

Abstract
In SOD1G93A transgenic rat model of ALS, breathing capacity is preserved until late in disease progression despite profound respiratory motor neuron (MN) cell death. To explore mechanisms preserving breathing capacity, we assessed inspiratory EMG activity in diaphragm and external intercostal T2 (EIC2) and T5 (EIC5) muscles in anesthetized SOD1G93A rats at disease end-stage (20% decrease in body mass). We hypothesized that despite significant phrenic motor neuron loss and decreased phrenic nerve activity, diaphragm electrical activity and trans-diaphragmatic pressure (Pdi) are maintained to sustain ventilation. We alternatively hypothesized that EIC activity is enhanced, compensating for impaired diaphragm function. Diaphragm, EIC2 and EIC5 muscle EMGs and Pdi were measured in urethane-anesthetized, spontaneously breathing female SOD1G93A rats versus wild-type littermates during normoxia (arterial pO2 ~90mmHg, pCO2 ~45mmHg), maximal chemoreceptor stimulation (MCS: 10.5% O2/7% CO2), spontaneous augmented breaths and sustained tracheal occlusion. Phrenic MNs were counted in C3/5; T2 and T5 ventrolateral MNs were counted. In end-stage SOD1G93A rats, 29% of phrenic MNs survived (vs. wild-type), yet integrated diaphragm EMG amplitude was normal. Nevertheless, maximal Pdi decreased ~30% vs. wild type (p<0.01) and increased esophageal to gastric pressure ratio (p<0.05), consistent with persistent diaphragm weakness. Despite major T2 and T5 MN death, integrated EIC2 (100% greater than wild type) and EIC5 (300%) EMG amplitudes were increased in mutant rats during normoxia (p<0.01), possibly compensating for decreased Pdi. Thus, despite significant phrenic MN loss, diaphragm EMG activity is maintained; in contrast, Pdi was not, suggesting diaphragm dysfunction. Presumably, increased EIC EMG activity compensated for persistent diaphragm weakness. These adjustments contribute to remarkable preservation of breathing ability despite major respiratory motor neuron death and diaphragm dysfunction.
Keywords: Respiratory motor neuron, Plasticity, Respiratory Muscles, Phrenic, Breathing
Graphical Abstract
Introduction
Amyotrophic lateral sclerosis (ALS) is a severe neuromuscular disease leading to progressive motor neuron cell death. Mutations on the copper-zinc superoxide dismutase 1 (SOD1) gene are observed in ~20% of the familial and ~1% of sporadic ALS patients (Pasinelli and Brown, 2006). Early rodent ALS models involved over-expression of human G93A-mutant SOD1 (SOD1G93A; (Gurney et al., 1994; Howland et al., 2002; Rosen et al., 1993).
Despite substantial respiratory motor neuron cell death in SOD1G93A mutant rodents, breathing capacity is preserved until late in disease progression. For example, in SOD1G93A mice, eupneic and hypercapnic (5% CO2) tidal volume generating ability is preserved until terminal stages of the disease (~18 weeks of age); breathing then precipitously declines over two days (Tankersley et al., 2007). In this study, respiratory motor neuron death/survival was not reported. At a defined disease end-stage (20% decrease in body mass), SOD1G93A rats retain the ability to generate tidal volume during room air breathing and with maximal chemoreflex stimulation (MCS: 7% CO2 and 10.5% O2) despite major loss of phrenic (~80%) and T5 (~40%) inspiratory motor neurons (Nichols et al., 2013a; Nichols et al., 2014; Nichols et al., 2015). Complete preservation of breathing capacity despite profound respiratory motor neuron death demonstrates a remarkable capacity for compensatory plasticity (Johnson and Mitchell, 2013; Nichols et al., 2013b). Although mechanisms giving rise to this plasticity are not known, electrical activity in the phrenic nerve is relatively preserved (55%) with respect to phrenic motor neuron survival (~15–30%; Nichols et al., 2013a; Nichols and Mitchell, 2016). Thus, CNS mechanisms contribute to preservation of phrenic motor output and breathing ability with disease progression. Nevertheless, additional mechanisms must contribute to enable full preservation of tidal volume generating ability (Nichols et al., 2013a,b).
One way to preserve breathing ability despite respiratory motor neuron death and diminished phrenic motor output is to preserve diaphragm function. However, it is not known if residual phrenic nerve activity in end-stage SOD1G93A rats is further amplified during transmission to diaphragm electrical activation and/or force generation. Thus, one major goal of this study was to test the hypothesis that diaphragm electrical activity and force generation are preserved in end-stage SOD1G93A rats.
Failure to preserve diaphragm function would suggest that alternative mechanisms preserve/restore breathing ability. Since thoracic respiratory MNs are less affected than phrenic MNs in end-stage SOD1G93A rats, respiratory muscle utilization might shift from more affected (phrenic) to less affected respiratory motor pools, such as the external intercostal muscles (EIC). We recently demonstrated that rats primarily activate T2, but not T5 EICs during normal or chemoreflex activated breathing (Navarrete-Opazo et al., 2014). Nevertheless, T5 EICs may make a greater relative contribution to breathing during disease states. Thus, a second major goal of this study was to assess the relative activation/contributions of T2 and T5 EICs in end-stage SOD1G93A rats.
Activation of individual inspiratory muscles has not been reported in end-stage SOD1G93A rats in a range of conditions, such as during eupnea, maximal chemoreflex activation or airway protective reflexes. Here, we explored inspiratory muscle activation in end-stage SOD1G93A rats to test the following hypotheses: 1) despite substantial phrenic MN death and reduced phrenic nerve activity, diaphragm electrical activity and transdiaphragmatic pressure (Pdi) generation are preserved; and/or 2) if diaphragm function is compromised, EIC muscles make a greater contribution to tidal volume generation, shifting contributions from more affected (phrenic/diaphragm) to less affected (EIC) motor pools/muscles. During these experiments on anesthetized rats, we measured and controlled arterial blood gases so that differences in chemoreceptor feedback between groups could not account for experimental results.
Methods
Animals
Studies were conducted on 22 adult female Taconic Sprague-Dawley rats (Taconic Laboratories, Germantown, NY) overexpressing the human SOD1G93A gene (SOD1G93A). SOD1G93A expression was detected with polymerase chain reaction of tail DNA using hSOD1 primers. Disease progression in female mutant (MT) rats is characterized by progressive weight loss, accompanied shortly by hindlimb and/or forelimb paralysis. Experiments were conducted when MT rats lost 20% from their peak body mass (“end-stage;” 172±3 days); at this stage, rats exhibited limb paralysis. Measurements were compared between MT and wild-type littermates (WT). Average body weights of rats on experimental days were 293±3 for WT and 230±11g for MT. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison, and were in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals.
Surgery and Electrode Implantation
Anesthesia was induced with 4% isoflurane, and maintained at 2–2.5% isoflurane throughout surgical procedures, initially with a nose cone and then via tracheal cannula (50% O2, balance N2). Body temperature was maintained at ~37°C with a custom-made heating table and a rectal thermometer (Physitemp, model 700 1H). The right femoral vein was catheterized to enable intravenous (i.v.) fluid delivery. Lactated Ringer’s solution with sodium bicarbonate was given as necessary to ensure fluid and acid-base balance. Blood gases and pH were measured in samples taken from a femoral arterial catheter (~0.3 ml; ABL800, Radiometer, Copenhagen, Denmark). Following laparotomy, diaphragm muscle was implanted with a pair of Teflon-coated, multistrand stainless-steel electrodes (AS631, Cooner Wire, Chatsworth, CA) uninsulated for ~2 mm at the point of contact with in the mid-costal region. In addition, the 2nd and 5th external intercostal muscles (EIC2 and EIC5) were implanted with EMG electrodes. Once surgery was complete, anesthesia was gradually converted to urethane (1.6 g/kg, i.v.) while isoflurane was slowly withdrawn. After conversion, the depth of anesthesia was confirmed by the absence of tail- and toe-pinch reflexes. Rats were given ample time to clear isoflurane from the body before experiments commenced (the same amount of time that they were on isoflurane).
EMG and Transdiaphragmatic Pressure Recordings
EMG signals were amplified (1000x) and band-pass filtered (10–1000 Hz) using an analog amplifier (Model 1800, A-M Systems, Carlsborg, WA, USA). They were digitized at 2 kHz and recorded (Powerlab, AD Instruments, Colorado, United States). EMG amplitude was estimated using root-mean-squared (RMS) EMG calculated over a 50-ms sliding window. RMS EMG value is a measure of the power of a signal and well correlated with force generated by various muscles including diaphragm muscle (Fuglevand et al., 1993; Lawrence and De Luca, 1983; Mantilla et al., 2010).
To estimate pressure differences generated by diaphragm contractions in vivo, transdiaphragmatic pressure (Pdi) was measured as the difference between esophageal (Peso) and gastric (Pgas) pressures (BD RT2000, BD, New Jersey, United States). Esophageal and gastric pressures were measured using dedicated catheters passing through the mouth. Initially, the catheter depth was estimated via the approximate positions of stomach and esophagus; the same length was marked on the catheters. After insertion, each transducer was adjusted to obtain consistent recordings. Gastric placement was further verified by a positive pressure in response to a gentle abdominal finger poke.
During inspiratory activity, esophageal recordings consistently display negative pressure swings; abdominal recordings display positive pressure swings. Reported Pdi values are the pressure difference at the peak of inspiration between esophageal and gastric values (Pgas − Peso). Pressure signals were low-pass filtered (cut-off frequency: 100 Hz; Model P122, Grass Technologies, Rhode Island, United States) and digitally sampled at 200 Hz. Others have proposed that the ΔPeso/ΔPgas ratio enables dissection of the relative contributions of diaphragm versus thoracic accessory muscles (i.e., such as intercostal muscles) to breathing (Macklem et al., 1978). Accordingly, diaphragm dysfunction reduces ΔPgas; however, ΔPeso may be compensated by recruitment of accessory muscles. In that case, ΔPeso/ΔPgas ratio increases, consistent with increased intercostal contributions. In the case of complete diaphragm paralysis, diaphragm moves in rostral direction (i.e., paradoxical diaphragm movement) leading to negative ΔPgas. Thus, ΔPeso/ΔPgas ratio changes its sign and becomes negative.
Experimental Protocol
Spontaneous EMG activities of diaphragm, EIC2 and EIC5 muscles were recorded across all experimental conditions in spontaneously breathing rats in a supine position. Before starting experiments, arterial blood samples were taken to monitor and correct blood gases, pH and base excess. The rats were tested under the following conditions: (1) Normoxia (Nx): Blood gas values were kept at >85 mmHg and ~45 mmHg for PO2 and PCO2, respectively. Standard base excess was kept between −3 and 3 mEq/L. In these anesthetized, spontaneously breathing rats, slightly elevated inspired O2 (21–45% O2) was necessary to attain normoxia; (2) Maximum chemoreceptor stimulation (MCS): Breathing with combined hypercapnia (7% CO2) and hypoxia (10.5% O2) for 3 min. (3) Spontaneous augmented breaths (AB): Spontaneous deep breaths (sighs) observed during MCS (RMS EMG amplitudes were more than twice normoxic RMS EMG amplitude, and typically greater than the MCS amplitude). Sighs were often followed by a post-sigh apnea; (4) Tracheal occlusion (TOCC): measured during the last 5 seconds of ~40s sustained TOCC. Spontaneous sighs and the response to TOCC were monitored as an indication of near-maximal respiratory muscle activation (Mantilla et al., 2011; Seven et al., 2014).
Histology and Motor Neuron Counts
After experiments, 6 rats per group were perfused and tissues fixed with 4% paraformaldehyde in 0.1M phosphate buffered saline and harvested (Lovett-Barr et al., 2012). The tissues were post-fixed overnight in 4% paraformaldehyde in 0.1M phosphate buffered saline and cryoprotected in 30% sucrose solution at 4°C. Specifically, cervical (C3–C5) and thoracic (T2 and T5) spinal segments were sectioned in transverse plane (40 μm thickness) using a cryostat. Sections were mounted on positively charged slides (Fisherbrand, Fisher Scientific, Pittsburgh, PA) and dried. Sections were stained using 0.1% cresyl-violet and dehydrated through graded alcohol (70–100%), cleared in Histoclear (National Diagnostics, Atlanta, GA) and cover-slipped (Eukitt, Electron Microscope Science, PA). Sections on positively charged slides were imaged at 10x zoom level (BZ-X710, Keyence Co., Osaka, Japan)
Putative phrenic motor neurons were counted as a cluster of large, medio-lateral neurons in the cervical ventral horn (Boulenguez et al., 2007; Mantilla et al., 2009; Watson et al., 2009). Similarly, putative intercostal motor neurons were counted as large, ventro-lateral neurons at T2 and T5 segments (Watson et al., 2009). Only intact motor neurons with an intact soma and nucleus were counted. Six sections were uniformly selected per segment to count neurons at cervical, T2 and T5 levels. The number of motor neurons in each spinal segment was summarized per slice and side by a blind investigator. Motor neuron counts were then averaged within WT and MT groups, and then statistical inferences were made across experimental groups. The number of phrenic motor neurons in the C3/5 segment and intercostal motor neurons in the T2 and T5 segments were extrapolated from the 6 sections as described previously (Nichols et al., 2013a).
Data Analysis and Statistics
Raw and AB-normalized RMS EMG amplitudes were calculated during Nx and MCS were analyzed for 1-minute period excluding ABs. For each rat, 4–8 ABs were analyzed. During TOCC, EMG bursts in the last 5 s of 40-s TOCC were analyzed. Rats were given ~5 min to recover from each experimental condition.
Data during each experimental condition for each animal were averaged to represent the sample for that experimental condition per animal. Statistical analyses were performed with 2-way repeated-measures ANOVA for EMG and Pdi analyses, and with a one-way ANOVA for analyses of histology. When significant differences were observed, Tukey-Kramer Honestly Significant Difference test was used as post hoc analysis. Data were summarized as mean ± SE. Statistical calculations were performed using JMP 11.0 (SAS Institute, Cary, NC). A significance level of 0.05 was set for all statistical comparisons.
Results
Respiratory Motor Neuron Loss in SOD1-Mutant Rats
Representative images are shown at three spinal segments in WT (Figure 1A, 1C and 1E) and SOD1G93A MT rats (Figure 1B, 1D and 1F). Phrenic (~71% loss), T2 intercostal (~64% loss) and T5 intercostal (~54% loss) motor neuron numbers were substantially reduced in late stage MT rats versus aged-matched WT littermates (p<0.0001; Figure 1G). Since phrenic motor neuron loss was more pronounced versus T5 intercostal motor neurons (p<0.05), there is potential for greater compensation from intercostal motor neurons throughout disease progression.
Figure 1.

Motor neuron survival in wild-type and SOD1G93A mutant rats. Images of spinal cord sections (10x) from C4 (A and B), T2 (C and D) and T5 (E and F) segments depicting putative phrenic and intercostal motor neurons for wild-type and SOD1G93A mutant rats. G. Motor neuron numbers were significantly reduced in all segments in MT rats (closed bars) versus WT (open bars; p<0.01). *p<0.01 Wild type vs. Mutant.
EMG Activities of Diaphragm and Intercostal Muscles
Representative recordings of diaphragm, EIC2 and EIC5 EMGs during Nx, MCS, AB and TOCC are shown in Figure 2. In general, both raw and normalized EMG amplitudes increased during MCS versus Nx across all muscles tested. The increase in RMS EMG increases more than MCS during spontaneous ABs and TOCC versus Nx and MCS. In MT rats, tonic firing motor units were sometimes observed during expiration.
Figure 2.

Representative EMG traces for diaphragm, 2nd and 5th external intercostal muscles during normoxia, maximum chemoreceptor stimulation, spontaneous augmented breaths (pointed by arrows) and tracheal occlusion in WT and SOD1G93A MT rats.
Raw Amplitudes
Diaphragm RMS EMG amplitude was consistently the largest, whereas EIC5 amplitude was consistently the smallest within each rat group (Diaphragm>EIC2>EIC5). In diaphragm muscle, the coefficient of variation for raw RMS EMG amplitude was ~30% during Nx across rats. Overall, despite significant motor neuron loss, there were no significant differences between WT and MT rats in raw diaphragm EMG amplitude (p>0.05; Figure 3). Similarly, there were no significant differences during ABs and TOCC.
Figure 3.

Raw root-mean-squared EMG amplitudes (time constant: 50 ms): A. Diaphragm EMG amplitudes were comparable between WT and MT rats across conditions. B. 2nd external intercostal EMG amplitude more than doubled in SOD1G93A versus WT rats during normoxia (p<0.05) C. 5th external intercostal EMG amplitude quadrupled in SOD1G93A MT versus WT rats in normoxia (p<0.05). *p<0.05 Wild type vs. Mutant. 1p<0.05 compared to normoxia. 2p<0.05 compared to 10.5% O2-7% CO2.
RMS EMG amplitudes of both EIC2 (more than doubled; p<0.05) and EIC5 (~quadrupled) muscles were significantly higher in MT (p<0.05) vs WT during Nx. EIC5 showed very little activity in WT rats, but exhibited robust respiratory activity in MT rats. Thus, MT animals recruit EIC muscles to help maintain resting breathing. During MCS, there were no significant differences in EMG activity in MT rats, although there was a tendency towards increased EMG activity.
Normalized Amplitudes
Normalization of RMS EMG amplitudes to near maximal behaviors such as ABs was suggested as a method of reducing the variability (Mantilla et al., 2011), although this normalization risks obscuring meaningful differences across groups in pathological states. When normalized to ABs, there were marginal increases in diaphragm RMS EMG amplitudes in MT rats versus WT during Nx (~50%; p=0.052) and MCS (~20%, p=0.07; Figure 4).
Figure 4.

Root-mean-squared EMG amplitudes normalized to augmented breaths during 10.5% O2/7% CO2: Diaphragm EMG amplitude was slightly higher in SOD1G93A rats (closed bars) versus WT rats (open bars) in normoxia and MCS (A). Normalized 2nd(B) and 5th(C) external intercostal EMG amplitudes were significantly higher in SOD1G93A during normoxia and MCS (p<0.05). *p<0.05 Wild type vs. Mutant. 1p<0.05 compared to normoxia. 2p<0.05 compared to 10.5% O2-7% CO2.
EIC2 and EIC5 activities were greater during both Nx and MCS (p<0.05). During Nx, normalized EIC2 and EIC5 RMS EMG amplitude in MTs were more than 2-fold and more than 4-fold above WT rats, respectively (p<0.05). Similarly, EIC2 and EIC5 EMG amplitudes during MCS were ~50% and ~70% higher in MT versus WT rats (p<0.05).
Diminished Transdiaphragmatic Pressure in Mutant Rats
Although diaphragm EMG amplitudes were comparable for raw electrical activity, and even higher in AB-normalized values in MT rats, Pdi was lower by ~29% during Nx (Figure 5, p<0.05). Pdi was also 30% lower in MT rats during TOCC (p<0.05). ΔPeso/ΔPgas ratio was significantly higher in MT rats during Nx and TOCC versus WT rats, suggesting greater accessory versus diaphragm muscle contributions (p<0.05).
Figure 5.
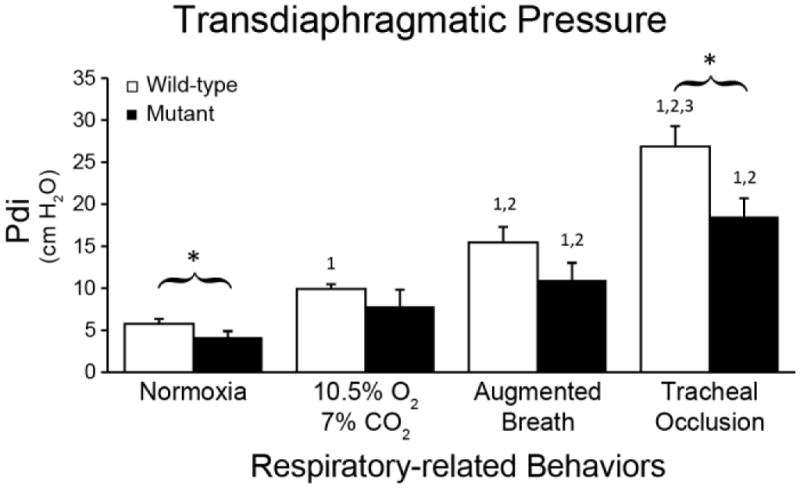
Transdiaphragmatic pressures during normoxia and tracheal occlusion were lower in end-stage SOD1G93A mutant (closed bars) versus wild-type rats (p<0.05). *p<0.05 Wild type vs. Mutant. 1p<0.05 compared to normoxia. 2p<0.05 compared to 10.5% O2-7% CO2. 3p<0.05 compared to augmented breath.
Respiratory and Augmented Breath Frequencies during MCS in Mutants
During Nx, respiratory and AB frequencies were comparable (p>0.05). However, during MCS, respiratory frequency in MT was ~20% lower than WT rats (p<0.05; Figure 6A). On the other hand, ABs frequency was increased ~70% (p<0.05; Figure 6B).
Figure 6.

Respiratory and augmented breath frequencies were comparable between WT (open bars) and end-stage SOD1G93A rats (closed bars) during normoxia. However, during maximum chemoreceptor stimulation (10.5% O2-7% CO2), respiratory burst and augmented breath frequencies were significantly higher in MT rats (p<0.05). *p<0.05 Wild type vs. Mutant. 1p<0.05 compared to normoxia.
Discussion
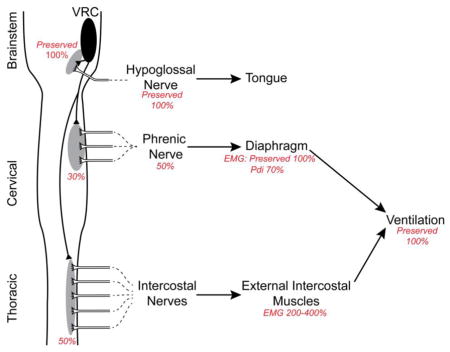
Since breathing is essential for life, powerful mechanisms of compensatory plasticity exist that compensate for life challenges, including neuromuscular diseases (Mitchell and Johnson, 2003; Mitchell, 2007; Johnson and Mitchell, 2013). Compensation may occur in sensory, central integrating and/or motor components of respiratory control. In the motor component, compensation may occur via: 1) amplified transmission of central input to the phrenic nerve activity at the spinal cord; 2) amplified transmission of phrenic nerve activity to the diaphragm activity; and/or 3) enhanced recruitment of accessory respiratory motor pools (Figure 7; Johnson and Mitchell, 2013). Spinal plasticity is demonstrated by the modest reduction in phrenic activity (~50–60% of normal) despite much greater losses of phrenic motor neurons (~20% spared) in end-stage SOD1G93A rats (Nichols et al., 2013a; Nichols and Mitchell, 2016). Here, EMG and Pdi measurements were used to demonstrate: 1) preserved diaphragm electrical activity despite reduced phrenic activity, demonstrating amplified transmission of electrical activity from phrenic nerve to the diaphragm muscle; 2) diminished diaphragm force generation and relative contribution to respiratory efforts despite normal electrical activity; and 3) increased contributions from inspiratory intercostal muscles. Collectively, these adjustments preserve breathing ability despite remarkable respiratory motor neuron death and diaphragm dysfunction.
Figure 7.

Schematic of postulated three-stage compensatory respiratory plasticity in end-stage SOD1G93A rats; the figure is based on current and previous findings (Nichols et al., 2013a; Nichols et al., 2014; Nichols and Mitchell, 2016; Nichols et al., 2015). Phrenic motor neuron death is compensated by two distinct mechanisms: 1) spinal synaptic plasticity, amplifying translation of descending respiratory drive into the activity of the spared phrenic motor neurons; and 2) amplification of phrenic-diaphragm electrical transmission, preserving diaphragm electrical activity via unknown mechanisms that may include motor neuron sprouting and/or other mechanisms of enhanced neuromuscular junction transmission. Because of diaphragm dysfunction (reflected in a reduced Pdi), the third step is to increase the relative contributions of accessory inspiratory muscles, despite (lesser) intercostal motor neuron death. In concert, these forms of plasticity preserve breathing ability.
Significant Respiratory Motor Neuron Loss in Late-Stage SOD1G93A rats
In the female rats studied here, we confirmed respiratory motor neuron death as found in end-stage male SOD1G93A rats (Nichols et al., 2013a; Nichols et al., 2014; Nichols et al., 2015). The number of surviving phrenic motor neurons in females (~29%; Figure 1) was slightly higher than previous estimates from male SOD1G93A rats at a similar end-stage (~20%), consistent with the somewhat slower disease progression reported in females (Suzuki et al., 2007). On the other hand, thoracic motor neuron loss at T5 was slightly higher in the females studied here (54%; Figure 1) versus previous studies on males (~40%; Nichols et al., 2013). Female SOD1G93A rats reached end-stage at a slightly older age (172 days) versus female and male SOD1G93A rats studied previously (162 days; (Nichols and Mitchell, 2016; Suzuki et al., 2007), most likely reflecting diminished copy number in successive generations of SOD1G93A rats.
Preserved Diaphragm Muscle EMG Activity Despite Phrenic Motor Neuron Loss
End-stage SOD1G93A rats maintain breathing capacity during room air breathing and MCS, despite ~80% phrenic motor neuron loss (Nichols et al., 2013a; Nichols et al., 2014). At this same end-stage, phrenic nerve activity of MT rats is reduced by only 45–50% during MCS, suggesting at least some compensatory plasticity in synaptic inputs to spared phrenic motor neurons, amplifying their individual activity (Nichols et al., 2013a; Nichols and Mitchell, 2016; Nichols et al., 2013b). Here, we demonstrate additional compensatory plasticity in the translation from spared phrenic motor neurons to diaphragm EMG activity in SOD1G93A rats (Figures 2–4); this observation is consistent with improved transmission of electrical activity from phrenic nerve to the diaphragm muscle. One potential mechanism of enhanced electrical transmission is an increase in motor unit innervation ratio (i.e., number of muscle fibers innervated per spared motor neuron). Increased motor unit innervation ratio is possible via axon terminal sprouting and re-innervation of motor end plates vacated by degenerating motor neurons (Schaefer et al., 2005). In tibialis anterior muscle of pre-symptomatic SOD1G93A mice, a 44% increase in the innervation ratio was reported (Hegedus et al., 2008). The motor unit innervation ratio may further increase with more advanced ALS. With an increasing innervation ratio (i.e. increased motor unit size), more muscle fiber action potentials will be represented in a single motor unit action potential. Therefore, the contribution of each motor unit to the compound EMG amplitude would be increased.
We also used EMG to evaluate individual accessory respiratory muscle activities, particularly the T2 and T5 EICs. There are no published data concerning the impact of motor neuron disease on EIC nerve activity. In studies of neuronal activity, the muscles must be paralyzed, precluding concurrent EMG assessment. Since diaphragm paralysis augments inspiratory intercostal muscle activity (De Troyer and Farkas, 1989; De Troyer and Kelly, 1982; Ninane et al., 1989; Nochomovitz et al., 1981), assessment of inspiratory intercostal function in vivo requires non-paralyzed preparations. In contrast to nerve recordings, EMGs are also affected by neuromuscular transmission, including changes in the neuromuscular junction due to sprouting, neuromuscular junction strength, and muscle fiber properties. Since we investigated phrenic activity in multiple previous studies on end-stage SOD1G93A rats (Nichols et al., 2013a; Nichols et al., 2014), the EMG recordings reported here allowed us to make inferences about changes in the transmission of nerve electrical activity to muscle activity. Further studies are necessary to assess contributions of each component to preserving diaphragm activity.
Reduced Transdiaphragmatic Pressure
Although diaphragm EMG activity was preserved in SOD1G93A rats, both Pdi and ΔPeso/ΔPgas ratio decreased, suggesting reduced contribution of diaphragm muscle to ventilatory and airway protective motor functions. This reduction is most likely due to diaphragm weakness (Figure 5 and Table 1). Mechanisms underlying persistent diaphragm weakness in this case have not been elucidated. Diaphragm weakness may result from reduction in muscle force generating capacity, or in other complications such as changes in chest wall mechanics. In pre-symptomatic SOD1G93A mice, force per muscle fiber decreased ~60% versus WT mice (Hegedus et al., 2008). One potential mechanism for this drop in muscle fiber force may be muscle fiber atrophy during the process of denervation and reinnervation (Atkin et al., 2005; Hegedus et al., 2008). In end stage SOD1G93A mice, diameters of slow- and fast-twitch muscle fibers are ~15% and ~35% smaller (Atkin et al., 2005). Moreover, maximum force per cross-sectional area elicited by Ca2+ in skinned flexor digitorum longus muscle fibers (fast-twitch) was reduced 74% (Atkin et al., 2005). It is not currently known how these changes occur in rat models of ALS. However, it is likely that similar pathologies occur in rats. Thus, the same level of muscle activation elicits reduced diaphragm force and Pdi, necessitating increased contributions from accessory respiratory muscles to preserve breathing capacity.
Table 1.
ΔPeso/ΔPgas ratio: Ratio of esophageal pressure to the gastric pressure during normoxia and sustained airway occlusion.
p<0.05 compared to wild type,
p<0.05 compared to normoxia)
Enhanced External Intercostal Muscle Recruitment
Our findings suggest increased EIC involvement (Figures 2–4) compensates for emerging diaphragm dysfunction, especially during normoxia. EIC EMG activity during MCS was only slightly increased since it was already near-maximal values when breathing room air (~80%; Figures 3 & 4). Our findings are in parallel with previous studies showing that accessory muscle recruitment is a distinct compensatory response utilized during chemoreflex activation (Megirian et al., 1987), physical exercise or pathological conditions such as diaphragm paralysis (Brichant and De Troyer, 1997; Katagiri et al., 1994; Maskrey et al., 1990; Ninane et al., 1989; Sherrey and Megirian, 1990; Teitelbaum et al., 1993). In ALS patients, accessory respiratory muscle recruitment may compensate for loss of diaphragm contractility, such as the sternocleidomastoid muscle (Arnulf et al., 2000). Importantly, accessory respiratory muscle weakness is associated with a poor prognosis in ALS patients; for example, sternocleidomastoid muscle weakness in ALS patients is associated with lower maximum inspiratory and sniff pressures (Pinto and de Carvalho, 2008). Indeed, neck muscle weakness is the most significant prognostic factor of the need for tracheostomy and positive pressure ventilation or patient death (Nakamura et al., 2013).
Overview of Compensatory Mechanisms in SOD1G93A Rats
Johnson and Mitchell (2013) suggested that compensatory respiratory plasticity in neuromuscular diseases that compromise breathing ability arises from a suite of mechanisms, each partially contributing to preservation of breathing capacity including: 1) spinal synaptic plasticity onto spared respiratory motor neurons, amplifying their relative contribution to respiratory muscle contractions and breathing; 2) neuromuscular junction plasticity via motor neuron terminal sprouting, increasing motor unit innervation ratio and enhancing neuromuscular transmission; and 3) shifting respiratory muscle utilization from more affected to less affected respiratory motor pools (e.g. enhanced accessory inspiratory muscle function). Figure 7 summarizes our current understanding of mechanisms compensating for respiratory motor neuron death in end-stage SOD1G93A rats, incorporating information from the present study with previous results from our group (Nichols et al., 2013a; Nichols et al., 2014; Nichols and Mitchell, 2016; Nichols et al., 2015). All three mechanisms outlined by Johnson and Mitchell (2013) appear to compensate for phrenic motor neuron loss (>70%), including: 1) spinal plasticity, amplifying the translation of descending respiratory drive to the nerve activity in spared phrenic motor neurons (Romer et al., 2017); 2) further amplification of residual respiratory nerve activity to preserve EMG electrical activity, possibly via motor neuron sprouting and/or enhanced neuromuscular junction transmission; and 3) persistent deficits in Pdi are compensated by increased inspiratory intercostal muscle contributions. We cannot comment on the roles of other accessory muscles in SOD1G93A rats such as the sternocleidomastoid or scalenus muscles at this time.
Methods of investigation used thus far do not allow us to make conclusions regarding the respective contributions of motor neuron sprouting vs other mechanisms of enhanced neuromuscular transmission. Further studies are needed to understand these mechanisms as well as the diaphragm muscle pathology associated with ALS.
Similar Compensatory Mechanisms in Multiple Neuromuscular Disorders
Utilization of multiple strategies to compensate for profound loss of nerve/muscle function is critical in many neuromuscular disorders that compromise breathing. Eventually, these compensatory strategies will be exhausted, leading to decompensation, ventilator dependence and/or death. Indeed, the most common cause of death in neuromuscular disorders that compromise movement is respiratory failure. Despite its importance to preserving breathing, little attention has been devoted to understanding how the respiratory system compensates for severe pathology, or what limits that compensation process (Johnson and Mitchell, 2013; Mitchell, 2007). In the neuromuscular disorder Pompe Disease both respiratory motor neurons and muscles are compromised (DeRuisseau et al., 2009). Patients with the worst respiratory function are those with the most severe intercostal muscle impairment (Carlier et al., 2011). Similarly, in Duchenne muscular dystrophy, diaphragm weakness is associated with hypoventilation during REM sleep, when intercostal muscles experience atonia (Barbé et al., 1994). After spinal contusion injuries in rats, muscle fiber reinnervation increases the motor unit innervation ratio ~2 several weeks post-injury (Nicaise et al., 2012). Collectively, these findings are consistent with the idea that common mechanisms of compensatory plasticity preserve breathing capacity despite impressive neural (motor neuron) and muscle pathology in diverse neuromuscular disorders that compromise movement (Johnson and Mitchell, 2013).
Cellular Mechanisms of Compensatory Respiratory Plasticity
Cellular mechanisms giving rise to compensatory respiratory plasticity are not known. In end-stage SOD1G93A rats, treatments expected to block known mechanisms of respiratory plasticity have little impact on breathing capacity, including: 1) systemic serotonin depletion or receptor antagonism; 2) adenosine 2A receptor inhibition; 3) NADPH oxidase inhibition; and 4) treatment combinations for 3 days (Nichols et al., 2014). However, once compensatory plasticity is established, it may not be possible to block the process with these treatments (i.e. they induce but not maintain plasticity).
Structural and/or functional plasticity may be induced during pre-symptomatic phases of disease progression. For example, in pre-symptomatic SOD1G93A mice, a 44% increase in motor unit innervation ratio was observed in tibialis anterior motor units (Hegedus et al., 2008). Therefore, compensatory plasticity in ALS rats may be a gradual process, requiring time-dependent assessments of suspected mechanisms throughout the life span of SOD1G93A rats.
Highlights.
Despite significant (~70%) phrenic motor neuron loss and decreased phrenic nerve activity, SOD1G93A rats preserve their ventilatory capacity until late disease stage.
Diaphragm electrical activity is maintained suggesting amplified transmission of phrenic nerve activity to the diaphragm activity.
Diaphragm muscle dysfunction is manifested via decreased transdiaphragmatic pressure and reduced contribution from diaphragm muscle relative to thoracic muscles to breathing.
Diaphragm muscle dysfunction is compensated by enhanced external intercostal activity.
Collectively, these mechanisms of compensatory plasticity preserve breathing ability despite remarkable respiratory motor neuron death and diaphragm dysfunction.
Acknowledgments
Supported by NIH 69064 (GSM), K99/R00 (NN), and HL119606 (GSM). The authors thank Kalen Nichols for assistance with blood-gas analysis, and Anna Emery and Jonathan Van Dyke for maintenance and genotyping of the ALS colony.
Footnotes
Author Contributions:
Conception and design of research: YBS, NLN, GSM
Performed experiments and analyzed data: YBS, NLN, MNK, ORH, IS
Interpreted results of experiments: YBS, IS, GSM
Prepared figures and drafted manuscript: YBS, NLN, GSM
Approved final version of manuscript: YBS, NLN, MNK, ORH, IS, GSM
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnulf I, Similowski T, Salachas F, Garma L, Mehiri S, Attali V, Behin-Bellhesen V, Meininger V, Derenne JP. Sleep disorders and diaphragmatic function in patients with amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2000;161:849–856. doi: 10.1164/ajrccm.161.3.9805008. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Scott RL, West JM, Lopes E, Quah AK, Cheema SS. Properties of slow- and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul Disord. 2005;15:377–388. doi: 10.1016/j.nmd.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Barbé F, Quera-Salva MA, McCann C, Gajdos P, Raphael JC, de Lattre J, Agustí AG. Sleep-related respiratory disturbances in patients with Duchenne muscular dystrophy. Eur Respir J. 1994;7:1403–1408. doi: 10.1183/09031936.94.07081403. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Gestreau C, Vinit S, Stamegna JC, Kastner A, Gauthier P. Specific and artifactual labeling in the rat spinal cord and medulla after injection of monosynaptic retrograde tracers into the diaphragm. Neurosci Lett. 2007;417:206–211. doi: 10.1016/j.neulet.2007.02.047. [DOI] [PubMed] [Google Scholar]
- Brichant JF, De Troyer A. On the intercostal muscle compensation for diaphragmatic paralysis in the dog. J Physiol. 1997;500(Pt 1):245–253. doi: 10.1113/jphysiol.1997.sp022014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier RY, Laforet P, Wary C, Mompoint D, Laloui K, Pellegrini N, Annane D, Carlier PG, Orlikowski D. Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: Involvement patterns. Neuromuscul Disord. 2011;21:791–799. doi: 10.1016/j.nmd.2011.06.748. [DOI] [PubMed] [Google Scholar]
- De Troyer A, Farkas GA. Inspiratory function of the levator costae and external intercostal muscles in the dog. J Appl Physiol (1985) 1989;67:2614–2621. doi: 10.1152/jappl.1989.67.6.2614. [DOI] [PubMed] [Google Scholar]
- De Troyer A, Kelly S. Chest wall mechanics in dogs with acute diaphragm paralysis. J Appl Physiol Respir Environ Exerc Physiol. 1982;53:373–379. doi: 10.1152/jappl.1982.53.2.373. [DOI] [PubMed] [Google Scholar]
- DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Mah C, Reier PJ, Byrne BJ. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci U S A. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuglevand AJ, Winter DA, Patla AE. Models of recruitment and rate coding organization in motor-unit pools. J Neurophysiol. 1993;70:2470–2488. doi: 10.1152/jn.1993.70.6.2470. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Mitchell GS. Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respir Physiol Neurobiol. 2013;189:419–428. doi: 10.1016/j.resp.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri M, Young RN, Platt RS, Kieser TM, Easton PA. Respiratory muscle compensation for unilateral or bilateral hemidiaphragm paralysis in awake canines. J Appl Physiol (1985) 1994;77:1972–1982. doi: 10.1152/jappl.1994.77.4.1972. [DOI] [PubMed] [Google Scholar]
- Lawrence JH, De Luca CJ. Myoelectric signal versus force relationship in different human muscles. J Appl Physiol Respir Environ Exerc Physiol. 1983;54:1653–1659. doi: 10.1152/jappl.1983.54.6.1653. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklem PT, Gross D, Grassino GA, Roussos C. Partitioning of inspiratory pressure swings between diaphragm and intercostal/accessory muscles. J Appl Physiol Respir Environ Exerc Physiol. 1978;44:200–208. doi: 10.1152/jappl.1978.44.2.200. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB, Hurtado-Palomino JN, Zhan WZ, Sieck GC. Chronic assessment of diaphragm muscle EMG activity across motor behaviors. Respir Physiol Neurobiol. 2011;177:176–182. doi: 10.1016/j.resp.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol. 2010;173:101–106. doi: 10.1016/j.resp.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskrey M, Megirian D, Sherrey JH. Alteration in breathing of the awake rat after laryngeal and diaphragmatic muscle paralysis. Respir Physiol. 1990;81:203–212. doi: 10.1016/0034-5687(90)90046-2. [DOI] [PubMed] [Google Scholar]
- Megirian D, Pollard MJ, Sherrey JH. The labile respiratory activity of ribcage muscles of the rat during sleep. J Physiol. 1987;389:99–110. doi: 10.1113/jphysiol.1987.sp016648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic Basis for Respiratory Control Disorders. Springer Publishing Company; New York: 2007. pp. 291–311. [Google Scholar]
- Nakamura R, Atsuta N, Watanabe H, Hirakawa A, Ito M, Senda J, Katsuno M, Tanaka F, Izumi Y, Morita M, Ogaki K, Taniguchi A, Aiba I, Mizoguchi K, Okamoto K, Hasegawa K, Aoki M, Kawata A, Abe K, Oda M, Konagaya M, Imai T, Nakagawa M, Tsuji S, Kaji R, Nakano I, Sobue G. Neck weakness is a potent prognostic factor in sporadic amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2013;84:1365–1371. doi: 10.1136/jnnp-2013-306020. [DOI] [PubMed] [Google Scholar]
- Nicaise C, Hala TJ, Frank DM, Parker JL, Authelet M, Leroy K, Brion JP, Wright MC, Lepore AC. Phrenic motor neuron degeneration compromises phrenic axonal circuitry and diaphragm activity in a unilateral cervical contusion model of spinal cord injury. Exp Neurol. 2012;235:539–552. doi: 10.1016/j.expneurol.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013a;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Johnson RA, Satriotomo I, Mitchell GS. Neither serotonin nor adenosine-dependent mechanisms preserve ventilatory capacity in ALS rats. Respir Physiol Neurobiol. 2014;197:19–28. doi: 10.1016/j.resp.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Mitchell GS. Quantitative assessment of integrated phrenic nerve activity. Respir Physiol Neurobiol. 2016;226:81–86. doi: 10.1016/j.resp.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Satriotomo I, Harrigan DJ, Mitchell GS. Acute intermittent hypoxia induced phrenic long-term facilitation despite increased SOD1 expression in a rat model of ALS. Exp Neurol. 2015;273:138–150. doi: 10.1016/j.expneurol.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Van Dyke J, Nashold L, Satriotomo I, Suzuki M, Mitchell GS. Ventilatory control in ALS. Respir Physiol Neurobiol. 2013b;189:429–437. doi: 10.1016/j.resp.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninane V, Farkas GA, Baer R, de Troyer A. Mechanism of rib cage inspiratory muscle recruitment in diaphragmatic paralysis. Am Rev Respir Dis. 1989;139:146–149. doi: 10.1164/ajrccm/139.1.146. [DOI] [PubMed] [Google Scholar]
- Nochomovitz ML, Goldman M, Mitra J, Cherniack NS. Respiratory responses in reversible diaphragm paralysis. J Appl Physiol Respir Environ Exerc Physiol. 1981;51:1150–1156. doi: 10.1152/jappl.1981.51.5.1150. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pinto S, de Carvalho M. Motor responses of the sternocleidomastoid muscle in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2008;38:1312–1317. doi: 10.1002/mus.21109. [DOI] [PubMed] [Google Scholar]
- Romer SH, Seedle K, Turner SM, Li J, Baccei ML, Crone SA. Accessory respiratory muscles enhance ventilation in ALS model mice and are activated by excitatory V2a neurons. Exp Neurol. 2017;287:192–204. doi: 10.1016/j.expneurol.2016.05.033. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Seven YB, Mantilla CB, Sieck GC. Recruitment of rat diaphragm motor units across motor behaviors with different levels of diaphragm activation. J Appl Physiol (1985) 2014;117:1308–1316. doi: 10.1152/japplphysiol.01395.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrey JH, Megirian D. After phrenicotomy the rat alters the output of the remaining respiratory muscles without changing its sleep-waking pattern. Respir Physiol. 1990;81:213–225. doi: 10.1016/0034-5687(90)90047-3. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Tork C, Shelley B, McHugh J, Wallace K, Klein SM, Lindstrom MJ, Svendsen CN. Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph Lateral Scler. 2007;8:20–25. doi: 10.1080/17482960600982447. [DOI] [PubMed] [Google Scholar]
- Tankersley CG, Haenggeli C, Rothstein JD. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J Appl Physiol (1985) 2007;102:926–932. doi: 10.1152/japplphysiol.00193.2006. [DOI] [PubMed] [Google Scholar]
- Teitelbaum J, Borel CO, Magder S, Traystman RJ, Hussain SN. Effect of selective diaphragmatic paralysis on the inspiratory motor drive. J Appl Physiol (1985) 1993;74:2261–2268. doi: 10.1152/jappl.1993.74.5.2261. [DOI] [PubMed] [Google Scholar]
- Watson C, Paxinos G, Kayalioglu G. The Spinal Cord: A Christopher and Dana Reeve Foundation Text and Atlas. 1. Academic Press; 2009. [Google Scholar]