

Abstract
Acute chest syndrome (ACS) of sickle cell disease (SCD) is characterized pathologically by vaso-occlusive processes that result from abnormal interactions between sickle red blood cells (RBCs), white blood cells (WBCs) and/or platelets, and the vascular endothelium. One potential mechanism of vascular damage in ACS is by generation of oxygen-related molecules, such as superoxide (O2-), hydrogen peroxide (H2O2), peroxynitrite (ONOO-), and the hydroxyl (•OH) radical. The present review summarizes the evidence for alterations in oxidant stress during ACS of SCD, and the potential contributions of RBCs, WBCs and the vascular endothelium to this process.
Keywords: acute chest syndrome (ACS), endothelium, hemoglobin, nitric oxide (NO), oxidant stress
Introduction
ACS is an important cause of morbidity and mortality in SCD, occurring in up to 45% of patients and recurring in up to 80% of those afflicted [1,2]. The hallmark pathologic event during ACS is vaso-occlusion, the etiology of which is probably multifactorial. One of the mechanisms responsible for vaso-occlusion is abnormal adherence of sickle RBCs, WBCs, and/or platelets to the vascular endothelium. Although the factors that lead to increased cellular adhesion and vascular damage are unclear, one possible explanation is that, during local vaso-occlusion, areas of ischemia/reperfusion develop. During periods of reperfusion, there is increased production of oxidizing molecules such as O2-, H2O2, •OH radical and ONOO - [3]. These compounds lead to the activation of second messengers such as nuclear factor-κB (NF-κB), resulting in upregulation of endothelial adhesion molecules. Adhesion molecules, such as vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1, facilitate binding of sickle RBCs and WBCs to the vascular endothelium, and thus may play a role in the development of vaso-occlusion [4,5,6,7]. In addition, oxygen-related species can directly injure the endothelium by peroxidation of the lipid membrane and/or DNA fragmentation, potentially leading to cellular apoptosis [8,9].
There is a growing body of literature that suggests that patients with SCD are subjected to increased oxidative stress, particularly during vaso-occlusive crises (VOCs) and ACS. Osarogiagbon et al [10] demonstrated that transgenic sickle cell mice had higher levels at baseline of markers of oxidative stress, such as ethane excretion and •OH radical generation, than did their normal counterparts. During exposure to hypoxia, this sickle cell mouse exhibits evidence of ischemia/reperfusion injury, which is characterized by increased oxygen radical formation, and leukocyte adherence and emigration [5,10]. In addition, ONOO- formation occurs within the renal tubular epithelium with associated cellular apoptosis [11].
In human studies [12,13], levels of thiobarbituric acid-reactive substances (TBARSs) indicated that lipid peroxidation occurs in sickle erythrocytes at baseline. In addition, we observed a ninefold increase in the plasma levels of F2 isoprostanes, a stable marker of lipid peroxidation, in the plasma of ACS patients as compared with that of normal volunteers (Klings ES et al, unpublished data). These findings suggest, both in humans and in mouse models of SCD, that there is increased oxidative burden and that alterations in the redox state may play a role in the development of vaso-occlusion. The source of oxygen radicals in these patients is probably multifactorial, because RBCs, WBCs, and the endothelium could each contribute to their aberrant metabolism. We review the role of each of these cell types in the generation of oxygen radicals, and the effects that these molecules have on cellular metabolism in SCD.
Role of sickle red blood cells in oxidant production
Hemoglobin S as a source of oxidants
The RBC is an important source of oxygen-related radicals in SCD. Hebbel et al [14], in 1982, demonstrated that sickle RBCs produce greater quantities of O2-, H2O2 and •OH than do normal RBCs. Additionally, sickle RBCs at baseline exhibit increased levels of TBARSs [12,13], suggesting that they are targets for oxidative stress. Although an evaluation of oxidant production by RBCs has not been conducted in SCD patients with ACS, data from mouse models of ACS [5,10] suggest that ischemia/reperfusion injury can occur in this setting.
Within RBCs, one of the mechanisms of O2- formation is via the deoxygenation of hemoglobin. During deoxygenation, there is a transfer of electrons between Fe and O2, leading to the production of O2-. Auto-oxidation of hemoglobin, which occurs to a small extent physiologically, leads to the production of methemoglobin and O2- [15,16]. Because hemoglobin S auto-oxidizes at 1.7 times the rate of hemoglobin A, SCD patients may have a higher propensity for oxidant production [12]. Once hemoglobin is subjected to oxidant damage, it denatures and precipitates; these events increase its susceptibility to auto-oxidation [17]. Because of these findings, it has been hypothesized that the production of oxidants by RBCs would be greater than that observed at baseline.
Effects of oxidant production on red blood cells
Within the RBC, one of the targets of oxidant damage is the plasma membrane. In the presence of an O2- generating system Fe(III) is reduced to Fe(II), with subsequent formation of •OH from H2O2 [16]. The hydroxyl radical oxidizes unsaturated esterified membrane lipids, resulting in changes in fluidity of the bilayer. Additionally, there is increased ion permeability, inactivation of membrane enzymes and receptors, and covalent cross-linking of lipid and protein membrane constituents [18]. Membrane lipid peroxidation, measured by TBARS production, is elevated in sickle RBCs at baseline [13,19,20]. In addition to being markers for oxidative stress, lipid peroxidation products such as malondialdehyde have additional toxic effects because of their ability to react with proteins, nucleic acids, and lipids [16].
Once molecules such as O2- and H2O2 are formed, they are metabolized by antioxidant enzyme systems, such as superoxide dismutase, catalase, and glutathione peroxidase (GPx), to O2 and H2O (Fig. 1) [16,21,22]. Schacter et al [23] and Gryglewksi et al [24] demonstrated that superoxide dismutase and catalase levels and activity are diminished in sickle RBCs at baseline; other investigators have found that GPx activity is reduced [25]. Together, these findings suggest that oxidants formed by sickle RBCs are less likely to be removed effectively. The activities of these antioxidant enzyme systems have never been directly studied during ACS, however. Nevertheless, it is hypothesized that a decrease in antioxidant defense mechanisms combined with increased production of oxygen-related molecules in sickle RBCs, at baseline and particularly during crisis, is responsible for the increased oxidant burden observed in these erythrocytes.
Figure 1.
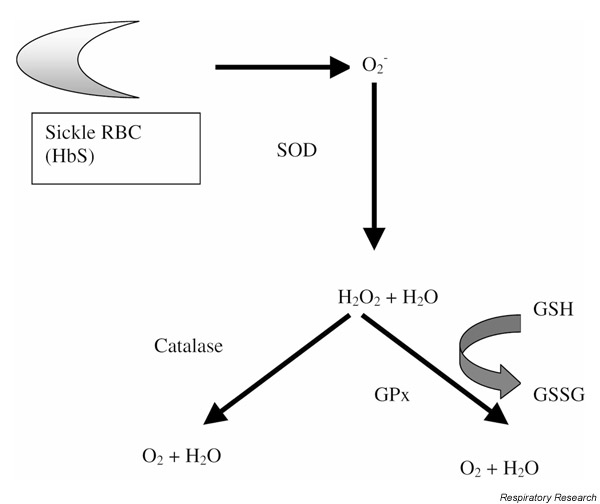
Mechanisms of oxidant production in sickle RBCs. Sickle RBCs, through the auto-oxidation of hemoglobin (Hb)S, produce O2-, which is metabolized to H2O2 by superoxide dismutase (SOD). H2O2 is then metabolized to O2 and H2O by catalase and GPx. Deficiencies in SOD, catalase, and GPx in sickle RBCs lead to increased O2- and H2O2 production. GSSG, oxidized glutathione.
Role of white blood cells in oxidant production in acute chest syndrome
Although SCD is a genetic disorder of the hemoglobin molecule, there is a growing body of evidence that suggests that WBCs, particularly polymorphonuclear leukocytes (PMNs), are abnormal in this disease as well. Peripheral WBC counts are elevated in VOC and ACS, and WBC counts greater than 15,000 are associated with increased mortality [26,27]. Additionally, sickle PMNs at baseline have increased expression of the high-affinity Fc receptor CD64; this receptor is even more pronounced during crisis [28,29]. In addition to being a marker of PMN activation, CD64 appears to play a role in the adherence of sickle PMNs to the vascular endothelium [28]. When PMNs become activated or adherent to the endothelium, they can produce oxidants such as O2- and H2O2 through the activation of enzymes such as myeloperoxidase [30]. In a transgenic sickle cell mouse model, induction of acute lung injury was associated with increased myeloperoxidase activity as compared with wild-type mice, suggesting that the development of ACS in SCD may be accompanied by PMN infiltration into the lungs [31].
SCD patients have increased levels of the PMN chemokine IL-8 during VOC, suggesting that during crisis there is increased PMN recruitment [32]. Additionally, PMNs from SCD patients at baseline have increased myeloperoxidase activity [33]. Although not directly measured in human ACS, demonstration of increased myeloperoxidase activity in sickle WBCs at baseline and in transgenic mice during crisis suggests that this enzyme may play a role in oxidant generation by PMNs in ACS. In addition, sickle PMNs generate nitric oxide (NO) and O2- during crisis [34], and these molecules can react to form ONOO-. These data suggest that, in addition to increased activation, PMNs in SCD patients may have a greater propensity toward oxidant generation. Finally, when incubated with sickle RBCs, PMNs exhibit increased adherence to these RBCs with a resultant increase in production of oxidants, as measured by 2',7'-dichlorofluorescein diacetate fluorescence [35].
Role of the endothelium in oxidant production during acute chest syndrome
In addition to being a potential target for oxidative damage, the vascular endothelium may play a primary role in the generation of oxidants. Sultana et al [6] demonstrated that coincubation of sickle RBCs with human umbilical vein endothelial cells resulted in lipid peroxidation, as measured by TBARSs, and transendothelial migration of monocytes. Additionally, we demonstrated that coincubation of plasma from ACS patients with bovine pulmonary artery endothelial cells resulted in formation of ONOO- within the vascular endothelium [36]. We also demonstrated [36] that there are decreases in the antioxidant thiols and glutathione reductase system in bovine pulmonary artery endothelial cells after coincubation with plasma from ACS patients. These findings suggest that the endothelium is more susceptible to oxidant-related damage during ACS.
These in vitro findings may be biologically relevant, because endothelial production of oxidants has been demonstrated in vivo using a transgenic mouse model of SCD [5]; exposure of these mice to hypoxia resulted in formation of oxidants within the vascular endothelium of the cremaster muscle. Although endothelial production of oxidants is difficult to measure directly in human disease, these in vitro and in vivo data suggest that the endothelium of the pulmonary arteries may be involved in oxidant production during ACS.
Vascular endothelium as a target for oxidant damage
Although there appears to be increased oxidant production and decreased antioxidant defense mechanisms in SCD, how this relates to the pathophysiology of ACS has not been elucidated. One mechanism responsible for vaso-occlusion is the adhesion of sickle RBCs to the vascular endothelium. It is hypothesized that, in part, this occurs secondary to endothelial damage and increased adhesion molecule expression resulting from increased oxidant burden. The vascular endothelium appears uniquely sensitive to damage from oxidizing molecules produced during VOC and ACS. One mechanism by which this may occur is via deactivation of NO (Fig. 2). Several studies have suggested that alterations in NO metabolism occur during SCD, particularly during VOC or ACS.
Figure 2.
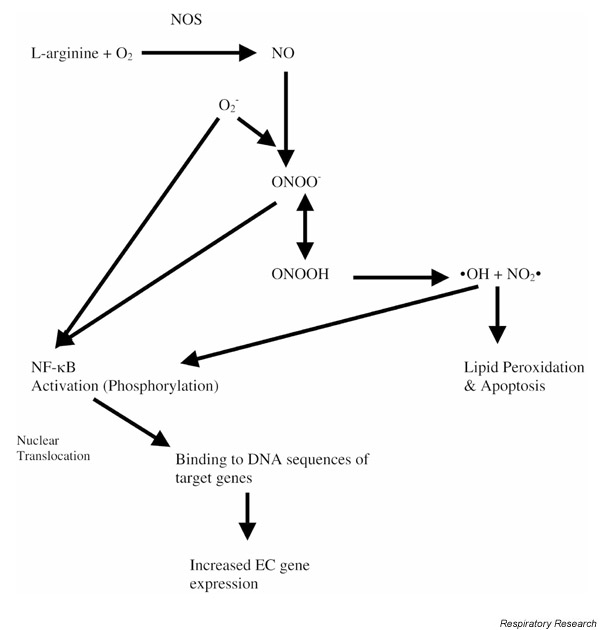
Mechanism by which free radicals alter NO bioavailability and endothelial cell biology. Under conditions of increased O2- production, NO preferentially forms ONOO-. Both O2- and ONOO- can alter endothelial cell (EC) gene expression via activation and nuclear translocation of second messengers such as NF-κB.
It was demonstrated that plasma NO levels are decreased in VOC and correlate with decreases in L-arginine [37] and increases in soluble VCAM-1 [38], a molecule that is implicated in adhesion of sickle RBCs to the endothelium. Rees et al [39] found that, compared with control individuals, plasma nitrite (NO2-) levels are elevated in SCD patients during crisis; however, these levels were not significantly different from those in SCD patients at baseline. Lopez and coworkers [40,41] demonstrated that, although initial NO levels in VOC patients correlate inversely with pain score, sequential values are not predictive of clinical course. Additionally, interactions between sickle RBCs and the endothelium lead to abnormal vascular responses to NO in SCD. In two models of SCD, aortic vascular strips failed to relax when stimulated with acetylcholine [42] and infusion of the nitric oxide synthase (NOS) inhibitor NG-nitro-L-arginine methyl ester resulted in decreased cerebral blood flow [43].
One possible explanation for the conflicting results regarding NO metabolism is that the NO that is produced is subsequently deactivated. NO, an endogenous vasodilator and inhibitor of platelet aggregation, is produced by a variety of cell types, including vascular endothelium, neurons, macrophages, and smooth muscle cells [44,45,46]. Additionally, by preventing metal catalyzed lipid oxidation, NO can act as an antioxidant [44]. It is formed from L-arginine by a family of enzymes termed the NOSs. Three isoforms of this enzyme exist [46,47]: constitutive neuronal NOS, endothelial NOS, and inducible NOS. Unfortunately, the beneficial actions of NO are often mitigated because of preferential shunting toward toxic metabolites such as NO2-, nitrate (NO3-), and ONOO- [47,48,49]. This occurs rapidly in the presence of oxygen and oxygen-related molecules such as O2- and H2O2; specifically, NO reacts with O2- rapidly to form ONOO-, which exists in equilibrium with peroxynitrous acid (ONOOH). Because the half-life of ONOO- is very short (approximately one second), most of its toxic effects occur via reaction products of ONOOH, specifically •OH and NO2•, molecules that are implicated in fatty acid oxidation and nitrosative stress via nitration of aromatic amino acids [50]. Production of oxygen radicals results in peroxidation of the lipid membrane, and may predispose the endothelial cell to apoptosis [9].
In addition to decreasing the bioavailability of NO, free radicals can contribute to vaso-occlusion through production of the potent vasoconstrictor endothelin-1. Exposure of cultured human endothelial cells to RBCs sickled in vitro results in a fourfold to eightfold induction of endothelin-1 mRNA [51]. Similarly, we demonstrated increased endothelin-1 mRNA and protein levels in endothelial cells exposed to plasma from ACS patients as compared with those exposed to plasma from SCD patients at baseline [52]. Endothelin-1 transcription may be induced by activation of the redox-sensitive NF-κB or activating protein (AP)-1 by reactive oxygen radicals generated during ACS [12]. These findings suggest that, in addition to direct toxicity to the vascular endothelium, reactive oxygen species may contribute to vaso-occlusion through alteration in vascular tone.
Additionally, reactive oxygen species may act as second messengers to alter endothelial cell gene expression via activation of the redox-sensitive transcription factor NF-κB. In unstimulated cells, NF-κB is sequestered in an inactive state within the cytoplasm. When exposed to oxygen radicals NF-κB is activated by phosphorylation and translocates to the nucleus, where it affects gene expression [53]. Among the genes that are upregulated by NF-κB activity are those that encode the adhesion molecules VCAM-1 and ICAM-1, which can facilitate binding of sickle RBCs and WBCs to the endothelium. In this way, free radical generation in SCD may contribute to the propagation of vaso-occlusion.
Antioxidant defense mechanisms in sickle cell disease
Enzymatic antioxidants
There are several mechanisms by which aerobic organisms protect themselves from oxidative stress. On a cellular level, this occurs primarily through the actions of the enzymes superoxide dismutase, catalase, and GPx. As noted above, sickle RBCs at baseline are deficient in each of these enzyme systems [23,24,25]. Moreover, the reduced glutathione (GSH) level in sickle RBCs was approximately 50% lower than that observed in hemoglobin A RBCs [54]. Additionally, we demonstrated that, compared with plasma from normal volunteers, exposure to plasma from SCD patients at baseline decreases levels of GSH, which is an essential cofactor for GPx activity in cultured endothelial cells; there is a greater decrease in endothelial cell reduced GSH on exposure to plasma from ACS patients. Similarly, exposure to ACS plasma resulted in a decreased level of the endothelial cell antioxidant thiols [36]. These findings suggest that, in SCD patients, particularly during ACS, there is a decreased capacity to scavenge free radicals, making such persons more susceptible to oxidant-related damage.
Vitamins A, C, and E
Other cellular antioxidants include α-tocopherol (vitamin E), ascorbic acid (vitamin C), and β-carotene (vitamin A).α-Tocopherol and β-carotene scavenge free radicals to prevent lipid peroxidation [12]; of note, SCD patients have approximately a 40% reduction in plasma carotene levels and a 30% reduction in vitamin E levels [25,55]. Additionally, there is an inverse correlation between vitamin E levels and the percentage of irreversibly sickled RBCs [56].
Although it appears that vitamin E depletion may play a role in the development of vaso-occlusion, the effect of vitamin E supplementation is unclear. In two small prospective studies that evaluated the effect of vitamin E supplementation in SCD patients [57,58], there was a decrease in the number of irreversibly sickled cells, but this did not correlate with a reduction in the number of occurrences or severity of VOC. Measurement of ascorbic acid levels in SCD has produced conflicting results; several studies [54,59,60,61,62] have demonstrated reduced levels in the plasma and WBCs of SCD patients, but others have found no significant difference. This disparity may reflect differences in the populations studied, but makes it less likely that vitamin C represents an important antioxidant in this population. To date, no clinical trials have been conducted to evaluate the efficacy of either vitamin A or C supplementation in the SCD population.
Homocysteine
Homocysteine, a sulfur-containing amino acid that is produced during methionine metabolism, can produce reactive oxygen species via auto-oxidation [12]. Although no clear link with ACS has been demonstrated, hyperhomocysteinemia is associated with increased risk for cere-brovascular disease in SCD patients [63], suggesting an increased propensity toward vascular disease. Additionally, serum levels of key cofactors in homocysteine metabolism (ie folate, and vitamins B12 and B6) are depressed in SCD patients, suggesting that they are more prone to hyperhomocysteinemia. Further work is needed to elucidate the role of homocysteine in vaso-occlusion.
Conclusion
Reactive oxygen species may play an important role in the vascular dysfunction that is observed during ACS and VOC of SCD. Currently, however, there is little direct information available to confirm this hypothesis. In addition to being directly toxic to the endothelium via peroxidation of the lipid membrane, reactive oxygen species can upregulate expression of molecules such as VCAM-1, ICAM-1, and endothelin-1. The adhesion molecules VCAM-1 and ICAM-1 facilitate interaction between sickle RBCs and WBCs and the endothelium, thereby promoting vaso-occlusion. Endothelin-1 is a potent vasoconstrictor and an important mediator of vascular tone. By upregulating endothelin-1 expression and deactivating the vasodilator NO, oxygen radicals modulate vascular tone, and thereby could increase vaso-occlusion.
Although the genetic hemoglobinopathy of SCD is responsible for a percentage of the oxygen radicals that are produced, deficiencies in antioxidant defense mechanisms and the presence of other sites of oxidant production suggest that other genetic polymorphisms exist within the SCD population. It is possible that both genetic and dietary heterogeneity exist among SCD patients, and that this is in part responsible for the clinical variability in disease course. Further work is necessary to define more clearly the oxidant/antioxidant profile of SCD patients at baseline and during VOCs, including ACS, and to examine the clinical effect of pharmacologic intervention to reduce oxidant production in SCD. This will hopefully lead to a better understanding of the role of reactive oxygen species in the pathogenesis of vaso-occlusion.
Abbreviations
ACS = acute chest syndrome; GPx = glutathione peroxidase; GSH = reduced glutathione; ICAM = intercellular adhesion molecule; NF-κB = nuclear factor-κB; NO = nitric oxide; NOS = nitric oxide synthase; PMN = polymorphonuclear leukocyte; RBC = red blood cell; SCD = sickle cell disease; TBARS = thiobarbituric acid-reactive substance; VCAM = vascular cell adhesion molecule; VOC = vaso-occlusive crisis; WBC = white blood cell.
Acknowledgments
Acknowledgements
This work was funded by an American Lung Association Research Training Fellowship (RT-030-N; ESK) and by an American Heart Association Grant-In-Aid (0150155N; HWF).
References
- Johnson CS, Verdigem TD. Pulmonary complications of sickle cell disease. Semin Respir Med. 1988;9:287–296. [Google Scholar]
- Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, Nickerson B, Orringer E, McKie V, Bellevue R, Daeschner C, Manci EA. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med. 2000;342:1855–1865. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res. 2000;47:446–456. doi: 10.1016/S0008-6363(00)00078-X. [DOI] [PubMed] [Google Scholar]
- Gee BE, Platt OS. Sickle erythrocytes adhere to VCAM-1. Blood. 1995;85:268–274. [PubMed] [Google Scholar]
- Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle cell mice but not in normal mice. J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood. 1998;92:3924–3935. [PubMed] [Google Scholar]
- Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlman TH, Harlan JM. Adaptive responses of the endothelium to stress. J Surg Res. 2000;89:85–119. doi: 10.1006/jsre.1999.5801. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of nitric oxide and superoxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314–320. [PubMed] [Google Scholar]
- Bank N, Kiroycheva M, Ahmed F, Anthony GM, Fabry ME, Nagel RL, Singhal PC. Peroxynitrite formation and apoptosis in transgenic sickle cell mouse kidneys. Kidney Int. 1998;54:1520–1528. doi: 10.1046/j.1523-1755.1998.00148.x. [DOI] [PubMed] [Google Scholar]
- Aslan M, Thornley-Brown D, Freeman BA. Reactive species in sickle cell disease. Ann NY Acad Sci. 2000;899:375–391. doi: 10.1111/j.1749-6632.2000.tb06201.x. [DOI] [PubMed] [Google Scholar]
- Jain SK, Shohet SB. A novel phospholipid in irreversibly sickled cells: evidence for in vivo peroxidative membrane damage in sickle cell disease. Blood. 1984;63:362–367. [PubMed] [Google Scholar]
- Hebbel RP, Eaton JW, Balasingam M, Steinberg MH. Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Invest. 1982;70:1253–1259. doi: 10.1172/JCI110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell WR, Winterbourn CC, Rachmilewitz EA. Activated oxygen and haemolysis. Br J Haematol. 1975;30:259–264. doi: 10.1111/j.1365-2141.1975.tb00540.x. [DOI] [PubMed] [Google Scholar]
- Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–412. [PubMed] [Google Scholar]
- Chui D, Lubin B. Oxidative hemoglobin denaturation and RBC destruction: the effect of heme on red cell membranes. Semin Hematol. 1989;26:128–135. [PubMed] [Google Scholar]
- Buettner GR. The pecking order of free radicals and anti-oxidants: lipid peroxidation, α-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Evans CR, Omorphos SC, Baysal E. Sickle cell membranes and oxidative damage. Biochem J. 1986;237:265–269. doi: 10.1042/bj2370265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Nair RC. Superoxide dismutase, glutathione peroxidase, catalase and lipid peroxidation of normal and sickled erythrocytes. Br J Haematol. 1980;44:87–92. doi: 10.1111/j.1365-2141.1980.tb01186.x. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. Ann Rev Biochem. 1975;44:157–174. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- Cohen G, Hochstein P. Glutathione peroxidase: the primary agent for the elimination of H2O2 in erythrocytes. Biochemistry. 1963;2:1420–1430. doi: 10.1021/bi00906a038. [DOI] [PubMed] [Google Scholar]
- Schacter L, Warth JA, Gordon EM, Prasad A, Klein BL. Altered amount and activity of superoxide dismutase in sickle cell anemia. FASEB J. 1988;2:237–243. doi: 10.1096/fasebj.2.3.3350236. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdown of endothelial derived relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- Chui D, Lubin B. Abnormal vitamin E and glutathione peroxidase levels in sickle cell anemia. J Clin Med. 1979;94:542–548. [PubMed] [Google Scholar]
- Boggs DR, Hyde F, Srodes C. An unusual pattern of neutrophil kinetics in sickle cell anemia. Blood. 1973;41:59–65. [PubMed] [Google Scholar]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, Fishlock K, Brown KA. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91:266–274. [PubMed] [Google Scholar]
- Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66:411–415. doi: 10.1002/jlb.66.3.411. [DOI] [PubMed] [Google Scholar]
- Varani J, Ginsburg I, Schuger L, Gibbs DF, Bromberg J, Johnson KJ, Ryan US, Ward PA. Endothelial cell killing by neutrophils: synergistic interaction of oxygen products and proteases. Am J Pathol. 1989;135:435–438. [PMC free article] [PubMed] [Google Scholar]
- Hsu L, McDermott T, Brown L, Aguayo SM. Transgenic HbS mouse neutrophils in increased susceptibility to acute lung injury. Chest. 1999;116:92S. doi: 10.1378/chest.116.suppl_1.92S. [DOI] [PubMed] [Google Scholar]
- Duits AJ, Schnog JB, Lard LR, Saleh AW, Rojer RA. Elevated IL-8 levels during sickle cell crisis. Eur J Haematol. 1998;61:302–305. doi: 10.1111/j.1600-0609.1998.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Mohamed AO, Hashim MS, Nilsson UR, Venge P. Increased in vivo activation of neutrophils and complement in sickle cell disease. Am J Trop Med Hyg. 1993;49:799–803. doi: 10.4269/ajtmh.1993.49.799. [DOI] [PubMed] [Google Scholar]
- Dias-Da-Motta P, Arruda VR, Muscara MN, Saad ST, De Nucci G, Costa FF, Condino-Neto A. The release of nitric oxide and superoxide anion by neutrophils and mononuclear cells from patients with sickle cell anemia. Br J Haematol. 1996;93:333–340. doi: 10.1046/j.1365-2141.1996.4951036.x. [DOI] [PubMed] [Google Scholar]
- Hofstra TC, Kalra VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996;87:4440–4447. [PubMed] [Google Scholar]
- Hammerman SI, Klings ES, Hendra KP, Upchurch GR, Jr, Rishikof DC, Loscalzo J, Farber HW. Endothelial cell nitric oxide production in acute chest syndrome. Am J Physiol. 1999;277:H1579–H1592. doi: 10.1152/ajpheart.1999.277.4.H1579. [DOI] [PubMed] [Google Scholar]
- Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in sickle cell disease patients with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22:515–520. doi: 10.1097/00043426-200011000-00009. [DOI] [PubMed] [Google Scholar]
- Stuart MJ, Setty BN. Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood. 1999;94:1555–1560. [PubMed] [Google Scholar]
- Rees DC, Cervi P, Grimwade D, O'Driscoll A, Hamilton M, Parker NE, Porter JB. The metabolites of nitric oxide in sickle cell disease. Br J Haematol. 1995;91:834–837. doi: 10.1111/j.1365-2141.1995.tb05397.x. [DOI] [PubMed] [Google Scholar]
- Lopez BL, Barnett J, Ballas SK, Christopher TA, Davis-Moon L, Ma X. Nitric oxide metabolites in acute vasoocclusive crisis patients in the emergency department. Acad Emerg Med. 1996;3:1098–1103. doi: 10.1111/j.1553-2712.1996.tb03367.x. [DOI] [PubMed] [Google Scholar]
- Lopez BL, Davis-Moon L, Ballas SK, Ma XL. Sequential nitric oxide measurements during the emergency room treatment of acute vasoocclusive sickle cell crisis. Am J Hematol. 2000;64:15–19. doi: 10.1002/(SICI)1096-8652(200005)64:1<15::AID-AJH3>3.3.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Mosseri M, Bartlett-Pandite AN, Wenc K, Isner JM, Weinstein R. Inhibition of endothelium-dependent vasorelaxation by sickle erythrocytes. Am Heart J. 1993;126:338–346. doi: 10.1016/0002-8703(93)91049-k. [DOI] [PubMed] [Google Scholar]
- French JA, II, Kenny D, Scott JP, Hoffmann RG, Wood JD, Hudetz AG, Hillery CA. Mechanisms of stroke in sickle cell disease: sickle erythrocytes decrease cerebral blood flow in rats after nitric oxide inhibition. Blood. 1997;89:4591–4599. [PubMed] [Google Scholar]
- Rubbo H, Darley-Usmar V, Freeman BA. Nitric oxide regulation of tissue free radical injury. Chem Res Toxicol. 1996;9:809–820. doi: 10.1021/tx960037q. [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs EA. Endogenous nitric oxide: physiology, pathology, and clinical relevance. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- Mayer B, Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci. 1997;22:477–481. doi: 10.1016/S0968-0004(97)01147-X. [DOI] [PubMed] [Google Scholar]
- Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest. 1997;100:2146–2152. doi: 10.1172/JCI119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Lewis RS, Tamir S, Tannebaum SR, Deen WM. Kinetic analysis of the fate of nitric oxide synthesized by macrophages in vitro. J Biol Chem. 1995;270:29350–29355. doi: 10.1074/jbc.270.49.29350. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96:1145–1151. doi: 10.1172/JCI118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman SI, Kourembanas S, Conca TJ, Tucci M, Brauer M, Farber HW. Endothelin-1 production during the acute chest syndrome of sickle cell disease. Am J Respir Crit Care Med. 1997;156:280–285. doi: 10.1164/ajrccm.156.1.9611085. [DOI] [PubMed] [Google Scholar]
- Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- Tatum VL, Chow CK. Antioxidant status and susceptibility of sickle erythrocytes to oxidative and osmotic stress. Free Radic Res. 1996;25:133–139. doi: 10.3109/10715769609149918. [DOI] [PubMed] [Google Scholar]
- Jain SK, Ross JD, Duett J, Herbst JJ. Low plasma pre-albumin and carotenoid levels in sickle cell disease patients. Am J Med Sci. 1990;299:13–15. doi: 10.1097/00000441-199001000-00004. [DOI] [PubMed] [Google Scholar]
- Ndombi IO, Kinoti SN. Serum vitamin E and the sickling status in children with sickle cell anaemia. East Afr Med J. 1990;67:720–725. [PubMed] [Google Scholar]
- Phillips G, Tangney CC. Relationship of plasma alpha tocopherol to index of clinical severity in individuals with sickle cell anemia. Am J Hematol. 1992;41:227–231. doi: 10.1002/ajh.2830410402. [DOI] [PubMed] [Google Scholar]
- Natta C, Machlin L, Brin M. A decrease in irreversibly sickle erythrocytes in sickle cell anemia patients given vitamin E. Am J Clin Nutr. 1980;33:968–971. doi: 10.1093/ajcn/33.5.968. [DOI] [PubMed] [Google Scholar]
- Tangney CC, Phillips G, Bell RA, Fernandes P, Hopkins R, Wu SM. Selected indices of micronutrient status in adult patients with sickle cell anemia (SCA). Am J Hematol. 1989;32:161–166. doi: 10.1002/ajh.2830320302. [DOI] [PubMed] [Google Scholar]
- Essien EU. Plasma levels of retinol, ascorbic acid, and alpha-tocopherol in sickle cell anemia. Cent Afr J Med. 1995;41:48–50. [PubMed] [Google Scholar]
- Akinkugbe FM, Ette SI. Ascorbic acid in sickle cell disease: results of a pilot therapeutic trial. East Afr Med J. 1983;60:683–687. [PubMed] [Google Scholar]
- Delekan DA, Thurham DI, Adekile AD. Reduced antioxidant capacity in paediatric patients with homozygous sickle cell disease. Eur J Clin Nutr. 1989;43:609–614. [PubMed] [Google Scholar]
- Houston PE, Rana S, Sekhsaria S, Perlin E, Kim KS, Castro OL. Homocysteine in sickle cell disease: relationship to stroke. Am J Med. 1997;103:192–196. doi: 10.1016/S0002-9343(97)00129-0. [DOI] [PubMed] [Google Scholar]