided by an Executive Committee comprised of the Core chairs and administrative personnel of the Administrative Core and the Phenotyping Core. EPGP is overseen by an Observational Study Monitoring Board (OSMB) appointed by NINDS and an Advisory Committee selected by the EPGP PIs. The OSMB is responsible for certifying to NINDS on a biannual basis that the study is proceeding according to plan and for approving any proposed modifications in study design made by the investigators. The Advisory Committee meets with the EPGP leadership annually and is used primarily as a ‘sounding board’ when problems arise and external perspectives may be of benefit in devising solutions. Coordinating the activities of the five scientific cores, 27 Clinical Centers, and two advisory bodies required a strong Administrative Core, the central operations group for the network. The Informatics Core manages all aspects of the software and hardware related to the collection, storage, and retrieval of phenotype data, collection and sending of blood for DNA banking, and design and maintenance of the study website. The Phenotyping Core oversees all phenotyping activities, including careful tracking of progress at each Clinical Center. The EEG Core oversees and reviews all electroencephalography-related data, and the Imaging Core oversees and reviews all imaging (primarily MRI) data. The Pharmacogenomics Core reviews and validates the data related to AED response and adverse effects. A Data Review Core, implemented in the fourth year of the project, assesses the accuracy of phenotyping data through reviews of both randomly and specifically selected study participants.
The Administrative Core coordinates and tracks all activities of the study and oversees the work of the other Cores and Clinical Centers, communications, national and site-specific recruitment, publicity, financials, and interactions with NINDS and oversight bodies. A full-time Project Director handles the overall implementation of the project, tracks deliverables from sites and cores, maintains study documentation and reports, and generates and tracks budgetary and phenotyping costs and metrics. A full-time Phenotyping Director trains and monitors clinical centers and handles all day-to-day eligibility and protocol questions. A full-time Recruitment Director was added to the Administrative Core to study clinical center recruitment trends and to manage a national campaign for participant recruitment. Later in the study, a full-time Statistician was hired to handle the new Data Review Core and data queries and to generate datasets for publication.
During the study initiation phase, the Administrative Core made three critical decisions: to integrate the study timeline; to put energy into frequent, standardized study communications; and to focus on robust financial tracking systems.
Study timeline
The Administrative Core tracked the Clinical Centers and Cores against a robust study timeline (see Supplemental Materials, Figure 1), which illustrated the ways in which the efforts of the individual cores and centers affected each other and the study's success overall. The timeline was developed by the Project Director, agreed upon by the network at the various progress meetings, and updated every 3–6 months, depending on the level of change being experienced by the network. Deviations from the timeline were identified early, and interventions could be applied rapidly to minimize the time and resources lost to the study.
Communications
To organize the efforts of more than 100 personnel working on the project, teleconferences were established at fixed times every week (later, every 2 weeks or every month), and these proved invaluable. The Administrative and Informatics Cores met internally at least once per week, and the study PIs met with the Administrative Core and Informatics Director weekly. At study start, the leaders of each scientific core met weekly with the Administrative Core, Informatics Core, and the Executive Committee to closely track progress and discuss challenges, and these meetings were scaled back to a monthly schedule as the study matured through the study expansion or establishment phase. Additionally, the staff from all Clinical Centers met at a set time each week for the EPGP ‘All Sites Call’ with the Administrative Core and Executive Committee. These meetings enabled the study administrators to check in on study deliverables, such as first recruitment and progress of IRB approvals, but more importantly, they created a forum for the investigators and coordinators of the Clinical Centers to describe both challenges and innovations. For example, numerous ideas were exchanged for maintaining awareness by faculty and trainees at Clinical Centers about EPGP and the need to identify potential participants.
Separate teleconferences were conducted monthly for study coordinators, and these were frequently utilized to review details of the study workflow when problems had been identified, such as best practices for the administration of the participant interviews. The network scheduled at least one face-to-face meeting each year and structured these annual meetings to meet the needs of the group at the time, including training, expansion of the network, data integrity issues, or planning for the future. Finally, agendas were followed closely and all meetings were followed by emails in a standardized newsletter template (see Supplemental Materials, Figure 2), so that all members of the study could keep track of critical information efficiently.
Key aspects of EPGP communications were transparency, regular reminders of the value of the collaborative and study achievements, and team-building. Updated tables and graphs of enrollment and phenotyping activities completion by each site were circulated before meetings, highlighting both high-performing and poor-performing sites, and this typically led to an exchange of information between sites as to how to improve performance. Important successes, such as recognition of the top-performing sites, enrollment of the 1000th participant, approvals by the OSMB, and acceptance of abstracts at national meetings, were highlighted at teleconferences and in the newsletter. Finally, in all communications, the study team focused on the contributions of the study coordinators at the Clinical Centers by highlighting coordinators who had achieved high recruitment or phenotyping in a month, by remembering birthdays, and by creating team-building materials, such as trivia booklets, about the coordinator team.
The study's website (http://www.epgp.org) has also proved to be a critical communications tool for all aspects of the project. The public site provides information to potential participants regarding the goals and design of the study, eligibility criteria, and a phone number and email address to inquire further about the study and express interest in enrollment. The ‘members only’ area contains all the tools required by study personnel, including the applications for enrollment, phenotyping, specimen delivery and follow-up, study documentation, and training material.
Pay-for-performance financial model
The funding agency, NINDS, required a per-activity funding model for EPGP from the start. This proved to be a powerful study management tool, since the Clinical Centers were strongly incentivized to identify, enroll, and fully complete the required study procedures for as many participants as possible. Based on our prior experience, there is no doubt this was far more effective than the traditional model of committing to a ‘percent effort’ for site investigators and study coordinators, which is disconnected from site performance. However, this model was initially difficult to implement because of the way in which U01 awards (the National Institutes for Health (NIH) designation for grants that are cooperative agreements and include substantial oversight by the funding agency) are handled by most institutions: they are treated as cost-reimbursement contracts, and provisions do not currently exist to establish performance contracts on a U01 award. Nonetheless, in the first year of the project, the Project Director established tools to supplement university systems, so that the Administrative Core could effectively execute contracts, update budgets, track spending, and monitor the correctness of Clinical Center invoices within budget categories of each subcontract.
Recruitment and phenotyping
With the structure of the project established, the study entered its pilot period and we quickly realized that a major challenge was overcoming recruitment difficulties: the participants specified by the protocol were, in all but the IS category, more difficult to find than initially estimated. We also discovered that despite a considerable effort to finalize all of the phenotyping procedures, there were many changes needed to make them acceptable to both the study participants and personnel. In response to these challenges, we revised the plan for participant recruitment and streamlined the phenotyping instruments, which led to a major improvement in virtually all aspects of project execution.
Participant recruitment
After 1 year of participant enrollment, more than 10,000 potential families had been screened for eligibility and 782 individual participants had enrolled (this was 45% of our goal for that time point). Eligible sibling pairs were more difficult to identify than was originally estimated, probably due to differences between epidemiological estimates of the prevalence of sibling pairs (which were used to predict enrollment numbers) and the observed prevalence of sibling pairs in academic epilepsy centers. In addition, obtaining the signed consent document from both family members proved to be a challenge. Often, one of the family members was not seen at an EPGP Clinical Center, and so completing the informed consent process by phone and mail was cumbersome for participants. It is possible that these family members were less inclined to participate because they did not feel a close affiliation with the PI. To close the enrollment gap, the Administrative Core hired a full-time Recruitment Director at the end of the first year. The Recruitment Director improved recruitment at the EPGP Clinical Centers by working with sites to tailor recruitment plans for their needs, establishing EPGP Referral Centers that identified participants to be directly enrolled at nearby EPGP Clinical Centers, recognizing monthly achievements with coordinator awards and acknowledgments, providing standardized recruitment materials that were visually appealing and easily understandable (such as participant brochures, posters, and pocket eligibility cards for clinicians), establishing a recruitment probation plan for sites with monthly enrollment expectations and interventions if goals were not met, and sharing best practices at all group meetings. The best practices included tips for consenting participants via phone and mail, how to convey complicated information about genetics and results during the consent process, and how to establish a mechanism at each site to ensure that all clinic patients were screened for the study. Prior to this intervention, many sites relied on the family history in the medical chart for screening. However, this information was often not complete or updated, and sites incorporated basic EPGP eligibility questions into every patient visit to ensure that all eligible families were identified.
The Recruitment Director also launched a national recruitment campaign to identify potential study participants from outside of the EPGP Clinical Centers. These participants were screened for eligibility by central study staff and then referred to Clinical Centers for enrollment. The national campaign included sending out ‘Dear Colleague’ letters and study update newsletters, collaborating with patient advocacy groups and other epilepsy research studies, cost-effective advertising directed at a targeted audience, establishing an online presence via websites and social networking sites, developing creative strategies to motivate attendees at national professional conferences to refer patients, creation of the EPGP Community Referral Network (CRN) (consisting of hundreds of neurologists and healthcare professionals who referred families), and generating additional recruitment materials (such as pens, coffee mugs, post-it pads, and magnets with eligibility criteria) to increase visibility of project in clinics around the country. The CRN, the most effective of these strategies, identified almost 100 eligible families who subsequently enrolled in EPGP.
Phenotyping workload
In the second year of the grant, we recognized that the recruitment and phenotyping workload of the Clinical Centers exceeded human resources for the projected timelines. The research team studied the phenotyping metrics across the sites, modeled the workload through the end of the study, and in collaboration with the OSMB, worked to streamline the EPGP dataset without sacrificing phenotyping depth. For example, the Diagnostic Interview underwent several revisions that reduced its average length from 2 h to 45 min, mainly by reducing redundancy and consolidating question sets. The original version of this form had been modified from an instrument used in a single-site genetic study, in which the interview length averaged 60 min. However, both the phenotyping staff and the method of administration differed in our study from those in the original study from which the instrument was derived. In the original study, the form was administered on paper by closely supervised staff dedicated to working exclusively on that study. Budgetary considerations, as well as the projected rate of enrollment at each site, precluded use of dedicated phenotyping staff in our study. Instead, we used clinical staff engaged in multiple tasks in addition to their work on the study, who had to administer a new instrument using an unfamiliar electronic platform. In addition, our large team of expert diagnosticians had expanded the previously used form before we initiated data collection, and this new form was not sufficiently pilot-tested to ensure its length was manageable. In retrospect, a longer pilot phase would have been advisable.
Additional changes included elimination of the 2-to 3-h pharmacogenomics abstraction for severe epilepsies (triads), since nearly all these participants were pharmacoresistant. A cap was set for the number of times a study coordinator should attempt to get medical records from referring institutions, and an Adjudications Core, managed by the Phenotyping Director, was established to review such cases. The adjudications process was later expanded to review a wide range of eligibility questions, in order to retain as many eligible participants as possible and limit the dropout rate. Each case was carefully scrutinized by the Phenotyping Director and the Adjudications Core. Finally, in the third year, we allocated an American Recovery and Reinvestment Act (ARRA)-funded administrator to each of the 13 actively enrolling Clinical Centers to close out the phenotyping backlog that developed in the second year.
Expanding the network
By the end of the second year of the grant, the study was fully established. All Clinical Centers had overcome their recruitment and phenotyping obstacles, or had been dropped. However, we were projecting we would end the study with approximately 50% of our cohort if enrollment continued at the same rate. The study team therefore considered three options: further reduce phenotyping from the protocol (to allow sites to focus even more on recruitment), increase the size of the network, or both. The Administrative Core used real-time study data to generate models that projected the impact of each change financially, to recruitment and phenotyping. These analyses suggested that the best strategy was to immediately expand the network. The Recruitment Director oversaw the addition of 14 new Clinical Centers, using a selection process designed to yield the most promising sites. The Phenotyping Director scheduled and conducted extensive on-site training with each new center's PI and study staff, to ensure that all sites were well-equipped to complete the numerous study procedures. Not surprisingly, the effort to successfully bring new sites on-board and have them fully functional benefitted from the experience gained during the initial phase of the study.
Addition of parent–child pairs with IGE/LRE
To further improve recruitment, and after consultation and review by our OSMB, the decision was made to expand the potentially eligible IGE/LRE cohort to include parent–child pairs in addition to the sibling pairs. The scientific drawback of including parent–child pairs (which could be included in genetic association studies but not genetic linkage studies) was felt to be worth the considerable advantage of enrolling as many familial cases as possible.
This expansion and change in inclusion criteria led to a marked upswing in enrollment, such that we now project we will reach at least 80% of our target recruitment by the end of the sixth year.
Data validation
In year 3 of the grant, when the initial 13 centers were fully operational and phenotype data were flowing in steadily, the research team began the process of review of interview, EEG, MRI, AED response, and final diagnosis data. Two in-person data review meetings were convened at which Phenotyping Core members reviewed samples of records from the Clinical Centers. From these meetings emerged the recognition that despite the in-depth interviews, medical record documentation, and laboratory data, there were still aspects of phenotyping that were open to interpretation, especially in the area of seizure semiology and classification. This led to extra phenotyping instructions and training for study personnel and more data checks being programmed into the informatics system. In addition, the data review meetings prompted the EPGP Administrative Core to create a Data Review Core, managed by the EPGP Statistician. The Data Review Core devised a protocol in which participant data are reviewed independently by two reviewers, potential discrepancies are reported back to the site, additional input is forwarded back to the reviewers, and a final adjudication is made based on all the available information. A custom module was designed by the Informatics Core that allows for this entire workflow to take place on a web-based platform (Figure 3).
Figure 3.
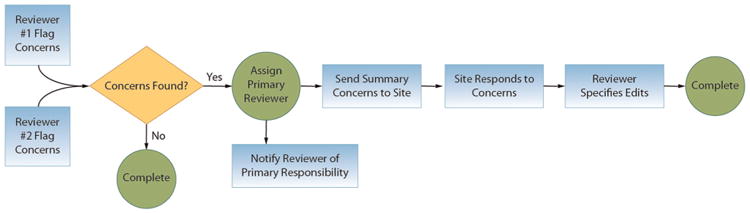
Workflow for the review of participant data by the EPGP Data Review Core.
EPGP: Epilepsy Phenome/Genome Project.
As a start, the Data Review Core reviewed the first three cases from each Clinical Center, with the plan to review an additional case from each center for any that had an error and eventually at least 6% of the EPGP dataset in detail (i.e., approximately 225 of the total number of expected probands). However, it quickly became apparent that in keeping with the results of the initial face-to-face data reviews, there were discrepancies in the dataset, although the vast majority were very minor in terms of the main scientific goals of the project. For example, in a review of 351 participants with LRE, errors have been found for 210 participants (60%). Importantly, most of the errors are related to seizure semiology (which is often open to interpretation by patients, family members, and epileptologists), and there have been only three cases with a major error such as an incorrect epilepsy diagnosis that affected eligibility. Nonetheless, this experience has emphasized the critical importance of added data validation in the study of a disorder such as epilepsy, and our plans are now to carry out this additional data review on all participants with LRE and a random subset of the remaining participants.
Conclusions
EPGP is a large-scale, multicenter, collaborative effort that is well on the way to completing the goals of creating the following: (1) a detailed phenotype database on a large number of very carefully selected individuals with specific forms of epilepsy, along with first-degree relative controls and (2) a permanent repository of DNA and cell lines linked to the phenotype data. As of June 2013, 5,535 participants have been consented, with 4,223 currently enrolled. In addition to the individuals identified directly through the Clinical Centers, more than 2000 families have contacted the national recruitment office for information and eligibility screening as a result of the National Recruitment Campaign; 328 of these families (16%) have been enrolled in EPGP.
In working toward these goals, we developed innovative approaches for carrying out a phenotype–genotype project of this scale, including solutions for some of the unexpected challenges that inevitably arise in large studies. These include new methods for standardized data collection and phenotyping (including clinical, electrophysiologic, and neuroimaging characteristics, and the creation of a schema for assessing AED response), novel web-based informatics tools for data collection and storage in a common repository, automated systems for shipment and receipt of blood samples, a national recruitment campaign to identify potential participants, and effective communication systems for managing the activities of the entire organization. We also benefitted from a variety of mechanisms for oversight and continuous quality improvement, which enabled us to rapidly identify and efficiently address problems such as lower than projected enrollment, an excessive phenotyping workload, and the need for further verification of phenotype data.
Supplementary Material
Acknowledgments
We would like to acknowledge the contributions to epilepsy research made by the staff, patients, and family members who are participating in EPGP, as well as the recruitment efforts of the EPGP Community Referral Network (CRN). The CRN consists of healthcare professionals not paid by the EPGP grant, who refer eligible families to EPGP. A list of individual contributors can be found at http://www.epgp.org.
Funding: This work is supported by National Institute of Neurological Diseases and Stroke (NINDS) grant U01 NS053998, as well as planning grants from the Finding a Cure for Epilepsy and Seizures (FACES) Foundation and the Richard Thalheimer Philanthropic Fund.
Administrative Core Members:
- Kathleen McGovern, Recruitment Assistant
- Nora Stillman, Recruitment Assistant
Informatics Core Members:
Kevin McKenna, Database Manager
- Vickie Mays, Data Coordinator
- Michael Williams, Informatics
- Alan Carpenter, Programmer Analyst
- Kevin Miller, Programmer Analyst
Study Coordinators:
- Prashant Agarwal, Study Coordinator, The Children's Hospital of Philadelphia
- Jennifer Ayala, Study Coordinator, Albert Einstein College of Medicine
- Cate Bakey, Study Coordinator, The Children's Hospital of Philadelphia
- Thomas Borkowski, PhD, Study Coordinator, Albert Einstein College of Medicine
- Riann Boyd, CCRP, Study Coordinator, Mayo Clinic College of Medicine Jacksonville, Florida
- Alicia Camuto, CCRP, Study Coordinator, The Children's Hospital Denver
- Cendy Carrasco, Study Coordinator, University of California, San Francisco
- Jennifer Cassarly, Study Coordinator, Cincinnati Children's Hospital Medical Center
- Yong Collins, RN, Study Coordinator, The Children's Hospital of Philadelphia
- Kevin Collon, Study Coordinator, University of Michigan
- Sean Collon, Study Coordinator, University of Michigan
- Heather Eckman, Study Coordinator, New York University School of Medicine
- Susan Fogarty, RN, Study Coordinator, University of Texas Health Science Center at Houston
- Dolores González Morón, MD, Study Coordinator, Hospital General de Agudos José Maria Ramos Mejía
- La June Grayson, Study Coordinator, Washington University in St. Louis
- Samantha Hagopian, RN, Study Coordinator, The Children's Hospital of Philadelphia
- Emily Hayden, Study Coordinator, University of Michigan
- Kristin Heggeli, Study Coordinator, Mayo Clinic College of Medicine Jacksonville, Florida
- Rachel Hennessy, Study Coordinator, New York University School of Medicine
- Jody Hessling, RN, Study Coordinator; Cincinnati Children's Hospital Medical Center
- Emily Hirschfield, Study Coordinator, Cincinnati Children's Hospital Medical Center
- Jennifer Howell, BA, BS, Study Coordinator, University of Alabama at Birmingham School of Medicine
- Sherry Klingerman, Study Coordinator, Mayo Clinic College of Medicine Rochester, Minnesota
- Maritza Lopez, RN, Study Coordinator, University of California, San Francisco
- Heather Marinelli, Study Coordinator, The Johns Hopkins University School of Medicine
- Brandy Maschhaupt, Study Coordinator, Mayo Clinic College of Medicine Scottsdale, Arizona
- Jennie Minnick, Study Coordinator, The Children's Hospital of Philadelphia
- Jade Misajon, RN, Study Coordinator, St. Barnabas Health Care System
- Jennifer Monahan, RN, Study Coordinator, Children's Hospital of Pittsburgh of University of Pittsburgh Medical Center
- Karen Oliver, Study Coordinator, The University of Melbourne
- Isha Parulkar, Study Coordinator, Children's Hospital Boston
- Laura Przepiorka, RN, Study Coordinator, Rush University Medical Center
- Paula Pyzik, BA, CRT, Study Coordinator, The Johns Hopkins University School of Medicine
- Brigid Regan, Study Coordinator, The University of Melbourne
- Catherine Shain, Study Coordinator, Children's Hospital Boston
- Lexie Slingerland, Study Coordinator, The University of Melbourne
- Caitlin Stanton, MPH, Study Coordinator, Seattle Children's Hospital
- Kelly Taylor, MS, CGC, Study Coordinator, Vanderbilt University Medical Center
- Stacy Thompson, RN, BSN, CCRC, Study Coordinator, University of Virginia Health System
- Jennifer Turczyk, Study Coordinator, Cleveland Clinic
- Alexander Vara, Study Coordinator, University of Texas Health Science Center at Houston
- Cindy Wesolowski, RN, NP, Study Coordinator, Cincinnati Children's Hospital Medical Center
- Andrew Yourich, Study Coordinator, University of Alabama at Birmingham School of Medicine
Footnotes
Conflict of interest: None declared.
References
- 1.Linehan C, Kerr M. Epidemiology of epilepsy in developed countries. In: Benbadis SR, Beran RG, Berg AT, et al., editors. Atlas of Epilepsies. Springer; London: 2010. pp. 51–56. [Google Scholar]
- 2.Diagana M, Bhalla D, Ngoungou E, Preux PM. Epidemiology of epilepsy in resource poor countries. In: Benbadis SR, Beran RG, Berg AT, et al., editors. Atlas of Epilepsies. Springer; London: 2010. pp. 57–63. [Google Scholar]
- 3.Hesdorffer DC, Logroscino G, Benn EK, et al. Estimating risk for developing epilepsy: A population-based study in Rochester, Minnesota. Neurology. 76(1):23–27. doi: 10.1212/WNL.0b013e318204a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Begley CE, Famulari M, Annegers JF, et al. The cost of epilepsy in the United States: An estimate from population-based clinical and survey data. Epilepsia. 2000;41(3):342–51. doi: 10.1111/j.1528-1157.2000.tb00166.x. [DOI] [PubMed] [Google Scholar]
- 5.Berg AT. Understanding the delay before epilepsy surgery: Who develops intractable focal epilepsy and when? CNS Spectr. 2004;9(9):136–44. doi: 10.1017/s109285290000849x. [DOI] [PubMed] [Google Scholar]
- 6.Wirrell EC, Camfield CS, Camfield PR, et al. Long-term psychosocial outcome in typical absence epilepsy. Sometimes a wolf in sheeps' clothing. Arch Pediatr Adolesc Med. 1997;151(2):152–58. doi: 10.1001/archpedi.1997.02170390042008. [DOI] [PubMed] [Google Scholar]
- 7.Annegers JF, Hauser WA, Anderson VE, Kurland LT. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology. 1982;32(2):174–79. doi: 10.1212/wnl.32.2.174. [DOI] [PubMed] [Google Scholar]
- 8.Ottman R, Annegers JF, Risch N, Hauser WA, Susser M. Relations of genetic and environmental factors in the etiology of epilepsy. Ann Neurol. 1996;39(4):442–49. doi: 10.1002/ana.410390406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi A, Viaggi S, Chiossi E. Family study of epilepsy in first degree relatives: Data from the Italian Episcreen Study. Seizure. 2003;12(4):203–10. doi: 10.1016/s1059-1311(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 10.Hemminki K, Li X, Johansson SE, Sundquist K, Sundquist J. Familial risks for epilepsy among siblings based on hospitalizations in Sweden. Neuroepidemiology. 2006;27(2):67–73. doi: 10.1159/000094976. [DOI] [PubMed] [Google Scholar]
- 11.Vadlamudi L, Andermann E, Lombroso CT, et al. Epilepsy in twins: Insights from unique historical data of William Lennox. Neurology. 2004;62(7):1127–33. doi: 10.1212/01.wnl.0000118201.89498.48. [DOI] [PubMed] [Google Scholar]
- 12.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: Genetics of the major epilepsy syndromes. Ann Neurol. 1998;43(4):435–45. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- 13.Corey LA, Berg K, Pellock JM, et al. The occurrence of epilepsy and febrile seizures in Virginian and Norwegian twins. Neurology. 1991;41(9):1433–36. doi: 10.1212/wnl.41.9.1433. [DOI] [PubMed] [Google Scholar]
- 14.Kjeldsen MJ, Corey LA, Christensen K, Friis ML. Epileptic seizures and syndromes in twins: The importance of genetic factors. Epilepsy Res. 2003;55(1–2):137–46. doi: 10.1016/s0920-1211(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 15.Ottman R, Hirose S, Jain S, Lerche H, Lopes-Cendes I, et al. Genetic testing in the epilepsies – Report of the ILAE Genetics Commission. Epilepsia. 2010;51(4):655–70. doi: 10.1111/j.1528-1167.2009.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87(1):41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: Unravelling the genetics of the epilepsies. Lancet Neurol. 2008;7(3):231–45. doi: 10.1016/S1474-4422(08)70039-5. [DOI] [PubMed] [Google Scholar]
- 18.Ottman R. Analysis of genetically complex epilepsies. Epilepsia. 2005;46(Suppl. 10):7–14. doi: 10.1111/j.1528-1167.2005.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakravarti A. To a future of genetic medicine. Nature. 2001;409(6822):822–23. doi: 10.1038/35057281. [DOI] [PubMed] [Google Scholar]
- 20.Lander ES. The new genomics: Global views of biology. Science. 1996;274(5287):536–39. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 21.Singleton AB, Hardy J, Traynor BJ, Houlden H. Towards a complete resolution of the genetic architecture of disease. Trends Genet. 2010;26(10):438–42. doi: 10.1016/j.tig.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavalleri GL, Weale ME, Shianna KV, et al. Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: A case-control study. Lancet Neurol. 2007;6(11):970–80. doi: 10.1016/S1474-4422(07)70247-8. [DOI] [PubMed] [Google Scholar]
- 23.Kasperaviciute D, Catarino CB, Heinzen EL, et al. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain. 2010;133(Pt 7):2136–47. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360(17):1696–98. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 25.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinzen EL, Radtke RA, Urban TJ, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86(5):707–18. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41(2):160–62. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6(5):e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheffer IE, Berkovic SF. Copy number variants – An unexpected risk factor for the idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):7–8. doi: 10.1093/brain/awp332. [DOI] [PubMed] [Google Scholar]
- 31.Kearney JA, Wiste AK, Stephani U, et al. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr Neurol. 2006;34(2):116–20. doi: 10.1016/j.pediatrneurol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–32. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pereira S, Vieira JP, Barroca F, et al. Severe epilepsy, retardation, and dysmorphic features with a 2q deletion including SCN1A and SCN2A. Neurology. 2004;63(1):191–92. doi: 10.1212/01.wnl.0000132844.20654.c1. [DOI] [PubMed] [Google Scholar]
- 34.Villard L, Nguyen K, Cardoso C, et al. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet. 2002;70(4):1003–08. doi: 10.1086/339433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansen A, Andermann E. Genetics of the polymicrogyria syndromes. J Med Genet. 2005;42(5):369–78. doi: 10.1136/jmg.2004.023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leeflang EP, Marsh SE, Parrini E, et al. Patient with bilateral periventricular nodular heterotopia and polymicrogyria with apparently balanced reciprocal translocation t(1;6)(p12;p12.2) that interrupts the mannosidase alpha, class 1A, and glutathione S-transferase A2 genes. J Med Genet. 2003;40(12):e128. doi: 10.1136/jmg.40.12.e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–88. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 38.Mencarelli MA, Spanhol-Rosseto A, Artuso R, et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet. 2010;47:49–53. doi: 10.1136/jmg.2009.067884. [DOI] [PubMed] [Google Scholar]
- 39.Bahi-Buisson N, Kaminska A, Boddaert N, et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 2008;49:1027–37. doi: 10.1111/j.1528-1167.2007.01520.x. [DOI] [PubMed] [Google Scholar]
- 40.Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): One gene leads to many phenotypes. Curr Opin Pediatr. 2003;15(6):567–71. doi: 10.1097/00008480-200312000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Nesbitt G, McKenna K, Mays V, et al. The Epilepsy Phenome/Genome Project (EPGP) informatics platform. Int J Med Inform. 2012 doi: 10.1016/j.ijmedinf.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.